

Abstract
Studying parallel evolution of similar traits in independent within-species lineages provides an opportunity to address evolutionary predictability of molecular changes underlying adaptation. In this study, we monitored biofilm forming capabilities, motility, and virulence phenotypes of a plethora of phylogenetically diverse clinical isolates of the opportunistic pathogen Pseudomonas aeruginosa. We also recorded biofilm-specific and planktonic transcriptional responses. We found that P. aeruginosa isolates could be stratified based on the production of distinct organismal traits. Three major biofilm phenotypes, which shared motility and virulence phenotypes, were produced repeatedly in several isolates, indicating that the phenotypes evolved via parallel or convergent evolution. Of note, while we found a restricted general response to the biofilm environment, the individual groups of biofilm phenotypes reproduced biofilm transcriptional profiles that included the expression of well-known biofilm features, such as surface adhesive structures and extracellular matrix components. Our results provide insights into distinct ways to make a biofilm and indicate that genetic adaptations can modulate multiple pathways for biofilm development that are followed by several independent clinical isolates. Uncovering core regulatory pathways that drive biofilm-associated growth and tolerance towards environmental stressors promises to give clues to host and environmental interactions and could provide useful targets for new clinical interventions.
Subject terms: Biofilms, Next-generation sequencing
Introduction
The ecological success of many opportunistic bacterial pathogens is based on their remarkable capability to adapt to, and survive in a broad range of diverse and challenging habitats.1,2 The ability to fine-tune the activity of a plethora of transcriptional regulators enables versatile lifestyles and flexible changes in bacterial behavior.3–5 This environment-driven transcriptional regulation increases the fitness of the individual bacterium.6–9 Additionally, specific genomic mutations help bacterial populations shape their behavior in distinct habitats.10–12
Pseudomonas aeruginosa is an environmental bacterium and opportunistic pathogen, which plays a dominant role as a causative agent of acute and chronic, often biofilm-associated, infections.13–18 Especially in cystic fibrosis (CF) patients, P. aeruginosa adopts a biofilm mode of growth, which contributes to high antibiotic tolerance and the recalcitrant nature of these infections.19–24 P. aeruginosa adapts to the hostile habitat of the chronically infected lung, producing a robust transcriptional response.25–27 Furthermore, ongoing inflammation generates very strong selective pressures, driving evolution and genotypic diversification of the infecting strain(s).28–33 Although the evolution of infecting bacteria present in advanced stages of CF infections has been extensively described,34–36 much less is known on how the observed genetic diversity is translated into better adapted phenotypes.33,37
In a previous study, we demonstrated that the variation width of the transcriptional profile (reaction norm) of one P. aeruginosa strain grown in different environmental conditions was much larger than the transcriptional variation among 151 genetically diverse clinical strains grown under standard laboratory conditions.38 However, various genotypes within a single species may also show differing reaction norms in different environments.39 Clinical P. aeruginosa isolates that have acquired a diverse array of adaptive mutations during an infection may produce more divergent transcriptional profiles when examined under infection-relevant conditions as opposed to standard laboratory conditions.
In this study, we recorded the transcriptional profiles of a diverse array of clinical P. aeruginosa isolates grown in planktonic and biofilm conditions. The latter is expected to more closely mimic conditions encountered during a chronic infection process. We demonstrate that while the transcriptional profiles elucidated in planktonic growth conditions were quite similar, more divergent transcriptional profiles were recorded when the isolates were grown in biofilm conditions. We also found that distinct groups of clinical isolates appear to follow parallel evolutionary paths and produce similar phenotypes. This convergence of organismal phenotypes was observed for multiple traits that included the formation of distinct biofilm structures characterized by specific transcriptional signatures, as well as virulence and motility phenotypes. Our results shed light on the enormous complexity that exists in the interrelationships between genetic and environmental factors in determining P. aeruginosa pathogenicity traits. As multiple genes interact with multiple environmental variables to produce a given phenotype, a holistic view of environmental and genetic effects on the bacterial phenotype will be critical to understand the intricate connection between genotypes and phenotypes.
