

Li-ion conductivity increases with introduction of an interfacial ice layer in nano-SCE.
Abstract
The transition to solid-state Li-ion batteries will enable progress toward energy densities of 1000 W·hour/liter and beyond. Composites of a mesoporous oxide matrix filled with nonvolatile ionic liquid electrolyte fillers have been explored as a solid electrolyte option. However, the simple confinement of electrolyte solutions inside nanometer-sized pores leads to lower ion conductivity as viscosity increases. Here, we demonstrate that the Li-ion conductivity of nanocomposites consisting of a mesoporous silica monolith with an ionic liquid electrolyte filler can be several times higher than that of the pure ionic liquid electrolyte through the introduction of an interfacial ice layer. Strong adsorption and ordering of the ionic liquid molecules render them immobile and solid-like as for the interfacial ice layer itself. The dipole over the adsorbate mesophase layer results in solvation of the Li+ ions for enhanced conduction. The demonstrated principle of ion conduction enhancement can be applied to different ion systems.
INTRODUCTION
Solid-state electrolytes are expected to provide the next boost for Li-ion batteries to exceed the practical ceiling of 800 W·hour/liter or 300 W·hour/kg imposed on currently available cathode and anode chemistries. The expected increase in energy density for solid-state batteries comes from several contributions, all targeting increasing the volume percent of active material in the cell. The most publicized is the introduction of lithium metal to replace graphite and graphite/silicon as an anode. Pure lithium metal has the highest energy density possible and thus would require the least space. However, many issues still need to be resolved, such as the irreversible reaction (and thus consumption) of lithium metal, dendrite formation, the increase in effective current density for planar lithium foils compared to the porous graphite (silicon) electrodes, and, last but not least, the “disappearance” of lithium during discharge (deplating) and thus loss of contact with the solid electrolyte. The mechanically rigid nature of ceramic solid electrolytes indeed has zero compliance, and extreme pressures need to be applied to press the lithium firmly against the solid electrolyte component. The discrete pressure points lower the effective surface area even more, leading to local dendrite formation and spongy deposits. Polymer electrolytes are more mechanically compliant but do not yet exhibit high enough ionic conductivity at room temperature. Very interesting new materials in this regard are silica gel electrolytes, which have also been referred to as “ionogels,” where an ionic liquid electrolyte (ILE) is confined in a nanoporous silica matrix (1). The extremely high porosity of the silica matrix (70 to 90%) gives these nanocomposite electrolyte materials a gel-like consistency and thus making them mechanically compliant similar to polymer electrolytes. These silica gels are sometimes indicated as hybrid solid electrolytes, as they contain a liquid. However, for the silica nanocomposites, as those described in this paper, the ionic “liquid” electrolyte becomes solid-like when confined in the tens of nanometer-sized channels both by the increase in viscosity and by the adsorption on the silica wall confining the channel. If the silica matrix would act merely as a porous separator, then the increase in viscosity for the confined liquid electrolyte would lead to a decrease in ionic conductivity. Instead, the interaction between the ILE molecules and the silica pore wall make the properties of the nanocomposite different from the sum of its individual components. Adsorption of ionic liquids on oxides with formation of solid mesophase layers up to a few nanometers in thickness has been shown on planar surfaces with atomic force microscopy (2). The selective adsorption of ionic liquid anions and cations on oxide surfaces can lead to enhanced Li+ conductivity along these interfaces. Of course, the enhancement along the oxide interfaces has to compensate for or even exceed the decreased conductivity through the ILE confined in the core of the pores. Hence, smaller pore size and high surface-to-volume ratios are desired. Thus far, ionogels with ion conductivities approaching that of the ILE itself have been demonstrated by optimization of the mesoporous structure (3). This means that interface enhancement was already present but not to the extent of exceeding bulk conductivity.
The preparation of ionogels starts from a homogeneous liquid mixture, in which an ILE is added to a sol-gel precursor solution for the synthesis of the oxide matrix (4, 5). In this method, the ILE and matrix form a composite in an “in situ” manner: The precursors in the solution react to form an oxide matrix around the ionic liquid template, encapsulating it in the process. Under certain synthesis conditions, the prepared ILE-SCE (solid composite electrolyte) can be in the form of a monolith with the ILE embedded in a continuous mesoporous inorganic oxide network. So far, mostly silica-based ILE-SCEs have been prepared this way, although examples have also been made with alumina (6), titania (7), and even tin oxide (8). Most reported sol-gel formulations contain an ILE, an alkyl-silicate such as tetraethyl orthosilicate (TEOS) as silica precursor, and formic acid as reagent and solvent (9, 10). According to the proposed mechanism (11) for this sol-gel process, silica is mainly produced by the reaction between TEOS and formic acid, although water is generated during the sol-gel process. Besides these formic acid–based “nonaqueous” mixtures, aqueous sol-gel formulations with HCl as a catalyst and H2O as a reagent (plus organic solvent) have also been described, however, in this particular case for the synthesis of a silica composite with ionic liquid only (12–15).
Typically, ionogels show ion conductivity lower than that of the ILE reference. The first generation of ionogels had room temperature conductivities typically only about 30 to 50% of the bulk ILE value, although some examples reaching up to 80% have been reported (9, 10, 16, 17). The effect of ILE content and resulting pore morphology on ionogel conductivity has already been investigated in detail (3); however, no systematic study of interface enhancement effects is known. Wu et al. (18) recently reported on an in situ functionalized ionogel, which also gave conductivity enhancement compared to the bulk ILE. The enhancement was attributed to the interaction between the anion and the 3-glycidyloxypropyl functional group on the silica surface. This finding supports the idea that surface functionalization can indeed enhance the interface conduction promotion.
In this work, we demonstrate the in situ formation of a solid ice water layer on the silica and detail the mechanism of interfacial Li-ion conduction by the increased dipole interaction between the surface ice functional layer and the adsorbed ionic liquid mesophase layer. By combination of high internal surface area and dense ice functional layer, solid nanocomposite electrolytes (nano-SCE) with 200% higher Li-ion conductivity than the bulk ILE reference were achieved. The silica matrix is shown to have a true monolithic mesoporous structure with pore volumes and surface areas up to 90% and 1400 m2/g, thus providing extreme surface-to-volume ratios allowing large contribution of conduction enhancement along these interfaces. By optimized functionalization of the silica surface combined with maximizing the surface-to-volume ratio, nano-SCE with ion conductivities well exceeding 10 mS/cm could potentially be engineered and thus are very attractive for large-capacity batteries for automotive applications.
The focus of our paper is on the mechanism of enhanced interface conductivity through the formation of a mesophase layer with evidence from Raman, Fourier transform infrared (FTIR), and nuclear magnetic resonance (NMR) spectroscopy. The interface stability of our nano-SCE material at high voltages is demonstrated using thin-film lithium manganese oxide (LMO) electrodes. In this way, focus maintains on the material rather than on the electrode integration and cell assembly issues. Similarly, the electrochemical window and stability against lithium metal foils are fully characterized. Functionality and integration of our nano-SCE is demonstrated through assembly and rate performance tests of lithium iron phosphate (LFP) and lithium titanate (LTO) cells. The stability of our electrolyte and the electrochemical inactivity of the ice water were shown through long-term cycling of symmetric Li-SCE-Li cells. Optimization of energy density, rate performance, and cycling performance of fully assembled cells will be the focus of follow-up papers (19, 20).
Interfacial ion conduction in composite electrolytes
Interfacial ion conductivity promotion in two-phase composite systems has been known for almost 90 years (21). For example, up to four orders of increase in ionic conductivity has been shown for composites of simple lithium salt such as lithium iodide with mesoporous oxide particles such as silica or alumina as compared to the ion conductivity of the pure lithium salt electrolyte (22). The ions in these SCEs can diffuse much faster along the Li ion–depleted (or vacancy-rich) electrical double layer formed at the oxide/electrolyte interface. Unfortunately, the ion conductivity obtained in these simple two-component inorganic solid-solid composites (1) has not exceeded the 1-mS/cm2 threshold needed to bridge the few hundred–micrometer distance between the current collector plates in the Li-ion battery. The concept of heterogeneous doping with an oxide matrix to engineer the ionic conductivity has also been explored for polymer electrolytes (23) and ILEs (24), which have a higher intrinsic ionic conductivity to start with. In addition, the rich molecular (stereo)chemistry of the third component opens up additional ion conduction mechanisms, as the (di)polar solvent-like molecules may participate in the formation of the electrical double layer. While the solvating action of the ether groups in polyethylene oxide polymer electrolytes provides solid-state ion conductivities of ~10−6 S/cm for LiClO4 to ~10−5 S/cm for LiN(SO2CF3)2, their composites with silica, alumina, or titania nanoparticles can indeed provide more than 10-fold enhancement in measured ion conductivity (25), unfortunately, still well below the room temperature threshold of 1 mS/cm. ILE solutions are mixtures of a Li-salt solute and an ionic liquid solvent, which can already have high intrinsic ionic conductivities between 0.1 and 10 mS/cm (26, 27). Several attempts have been made to enhance the ion conductivity by mixing or gelling it with oxide nanoparticles or to confine the ILE in mesoporous microparticles (9, 16, 28, 29). However, so far, no enhancement of the ion conductivity has been observed for the three-component Li-salt/ionic liquid/oxide composites (fig. S1). Although the use of mesoporous silica microparticles does result in higher conductivity as compared to composites with solid nanoparticles, the interfacial surface area and ion conduction promotion are not sufficient to exceed bulk ILE conductivity.
