

Abstract
PmoD, a recently discovered protein from methane-oxidizing bacteria, forms a homodimer with a dicopper CuA center at the dimer interface. Although the optical and electron paramagnetic resonance (EPR) spectroscopic signatures of the PmoD CuA bear similarities to those of canonical CuA sites, there are also some puzzling differences. Here we have characterized the rapid formation (seconds) and slow decay (hours) of this homodimeric CuA site to two mononuclear Cu2+ sites, as well as its electronic and geometric structure, using stopped-flow optical and advanced paramagnetic resonance spectroscopies. PmoD CuA formation occurs rapidly and involves a short-lived intermediate with λmax of 360 nm. Unlike other CuA sites, the PmoD CuA is unstable, decaying to two type 2 Cu2+ centers. Surprisingly, nuclear magnetic resonance (NMR) data indicate that the PmoD CuA has a pure σu* ground state (GS) rather than the typical equilibrium between σu* and πu of all other CuA proteins. EPR, ENDOR, ESEEM, and HYSCORE data indicate the presence of two histidine and two cysteine ligands coordinating the CuA core in a highly symmetrical fashion. This report significantly expands the diversity and understanding of known CuA sites.
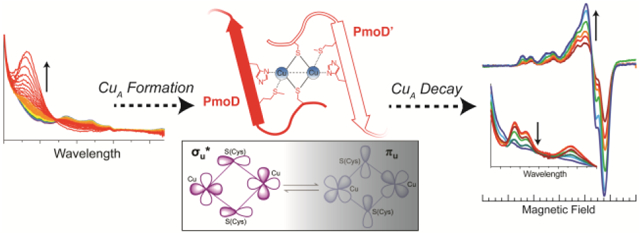
Graphical Abstract
INTRODUCTION
Biological copper centers play key roles in many enzymes and proteins, both catalyzing chemical reactions and mediating electron transfer (ET). Dicopper CuA centers are ET sites found in enzymes such as cytochrome c oxidase (CcO) and nitrous oxide reductase (N2OR), which are terminal electron acceptors for aerobic and anaerobic respiration, respectively.1 Initially a source of controversy,2 CuA sites are now well known to contain a Cu2[S(Cys)]2 core with two Cu ions bridged by two Cys side chains as well as one in-plane His side chain ligand per Cu. In addition, there is a weak axial Met side chain ligand on one Cu and an axial backbone carbonyl oxygen ligand on the other that induce a degree of structural asymmetry at the metal site. During ET, a CuA site cycles between the reduced [2Cu]2+ and oxidized [2Cu]3+ (often stylized as Cu1.5+-Cu1.5+ to reflect the electronic equivalence of the two Cu ions) states as it shuttles an electron to a metallocofactor.1,3 The amino acid ligands to the CcO and N2OR CuA sites are found in a Hx34Cx3Cx3Hx2M motif,2 derived from a single polypeptide with a cupredoxin fold.4-6
We recently discovered a new type of CuA site in some homologs of PmoD/AmoD, a protein encoded exclusively in the genomes of methane- and ammonia-oxidizing bacteria.7 This CuA forms in PmoD proteins encoded within methanotroph pmo operons. These operons also contain the genes encoding the three subunits of particulate methane monooxygenase (pMMO),7 a copper-dependent inåtegral membrane enzyme that converts methane to methanol.8 In Methylosinus trichosporium OB3b, the pmoD gene in the pmo operon is co-regulated with the pMMO genes,9 and its disruption results in a severe copper-dependent growth defect.7 Biochemical, structural, and spectroscopic data indicate that the N-terminal periplasmic domain of PmoD from Methylocystis species strain Rockwell adopts a cupredoxin-like fold and forms a mixed-valence, delocalized Cu2[S(Cys)]2 CuA core at the interface of a PmoD homodimer utilizing a Cx7MxH binding motif rather than the typical CuA Hx34Cx3Cx3Hx2M motif (Figure 1A). In contrast to canonical CuA sites, each PmoD monomer is proposed to contribute one Cys41 ligand to the Cu2[S(Cys)]2 core as well as one Met49 and one His51 from each Cx7MxH motif. Mutation of these residues prevents CuA formation, and the Cx7MxH motif is highly conserved in PmoD homologs from pmo operons in alpha-proteobacterial methanotrophs.7
Figure 1.

The PmoD CuA site. (A) Homodimeric molecular model. (B) CW X-band (~9.5 GHz) EPR spectrum of the PmoD CuA. Bracket defines the hyperfine splitting Az (adapted from 7).
Copper-loaded PmoD exhibits an electron paramagnetic resonance (EPR) spectrum with a seven hyperfine-line splitting (Az) along gz,7 defining its CuA as a Robin-Day class III fully valence-delocalized [2Cu]3+ dicopper center (Figure 1B). Similar to other CuA sites, the PmoD gz and Az values are less than those observed for “normal” type 2 monocopper sites, in part due to the highly covalent Cu-S(Cys) bonding, which leads to very large ρS(Cys) (where ρX is the spin density on atom X) and small ρCu (for example, for CuAAz, total ρS(Cys) = 46%, total ρCu = 44%).10,11 The PmoD optical spectrum is dominated by the two typical S(Cys)-Cu ligand-to-metal charge transfer (LMCT) bands at 475 and 530 nm and a ψ→ψ* intervalence band in the near-IR (770 nm).7 The intensity of these LMCT bands is consistent with large Cu-S(Cys) covalence that gives rise to a large ρS(Cys). The energy of the near-IR optical transition of a CuA center is inversely correlated with the Cu-Cu distance and reflects the relative strength of the Cu-Cu and Cu-N(His) bonds; a high energy transition corresponds to strong Cu-Cu and Cu-N(His) bonding.12-14 The PmoD near-IR optical transition is blue-shifted with respect to those observed for Pseudomonas nautica N2OR (PnN2OR, 800 nm)15 and Thermus thermophilus CcO (TtCcO, 790 nm)16, and is comparable to that reported for the semisynthetic CuA-containing azurin (CuAAz, 765 nm)17. This shift suggests strong Cu-Cu bonding in PmoD, consistent with the short Cu-Cu distance determined from extended X-ray absorption fine structure (EXAFS) data (2.41 Å for PmoD, compared to 2.43 Å for TtCcO, 2.43 Å for PnN2OR, and 2.39 Å for CuAAz).7,14,18,19
However, a variety of the characteristics of the PmoD CuA are not typical for a CuA center. The energy of the near-IR transition is highly sensitive to the CuA electronic structure and resulting spin density distribution, and CuA proteins with similar near-IR transition energies typically have similar EPR spectra.12,13,20 Although the energies of the near-IR transitions of CuA-containing PmoD and CuAAz are essentially the same, their Cu hyperfine couplings are appreciably different (PmoD Az = 141 MHz; CuAAz Az = 167 MHz).7,14 The CuA Az is proportional to ρCu,21 and thus the decreased PmoD CuA Az value indicates anomalously small ρCu, and by extension, correspondingly large spin density on the ligands. Likewise, another PmoD spin-Hamiltonian parameter, gz, has the smallest value of any CuA site (PmoD gz = 2.13, CuAAz gz = 2.17, and SoxM gz = 2.20 (the largest gz for a biological CuA)).1,7,14,22
To understand the unusual spectroscopic characteristics of the PmoD CuA site and to gain insight into its possible functions, we have studied its rapid (seconds) formation as Cu2+ is added to PmoD, and its subsequent slow decay (hours) to two mononuclear Cu2+ sites. We additionally probed the electronic and geometric structure of the PmoD CuA by nuclear magnetic resonance (NMR) and advanced paramagnetic resonance spectroscopies (EPR, ENDOR, ESEEM, and HYSCORE).