Results
Biofilm phenotypes of clinical P. aeruginosa isolates fall into distinct clusters
We have previously shown that clinical P. aeruginosa isolates from various patients and infectious sites exhibit very different in vitro biofilm structures.40,41 In this study, we recorded the biofilm phenotypes of 414 clinical P. aeruginosa isolates by the use of confocal laser scanning microscopy (CLSM) following live/dead staining. Clinical isolates exhibited a large diversity of biofilm phenotypes; however, on the basis of visual inspection of microscopy images, we identified clusters of strains that share common structural characteristics (Fig. 1a). We found that 138 (=33%) of the tested clinical isolates produced biofilm structures that could be grouped into three major clusters according to their biofilm phenotype: cluster A contained 59 strains, cluster B 38 strains and cluster C 47 strains. Figure 1b depicts exemplary CLSM images of P. aeruginosa isolates from each of these three biofilm clusters. The biofilms of clinical isolates belonging to cluster A showed overall weak fluorescence and were unstructured, or poorly structured. In contrast, cluster B isolates produced tall, highly structured and filamentous biofilms. Cluster C comprised isolates characterized by small microcolony-like aggregates in flat structures. In addition, minor clusters with <15 strains per group could be identified. Those consisted of isolates forming honeycomb-like biofilm structures (cluster D) or flat, coarsely cross-linked biofilms (cluster E) (Fig. 1a). Clinical isolates, which exhibited individual variations in the features represented in the major clusters or which exhibited very unique structural biofilm features, were categorized as unassigned (other).
Fig. 1. Biofilms of clinical P. aeruginosa isolates fall into three major clusters independent of their phylogenetic background.

a Despite a large structural diversity in biofilms of 414 clinical isolates, groups of strains that share structural characteristics were identified by visual inspection of biofilm microscopy images. Biofilms were grown for 48 h in a microtiter plate-based in vitro biofilm assay; images were acquired using confocal laser scanning microscopy (CLSM) following live/dead staining. Living cells are displayed in green (Syto9); dead cells in red (propidum iodide: PI). 3D reconstructions were generated with the Imaris Software. The scale bar represents 50 µm. b Representative biofilm images of selected clinical isolates show exemplarily the structural characteristics of the three major biofilm clusters, of which each contains 59 strains (cluster A), 38 strains (cluster B), and 47 strains (cluster C), respectively. c The phylogenetic relationship of 33 representative clinical isolates is displayed in a phylogenetic tree based on 3524 genes that are present in the DNA sequences of 414 clinical isolates and 5 reference strains. The color code of the strain names represents the affiliation to a certain biofilm cluster: Red—cluster A; green—cluster B; blue—cluster C. Reference strains are displayed in black. The proportion of PAO1-like strains is highlighted in light gray; PA14-like strains are highlighted in dark gray. d Crystal violet quantification was performed for 33 representative clinical strains to assess air–liquid biofilm formation on a PVC surface after 24 h. Each datapoint represents one clinical isolate. Statistical significance was calculated using Tukey’s HSD (honest significant difference) following analysis of variance (ANOVA) and is displayed as *p < 0.05.
Of note, the distinct phenotypes of the three major biofilm clusters were independent of the genetic background of the clinical isolates (Fig. 1c, Supplementary Fig. 1). Furthermore, the in vitro biofilm structures, as recorded by confocal microscopy, could not be deduced from crystal violet biofilm test assays. We observed large variations in the quantified surface-bound biomass within 10–12 isolates of each cluster (Fig. 1d). No significant differences between cluster A and B isolates were documented, while cluster C isolates exhibited an overall increased surface-bound biomass.
Environment-dependent divergence of transcriptional profiles of clinical P. aeruginosa isolates
We next recorded the transcriptional profiles of a subgroup of 77 clinical P. aeruginosa isolates under planktonic (in a previous study42) and biofilm growth conditions (this study) (Fig. 2a). The selected 77 strains (Supplementary Table 1) represent the phylogenetic diversity of our entire collection of 414 strains and included strains of the three major biofilm clusters as well as strains exhibiting various other biofilm characteristics. As previously described,38 the transcriptional profiles of the majority of the clinical isolates were similar when grown under planktonic conditions. However, transcriptional profiles exhibited significantly higher diversity when biofilm-associated conditions were recorded. Measurements of the pairwise Euclidean distances revealed higher values among the biofilm samples than among the planktonic samples (Fig. 2b). We also found a higher pairwise Pearson’s distance among the biofilm-associated transcriptional profiles (Fig. 2c). Our results clearly show that the genomic make-up of the clinical isolates has a higher impact on the transcriptional profile under biofilm growth conditions than in planktonic cultures.
Fig. 2. Transcriptional profiles recorded for biofilm-associated conditions exhibit a higher diversity than those recorded for planktonic conditions.