Mesoporous silica is a well-known material used in catalysis. It is typically made by hydrothermal or simple sol-gel synthesis. Hydrothermal processes typically lead to mesoporous powders, but with careful control of the room temperature sol-gel process, large porous glass monoliths or aerogels have also been produced. The silica matrix is formed through hydrolysis and condensation reactions of tetra-alkyl orthosilicates (30). Key in control of the pore structure is the use of templates, for example, a surfactant-type micelle, around which the silica matrix is formed. When an ionic liquid is added as templating molecule, the hydrated silica matrix interacts with the ionic liquid, forming a gel, and after curing and drying, the ionic liquid is confined within the solid nanoporous silica matrix (13). When lithium salt is added as a third component, the ILE confined in the silica matrix forms a silica gel electrolyte, which has also been referred to as ionogel (24). However, so far, these silica gel electrolytes show conductivities approaching that of the bulk ILE but not exceeding it, except for one case where the silica had been chemically functionalized (see Introduction) (18).
Here, we show, systematic promotion of the Li-ion conductivity of the nanocomposite well beyond that of the pure ILE. The example of 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide (BMP-TFSI) is used here. It is postulated that the adsorption of the ionic liquid molecules on the OH-terminated silica surface is promoted by the presence of an interfacial ice water layer. Strong hydrogen bonding between the ice water and the TFSI− anion induces molecular ordering of the ionic liquid, similar to the ordered domains that spontaneously form in ionic liquids (31). The key difference with the randomly formed domains in bulk ILE is that the ice layer acts as a functional layer that (i) induces the molecular ordering on the oxide surface and (ii) introduces strong enough H-bonding to induce dipoles to release free Li+ for enhanced conduction. Next to the increase in free Li+ concentration, we will show that the activation energy for diffusion is lower along the composite interface with the adsorbed ILE layer and ice water layer.
The few-monolayers-thick surface water layer on silica is a solid-like layer, as it is strongly bonded to silanol groups through H-bridges and therefore also referred to as ice layer (32). Its density and thickness (estimated up to three to four monolayers, with ~0.25 nm per ice monolayer) are in thermodynamic equilibrium with the partial water pressure [relative humidity (RH)] in the environment (fig. S2). We show that the ion conductivity increases with the thickness of the ice water layer as hydrogen bonding with the adsorbed ionic layers also increases. The ice water layer is stable similar to crystal water in chemical compounds. This is in stark contrast with the super concentrated aqueous electrolytes or so-called water in salt mixtures, where the electrochemical window is drastically widened but, eventually, the water is still electrochemically active (33).
RESULTS
Enhanced ion conductivity in the nano-SCE with interfacial ice layer
Different from typical formic acid–catalyzed ionogel recipes, we used a mild pH 5 mixture with large excess of water and PGME (1-methoxy-2-propanol) added to a TEOS precursor with Li-TFSI salt and BMP-TFSI ionic liquid. At this pH, the hydrolysis reactions are slow, while the condensation is favorable (30). The Li ions are believed to act as the catalyst for the hydrolysis reaction, as no gelation occurred in the absence of the lithium salt while both had the same pH of 5. The molar ratio of ionic liquid to TEOS (and thus silica moieties) is indicated as x value and was varied between 0.25 and 2. The molar ratio of BMP-TFSI to Li-TFSI was kept at 3 (corresponding to 1 M Li-ion solution). Slow drying was necessary to maintain the structural integrity of the monolith structure (see Materials and Methods). Figure 1A shows a photograph of a monolithic pellet obtained after vacuum drying. The 72-hour vacuum drying was sufficient to remove all the moisture down to a point where all free water was removed while the adsorbed ice water layer remained fully intact, as confirmed by FTIR. No vibrations for free water were detected at 1635 cm−1 in any of the samples after the vacuum drying step (Fig. 2). For comparison, the FTIR spectrum for a nano-SCE sample (x = 1.5) stored for 1 week in a N2 glove box at 60% RH is shown. In this case, a clear free water peak appears. All samples, on the other hand, did show a clear signal for silanol surface functionalization (Si─OH bending between 950 and 980 cm−1) and an adsorbed ice water layer (O─H stretching at ~3540 cm−1) bonded to the ─OH surface groups by H-bonding (more details below). The vials were weighted before and after the drying step to measure the water retained in the nano-SCE (table S1). Later, we will calculate the number of corresponding monolayers of surface-bound ice layers from the excess weight. The vacuum-dried pellets were brought into the glove box [<0.1-ppm (parts per million) H2O] and stored in closed vials to maintain the original water content. A small volume was taken from the pellet for further characterization.
Fig. 1. Macro- and microstructure of the nano-SCE.

(A) Picture of two nano-SCE pellets (left) synthesized in the vial; after gelation, a transparent pellet is obtained. Note that the pellet is fully transparent and was therefore given a blue hue for visibility. When the ILE is removed, a brittle white pellet remains for the highly porous silica matrix (right). (B) Scanning electron microscopy (SEM) image of the SiO2 matrix that remains after removal of the ILE. (C) Zoom of the picture shown in (B) depicting the mesoporous nature of the matrix material with some macropores. (D) Transmission electron microscopy (TEM) image showing a dense packing of 7- to 10-nm silica nanoparticles as the building blocks of the porous matrix material. (E) The porosity of the matrix structure plotted for different molar ratios of ILE with respect to SiO2 (x value). The dashed line gives the theoretical porosity determined from volume fraction of ILE and silica. The acetone-rinsed samples (black squares) were dried in air, which gives partial collapse of the structure for x > 0.5. Supercritical CO2 drying of ethanol-rinsed nano-SCE (green circles) prevents collapse up to x = 2 for extra slow removal of the CO2 (open circle). BET, Brunauer-Emmett-Teller. Photo credit: Fred Loosen, imec; Akihiko Sagara, Panasonic.
Fig. 2. Free versus ice water in the nano-SCE.
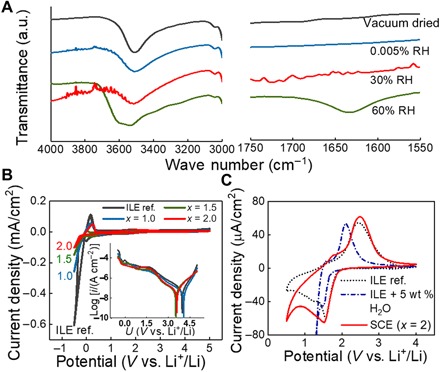
(A) IR spectra of the nano-SCE as dried in vacuum (black) and subsequently further dried in a glove box with 0.0005% RH for 9 days (blue) and exposed to 30% RH for 4 days (red) and to 60% RH for 8 days (green), respectively. a.u., arbitrary units. (B) Cyclic voltammograms of a Li/SCE/TiN stack with x values of 1.0 (blue), 1.5 (green), and 2.0 (red) and of ILE reference (black); the inset shows the current in logarithmic scale. (C) Cyclic voltammograms of Li/SCE (x = 2)/40-nm TiO2 stack (red), ILE (dotted black), and ILE spiked with 5 weight % (wt %) H2O (dash-dotted blue line); in (B) and (C), measurements with ILE and ILE with H2O were done in three-electrode configuration with TiN as a working electrode and Li as counter and reference electrodes. The SCE was dried for 2 days in the glove box after vacuum drying.
The ionic conductivity (σi) of our vacuum-annealed nano-SCE increased with volume fraction of ILE (x value) as for the particle composites (fig. S1). However, in this case, the ionic conductivity exceeded that of the pure ILE itself by more than 200% for the highest x values (Fig. 3). Furthermore, the temperature dependence of the nano-SCE with enhanced ion conductivity showed a different behavior than that of the pure ILE: Whereas the Li-TFSI in BMP-TFSI ILE shows a clear change in conductivity and activation energy (slope) around the melting point of the mixture at 29°C, the nano-SCE with enhanced conductivity does not. Instead, it shows a continuous variation in σi with temperature, indicating that a previously unidentified type of phase or mesophase is formed, which is then responsible for the enhanced conductivity. Moreover, the smaller slope and thus lower activation energy for diffusion for the nano-SCE compared to the ILE indicate different material properties (fig. S3). It is postulated that strong interaction between the ionic liquid molecules and the solid ice layer on the silica scaffold is responsible for the observed mesophase behavior, as will be discussed with the proposed model below.
Fig. 3. Conductivity of the nano-SCE.