EXPERIMENTAL SECTION
Expression and purification of PmoD.
The periplasmic domain of Methylocystis sp. str. Rockwell PmoD encoded within the pmo operon (locus tag Met49242_1452) was expressed in E. coli BL21* using the N-terminally His6-tagged constructs for the wildtype (WT) and Cys65Ser proteins and purification protocols described previously.7 Briefly, cells were grown in autoinduction media and harvested after overnight incubation. The cell pellets were resuspended in a lysis buffer composed of 20 mM imidazole, 20 mM Tris, pH 8, 500 mM NaCl, 1 mM DTT, 1 mg/mL DNaseI, and 1 mM PMSF. After lysis by sonication, cell debris was removed by centrifugation and the clarified lysate was purified on NiNTA resin. The His6 tag was then removed by overnight incubation with His-tagged TEV protease. After removal of the TEV protease using a second NiNTA column, the untagged PmoD was stored in 20 mM Tris, pH 7.0 and 1 mM DTT. The protein concentrations of all PmoD samples were determined by the Bradford assay using known concentrations of BSA to generate a standard curve.
Stopped-flow optical spectroscopy.
All stopped-flow experiments were conducted on a SX20 stopped-flow instrument (Applied Photophysics) at 6 °C. Purified PmoD samples were diluted to 200 μM in 20 mM Tris pH 7.5, 100 mM NaCl and loaded into one syringe. The second syringe was loaded with 400 μM CuSO4. To obtain full kinetic spectra for each sample, a photodiode array (PDA) detector was used to collect 1000 data points logarithmically over a 1000 s time interval. The rates of formation and decay of the CuA species were calculated using the data from these experiments at 475 nm. To determine the rates for the 360 nm intermediate, a PMT detector was used to allow for collection of sufficient data at the earliest time points. In these experiments, 1000 data points were collected logarithmically over a 5 s time interval. For the Cys65Ser variant, which exhibited slower kinetics for formation of the 360 nm species, data were collected over a 20 s time interval. Kinetic data were analyzed using GraphPad Prism.
Preparation and analysis of samples to monitor decay of CuA site.
For both the WT and Cys65Ser samples, the DTT-reduced protein was buffer exchanged into 20 mM Tris, pH 7.5, 100 mM NaCl. Two molar equivalents of CuSO4 were slowly added to the protein by pipetting. Unbound copper was removed immediately by loading the sample onto a PD10 desalting column (GE), eluting into 20 mM Tris, pH 7.5, 100 mM NaCl, and concentrating to 150 μM using a 10 kDa molecular weight cut-off centrifugal concentrator (Millipore). Samples were stored on ice and removed at specific time points ranging from 0 to 144 hr for further analysis by optical and X-band continuous wave (CW) EPR spectroscopy as well as inductively coupled plasma optical emission spectroscopy (ICP-OES). X-band EPR samples were prepared by transferring ~180 μL of protein solution to a Wilmad quartz EPR tube (Sigma Aldrich), which was then frozen in liquid nitrogen, where it was stored until analysis. Optical spectra were collected at room temperature on 100 μL protein in a quartz cuvette (Helma) using an Agilent 8453 spectrophotometer. Prior to conducting elemental analysis, each protein sample was first applied to a PD-10 column and eluted into 20 mM Tris, pH 7.5, 100 mM NaCl to ensure that the copper was not dissociating from the protein through the timecourse. The protein was then digested in 5% nitric acid in metal-free conical tubes (VWR). A dilution series of a custom multi-element standard (Inorganic Ventures) was also prepared in a similar fashion to generate a standard curve. The copper contents of the samples were determined by ICP-OES using a Thermo iCAP 7600 instrument in the Quantitative Bio-element Imaging Center (QBIC) core facility at Northwestern University.
Oligomerization analysis of PmoD pre- and post-EDTA treatment.
DTT-treated Cys65Ser PmoD was exchanged into reductant-free buffer prior to the addition of two molar equivalents of CuSO4. Excess copper was then removed by desalting on a PD10 column into 4 mL copper-free buffer. A 1 mL aliquot of this sample was immediately run on a Superdex 75 Increase column (GE). A second 1 mL aliquot was treated with 50 mM EDTA, pH 8 and incubated on ice for 3 hr prior to running on the Superdex 75 Increase column. The remaining sample was incubated on ice at 4 °C for 144 hr to allow the CuA site to decay and subjected to size exclusion chromatography and EDTA treatment as described for the earlier time points. The peak fractions from each column were pooled and concentrated using 10 kDa molecular weight cut-off centrifugal concentrators (Millipore). Approximately 60 μg of each sample was run on a denaturing gel in either the absence or presence of β-mercaptoethanol, and ICP-OES was used to confirm that the EDTA treatment effectively removed copper. The Cys41Ser sample was prepared as described for the Cys65Ser sample and run on a HiLoad 16/600 Superdex 75 column in the absence of reducing agents.
NMR spectroscopy.