a The multidimensional scaling plot (MDS) of transcriptional profiles of 77 clinical strains shows a higher divergence in biofilm conditions (BF; filled circles) as compared to planktonic conditions (PL; triangles). b Pairwise measurements of Euclidian distances and c the Pearson’s distance (Pearson correlation coefficient subtracted from 1 to describe the variance) between the samples of the two culture conditions (biofilm and planktonic) are depicted. Significance of the Wilcoxon’s rank sum-test is displayed as ****p < 0.0001. Boxplot elements are: center line—median; box limits—upper and lower quartiles; whiskers—1.5× interquartile range; points—outliers. d Clinical isolates show a broad range in the number of differentially expressed genes (biofilm versus planktonic). The number of core biofilm transcriptome genes present in each strain is displayed in dark red (upregulated in biofilms) and dark blue (downregulated in biofilms).
The core biofilm transcriptome
To analyze whether the individual clinical strains share a common biofilm-specific transcriptional signature, we identified all genes that were differentially expressed between biofilm and planktonic growth conditions in each of the 77 clinical isolates (Fig. 2d). The number of differentially expressed genes (DEGs) between the two conditions ranged from 246 to 2465 per isolate (Fig. 2d). More than half of the identified genes (51%) were differentially regulated in 1–6 of the 77 clinical isolates (Supplementary Fig. 2).
We also recorded the genes that were differentially regulated between the biofilm and planktonic growth states if the respective transcriptional profiles of the individual 77 clinical isolates were considered as replicates. This core biofilm transcriptome consisted of overall 143 genes (Table 1, Supplementary Fig. 3). Although not all of these 143 genes were also differentially regulated at least two-fold in all individual isolates, more than 90% of the genes (130 of the 143 genes) were regulated in a minimum of 45 out of the 77 isolates (Supplementary Figs 2 and 3).
Table 1.
The core biofilm transcriptome.
| Upregulated genes | Downregulated genes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus tag | Gene | Log 2 FC | Locus tag | Gene | Log 2 FC | Locus tag | Gene | Log 2 FC | |||
| PA14 | PAO1 | PA14 | PAO1 | PA14 | PAO1 | ||||||
| PA14_06090 | PA0466 | 2.9 | PA14_38850 | PA1983 | exaB | 6.8 | PA14_04650 | PA0355 | pfpI | −3.2 | |
| PA14_06180 | PA0472 | fiuI | 2.5 | PA14_38860 | PA1982 | exaA | 5.8 | PA14_06650 | PA0509 | nirN | −3.0 |
| PA14_07355 | PA0565 | 3.6 | PA14_38880 | PA1981 | 5.7 | PA14_06660 | PA0510 | nirE | −3.3 | ||
| PA14_09980 | PA4167 | dkgB | 2.7 | PA14_38900 | PA1980 | exaE | 2.9 | PA14_06670 | PA0511 | nirJ | −3.5 |
| PA14_10170 | PA4159 | fepB | 3.2 | PA14_38910 | PA1979 | exaD | 3.0 | PA14_06680 | PA0512 | nirH | −3.2 |
| PA14_10200 | PA4156 | 2.8 | PA14_38920 | — | 4.1 | PA14_06690 | PA0513 | nirG | −2.9 | ||
| PA14_10240 | PA4152 | acoC | 2.8 | PA14_38970 | PA1976 | 2.9 | PA14_06700 | PA0514 | nirL | −3.3 | |
| PA14_10250 | PA4151 | acoB | 3.0 | PA14_38990 | PA1975 | 3.1 | PA14_06710 | PA0515 | nirD | −3.3 | |
| PA14_10260 | PA4150 | acoA | 3.1 | PA14_39000 | PA1974 | 4.9 | PA14_06720 | PA0516 | nirF | −3.1 | |
| PA14_10270 | PA4149 | acoX | 3.5 | PA14_39050 | PA1971 | braZ | 2.5 | PA14_06730 | PA0517 | nirC | −3.9 |