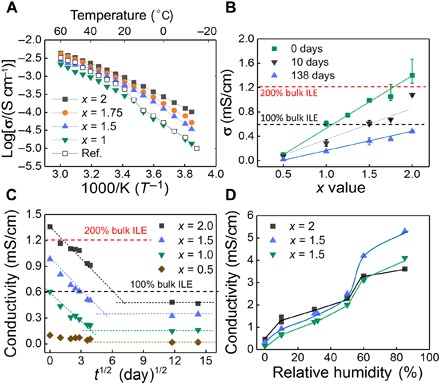
(A) Temperature dependence of the conductivity of nano-SCEs dried for 8 days in the glove box (GB) with x values of 2 (black squares), 1.75 (orange circles), 1.5 (blue triangles), and 1.0 (green triangles) and of ILE reference (open squares). (B) Conductivity of nano-SCEs additionally dried in GB for 0 days (green squares), 10 days (black triangles), and 138 days (blue triangles). (C) Conductivity versus square root of drying time of nano-SCE with x values of 2 (black squares), 1.5 (blue triangles), 1.0 (green triangles), and 0.5 (brown diamonds). (D) Conductivity of nano-SCE with x = 2 (black squares), 1.5 (blue triangles), and 1.0 (green triangles) exposed in a N2-filled humidity chamber.
The argon atmosphere in the glove box contains less than 0.1 ppm of water, which corresponds to 0.0005% RH, a partial water pressure of 0.01 Pa, or a dew point of −88°C. As the number of adsorbed water layers on silanol-terminated silica is in equilibrium with the partial pressure of water (fig. S2), the surface water will slowly diffuse out of the nano-SCE and sublimate at the edges. Figure 3C shows the change in conductivity for 23 μl of nano-SCE as a function of residence time in the glove box. The ion conductivity decreases with drying until it saturates at a value corresponding to the silica surface in equilibrium with the water partial pressure of 0.01 Pa in the glove box. Even under the extreme dry conditions of the glove box, at least, a partial monolayer of adsorbed water on silanol is present, as Raman spectroscopy still showed a signal at 3524 cm−1, which is specific for the first monolayer of adsorbed water on silanol (Fig. 4B). The ion conductivity under saturated conditions was well below that of the individual ILE in all cases. Hence, the enhancement is not sufficient to compensate for the loss in ionic conductivity of the confined ILE in the core of the pore.
Fig. 4. Model for the adsorbed interfacial layer.
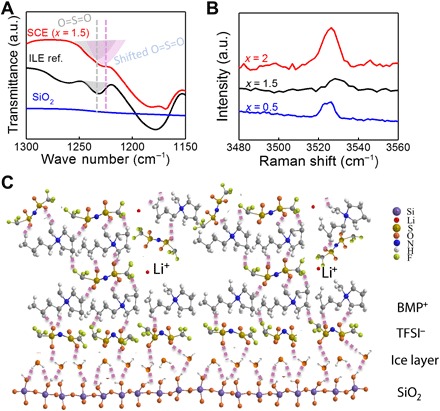
(A) IR spectra of nano-SCE with an x value of 1.5 (red), ILE reference (black), and SiO2 (blue), showing that the O═S═O group (1231 cm−1) is involved in the interaction with OH-groups on the silica surface. (B) Raman spectra of nano-SCE with x values of 2 (black), 1.5 (red), and 0.5 (blue), showing the presence of ice water bonded on silanol-terminated silica even for nano-SCE near saturation (0.0005% RH) in a glove box (30 days). (C) Proposed model for the interface interaction in the nano-SCE with dissociation Li-TFSI into free Li+ as the TFSI− anion shares part of its negative charge with the adsorbed ice-TFSI-BMP layer; the colors represent different elements with purple (silicon), red (lithium), dark yellow (sulfur), orange (oxygen), blue (nitrogen), white (hydrogen), and green (fluorine). The purple dashed lines represent the hydrogen bond between the O═S group of TFSI anion and the OH-groups of hydroxylated silica surface. The Li+ ions set free by the dipole over the adsorbed layer can migrate through subsequent mobile or diffuse ionic liquid layers above the interface layers. Note that depending on the strength of the hydrogen bonds and the equivalent charge on the silica, multiple adsorbed layer could be formed as well. Full spectra are shown in fig. S8.
An interesting observation is the linear relationship with the square root of drying time as shown in Fig. 3C, indicating that the conductivity change is directly proportional to the changes in amount of adsorbed ice water on the silica and that the removal of this surface water is diffusion limited. Note that the “drying” only occurs in an open environment where the RH is lower than for the equilibrium ice layer. The conductivity did not change notable, for example, in the closed coin cells used for temperature-dependent measurements.
The temperature dependence of the nano-SCE was measured for different times of drying in the glove box. As the conductivity of the dried nano-SCE approached that of the ILE, the continuous σi versus 1/T profiles for mesophase conductivity gradually changed to the profile for the ILE, again revealing the drop around its melting point (fig. S3). This observation further supports the assumption that the ice layer acts as a functional layer for interface interaction with the ILE, giving rise to the mesophase behavior in the nano-SCE. Hence, when the functional layer is removed, the ILE becomes merely confined in a mesoporous oxide membrane.
Measurements of the electrochemical stability window confirm that the ice water in the nano-SCE is stable, as no peaks for water reduction or oxidization were observed at inert TiN electrode (Fig. 2) nor at a TiO2 thin-film electrode, which otherwise acts as an electro-catalyst for water reduction. Instead, the electrochemical stability of the nano-SCE is very similar to that of the ILE and thus limited by the oxidation of TFSI− at electrode potentials >4.3 V and the reduction of TFSI− and BMP+ at potentials <1 V versus Li+/Li (33). For comparison, a voltammogram is shown for an ILE with 5 weight % (wt %) water added (similar content as for some nano-SCE; see table S1). In this case, a cathodic branch for water reduction is measured immediately following the Li-intercalation peak of anatase at 1.5 V versus Li+/Li.
The thermal and (electro)chemical stability of the nano-SCE is mostly determined by the ILE filler. Thermogravimetric analysis (TGA) showed thermal stability of the SCE and ILE up to 320°C, irrespective of the ILE-to-silica ratio (fig. S4). Above this temperature, the Li-TFSI and BMP-TFSI decompose completely to volatile components, and only the silica matrix remains around 450°C. The mass percentage remaining after thermal decomposition indeed matched very well with the fraction of silica in the SCE.
Characterization of the silica matrix and quantification of ice layer
The nano-SCE showed no clear microstructure in the scanning electron microscopy (SEM) except for a smooth surface with some silica patches peeking out (fig. S5). The specific density of the SCE was determined with a helium pycnometer and was around 1.5 g/cm3 for all x values (table S1). The full silica matrix was revealed by tedious extraction of the ILE in a solvent (see Materials and Methods). By carefully drying at the critical point of CO2, intact aerogel monoliths could be obtained like the one shown in Fig. 1A. SEM inspection shows a scaffold of mesoporous silica with 10- to 30-nm pore diameter, which is wrapped around larger macropores of 100 to 150 nm, as can be seen in Fig. 1 (B and C). High-resolution transmission electron microscopy (TEM) (Fig. 1D) further exposed a microstructure composed of closely packed silica nanoparticles. The average particle diameter ranged from 7 to 14 nm for x values between 0.5 and 1.5.
The specific surface area [Brunauer-Emmett-Teller (BET)], porosity, average pore size, and pore size distribution were determined with N2 adsorption/desorption measurements (table S1 and fig. S6). Partial collapse of the structure and uncomplete removal of adsorbed ILE may somewhat misrepresent the numbers. Careful extraction of the ionic liquid and slow drying using supercritical CO2 provided, however, reliable results close to the expected porosity calculated from the volume fraction of ILE to silica (Fig. 1). The BET surface area ranges between 800 and 1000 m2/g. The mean pore size obtained from the slope of the isotherm ranged between 7 and 16 nm. In addition, a smaller fraction of larger pores up to about 200 nm was measured (fig. S6), in accordance with SEM observations. The pore diameter corresponds very well with twice the equivalent thickness of the ILE layer obtained from the ILE volume fraction and the BET surface area, meaning that the mesopores are completely filled with ILE.
The reported BET surface area is for the mesopores and macropores only. For acetone-rinsed matrix, micropores (~0.6 nm) were also measured. The micropores are found between the individual silica nanoparticles making up the structure as those shown in the TEM image of Fig. 1D. A maximum additional surface area between 650 (x = 0.5) and 360 m2/g (x = 1.5) is estimated (table S1).
Both FTIR and Raman spectra show clear evidence for silanol groups with adsorbed ice water molecules on the high-porosity silica matrix with extreme effective surface areas exceeding 1400 m2/g when taking into account micropores, mesopores, and macropores. Between zero and three water monolayers are estimated from the excess water in the nano-SCE for x < 1.75. For planar silica, the first three monolayers of adsorbed water are indeed considered immobile and solid-like because of their strong hydrogen bonding to the OH-terminated surface (32) (see fig. S2). The O─H stretch associated with silanol hydrogen bonded to an ice water layer is found at 3540 cm−1 in the FTIR spectra. All nano-SCEs show, indeed, a distinct peak at 3540 cm−1 for ice water after vacuum drying and after further drying in the glove box (Fig. 2). Even for the equilibrated nano-SCE at 0.0005% RH (glove box), Raman spectroscopy still showed the presence of at least a partial monolayer (Fig. 4B). The fourth monolayer on planar silica is believed to be a transitional layer, meaning that it is still adsorbed and restricted but can have some mobility. From the fifth layer on, the water becomes mobile and liquid-like. Liquid-like water will show up at higher wave numbers in the FTIR spectrum due to the lower degree of H-bonding in liquid water. For the nano-SCE exposed to 60% RH, the 3540-cm−1peak indeed shows additional vibrations shifted to higher wave numbers because of the additional adsorbed liquid water layer. Interesting in this regard is the experiment where the sample was exposed to 30% RH, as no liquid water is yet expected on silica at this humidity (fig. S2). For this sample, only the 3540 cm−1 peak for ice water is seen in FTIR. In addition, no free water peak was detected at 1635 cm−1 even after 4 days at 30% RH. This means that water is not taken up by the hygroscopic Li-TFSI dissolved in the hydrophobic BMP-TFSI once the nano-SCE is dried by vacuum treatment. Therefore, any additional water in the SCE will be adsorbed on the OH-terminated silica surface. Hence, as for planar silica, the SCE silica matrix is in equilibrium with the partial pressure of water in the environment.