1H NMR spectra were recorded on a Bruker Avance II-600 NMR spectrometer. PmoD prepared in 100 mM NaCl, 20 mM Tris, pH 7.0 and 1 mM DTT was lyophilized using a FreeZone 4.5 Liter Cascade Benchtop Freeze Dry System (Keck Biophysics Facility, Northwestern University). Lyophilized samples of PmoD were dissolved in ultrapure water with 2 mM TCEP. CuSO4 was added to form the PmoD CuA site and excess copper was removed by desalting using PD minitrap desalting columns pre-equilibrated with 100 mM phosphate buffer, 100 mM KCl, 10% D2O, pH 7.2 before the acquisition. 16384 free induction decays were acquired with the use of a super-WEFT pulse sequence23 (inter-pulse delay: 100 ms; accumulation: 1024 points; spectral width: 200 ppm), Fourier transformed with the use of an exponential window (LB = 150 Hz), and baseline corrected using TopSpin NMR Software.
EPR, ESEEM, and ENDOR spectroscopy.
For the advanced spectroscopic studies of the PmoD CuA site, the CuA-containing dimeric species was isolated by size exclusion chromatography as described previously.7 This sample was concentrated to 300 μM in 20 mM Tris pH 7.0 and frozen in an X-band EPR tube. The sample was subsequently thawed on ice, and an aliquot was quickly transferred to a Q-band tube for Q-band measurements, after which point both EPR tubes were frozen in liquid nitrogen. X- and Q-band samples utilized Wilmad quartz EPR tubes (Sigma Aldrich) and custom quartz Q-band tubes, respectively. X-band EPR tubes were filled with ~180 μL of protein solution, while Q-band EPR tubes were filled with ~80 μL of protein solution. Samples were frozen in liquid nitrogen, where they were stored until analysis. All CW X-band EPR measurements were collected utilizing a Bruker ESP-300 spectrometer with a liquid helium flow Oxford Instruments ESR-900 cryostat. All wide scan (2400-3600 G) spectra were background corrected by subtraction of an EPR spectrum of 50 mM Tris, pH 8.0, 150 mM NaCl measured under identical conditions. For Cu2+ spin quantitation, the double integral of the experimental spectrum was compared to that of Cu2+-EDTA standards in 50 mM Tris, pH 8.0, 150 mM NaCl buffer containing 100-400 μM Cu2+.
X-band three pulse [π/2–τ–π/2–T–π/2–τ–echo] ESEEM and four pulse [π/2–τ–π/2–T1 π/2–T2–π/2–τ–echo] HYSCORE measurements were collected on a Bruker Elexsys E580-X utilizing split ring resonator (ER4118X-MS5). The temperature was maintained at 10 K using an Oxford Instruments CF935 continuous flow cryostat using liquid helium.
Pulsed Q-band EPR, ENDOR, and PESTRE measurements were conducted at ~2 K in a liquid helium immersion dewar on a spectrometer described elsewhere, with SpinCore PulseBlaster ESR_PRO 400MHz digital word generator and Agilent Technologies Acquiris DP235 500 MS/sec digitizer using SpecMan software.24,25 A Davies [π–TRF–π/2–τ–π–τ–echo] pulse sequence was employed for all ENDOR measurements, in which TRF denotes the interval during which the RF was applied.
RESULTS
Formation of the PmoD CuA center.
To probe formation of the PmoD CuA site, we monitored the reaction between reduced PmoD and Cu2+ by stopped-flow optical spectroscopy. CuA formation is preceded by the development of a transient intermediate characterized by an intense absorbance feature at 360 nm, consistent with a S(Cys) to Cu2+ LMCT transition (Figure 2A).26 The rate of formation for this intermediate is 660 s−1 and its rate of decay is 2.2 s−1 (Table S1). The rate of CuA formation (monitored at 475 nm; absorption bands at 475, 530, and 770 nm are characteristic of the CuA site) is similar to the rate of intermediate decay (Table S1). As the two share an isosbestic point, the 360 nm species directly converts to CuA. This mechanism resembles that observed for CuAAz and Thermus thermophilus cytochrome ba3 CuA, wherein the "capture complex" type 2 ‘red’ Cu2+ center, with λmax ~ 385 nm, converts into the CuA center,1,27 although the PmoD intermediate forms and decays on significantly shorter timescales and does not proceed through any of the additional intermediates observed in the other CuA systems.27-29 As the stopped-flow optical spectroscopy experiment involves adding Cu2+ to form a mixed valent, formally Cu2+-Cu1+ delocalized center, either or both of the cysteines in PmoD must be reducing Cu2+ to Cu1+ (with concomitant oxidation of cysteines to cystine). This Cu1+ is then used to produce the CuA as in other CuA metalation mechanisms.27-29
Figure 2.

Stopped-flow optical spectroscopy experiments monitoring formation of the PmoD CuA site at room temperature. The optical spectra were monitored over 1000 s after mixing 400 μM CuSO4 with 200 μM PmoD for (A) WT protein, (B) Cys65Ser variant, (C) Cys41Ser variant, (D) Met49Ala variant, (E) His51Ala variant, and (F) Cys41Ser/Cys65Ser variant. Red, 0.014 s-1.009 s; orange, 1.168-3.295 s; yellow, 3.634 s-8.183 s; green, 8.908 s-18.638 s; light blue, 20.184 s-40.976 s; blue, 44.29 s-190.867 s; purple, 206.014 s-409.204 s; dark purple, 441.588 s-1000 s.
In the CuAAz mechanism, a solvent exposed cysteine first binds Cu2+ to form the red Cu intermediate and positions it for eventual CuA formation (a methionine was also proposed to be a ligand to this intermediate in the T. thermophilus cytochrome ba3 CuA)29. To determine whether this also occurs in PmoD and to identify which cysteine residue(s) may bind Cu2+ initially, we monitored CuA formation by variants lacking either or both cysteine residues. The Cys65Ser PmoD variant (a variant with the only non-CuA-ligating cysteine replaced with serine) also forms a 360 nm species which directly converts to CuA (Figure 2B), but the rate of intermediate formation is decreased nearly 100-fold compared to wild-type (WT) PmoD, while the rate of intermediate decay and CuA formation are very close. Furthermore, more of the CuA species is formed relative to WT despite a lower intensity 360 nm intermediate absorbance (Table S1, Figure 2B). By contrast, the Cys41Ser variant does not form the CuA site (Figure 2C), though it does still form a transient 360 nm intermediate with similar kinetics to WT PmoD (Table S1). Thus, this intermediate species is not stable even when it does not convert to a CuA site. These results suggest that either Cys41 or Cys65 can bind Cu2+ to form a ~360 nm intermediate, but only the Cys41-Cu2+ 360 nm intermediate is used to form the CuA site. Thus, for the Cys65Ser variant, a greater amount of CuA is produced from a smaller amount of 360 nm intermediate as a result of less non-productive Cu2+ binding at the Cys65 site and more Cu2+ binding by Cys41.