| PA14_10280 | PA4148 | 3.6 | PA14_39260 | PA1952 | 2.7 | PA14_06740 | PA0518 | nirM | −4.8 | ||
| PA14_10400 | — | 3.2 | PA14_39590 | PA1927 | metE | 4.3 | PA14_06750 | PA0519 | nirS | −4.9 | |
| PA14_10410 | — | 2.9 | PA14_39720 | PA1918 | 2.6 | PA14_06770 | PA0520 | nirQ | −3.3 | ||
| PA14_11070 | PA4085 | cupB2 | 2.7 | PA14_39750 | PA1916 | 2.7 | PA14_06810 | PA0523 | norC | −5.9 | |
| PA14_19270 | PA3467 | 2.7 | PA14_39860 | PA1907 | 2.5 | PA14_06830 | PA0524 | norB | −4.5 | ||
| PA14_20010 | PA3408 | hasR | 3.3 | PA14_39980 | PA1898 | qscR | 2.6 | PA14_06840 | PA0525 | norD | −4.4 |
| PA14_20020 | PA3407 | hasAp | 5.1 | PA14_40040 | PA1893 | 2.7 | PA14_06860 | PA0526 | −3.9 | ||
| PA14_20030 | PA3406 | hasD | 3.1 | PA14_40050 | PA1892 | 2.7 | PA14_09610 | PA4200 | −2.5 | ||
| PA14_20040 | PA3405 | hasE | 3.0 | PA14_40060 | PA1891 | 3.0 | PA14_09660 | PA4198 | −2.6 | ||
| PA14_20050 | PA3404 | hasF | 2.7 | PA14_40270 | PA1873 | 2.8 | PA14_16640 | PA3691 | −2.9 | ||
| PA14_21530 | PA3287 | 3.1 | PA14_40520 | PA1855 | 2.7 | PA14_20180 | PA3394 | nosF | −2.8 | ||
| PA14_22320 | PA3237 | 4.0 | PA14_41510 | PA1783 | nasA | 2.8 | PA14_20190 | PA3393 | nosD | −2.8 | |
| PA14_27370 | PA2840 | deaD | 2.4 | PA14_41520 | PA1782 | ppkB | 2.6 | PA14_20200 | PA3392 | nosZ | −4.8 |
| PA14_28100 | PA2783 | 2.6 | PA14_41530 | PA1781 | nirB | 2.7 | PA14_20230 | PA3391 | nosR | −4.3 | |
| PA14_28360 | — | 2.7 | PA14_44190 | PA1569 | 2.8 | PA14_29640 | PA2664 | fhp | −3.8 | ||
| PA14_28620 | PA2746 | 3.2 | PA14_44520 | PA1541 | ydgF | 3.3 | PA14_29660 | PA2662 | −2.6 | ||
| PA14_29350 | PA2688 | pfeA | 2.6 | PA14_44530 | PA1540 | ygdE | 2.8 | PA14_40850 | PA1831 | gpmA | −2.4 |
| PA14_29480 | PA2678 | wzm | 2.7 | PA14_46850 | PA1347 | 2.8 | PA14_50520 | PA1074 | braC | −2.5 | |
| PA14_32080 | PA2518 | xylX | 3.3 | PA14_47380 | PA1302 | hxuC | 2.6 | PA14_52660 | PA0899 | aruB | −2.3 |
| PA14_32100 | PA2517 | xylY | 2.9 | PA14_47390 | PA1301 | 3.0 | PA14_52670 | PA0898 | astD | −2.2 | |
| PA14_33250 | PA2427 | 4.4 | PA14_47400 | PA1300 | 2.8 | PA14_52690 | PA0897 | aruG | −3.0 | ||
| PA14_33260 | PA2426 | pvdS | 3.0 | PA14_49690 | PA1137 | 3.8 | PA14_52720 | PA0895 | argD | −2.4 | |
| PA14_33270 | PA2425 | pvdG | 4.0 | PA14_50050 | PA1108 | 2.6 | PA14_56370 | PA4336 | −2.9 | ||
| PA14_33280 | PA2424 | pvdL | 3.8 | PA14_51460 | PA0993 | cupC2 | 3.2 | PA14_56780 | PA4366 | sodB | −2.6 |
| PA14_33500 | PA2413 | pvdH | 3.0 | PA14_52960 | PA0874 | 2.7 | PA14_57020 | PA4386 | groES | −2.7 | |
| PA14_33510 | PA2412 | 3.5 | PA14_53300 | PA0848 | 3.4 | PA14_60570 | PA4578 | −2.9 | |||
| PA14_33520 | PA2411 | 3.7 | PA14_55500 | PA0680 | hxcV | 2.6 | PA14_61650 | PA4661 | pagL | −2.7 | |
| PA14_33600 | PA2403 | fpvG | 3.1 | PA14_56770 | PA4365 | 2.5 | PA14_64480 | PA4876 | osmE | −2.7 | |
| PA14_33610 | PA2400 | 2.9 | PA14_57990 | PA4467 | 3.1 | PA14_64520 | PA4880 | −2.9 | |||
| PA14_33690 | PA2397 | pvdE | 3.3 | PA14_58000 | PA4468 | sodM | 3.1 | PA14_69770 | PA5285 | −2.6 | |
| PA14_33710 | PA2395 | pvdO | 3.0 | PA14_58010 | PA4469 | 3.5 | |||||
| PA14_33720 | PA2394 | pvdN | 3.2 | PA14_58030 | PA4470 | fumC | 3.3 | ||||
| PA14_33730 | PA2393 | 3.6 | PA14_58040 | PA4471 | 3.9 | ||||||
| PA14_33810 | PA2386 | pvdA | 4.5 | PA14_60480 | PA4570 | 4.1 | |||||