To test this hypothesis further, the ion conductivity of nano-SCE (x = 1, 1.5, and 2) was measured at different % RH; the samples were exposed to a controlled mixture of dry and moistened N2 gas in a glove box for 2 days to allow the adsorbed water coverage to reach equilibrium (Fig. 3D). For the points at ~0% RH, the conductivity for the equilibrated nano-SCE in the glove box was taken. Astonishingly, the ion conductivity versus RH(%) profile followed the expected behavior for water adsorption on planar silica (fig. S2). Between 0 and 30% RH, the conductivity increased with increasing RH. as expected for an increase in adsorbed ice layer density and thickness (corresponding with one to three ice layers on planar silica). Note that FTIR showed that no free water was present in the nano-SCE for several days at 30% RH. A transition is seen around 50% RH, corresponding with conditions where a transitional adsorbed water layer is expected for planar silica. Eventually, a distinct stepped increase in ion conductivity is found toward 60% and higher humidities where, in resemblance with planar silica, now, also a liquid-like water layer is likely formed at the interface between the silica and the embedded ILE. With FTIR, a liquid water layer on the ice layer is now detected by the shift of the silanol/ice/water vibrational peak to higher energies (Fig. 2A). The observed change in conductivity is reversible; thus, the nano-SCE can act as a humidity sensor and a Li-ion electrolyte. From Fig. 3D, the ion conductivity of the nano-SCE immediately after vacuum anneal corresponds with an equilibrium hydrated silica of ~10% RH. The ion conductivity for saturation in dry room conditions (~0.5% RH) would be around 0.6 mS/cm (for x = 2). This experiment manifestly demonstrates the effect of interfacial water on the ion conductivity. For RH > 60%, the higher ion conductivity could be explained by faster diffusion of solvated Li+ through the liquid-like layer. However, in the case of a solid ice layer, the Li+ ion diffusion would be a solid-state type diffusion and thus slower than through the ionic liquid itself. Instead, the enhancement is attributed to enhanced adsorption of the organic anions and cations of the Li-salt and ionic liquid molecules, as proposed in the model below.
Model and evidence for mesophase layer
We propose a model where the ionic liquid molecules are adsorbed on the silica surface via H-bridges with the immobile ice layer on the silanol groups (Fig. 4). The intrinsic nature of the hydrolysis condensation reaction provides the highest silanol density (4 × 1014 to 8 × 1014 cm−2, which matches well with the density of one monolayer of ice with ~8 × 1014 water molecules per cm2) (34). Evidence for molecular interactions between the O atoms of the TFSI anions and the silica is given by FTIR, which shows a doubling of the O═S═O peak for all nano-SCE as compared to the ILE reference (Fig. 4A; full spectra in fig. S8). The shift of the additional peak with about −5 cm−1 from 1231 cm−1 indicates bonding of the O═S═O groups for at least part of the TFSI anions. Therefore, H-bonding of the TFSI anions on the ice water layer is assumed. Subsequently, the large hydrophobic BMP cations associate with the first TFSI layer, completing the first adsorbed layer of ionic liquid molecules. As for the ice layer, the adsorbed BMP-TFSI molecules are thought to be mostly immobile, thus extending the solid-like ice layer on the silica surface. As the TFSI anion has a symmetric O═S═O group, one oxygen atom can interact with the hydroxylated silica surface while the other forms the sticking points for the BMP cations. The TFSI anion also has two O═S═O groups, assuring firm adsorption and dense ordering of the anion monolayer. Adsorption is most efficient in the case of a dense ice layer with the highest density of OH-groups as potential sticking points. In the presence of only silanol groups, the adsorption might not be strong enough to form a continuous adsorbate layer. In addition, an increasing number of ice monolayers are known to increase the strength of the hydrogen bond (35). Note that the molecular interactions between the BMP cation and the ordered TFSI monolayer will be different from that in the ionic liquid where the TFSI anion has rotational freedom and no polarization from an underlying surface. The charge of the large BMP cation is indeed distributed over the many atoms by polarization of the intrinsic bonds and by molecular interactions with its chemical environment and, specifically, the adsorbed TFSI anion. The H-bonding between the O-group of the TFSI anion and the OH-termination of the ice layer now introduces a dipole over the first adsorbed layer, inducing further molecular ordering by association. It is believed that at this point, the smaller Li-TFSI molecules adsorb on the molecular layer whereby the TFSI anion now compensates the residual positive dipolar charge of one or more of the BMP cations in the upper layer, hence loosening its association with its Li ion. In this way, the concentration of free Li+ is increased at this interface, leading to higher ion conductivity. Hence, denser and thicker ice layers then introduce a larger dipole with a higher residual charge to compensate, giving a proportionally higher free Li+ concentration and thus ion conductivity.
On top of the adsorbed ILE layer, either another ILE layer can adsorb similar to the ice multilayers on silica or the dipole pull of the ice layer is too weak and a lightly bound ILE is on top, which then can provide liquid-like conduction for the Li+ ions released in the lower adsorbed layer (Fig. 4C). The change in free Li+ ion concentration was corroborated by both NMR and Raman spectroscopy measurements. The Raman measurements indirectly show that a larger fraction of free Li+ ions are indeed present in the nano-SCE with more ice water layers bound to the silica (Fig. 5). The Raman measures the association of the cation with TFSI by probing the vibration of the N-group of the TFSI anion (36). In the pure BMP-TFSI ionic liquid, only a single peak at 741 cm−1 is seen. In the case of the pure ILE, an additional peak is seen at 746 cm−1 where two TFSI anions coordinate with a single Li+ ion [see density functional theory (DFT) calculations in Materials and Methods]. For all nano-SCEs, the peak intensity at 746 cm−1 is weaker than that for ILE, indicating a smaller fraction of associated Li-TFSI and, consequently, a larger fraction of nonassociated or free Li+ cations. The peak drastically decreases for those nano-SCE that shows the highest conductivity enhancement, i.e., those with the thickest ice layer. For the nano-SCE at equilibrium in the glove box, still, a fraction of free Li+ is measured although much smaller than for the vacuum-annealed samples. The ratio of the peak intensities for the 746 over 741 cm−1 Raman shifts then is a measure of the ratio of free to TFSI-associated Li-ions (Fig. 5B). The linear increase in free Li+ ion fraction with x value nicely follows the trend of conductivity enhancement with the x value in Fig. 3B, both for vacuum dried nano-SCE (day 0) and the SCE at equilibrium with the glove box dryness (day 138).
Fig. 5. Free Li+ ions in the nano-SCE.
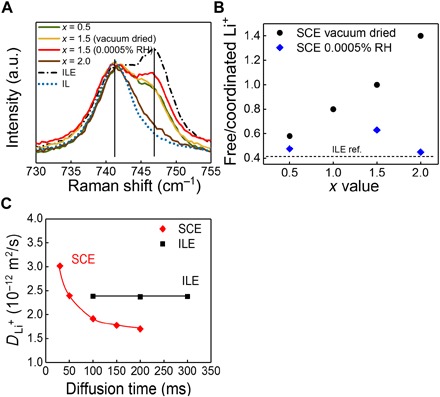
(A) Raman spectra of an ionic liquid (IL; dotted blue line) and ILE reference (ILE; dash-dotted line) of as prepared nano-SCE (vacuum dried) with x values of 0.5 (green), 1.5 (yellow), and 2 (brown) and of nano-SCE (x = 1.5) additionally dried in glove box for 30 days or near saturation at 0.0005% RH (red). The vertical lines label the Raman shift for TFSI with its N center coordinated to Li+ (746 cm−1) and not coordinated to Li+ (741 cm−1), respectively. (B) Ratio of free to coordinated Li+ of nano-SCE as synthesized (vacuum dried, black circles) and additionally dried in glove boxs with 0.0005% RH for 30 days (blue diamonds), corresponding to the ratio of the integrated intensity of the Raman peaks (746 cm−1 over 741 cm−1). (C) PFG-NMR–derived Li+ self diffusion coefficient of nano-SCE (red diamonds) and ILE ref. (black squares) as a function of the interval between the gradient magnetic field pulses. The theoretical peaks on Raman spectra were simulated using DFT calculation.