Consistent with the notion that the faster-forming/decaying Cys65-Cu2+ 360 nm intermediate cannot convert to CuA, both the Met49Ala and His51Ala variants form a 360 nm intermediate that forms and decays on the same timescale as WT PmoD, yet neither produces a CuA (Figures 2D, E, Table S1). Mutation of both Cys residues prevents formation of both the 360 nm intermediate and CuA (Figure 2F), as expected since this variant does not bind copper. These data indicate that the PmoD CuA site forms via a modified version of the CuAAz "capture complex" mechanism, wherein Cys41 recruits Cu2+ for CuA formation.
PmoD CuA decays to type 2 mononuclear Cu centers.
We also noticed that the PmoD CuA spectroscopic features decay slowly, on the timescale of hours at 4 °C under aerobic conditions (Figure 3A). All previously characterized CuA centers are quite stable at physiological pH, with the exception of a CuA site engineered into a coiled-coil scaffold that decayed over a time of approximately 6 hours.30 The PmoD CuA center decays to form a species with no CT bands in the optical spectrum, only a broad Cu2+ d-d transition at ~615 nm (Figure 3A). The CuA decay was monitored over several days at 4 °C via optical and continuous wave (CW) X-band EPR spectroscopy for WT PmoD. The CuA site decayed into two distinct type 2 Cu2+ centers, as evidenced by the appearance of two overlapping but distinguishable Cu2+ EPR signals with gz and Az consistent with type 2 Cu2+ (Cu1 gz = 2.23, Az = 610 MHz; Cu2 gz = 2.20, Az = 545 MHz, Figure 3B, Table 1).
Figure 3.

Decay of WT PmoD CuA at 4 °C observed in parallel by (A) optical and (B) CW X-band EPR spectroscopies. The inset in B depicts scans measured in the gz region for the WT PmoD t = 144 hr sample, where brackets define the hyperfine splitting Az of the two type 2 Cu2+ centers (the fourth hyperfine line is outside of the range shown). Conditions: (B inset) 9.364-9.365 GHz microwave frequency, 40 s scan rate, 320 ms time constant, 12.5 G modulation amplitude, temperature 20 K; (B bottom) 9.364-9.366 GHz microwave frequency, 90 s scan rate, 320 ms time constant, 12.5 G modulation amplitude, temperature 20 K. Spectra intensities were normalized to account for different gain settings. The protein concentration was 150 μM for all samples.
Table 1.
PmoD paramagnetic spectroscopic features.
| Paramagnet | gx, gy, gz |
Cu Az (MHz) |
14N1
Ax,y, Az (MHz)§ |
14N2*
Ax,y, Az (MHz)§ |
Cys-Cβ
1H1
Ax,y, Az∇ (MHz) |
Cys-Cβ
1H2
Ax,y,z (MHz) |
|---|---|---|---|---|---|---|
| CuA | 2.01, 2.05, 2.13 | 141 | +17, +16.5 | +16, +16.5 | +14.5, +17 | ~4.5 |
| Type 2 Cu1 | −, −, 2.23 | 610 | ||||
| Type 2 Cu2 | −, −, 2.20 | 545 | ||||
14N2 shows resolved quadrupole splitting at the high field extreme, 3P = 2.3 MHz.
14N hyperfine couplings assumed to be positive, as necessitated by the fact that they are directly-coordinated in-plane Cu ligands (with respect to the Cu2[S(Cys)]2 core).
Concomitant with the aerobic loss of the CuA optical and EPR spectroscopic features, the concentration of paramagnetic Cu becomes almost three-fold higher (Figure 3A, B). As CuA-[2Cu]3+ oxidation to two monocopper Cu2+ ions would at most double the concentration of paramagnetic Cu, the starting protein must also contain a substantial amount of mononuclear Cu+ or CuA-[2Cu]2+, which air oxidizes. As WT PmoD binds more than one Cu equivalent per monomer,7 this result indicates that some of this excess copper is mononuclear Cu+.
Although the CuA site bridges the monomer-monomer interface, PmoD remains a dimer even after CuA decay, as determined by size exclusion chromatography of the Cys65Ser PmoD variant, which only has one Cys per monomer (Figure S1). This variant decays to the same type 2 Cu2+ centers as WT PmoD (Figure S2) indicating that the decay process is the same for WT and mutant. The Cys65Ser PmoD dimer post CuA decay is maintained after treatment with EDTA (to remove copper) in denaturing gel electrophoresis experiments performed under nonreducing conditions, but not under reducing conditions (Figure S1), indicating that an intermolecular disulfide bond links the two monomers. The disulfide must therefore be formed by the two Cys41 residues as Cys41 is the only cysteine residue present in Cys65Ser PmoD. Moreover, the Cys41Ser/Cys65Ser variant is exclusively monomeric (Figure S3). This conclusion is reminiscent of other studies that have reported CuA destruction with concomitant disulfide formation when attempting to oxidize the [2Cu]3+ state.1
Nuclear magnetic resonance characterization of the PmoD CuA site.
To further assess the differences between the PmoD CuA site and previously characterized CuA sites, we examined PmoD using NMR. NMR can be applied to proteins with fast relaxing paramagnets like CuA centers and provides a wealth of information on both the ligation of the CuA site and the presence of thermally-accessible excited electronic state(s). At room temperature, typical CuA centers are in dynamic equilibrium between two states, a majority form with a low energy σu* ground electronic state (GS) and a minority form with a πu GS (Figure 4).12,20,33,34 The two states are proposed to provide distinct electron transfer pathways for CuA reactivity in vivo.20 In the typical CuA center, an equilibrium population of the πu GS leads to fast overall electron-spin relaxation times (10−11 s) that produce sharp signals in the NMR spectra of CuA ligands with sharp resonances. Surprisingly, the 1H NMR spectrum of PmoD instead showed a set of broad resonances (a-e) located between 50 and −10 ppm (Figure 5A). When the CuA site decays as discussed above, resonances b-e disappear, leaving only the broad signal a. Resonances b-e are thus attributed to the CuA center; signal a, which does not correspond to the CuA center, is attributed to a distinct Cu2+ site. The loss of the CuA signals confirm that it converts to type 2 Cu2+ sites, whose slow electron-spin relaxation prevents observation of NMR signals from ligand nuclei.