From pulsed-field gradient NMR (PFG-NMR), the self-diffusion coefficient of the different mobile Li-ion species was determined as a function of the interval between the gradient magnetic field pulses ∆ for the ILE liquid reference and for a nano-SCE (x = 1.5) with the same ion conductivity of 0.6 mS/cm (Fig. 5C). The Li+ self-diffusion coefficient in the ILE reference was constant, indicating that only one or multiple Li species with very similar mobility are present in the liquid. For the nano-SCE, the self-diffusion coefficient varied with ∆ and exceeded that of ILE at short ∆, indicating the presence of fast-moving species that respond only at short intervals between magnetic field pulses. The gradient in self-diffusion coefficient suggests that next to the increase in free Li-ion concentration, as inferred from Raman spectroscopy, the activation energy for diffusion is lowered in the mesophase interface layer as well. This supports the conductivity enhancement introduced by the (more) free Li+ ions in the mesophase layer. At longer ∆, the self-diffusion coefficient was lower than that of the ILE reference. This corroborates the much lower ion conductivity for the glove box–saturated nano-SCE compared to ILE. The ILE confined in the core of the mesopores will have a higher viscosity due to the restriction of molecular movement. Hence, the enhancement by creation of much faster diffusing Li-ions at the silica/ice/ILE interface has to overcompensate the decrease in conductivity in the core of the pore. This explains the absence of enhancement in the particle-based systems where the interfaces do not provide sufficient ion conduction promotion (fig. S1).
Demonstration of functionality of nano-SCE as Li-ion electrolyte
The electrochemical stability of the nano-SCE against lithium metal was tested using a three-electrode setup (schematic of the setup is shown in fig. S7). The current-potential characteristic of Li/SCE (x = 1.5) and Li/ILE half-cell are shown in Fig. 6A. As for the electrochemical window in Fig. 2, the electrochemistry is limited by the ILE filler. Reversible lithium plating and stripping are observed. A stable solid electrolyte interphase (SEI) layer is formed at metallic lithium with an RSEI of about 0.9 kilo-ohm·cm2, responsible for the large IR drop in the i-U curve on both the cathodic and anodic sides. The cathodic current in the pure ILE solutions did not show any hysteresis down to −2.5 mA/cm2. However, the anodic dissolution showed a passivation peak with a steady-state anodic current of only 0.06 mA/cm2. The cathodic current branch at the solid-solid Li/SCE interface showed no hysteresis for cathodic currents less than −0.5 mA/cm2. The resistance of the SEI was, however, about double. Similarly, the anodic peak was lower and the steady-state current after the anodic passivation peak was 0.03 mA/cm2, only half that of the pure ILE solution. The formation of SEI and passivation layers in the pores of the SCE limits the current at lithium metal. Both the voltammograms for the Li/ILE and Li/SCE electrodes were reproducible upon multiple cycles, indicating that the anodic passivation layer and chemical SEI layer are reversible and stable. The slow dissolution kinetics at the Li/SCE interface severely limits the performance of half-cells made with Li metal anodes below.
Fig. 6. Functionality of nano-SCE as Li-ion electrolyte.
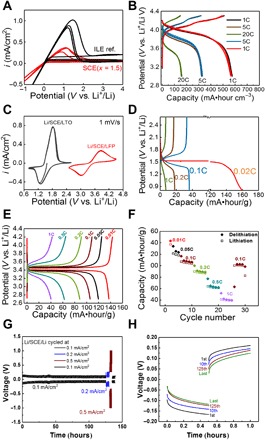
(A) Cyclic voltammogram of nano-SCE (x = 1.5, as synthesized after vacuum drying) (red) and ILE reference (black) measured in three-electrode configuration with Li as working, counter, and reference electrodes (SEI resistance estimated from IR drop on cathodic current is 0.9 and 1.8 kilo-ohm·cm2 for ILE and SCE, respectively). (B) Galvanic charge/discharge curves of Li/SCE (x = 1)/100-nm thin-film LiMn2O4 cell for five cycles at C-rates of 1C, 5C, and 20C. (C) Cyclic voltammograms of the Li/SCE/40-μm Li4Ti5O12 and Li/SCE/30-μm LiFePO4 powder electrode cells (1 mV/s). (D) Galvanic charge/discharge curves of Li/SCE/40-μm Li4Ti5O12 powder electrode at 1C, 0.1C, 0.2C, and 0.02C. (E) Galvanic charge/discharge curves of the Li/SCE/30-μm LiFePO4 powder electrode at 1C, 0.5C, 0.2C, 0.1C, 0.05C, and 0.01C. (F) Capacity (filled diamonds for delithiation and open squares for lithiation) versus cycle number of the Li/SCE/30-μm LiFePO4 powder electrode; the thickness of the SCE in the cells is about 280 μm. The density of the LFP and LTO cathode is about 1.9 and 11.0 mg/cm2, respectively. (G) Potential versus time curves of a Li/SCE/Li stack cycled at current densities of 0.1, 0.2, 0.5, and 0.1 mA/cm2. (H) The 1st, 10th, 125th, and last polarization of the Li/SCE/Li stack stressed at 0.1 mA/cm2, shown in (G). For (G) and (H), the SCE has a conductivity of 0. 34 mS/cm, and the thickness of the SCE pellet is 0.152 cm.
A 100-nm LiMn2O4 (LMO) thin-film was used as a model positive electrode to test both the stability of the nano-SCE and the electrode material while eliminating potential interface issues in particle composite electrodes (37). The cycling performance of the thin-film electrode/SCE stack demonstrates the stability of the interface between the electrode and electrolyte. In this model thin-film setup, only one single, well-defined, and planar interface contact is present between the electrolyte and electrode, i.e., it is an ideal platform to study the electrochemistry of the electrolyte/electrode interface without issues of volume change, etc. Also in this experiment, the rate performance is not limited by the Li-foil counter electrode, as the current density (6 μA/cm2 for 1C) is below that of the steady-state anodic current plateau for the lithium half-cell (0.03 mA/cm2). Reproducible and stable charge/discharge curves are obtained for a cutoff voltage at 4.3 V for C-rates between 1 and 20C for over 20 cycles (Fig. 6B). LMO is unstable in a liquid electrolyte for LiB. For example, a 50% capacity reduction was observed on a 100-nm LMO film charge-discharged for 10 cycles in a LiClO4/propylene carbonate electrolyte at 1C (37). Our results show that the nano-SCE is more compatible with LMO than a typical liquid electrolyte.
To demonstrate the integration of the nano-SCE, we also fabricated half-cells with Li4Ti5O12 (LTO) and LiFePO4 (LFP) powder electrodes. The precursor solution was drop-casted into the coin cell to impregnate the porous electrodes and left for further gelation before they were dried and vacuum-annealed similarly as for the nano-SCE pellets. The cells show characteristic lithiation/delithiation of the corresponding electrodes (Fig. 6C). The lower peak currents for LFP than LTO are due to the difference in coating thickness. The rate performance during charge/discharge measurements was now limited by the Li-foil counter electrode pressed on the nano-SCE layer formed on top of the 30- to 40-μm-thick electrode coatings (Fig. 6, D and E). The LTO/nano-SCE/Li cell reached its maximum capacity of 160 mA·hour/g only at low C-rate of 0.02C (Fig. 6D). The accessible capacity drops rapidly with C-rate with less than 10% for C-rates larger than 0.1C. Similarly, the LFP/SCE/Li cell reached its maximum capacity of about 140 mA·hour/g at 0.01C (Fig. 6E). Figure 6F shows the rate performance for a total of 30 cycles, demonstrating stable cell configuration. These experiments demonstrate the functionality of the nano-SCE as Li-ion electrolyte and the feasibility for integration in Li-ion cells.
The stability or cyclability of the nano-SCE was tested using Li/SCE/Li symmetric stack. It was cycled for more than 120 cycles at a current density of 0.1 mA/cm2 for 0.5 hours (Fig. 6G) without any issues or dendrite formation (Fig. 6H). The polarization voltage became smaller over time, indicating improvement of contact. Moreover, the cell was stressed up to current densities of 0.5 mA/cm2, without any formation of lithium dendrites or signs of deterioration of the nano-SCE or the interface (Fig. 6G). Metallic lithium is known to form a protective interphase layer or SEI on its surface in BMP-TFSI–based ILEs (27). This reaction also happens at the lithium/nano-SCE interface; as discussed under Fig. 6A, the SEI might grow somewhat inside the pores, explaining the higher SEI resistance for nano-SCE than ILE (see above). Proof for an SEI layer was obtained from IR spectra (fig. S9). Similar to an SEI coating in classical LiB, which screens the graphite electrode from the liquid electrolyte avoiding further reaction, we believe that the SEI here also protects the ice water layer from further reaction from the metallic lithium anode. Impedance spectra before and after polarization of the Li/nano-SCE (x = 1.5) for 10 hours did not show any change in bulk electrolyte resistance. Long cycling performance measurements will be needed to exclude slow drying of the nano-SCE by lithium metal, but these results already show its potential for excellent cyclability of the SCE in lithium metal–based solid-state batteries. Nevertheless, artificial interphase coatings can be considered to improve the interface impedance altogether.
DISCUSSION
We have shown that ion conduction promotion at silica interfaces can be achieved through the introduction of a chemisorbed water layer on OH-terminated silica surfaces. The TFSI anions chemisorb on this water functional layer through hydrogen bonding with the symmetric O═S═O group. The water surface layer is immobile and hence also pins the adsorbed TFSI layer to the surface. The large BMP cations associate to the TFSI monolayer, thus introducing molecular ordering of the TFSI-BMP on the surface. We believe that the slow gelation in the aqueous environment and the slow drying help in the concerted formation of the functional water layer and the organized layer of organic ions on top of it. As the first TFSI anion layer shares part of its negative charge with the hydroxylated silica, the BMP cation layer on top will seek association with another TFSI anion, whereby multiple BMP can share their uncompensated charge with one TFSI (presumably three to one as in the ratio of IL to Li-TFSI in the ILE). As the Li-TFSI salt molecules have the closest approach, the Li+ ions will dissociate and be set free for fast diffusion along this interface layer. For enhanced conduction, these free Li+ species need at least one additional ionic liquid layer to move through. For this reason, the nano-SCE with the low x value of 0.5 showed no enhanced conductivity, as the ILE volume/silica surface area is sufficient for only one closed monolayer.
It was further shown that the solid-like surface water or ice layer is not electrochemically active. At this point, we cannot exclude that the ice water in direct contact with the electrode surface is not reacting. However, we showed that the out-diffusion of the surface water is slow and thus kinetically negligible for detection. We realize that water contamination, even if it is small, will always be a concern, and only long life cycle tests can provide a definite answer on whether the water is sufficiently bound. However, other functional surface layers that give similar or even larger surface promotion can now be developed. In this regard, the group of Li has already shown the potential of a glycidyloxypropyl layer as a functional group (18). Ice water is native to silica and therefore ideally suited to study the effect of surface functionalization on ion conduction promotion systematically, as was demonstrated successfully here. In addition, the mesophase layer and its dipole will depend on the oxide and on the adsorbed organic molecules and thus can be tuned by both. In the laboratory, we have already shown large differences in ion conduction promotion for different ionic liquids. Furthermore, the principle shown is generic toward ion conduction and can also thus be applied for different ion systems suitable, for example, for sodium, magnesium, calcium, or aluminum ion batteries. In conclusion, the nanocomposite electrolyte with interface conduction shown here is a concept rather than a single material, which can be further (nano)engineered to desired properties of ion conduction, transport number, electrochemical window, safety, and cost for future battery cell generations.
MATERIALS AND METHODS
Synthesis of nano-SCE
The nano-SCE was prepared using a sol-gel method. Lithium bis(trifluoromethylsulfonyl)imide Li-TFSI; Sigma-Aldrich; 99.95%), 0.5 ml of deionized H2O, 0.5 ml of TEOS (Sigma-Aldrich; 99.0%), 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide (BMP-TFSI; Sigma-Aldrich; 98.5%), and 1 ml of PGME were mixed in a glass vial. The molar ratio, x, between [BMP][TFSI] and TEOS in the mixture was varied between 0.25 and 2. The molar ratio of Li[TFSI] and [BMP][TFSI] was fixed at 0.33:1. The amounts of Li[TFSI] and [BMP][TFSI] were determined from these ratios. For example, when x = 1, the added [BMP][TFSI] and Li[TFSI] in the solution were 0.97 and 0.22 g, respectively. The mixtures were shaken for 1 min to form monophasic solutions. These solutions were then stored in the closed vials without stirring to form gels in a temperature- and humidity-controlled chamber (SH-641, ESPEC Corp.) with the temperature and RH% set at 25°C and 50%, respectively. Dependent on the x, the mixtures took, on average, 5 to 9 days to form a clear gel. After gelation, the vials with 2.4- to 7.4-ml gel were first dried at 40°C for four full days at slightly reduced pressure (80 kPa) and then moved into a vacuum oven for 72 hours at 25°C. As the remaining moisture was removed, the vacuum gradually decreased from an initial pressure around 50 Pa to a final constant pressure of 5 Pa after about 1 day. Because of the large amount of water and PGME that had to be removed, the resulting SCE pellets had shrunk down from 20% (x = 0.5) to ~50% (x = 2) of the original gel volume. The weight of the resulting gels was measured with a semimicro balance (SM 1245Di-C, VWR).
TGA was performed on a Q5000 IR (TA Instruments, New Castle, DE, USA) under nitrogen. During the measurement, samples were heated to 700°C at a heating rate of 2°C/min. FTIR spectrometry was done using a Bruker Vertex 70 in the wave number ranging from 4000 to 400 cm−1 in a transmission mode. He pycnometry was done using a Micromeritics AccuPyc II 1340.
Ion conductivity measurements in an argon-filled glove box
To measure the ionic conductivity, a small volume of SCE was taken from the mother vial inside an Ar-filled glove box (0.1-ppm H2O and 0.1-ppm O2). Approximately 23 μl of SCE was filled in a polytetrafluoroethylene (PTFE) ring with a 4.34-mm inner diameter and 1.57-mm height, forming a pellet. The pellet in the ring was then sandwiched between two stainless steel (SS) disks (0.2 mm thick; MTI). Impedance measurements were done using PGSTAT302 (Metrohm), with an AC amplitude of 5 mV in a frequency range from 1 MHz to 1 Hz. The ion conductivity (σi) was determined from the high-frequency intercept with the real axis in the Nyquist plots. After the conductivity measurement, the nano-SCE pellet was allowed to further dry out in the glove box. For temperature dependence measurement, the SS/SCE/SS stacks were sealed in a coin cell. After sealing, the conductivity remained constant for several days (see fig. S3). The temperature of the coin cell is controlled with a thermal jacket with a thermal bath using H2O/ethylene glycol as working medium. The cells were first cooled to about −15°C and then step-wisely heated to 60°C.
Conductivity measurements under controlled humidity
From each nano-SCE pellet, approximately 23 μl was brought into a ring (4.34-mm inner diameter and 1.57-mm height) for electrical measurements directly inside a N2-filled glove box with controlled humidity. The ring with the SCE was then sandwiched between two SS disks (0.2 mm thick; MTI). Impedance measurements were done using PGSTAT302 (Metrohm) with an AC amplitude of 5 mV and frequency ranging from 1 MHz to 1 Hz controlled through Nova software. The samples were kept at each RH% value for 48 hours before the conductivity was monitored until stabilization. The stabilized ionic conductivity for a given RH% value (σi) was determined from the high-frequency intercept with the real axis in the Nyquist plots.
Electrochemical characterization
All electrochemical measurements and the related sample preparation were done in an argon-filled glove box (PureLab, PL-HE-4GB-1800; <1-ppm O2 and H2O levels) dedicated for electrochemical characterizations.
Imaging of the SCE/silica matrix
The morphology of the pellet with and without Li[BMP][TFSI] ILE was checked with SEM using a Thermo Fisher Scientific Apreo tool at 1.5 to 2.0 kV whereby operating in a dual-detector imaging mode using the T1 and T2 detector in parallel for live-image adjustments, and the T2 detector was used for recording the shown SEM images; the sample was fixed on carbon conductive tape. TEM was done using a Tecnai operating at 300 kV.
ILE removal from pellet
The ILE was removed from the SCE pellet in two different ways. One option to obtain the porous silica was done by immersing the SCE in acetone for 12 hours to extract the Li[BMP][TFSI] ILE. This rinse was repeated three times. The other option was by soaking the SCE in ethanol. In this case, the ethanol was removed by using a liquid CO2 critical point dryer.
Two different tools were used for the supercritical drying, namely, the Automegasamdri-916B, Tousimis (method 1) and a custom-built tool by JASCO Corporation (method 2). When using the first tool, the drying sequence started with a decrease in temperature down to 8°C. Subsequently, CO2 was purged through the chamber, increasing the pressure to 5.5 MPa. In the following step, the CO2 was heated to 41°C, increasing the pressure to 10 MPa, and kept as such for 5 min. To conclude, in the bleeding step, the pressure was lowered over a time span of 10 min. When using the custom built tool, a similar sequence was followed. However, the timing and pressures differed significantly. After the purging step, the pressure was increased to 12 MPa at a temperature of 70°C and remained as such for 5 to 6 hours. Subsequently, the pressure was decreased in intervals from 12 to 7 MPa, 7 to 3 MPa, and 3 to 0 MPa over time spans of 10, 60, and 10 min, respectively.
Nitrogen physisorption
Nitrogen physisorption isotherms were measured at T = 77 K using a Micromeritics 3Flex surface characterization analyzer. The obtained porous silica was then outgassed for 8 hours at 100°C under a 0.1-mbar vacuum. The porous silica derived from supercritical drying was outgassed for 18 hours at 120°C under a 0.1-mbar vacuum. Afterward, nitrogen physisorption isotherms were measured at T = 77 K using a Micromeritics TriStar 3000 automated gas adsorption analyzer.
Pulsed-field gradient NMR
PFG-NMR measurements were performed using a JEOL JNM-ECX400. The stimulated echo pulse sequence was used for the diffusion measurements. The normalized echo signal attenuation, E, is described in the equation (38)
| (1) |
where g is the strength of the gradient pulse, δ is the duration of the gradient pulse, ∆ is the interval between the leading edges of the gradient pulses, γ is the magnetogyric ratio, and D is the self-diffusion coefficient of the molecules. The self-diffusion coefficients were estimated by fitting the echo signals that were obtained by changing ∆ with Eq. 1. 7Li was selected to determine the diffusion coefficient of the lithium ion. All the measurements were done at 30°C.