Figure 4.

Schematic representation of the two alternative ground electronic states in the thermal equilibrium in typical CuA sites, σu* and πu.
Figure 5.

Paramagnetic NMR and electronic spectra suggest the absence of a thermally-accessible πu state in the PmoD CuA. (A) 600 MHz 1H NMR spectra of PmoD CuA recorded at 298 K in H2O. The broad signal a is observed after loss of the purple color, and is therefore attributed to a different Cu2+ binding site. Resonances b-e correspond to copper ligands of the PmoD CuA site. (B) Optical spectrum of PmoD CuA compared to those of subunit II of Tt CcO ba3 CuA and a loop mutant with a larger population of the πu state, Tt3L CuA.40
The line widths of resonances b-e from the CuA center resemble those of oxidized type 1 blue copper centers, which exhibit longer electron relaxation times of 10−10 s,35,36 and are much broader than those of other CuA sites. Since a 1% population of the πu GS is enough to provide an efficient relaxation pathway giving sharp NMR lines,20 we conclude that the πu GS in PmoD CuA is not thermally accessible, in contrast to all other known CuA sites. This conclusion is supported by the temperature dependence of the contact-shifted resonances in the PmoD NMR spectrum. In typical CuA NMR spectra, the chemical shifts show temperature dependences with large deviation from the Curie law, an effect of relaxation associated with occupation of the πu GS.33,37 In contrast, the temperature dependences of all 1H NMR resonances in PmoD CuA follow the Curie law, confirming that such a GS is not significantly populated (temperature dependence of isolatable signals c and d shown in Figure S4).
The conclusion from the NMR data that the PmoD CuA center has a pure σu* GS is also supported by optical spectroscopic characterization. The intensity ratio of the LMCT bands at 350 and 530 nm is another bona fide indicator of the relative population of these two GS,38 and the absence of a band at ca. 350 nm in PmoD CuA, in contrast to other CuA sites (Figure 5B), supports a null population of the πu level.
Finally, the unusually small gz value observed in the PmoD CuA EPR spectrum is entirely consistent with the conclusions derived from the NMR results. The gz values of CuA centers have been related to the energy gap between the σu* GS and the πu Franck-Condon excited state (ES) according to the equation34,39
| (1) |
in which ge is the g-factor for a free electron, α2 and β2 represent the Cu character of the σu* and πu states, respectively, and is the Cu2+ spin-orbit coupling constant for the 3d wave functions. By applying the parameters reported for other CuA centers34 (α2 = 0.44, β2 = 0.33) and the experimentally determined gz value, we calculate an approximation of the energy gap between the σu* GS and a πu ES as 6950 cm−1, the largest of any known CuA center, consistent with the observed NMR features and null population of the πu GS (Table S2).
ENDOR, ESEEM, and HYSCORE characterization of the PmoD CuA ligation sphere.
Previous mutagenesis data implicated His51 in PmoD CuA formation.7 To further investigate nitrogen ligation of the CuA site, we collected Q-band EPR (Figure 6A) and 14N-ENDOR (Figure 6B) spectra to detect and characterize directly coordinated nitrogenous ligands.
Figure 6.

Pulsed Q-band two pulse EPR and 14N, 1H Davies ENDOR of PmoD. (A) Two pulse echo-detected EPR. The Cu2+ region denoted by the dotted line, g ~ 2.2-2.15, is attributable to exclusively mononuclear Cu2+ resonance, while the region from g ~ 2.15 – 2.0 corresponds to predominantly CuA resonance (as well as the overlapping minor Cu2+ resonance). Field-swept spectrum with X-axis of magnetic field provided in Figure S8. (B) 14N Davies ENDOR measurements across the EPR envelope at g-values indicated, demonstrating the nearly equivalent hyperfine coupling of the two CuA 14N ligands. The region under the dotted brackets denotes resonance attributable to Cu2+−14N ligation, as confirmed in Figure S5A. The black goalpost width signifies twice the 14N Larmor frequency (2 x v14N, and the filled circle denotes one half the 14N hyperfine coupling (A/2). Additional splitting resolved at the high field edge (g = 2.02) of the 14N2 resonance is attributed to resolved quadrupole splitting 3P = 2.3 MHz. Only the higher frequency v+ peaks are well-resolved. (C) 1H Larmor-centered Davies ENDOR, where the triangle denotes the 1H Larmor frequency (v1H) and goalpost width defines the hyperfine coupling magnitude (A) to CuA Cys-Cβ 1H (black). The modestly large 1H response seen at lower fields (A ~ 8 MHz) is attributed to a 1H coupled to the underlying Cu2+ resonance, as confirmed in Figure S5B. EPR conditions: 34.649 GHz microwave frequency, 200 s scan, π = 80 ns, τ = 500 ns, 20 ms repetition time; 14N ENDOR conditions: 34.63-34.67 GHz microwave frequency, π = 80 ns, τ = 375 ns, TRF = 200 μs, 20 ms repetition time; 1H ENDOR conditions: 34.64-34.65 GHz microwave frequency, π = 200 ns, τ = 575 ns, TRF = 60 μs, 50 ms repetition time.
CuA centers typically exhibit two strongly-coupled N(His) hyperfine couplings corresponding to the two N(His) ligands.41-43 These N(His) hyperfine couplings (6 MHz ≲ CuA Ax,y,z(14N) ≲ 20 MHz) are substantially smaller than those of typical mononuclear Cu-N(His) ligands.41-43 Orientation-selective Davies ENDOR spectra of PmoD CuA reveal the presence of an effectively isotropic, strongly-coupled 14N ENDOR response with A(14N) ~ 16-17 MHz, arising from direct ligation of 14N to the CuA (Figure 6B). Additional 14N resonances observed at higher frequency are attributed to 14N ligand(s) of the underlying mononuclear Cu2+ signal evident in the Q-band EPR spectrum (Figure 6A, Figure S5A).