Raman scattering experiments
The Raman spectroscopy setup was a homemade system using an argon ion capable of being tuned to a 458-nm laser excitation light that was coupled into an inverted Olympus IX71 microscope, and back-scattered light was passed through a TriVista triple spectrometer setup (Princeton Instruments), which was used to disperse optical signals that are detected using a liquid nitrogen-cooled charge-coupled device camera. Given the high optical absorbance at these wavelengths, relatively low laser powers were used to avoid laser heating (<100 W·cm−2).
DFT calculations
DFT ground-state geometry optimization and analytical frequency calculations used the popular B3LYP hybrid functional and 6-311++G** basis set, with Grimme’s atom-pairwise dispersion correction (39) with the Becke-Johnson damping scheme (D3BJ), as implemented in ORCA 3.0.3 (40). Raman spectra were simulated using ORCA, and the visualization of molecular properties was achieved using the Avogadro software package (41) with the ORCA-supported update.
Three-electrode measurement on nano-SCE pellet
All electrochemical measurements and the related sample preparation were done in an argon-filled glove box (PureLab, PL-HE-4GB-1800; <1-ppm O2 and H2O levels) dedicated for electrochemical characterizations. The SCE pellet was placed on a Li ribbon (Sigma-Aldrich; 99.9%) supported on a copper plate as the counter electrode and two punched out Li disks (5-mm diameter) was placed on top of the SCE pellet for reference and working electrodes. The setup is shown in fig. S7. Gold pins were used for contacting the lithium reference and working electrodes. Cyclic voltammetry and impedance measurements were done using PGSTAT302 (Metrohm) controlled through Nova software. Cyclic voltammetry was done with a scan rate of 20 mV/s. Impedance measurements were done with an AC amplitude of 5 mV and frequency ranging from 1 MHz to 0.1 Hz.
Fabrication of half-cells with Li
A 40-nm anatase TiO2 thin-film electrode was deposited by atomic layer deposition (ALD) on a 300-mm silicon wafer with a 40-nm TiN underlayer also deposited by ALD. It is an excellent test electrode for the demonstration of Li-ion conductivity through the electrolytes, as TiO2 does not suffer from chemical degradation nor mechanical stress (no significant volume change) during cycling. To measure the Li/SCE/TiO2 cell, the ILE-SCEs were filled in a PTFE ring with a diameter of 4.3 mm and a thickness of 0.15 cm; then, the ring was sandwiched between a Li foil and the TiO2 film.
Nano-SCE/thin-film electrode half stacks, with the LMO electrode, were fabricated by synthesizing the nano-SCE film on the electrodes. A total of 150 μl of x = 1.5 solution, aged for 2 days, was drop-casted in a glass ring (diameter, 1.3 mm) mounted on the electrolyte films. The ring was then sealed with parafilm, and the solution was kept in such a sealed container to gel for 4 days. The formed gel/electrode stack as such was dried to form nano-SCE/electrode stacks. The thickness of the nano-SCE, determined using a micrometer, was 300 μm. Last, a lithium foil (1.75 mm thick, 99.9%; Sigma-Aldrich) was pressed on the nano-SCE/electrode stack as the anode. The 100-nm LiMn2O4 (LMO) thin-film electrode was deposited by radio frequency sputtering under Ar flow on a silicon wafer coated with 80-nm Pt (DC sputtering)/10-nm TiN (ALD) underlayers. This stack was annealed for 20 min at 800°C in oxygen atmosphere.
LiFePO4 (LFP) electrode films were prepared by blade coating. First, carbon black and LFP (2 to 3 μm) were added to an aqueous solution containing carboxymethylcellulose (CMC) to form a mixture that was afterward homogenized using a planetary mixer. Then, the homogenized product was mixed with deionized water and a fluorinated acrylic latex (JSR, TRD202A) in a vacuum mixer to form a slurry for electrode coating. The prepared slurry was cast on aluminum foils to deposit electrode films using a blade coater. These as-coated wet electrodes were immediately predried in an atmospheric oven with stagnant air at 70°C for 10 min and were further dried at 140°C for 4 hours in a vacuum oven. The dried electrode films consisted of 91 wt % LiFePO4, 3 wt % carbon black, 2 wt % CMC, and 4 wt % TRD202A. The film thickness is 30 μm (determined using a micrometer and scanning electron microscope).
Li4Ti5O12 (LTO) electrode films were made on copper foils in the same way. The composition of dried electrodes is 85 wt % Li4Ti5O12, 5 wt % carbon black, 5 wt % CMC, and 5 wt % fluorinated acrylic latex (TRD2001A). The film thickness is 40 μm.
The solution of SCE was drop-casted on the particle-based LFP and LTO electrode film. First, 100 μl of x = 1.5 solution, aged for 2 days, was drop-casted on an electrode film, with a diameter of 15 mm, placed in a coin cell (#2032, MTI). After the impregnated SCE was gelled, the film was dried at 25°C for 72 hours in a vacuum oven (<5 × 10−2 mbar) to make the nano-SCE and electrode stack. The nano-SCE thickness was 380 μm. Last, a lithium foil was pressed on the SCE/electrode stacks as the anode, and the coin cell was sealed. The electrochemical measurements were done using a Solartron 1470E potentiostat at room temperature.
Supplementary Material
Acknowledgments
Funding: Thermo Fisher Scientific is acknowledged for SEM tool support. K.G. acknowledges funding from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement no. 752716. J.A.S. acknowledges financial support from the Research Foundation-Flanders (FWO; grant no. 12Y7218N). Author contributions: P.M.V. wrote the paper, was the scientific lead of the project, codeveloped the model, and interpreted the results together with the researchers. X.C. came up with the original idea and the design of the water-based synthesis chemistry, made major contributions to the experimental work, discovered the enhanced conductivity, contributed to the development of the model, and assisted in writing the paper and making the figures. B.P., A.S., K.G., and J.A.S. made significant contribution to the experimental work, assisted in drafting of the paper and figures, and contributed to the analysis and interpretation of the results. M.Mu., H.Y., and T.H. contributed to the results with specialized techniques. M.R., M.T., H.A., and Y.K. were responsible for parts of the research project and assisted in the interpretation of the results and editing of the paper. M.S. gave scientific guidance and helped with interpretation of the results. M.Me. was the technical lead of the project, assisted in interpretation of the results, and assisted in drafting of the paper and figures. Competing interests: P.M.V., X.C., and M.Me. submitted a patent application titled “Solid nanocomposite electrolyte materials” with applicants IMEC VZW and KU Leuven, which was published on 1 November 2018 under publication number WO 2018 197073 A1. P.M.V., A.S., M.Mu., K.G., and M.Me. submitted patent applications (JP patent application filed on 12 June 2018, PCT patent application filed on 12 February 2019) titled “Solid nanocomposite electrolyte materials and method for fabrication” with applicants Panasonic Corporation and IMEC VZW. All authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaav3400/DC1
Table S1. Structural properties of silica matrix in the nano-SCE for increasing molar fraction of ionic liquid to silica (x value) determined from N2 adsorption/desorption or BET measurements and TEM observations.
Fig. S1. Normalized conductivity versus equivalent ILE thickness.
Fig. S2. The effect of ice-like and liquid-like adsorbed water on nano-SCE ion conductivity.
Fig. S3. Temperature dependence of the conductivity of the nano-SCE.
Fig. S4. TGA curves for nano-SCE.
Fig. S5. SEM images of the nano-SCE.
Fig. S6. N2 adsorption/desorption isotherm of the silica derived from nano-SCE.
Fig. S7. Solid-state three-electrode setup for cyclic voltammetry on nano-SCE pellets.
Fig. S8. IR spectra of nano-SCE (x = 1.5), ILE, and silica in the range of 4000 to 400 cm−1.
Fig. S9. IR spectra of 225-nm nano-SCE (x = 1.5) and Li/SCE thin-film stack.
REFERENCES AND NOTES
- 1.Chen N., Zhang H., Li L., Chen R., Guo S., Ionogel electrolytes for high-performance lithium batteries: A review. Adv. Energy Mater. 8, 1702675 (2018). [Google Scholar]
- 2.Yokota Y., Harada T., Fukui K., Direct observation of layered structures at ionic liquid/solid interfaces by using frequency-modulation atomic force microscopy. Chem. Commun. 46, 8627–8629 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Guyomard-Lack A., Said B., Dupré N., Galarneau A., Le Bideau J., Enhancement of lithium transport by controlling the mesoporosity of silica monoliths filled by ionic liquids. New J. Chem. 40, 4269–4276 (2016). [Google Scholar]
- 4.Vioux A., Viau L., Volland S., Le Bideau J., Use of ionic liquids in sol-gel; ionogels and applications. C. R. Chim. 13, 242–255 (2010). [Google Scholar]
- 5.Chen X., Vereecken P. M., Solid and solid-like composite electrolyte for lithium ion batteries: Engineering the ion conductivity at interfaces. Adv. Mater. Interfaces 6, 1800899 (2019). [Google Scholar]
- 6.Park H. S., Choi Y. S., Kim Y. J., Hong W. H., Song H., 1D and 3D ionic liquid-aluminum hydroxide hybrids prepared via an ionothermal process. Adv. Funct. Mater. 17, 2411–2418 (2007). [Google Scholar]
- 7.Verma Y. L., Tripathi A. K., Singh V. K., Balo L., Gupta H., Singh S. K., Singh R. K., Preparation and properties of titania based ionogels synthesized using ionic liquid 1-ethyl-3-methyl imidazolium thiocyanate. Mater. Sci. Eng. B. 220, 37–43 (2017). [Google Scholar]
- 8.Bellayer S., Viau L., Tebby Z., Toupance T., Le Bideau J., Vioux A., Immobilization of ionic liquids in translucent tin dioxide monoliths by sol–gel processing. Dalton Trans. 2009, 1307–1313 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Guyomard-Lack A., Abusleme J., Soudan P., Lestriez B., Guyomard D., Le Bideau J., Hybrid silica-polymer ionogel solid electrolyte with tunable properties. Adv. Energy Mater. 4, 1301570 (2014). [Google Scholar]
- 10.Wu F., Tan G., Chen R., Li L., Xiang J., Zheng Y., Novel solid-state Li/LiFePO4 battery configuration with a ternary nanocomposite electrolyte for practical applications. Adv. Mater. 23, 5081–5085 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Sharp K. G., A two-component, non-aqueous route to silica gel. J. Solgel Sci. Technol. 2, 35–41 (1994). [Google Scholar]
- 12.Zhou Y., Antonietti M., A series of highly ordered, super-microporous, lamellar silicas prepared by nanocasting with ionic liquids. Chem. Mater. 16, 544–550 (2004). [Google Scholar]
- 13.Zhou Y., Schattka J. H., Antonietti M., Room-temperature ionic liquids as template to monolithic mesoporous silica with wormlike pores via a sol–gel nanocasting technique. Nano Lett. 4, 477–481 (2004). [Google Scholar]
- 14.Zhang J., Ma Y., Shi F., Liu L., Deng Y., Room temperature ionic liquids as templates in the synthesis of mesoporous silica via a sol–gel method. Microporous Mesoporous Mater. 119, 97–103 (2009). [Google Scholar]
- 15.Wu C.-M., Lin S.-Y., Chen H.-L., Structure of a monolithic silica aerogel prepared from a short-chain ionic liquid. Microporous Mesoporous Mater. 156, 189–195 (2012). [Google Scholar]
- 16.Tan G., Wu F., Zhan C., Wang J., Mu D., Lu J., Amine K., Solid-state Li-Ion batteries using fast, stable, glassy nanocomposite electrolytes for good safety and long cycle-life. Nano Lett. 16, 1960–1968 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Le Bideau J., Ducros J.-B., Soudan P., Guyomard D., Solid-state electrode materials with ionic-liquid properties for energy storage: The lithium solid-state ionic-liquid concept. Adv. Funct. Mater. 21, 4073–4078 (2011). [Google Scholar]
- 18.Wu F., Chen N., Chen R., Wang L., Li L., Organically modified silica-supported ionogels electrolyte for high temperature lithium-ion batteries. Nano Energy 31, 9–18 (2017). [Google Scholar]
- 19.M. Debucquoy, M. Mees, A. Sagara, S. Remizov, X. Chen, K. Gandrud, B. Put, H. Arase, Y. Kaneko, P. M. Vereecken, Bioelectrochemistry and energy storage, presented at the Electrochemical Conference on Energy and the Environment, Glasgow, Scotland, 21 to 26 July 2019. [Google Scholar]
- 20.A. Sagara, K. Gandrud, X. Chen, M. Murata, M. Mees, Y. Kaneko, H. Arase, M. Shimada, P. M. Vereecken, presented at AABC, San Diego, CA, 2019. [Google Scholar]
- 21.Jande W., Neuere Forschungen über Diffusion und elektrische Leitfähigkeit fester Salze. Angew. Chem. 42, 462–467 (1929). [Google Scholar]
- 22.Yamada H., Bhattacharyya A. J., Maier J., Extremely high silver ionic conductivity in composites of silver halide (AgBr, AgI) and mesoporous alumina. Adv. Funct. Mater. 16, 525–530 (2006). [Google Scholar]
- 23.Manuel Stephan A., Nahm K. S., Review on composite polymer electrolytes for lithium batteries. Polymer 47, 5952–5964 (2006). [Google Scholar]
- 24.Le Bideau J., Viau L., Vioux A., Ionogels, ionic liquid based hybrid materials. Chem. Soc. Rev. 40, 907–925 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Long L., Wang S., Xiao M., Meng Y., Polymer electrolytes for lithium polymer batteries. J. Mater. Chem. A 4, 10038–10069 (2016). [Google Scholar]
- 26.Galiński M., Lewandowski A., Stępniak I., Ionic liquids as electrolytes. Electrochim. Acta 51, 5567–5580 (2006). [Google Scholar]
- 27.Lewandowski A., Świderska-Mocek A., Ionic liquids as electrolytes for Li-ion batteries—An overview of electrochemical studies. J. Power Sources 194, 601–609 (2009). [Google Scholar]
- 28.Li X., Li S., Zhang Z., Huang J., Yang L., Hirano S.-i., High-performance polymeric ionic liquid-silica hybrid ionogel electrolytes for lithium metal batteries. J. Mater. Chem. A 4, 13822–13829 (2016). [Google Scholar]
- 29.Shimano S., Zhou H., Honma I., Preparation of nanohybrid solid-state electrolytes with liquidlike mobilities by solidifying ionic liquids with silica particles. Chem. Mater. 19, 5216–5221 (2007). [Google Scholar]
- 30.Brinker C. J., Hydrolysis and condensation of silicates: Effects on structure. J. Non Cryst. Solids 100, 31–50 (1988). [Google Scholar]
- 31.Hayes R., Warr G. G., Atkin R., Structure and nanostructure in ionic liquids. Chem. Rev. 115, 6357–6426 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Asay D. B., Kim S. H., Evolution of the adsorbed water layer structure on silicon oxide at room temperature. J. Phys. Chem. B 109, 16760–16763 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Suo L., Borodin O., Gao T., Olguin M., Ho J., Fan X., Luo C., Wang C., Xu K., “Water-in-salt” electrolyte enables high-voltage aqueous lithium-ion chemistries. Science 350, 938–943 (2015). [DOI] [PubMed] [Google Scholar]
- 34.J. P. D. V. Buch, Thin Film Water on Insulator Surfaces (Springer, Berlin, 2003). [Google Scholar]
- 35.E. Papirer, Adsorption on Silica Surfaces (CRC Press, 2000). [Google Scholar]
- 36.Umebayashi Y., Mitsugi T., Fukuda S., Fujimori T., Fujii K., Kanzaki R., Takeuchi M., Ishiguro S.-I., Lithium ion solvation in room-temperature ionic liquids involving Bis(trifluoromethanesulfonyl) imide anion studied by Raman spectroscopy and DFT calculations. J. Phys. Chem. B 111, 13028–13032 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Put B., Vereecken P. M., Labyedh N., Sepulveda A., Huyghebaert C., Radu I. P., Stesmans A., High cycling stability and extreme rate performance in nanoscaled LiMn2O4 thin films. ACS Appl. Mater. Interfaces 7, 22413–22420 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Stejskal E. O., Tanner J. E., Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 42, 288–292 (1965). [Google Scholar]
- 39.Grimme S., Antony J., Ehrlich S., Krieg H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Neese F., The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 73–78 (2012). [Google Scholar]
- 41.Hanwell M. D., Curtis D. E., Lonie D. C., Vandermeerschd T., Zurek E., Hutchison G. R., Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Atsushi Unemoto I. H., Iwai Y., Mitani S., Baek S.-W., Ito S., Tomai T., Kawamura J., Electrical conductivity and dynamics of quasi-solidified lithium-ion conducting ionic liquid at oxide particle surfaces. Solid State Ion. 201, 11–20 (2011). [Google Scholar]
- 43.Unemoto A., Ogawa H., Ito S., Honma I., Electrical conductivity, self-diffusivity and electrolyte performance of a quasi-solid-state pseudo-ternary system, bis(trifluoromethanesulfonyl)amide-based room temperature ionic liquid-lithium bis(trifluoromethanesulfonyl)amide-fumed silica nanoparticle. J. Electrochem. Soc. 160, A138–A147 (2012). [Google Scholar]
- 44.N. Krawczyk, thesis, Justus-Liebig-Universität Gießen (2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaav3400/DC1
Table S1. Structural properties of silica matrix in the nano-SCE for increasing molar fraction of ionic liquid to silica (x value) determined from N2 adsorption/desorption or BET measurements and TEM observations.
Fig. S1. Normalized conductivity versus equivalent ILE thickness.
Fig. S2. The effect of ice-like and liquid-like adsorbed water on nano-SCE ion conductivity.
Fig. S3. Temperature dependence of the conductivity of the nano-SCE.
Fig. S4. TGA curves for nano-SCE.
Fig. S5. SEM images of the nano-SCE.
Fig. S6. N2 adsorption/desorption isotherm of the silica derived from nano-SCE.
Fig. S7. Solid-state three-electrode setup for cyclic voltammetry on nano-SCE pellets.
Fig. S8. IR spectra of nano-SCE (x = 1.5), ILE, and silica in the range of 4000 to 400 cm−1.
Fig. S9. IR spectra of 225-nm nano-SCE (x = 1.5) and Li/SCE thin-film stack.