Considering the number of 14N ligands that contribute to the PmoD ENDOR response, the observation of a seven hyperfine line splitting pattern in the EPR spectrum of PmoD requires that the two Cu ions must be in essentially equivalent environments. Therefore, there cannot be only one directly coordinated N ligand, as it would produce a highly asymmetric ligand field and valence localization (i.e. the EPR spectrum would resemble a mononuclear Cu2+ spectrum with four resolved Cu hyperfine lines) as seen in the His120Ala CuAAz mutant.11 Thus, the strongly-coupled 14N ENDOR response is assigned to two CuA 14N ligands with nearly identical, effectively isotropic hyperfine coupling, Table 1). This conclusion is consistent with the previous proposal that one His51 from each monomer in the CuA-containing PmoD homodimer serves as a ligand (Figure 1).7 While it is possible to form a CuA center with only one His ligand,11 CuA formation was not observed in the His51Ala PmoD variant,7 necessitating assignment of both CuA N(His) ligands to His51 side chains.
The near equivalence of the two isotropic couplings suggests two nitrogenous ligands bound to PmoD CuA in a very symmetrical fashion. As expected, ESEEM and hyperfine sublevel correlation (HYSCORE) spectroscopy measurements identified two weakly-coupled 14N nuclei (Figure S6) characteristic of the distal, non-coordinated nitrogen from two N(His) imidazole side chain ligands to the CuA. Thus, the two strongly-coupled, nearly identical 14N ENDOR responses correspond to two directly coordinated N(His) CuA ligands. The isotropic component of the strongly-coupled 14N hyperfine coupling (Aiso ~16-17 MHz for the two His) is proportional to the magnitude of ρN, and the sum of the two Aiso values is the largest of any CuA site characterized to date (Table S3). This implied additional delocalization in the PmoD CuA-N bonding is consistent with the optical spectroscopy, which also indicated enhanced ligand spin density relative to other CuA centers.
To further define the geometry of the PmoD CuA center, we examined past crystal structures and ENDOR studies of CuA-containing proteins to identify structural and spectroscopic correlation(s). We find that increasing colinearity of the two N(His)-Cu bond vectors of a CuA site with respect to the Cu-Cu vector correlates with increased sum of 14N(His)-Aiso of the two 14N(His) ligands (Table S3). Given the nearly identical hyperfine couplings of the two N(His) ligands, and the fact that the sum of Aiso for the CuA 14N(His) of PmoD is larger than for all previously characterized CuA centers, we deduce that the PmoD CuA His ligands coordinate the Cu2[S(Cys)]2 core in a very symmetrical fashion, with an essentially linear, N-Cu-Cu-N arrangement, strongly resembling CuAAz.6
We also characterized the two proposed S(Cys41) components of the Cu2[S(Cys)]2 core by ENDOR. CuA Cys ligands exhibit large isotropic hyperfine couplings to the Cys-Cβ 1H that arise from hyperconjugation to, and are proportional to the magnitude of the spin density on the S(Cys). In the PmoD CuA 1H ENDOR spectrum (Figure 6C), there are two well-resolved 1H responses that do not exchange in D2O (Figure S9) and exhibit large, isotropic coupling consistent with a CuA Cys-Cβ 1H: Aiso Cys–Cβ1H1 = 15 MHz and Aiso Cys–Cβ1H2 ≈ 4.5 MHz (Table 1). The Aiso Cys–Cβ1H1 = 15 MHz, and thus the spin density on S(Cys41), is similar to the largest reported to date for a CuA (CuA-containing soluble fragment of Tt CcO ba3, CuA Cys-Cβ 1H Aiso max = 15.4 MHz, where total ρS(Cys) = ~50-55%).21
DISCUSSION
We here provide an extensive spectroscopic characterization of the formation and characteristics of the PmoD CuA, finding both similarities and key differences relative to other CuA centers. Stopped-flow optical spectroscopy indicates that, like CuAAz, the PmoD CuA forms via a cysteine “capture complex” mechanism, in which a solvent exposed cysteine (Cys41) binds and positions the Cu for CuA formation. However, unlike all other biological or semisynthetic CuA centers, the PmoD CuA is unstable, in the presence of air slowly decaying to two type 2 Cu2+ centers. Moreover, through NMR we have shown that the PmoD CuA is unlike all other CuA centers in that in fluid solution it exclusively resides in a form that features a σu* GS without a contribution from a form with πu GS. Finally, advanced paramagnetic resonance characterization of the PmoD CuA and CuA ligands interpreted in the context of past work7 confirmed (1) that two His51 side chains ligate the CuA and (2) the CuA ligands feature anomalously large spin density relative to what would be expected from interpreting the optical spectroscopy (and particularly the similarity of the spectrum to CuAAz).
In addition to the two S(Cys41) and two symmetrically-placed N(His51) ligands, the ligation sphere of the PmoD CuA site is likely completed by two axial Met49 thioethers.7 Indeed, the observed spectroscopic characteristics of the PmoD CuA site are readily rationalized by considering the contributions for the two Met axial ligands. For previously characterized CuA sites, stronger axial ligation extends the Cu-Cu distance, weakening Cu-Cu and Cu-N(His) bonding while shifting spin density from the ligands onto the Cu.44,45 Conversely, weaker axial ligation shortens the Cu-Cu distance by strengthening Cu-Cu bonding.46 Furthermore, as a general rule, hard Lewis base axial CuA ligands bind more strongly than soft ones. Consequently, substitution of the typical CuA axial carbonyl ligand with the S of a Met side chain, which is a softer Lewis base, should increase ligand spin density at the expense of Cu spin density, and result in a very short Cu-Cu distance. Thus, the apparent discrepancy between the PmoD CuA and CuAAz optical and paramagnetic resonance properties, namely the failure of similar energies for the near-IR intervalence CT bands to correspond to similar spin density distributions, is rationalized as follows: the proteins both have strong Cu-Cu bonding and a short Cu-Cu distance, hence the same near-IR transition energy, but the two weak S(Met) ligands in PmoD CuA support increased covalency with the N(His) and S(Cys) ligands. This increased covalency causes PmoD to exhibit very small CuA Az and ρCu combined with large ligand hyperfine couplings and large ligand spin densities. In addition, in CuAAz, mutation of Met to stronger axial ligands decreases .12,20 Following this trend, the two weak axial Met ligands in PmoD are expected to increase the value of , consistent with the fact that the PmoD value is the largest of any CuA.
A symmetrical CuA site (D2h symmetry) with equivalent Met ligands would dramatically lower the intensity of the 530 nm S(Cys) to Cu LMCT band because this transition would be Laporte-forbidden.12 Due to the instability of the site, we could not determine extinction coefficients for the PmoD CuA optical features. Instead, the 475 nm LMCT band intensity is much less sensitive to changes in axial ligation,12 and consequently the intensity of the 530 nm LMCT band relative to the 475 nm LMCT band may be used as a surrogate for the 530 nm extinction coefficient. The ratio of 530 nm to 475 nm intensities is lower for PmoD CuA than for any other biological or engineered CuA (0.81 in PmoD, compared to 0.90 in CuAAz, 1.02 in N2OR, and 1.03 in CcO),1 indicating a very symmetrical site. However, the presence of a prominent 530 nm LMCT band indicates that the axial Met ligands do not bind the CuA equivalently and/or other noncovalent interaction(s) with the CuA center cause distortion from perfect symmetry.
PmoD is needed for copper-dependent growth of methanotrophs, but its specific function remains unknown.7 It is not clear whether the CuA site is formed in vivo when PmoD is tethered to the inner membrane or whether the site itself is linked to the observed growth defect in the absence of PmoD.7 However, it is tempting to speculate that PmoD is involved in electron transfer to pMMO, perhaps reducing the catalytic Cu center. If PmoD does shuttle electrons to an oxidant, its unique CuA site may offer some advantages. The πu GS has larger inner and outer sphere reorganization energies than the σu* GS;34,38 thus by not accessing the πu GS, the PmoD CuA would lower ET reorganization energy, resulting in faster ET. Additionally, in the CcO CuA, there are distinct ET pathways from the CuA center through both the S(Cys) and N(His) ligands to the target heme.3,12,20,34 In this way, ET may thus be optimized for a σu* GS. While additional work is needed to address how the unprecedented electronic structure of the PmoD CuA site relates to biological function, the current results show that it significantly expands the diversity of known CuA sites.
Supplementary Material
ACKNOWLEDGMENT
We thank Profs. Laura M. K. Dassama and Yi Lu for helpful discussions.
Funding Sources
This work was supported by Department of Energy grant DE-SC0016284 (A.C.R.), National Institutes of Health Award grants GM111097 (B.M.H.), 5T32GM008382 (M.O.R.), and F32GM119191 (O.S.F.), and Department of Energy grant DE-FG02-99ER14999 (M.R.W.). M.N.M is recipient of a postdoctoral fellowship from CONICET and A.J.V. is staff member from CONICET. The Quantitative Bio-element Imaging Center at Northwestern is supported by NASA Ames Research Center NNA06CB93G. The Keck Biophysics Facility at Northwestern is supported in part by NCI CCSG P30 CA060553.
Footnotes
Supporting Information. Size exclusion chromatography traces and nondenaturing gel electrophoresis probing the dimeric nature of the post-CuA-decay PmoD, optical and spectroscopic monitoring of the Cys65Ser PmoD CuA decay, ENDOR comparison of CuA resonance compared to on overlapping monocopper Cu2+ resonance, ESEEM and HYSCORE characterization of distal imidazole 14N from CuA-N(His) ligands, 1H ENDOR in H2O vs. D2O buffer, presentation of Q-band EPR with magnetic field as x-axis, PESTRE ACys–Cβ1H sign determination, stopped flow kinetics parameters, geometry and 14N isotropic hyperfine couplings from various CuA centers. The Supporting Information is available free of charge on the ACS Publications website.
No competing financial interests have been declared.
REFERENCES
- (1).Liu J; Chakraborty S; Hosseinzadeh P; Yu Y; Tian S; Petrik I; Bhagi A; Lu Y Chem. Rev 2014, 114, 4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Beinert H Eur. J. Biochem 1997, 245, 521. [DOI] [PubMed] [Google Scholar]
- (3).Solomon EI; Xie X; Dey A Chem. Soc. Rev 2008, 37, 623. [DOI] [PubMed] [Google Scholar]
- (4).Williams PA; Blackburn NJ; Sanders D; Bellamy H; Stura EA; Fee JA; McRee DE Nat. Struct. Mol. Biol 1999, 6, 509. [DOI] [PubMed] [Google Scholar]
- (5).Brown KR; Djinovic-Carugo K; Haltia T; Cabrito I; Saraste M; Moura JJG; Moura I; Tegoni M; Cambillau CJ Biol. Chem 2000, 275, 41133. [DOI] [PubMed] [Google Scholar]
- (6).Robinson H; Ang MC; Gao Y-G; Hay MT; Lu Y; Wang AH-J Biochemistry 1999, 38, 5677. [DOI] [PubMed] [Google Scholar]
- (7).Fisher OS; Kenney GE; Ross MO; Ro SY; Lemma BE; Batelu S; Thomas PM; Sosnowski VC; DeHart CJ; Kelleher NL; Stemmler TL; Hoffman BM; Rosenzweig AC Nat. Commun 2018, 9, 4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sirajuddin S; Rosenzweig AC Biochemistry 2015, 54, 2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kenney GE; Sadek M; Rosenzweig AC Metallomics 2016, 8, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).DeBeer George S; Metz M; Szilagyi RK; Wang H; Cramer SP; Lu Y; Tolman WB; Hedman B; Hodgson KO; Solomon EI J. Am. Chem. Soc 2001, 123, 5757. [DOI] [PubMed] [Google Scholar]
- (11).Xie X; Gorelsky SI; Sarangi R; Garner DK; Hwang HJ; Hodgson KO; Hedman B; Lu Y; Solomon EI J. Am. Chem. Soc 2008, 130, 5194. [DOI] [PubMed] [Google Scholar]
- (12).Tsai M-L; Hadt RG; Marshall NM; Wilson TD; Lu Y; Solomon EI Proc. Natl. Acad. Sci 2013, 110, 14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Slutter CE; Gromov I; Richards JH; Pecht I; Goldfarb DJ Am. Chem. Soc 1999, 121, 5077. [DOI] [PubMed] [Google Scholar]
- (14).Hay MT; Ang MC; Gamelin DR; Solomon EI; Antholine WE; Ralle M; Blackburn NJ; Massey PD; Wang X; Kwon AH Inorg. Chem 1998, 37, 191. [Google Scholar]
- (15).Prudêncio M; Pereira AS; Tavares P; Besson S; Cabrito I; Brown K; Samyn B; Devreese B; Van Beeumen J; Rusnak F; Fauque G; Moura JJG; Tegoni M; Cambillau C; Moura I Biochemistry 2000, 39, 3899. [DOI] [PubMed] [Google Scholar]
- (16).Slutter CE; Sanders D; Wittung P; Malmström BG; Aasa R; Richards JH; Gray HB; Fee JA Biochemistry 1996, 35, 3387. [DOI] [PubMed] [Google Scholar]
- (17).Hay M; Richards JH; Lu Y Proc. Natl. Acad. Sci 1996, 93, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Blackburn NJ; de Vries S; Barr ME; Houser RP; Tolman WB; Sanders D; Fee JA J. Am. Chem. Soc 1997, 119, 6135. [Google Scholar]
- (19).Charnock JM; Dreusch A; Körner H; Neese F; Nelson J; Kannt A; Michel H; Garner CD; Kroneck PM; Zumft WG Eur. J. Biochem 2000, 267, 1368. [DOI] [PubMed] [Google Scholar]
- (20).Abriata LA; Álvarez-Paggi D; Ledesma GN; Blackburn NJ; Vila AJ; Murgida DH Proc. Natl. Acad. Sci 2012, 109, 17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Epel B; Slutter CS; Neese F; Kroneck PMH; Zumft WG; Pecht I; Farver O; Lu Y; Goldfarb DJ Am. Chem. Soc 2002, 124, 8152. [DOI] [PubMed] [Google Scholar]
- (22).Komorowski L; Anemüller S; Schäfer GJ Bioenerg. Biomembr 2001, 33, 27. [DOI] [PubMed] [Google Scholar]
- (23).Inubushi T; Becker ED J. Magn. Reson 1983, 51, 128. [Google Scholar]
- (24).Davoust CE; Doan PE; Hoffman BM J. Magn. Reson. A 1996, 119, 38. [Google Scholar]
- (25).Epel B; Gromov I; Stoll S; Schweiger A; Goldfarb D Concepts Magn. Reson. Part B Magn. Reson. Eng 2005, 26, 36. [Google Scholar]
- (26).Savelieff MG; Wilson TD; Elias Y; Nilges MJ; Garner DK; Lu Y Proc. Natl. Acad. Sci 2008, 105, 7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wilson TD; Savelieff MG; Nilges MJ; Marshall NM; Lu YJ Am. Chem. Soc 2011, 133, 20778. [DOI] [PubMed] [Google Scholar]
- (28).Chakraborty S; Polen MJ; Chacón KN; Wilson TD; Yu Y; Reed J; Nilges MJ; Blackburn NJ; Lu Y Biochemistry 2015, 54, 6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chacón KN; Blackburn NJ J. Am. Chem. Soc 2012, 134, 16401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shiga D; Funahashi Y; Masuda H; Kikuchi A; Noda M; Uchiyama S; Fukui K; Kanaori K; Tajima K; Takano Y Biochemistry 2012, 51, 7901. [DOI] [PubMed] [Google Scholar]
- (31).Doan PE J. Magn. Reson 2011, 208, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Epel B; Manikandan P; Kroneck PMH; Goldfarb D Appl. Magn. Reson 2001, 21, 287. [Google Scholar]
- (33).Abriata LA; Ledesma GN; Pierattelli R; Vila AJ J. Am. Chem. Soc 2009, 131, 1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gorelsky SI; Xie X; Chen Y; Fee JA; Solomon EI J. Am. Chem. Soc 2006, 128, 16452. [DOI] [PubMed] [Google Scholar]
- (35).Bertini I; Fernández CO; Karlsson BG; Leckner J; Luchinat C; Malmström BG; Nersissian AM; Pierattelli R; Shipp E; Valentine JS; Vila AJ J. Am. Chem. Soc 2000, 122, 3701. [Google Scholar]
- (36).Donaire A; Jiménez B; Fernández CO; Pierattelli R; Niizeki T; Moratal J-M; Hall JF; Kohzuma T; Hasnain SS; Vila AJ J. Am. Chem. Soc 2002, 124, 13698. [DOI] [PubMed] [Google Scholar]
- (37).Bertini I; Bren KL; Clemente A; Fee JA; Gray HB; Luchinat C; Malmström BG; Richards JH; Sanders D; Slutter CE J. Am. Chem. Soc 1996, 118, 11658. [Google Scholar]
- (38).Zitare U; Alvarez-Paggi D; Morgada MN; Abriata LA; Vila AJ; Murgida DH Angew. Chem 2015, 127, 9691. [DOI] [PubMed] [Google Scholar]
- (39).Neese F; Zumft WG; Antholine WE; Kroneck PM J. Am. Chem. Soc 1996, 118, 8692. [Google Scholar]
- (40).Morgada MN; Abriata LA; Zitare U; Alvarez-Paggi D; Murgida DH; Vila AJ Angew. Chem 2014, 126, 6302. [DOI] [PubMed] [Google Scholar]
- (41).Lukoyanov D; Berry SM; Lu Y; Antholine WE; Scholes CP Biophys. J 2002, 82, 2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gurbiel RJ; Fann YC; Surerus KK; Werst MM; Musser SM; Doan PE; Chan SI; Fee JA; Hoffman BM J. Am. Chem. Soc 1993, 115, 10888. [Google Scholar]
- (43).Neese F; Kappl R; Hüttermann J; Zumft W; Kroneck PJ Biol. Inorg. Chem 1998, 3, 53. [Google Scholar]
- (44).Slutter CE; Gromov I; Epel B; Pecht I; Richards JH; Goldfarb DJ Am. Chem. Soc 2001, 123, 5325. [DOI] [PubMed] [Google Scholar]
- (45).Ledesma GN; Murgida DH; Ly HK; Wackerbarth H; Ulstrup J; Costa-Filho AJ; Vila AJ J. Am. Chem. Soc 2007, 129, 11884. [DOI] [PubMed] [Google Scholar]
- (46).Clark KM; Tian S; van der Donk WA; Lu Y Chem. Commun 2017, 53, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.