| PA14_33820 | PA2385 | pvdQ | 3.3 | PA14_68430 | PA5180 | fdhD | 2.9 | ||||
| PA14_35430 | PA2254 | pvcA | 2.7 | PA14_68440 | PA5181 | 2.5 | |||||
| PA14_35460 | PA2252 | 2.9 | PA2958.1 | PA2958.1 | rgsA | 3.7 | |||||
| PA14_35980 | — | 2.9 | PA3964-PA3965 | PA3964-PA3965 | P24 | 3.6 | |||||
| PA14_37380 | PA2097 | 2.4 | PA4704.1 | PA4704.1 | prrF1 | 3.3 | |||||
| PA14_38210 | PA2034 | 2.6 | PA4704.2 | PA4704.2 | prrF2 | 2.8 | |||||
| PA14_38220 | PA2033 | 2.6 | PA4704-PA4705 | PA4704-PA4705 | prrH | 3.1 | |||||
| PA14_38310 | PA2027 | 3.3 | |||||||||
The analysis of differentially expressed genes between transcriptional profiles of 77 clinical P. aeruginosa isolates recorded under static biofilm (BF) and planktonic (PL) growth conditions resulted in the identification of 143 genes. 103 genes were significantly upregulated; 40 genes were significantly downregulated (threshold: log 2 fold change [log 2 FC] ≤ −2 and ≥2 with a false discovery rate of FDR < 0.05). Additional information about the identified genes (e.g., FDR values, gene products, and pseudoCAP) are available in Supplementary Data 1
The core biofilm transcriptome consisted of 103 genes that were upregulated in biofilm growth conditions and 40 genes that were downregulated (Table 1). Among the 103 genes that were commonly upregulated in biofilms, we identified genes required for the biosynthesis of pyoverdine (pvdAEGHLNOPQR, pvdS),43 the small regulatory RNAs prrF1, prrF2, and prrH, and genes of the heme assimilation system (hasAp, hasD, hasE, hasF, hasR). Furthermore, genes encoding for a superoxide dismutase (sodM) and a fumarate hydratase (fumC) were upregulated, implying that cells within biofilms face iron-limiting conditions and oxidative stress. Moreover, genes involved in the central carbon catabolism and energy metabolism were identified among the upregulated genes in the core biofilm transcriptome. We found acoABCX, involved in the conversion of 2,3-butanediol into aldehydes,44 to be highly expressed under biofilm conditions as well as the ethanol oxidation genes (exaABDE), which catalyze the conversion of ethanol via acetaldehyde to acetate and subsequently to acetyl-CoA before its introduction into the glyoxylate cycle for further metabolism.44
Among the downregulated genes were genes involved in the denitrification pathway including genes of the nir, nor, and nos operons. Furthermore, genes of the arginine succinyltransferase (AST) pathway (aruBG encoding the AST; astD and argD) required for aerobic arginine catabolism were commonly downregulated.
Convergence of biofilm transcriptional profiles
We wondered whether the clinical strains that produced similar biofilm structures in our in vitro system (clusters A–C, Fig. 1b) also produce similar biofilm-associated transcriptional profiles. As demonstrated for the 77 clinical isolates (Fig. 2a), the transcriptional profiles of all 33 clinical isolates were very similar when grown under planktonic conditions, irrespective of their biofilm structure (Fig. 3). However, the biofilm-specific transcriptional profiles of the clinical isolates appeared to cluster according to their affiliation to a specific biofilm phenotype (Fig. 3, Supplementary Fig. 6).
Fig. 3. Biofilm clusters exhibit distinct transcriptional signatures in biofilm growth conditions.
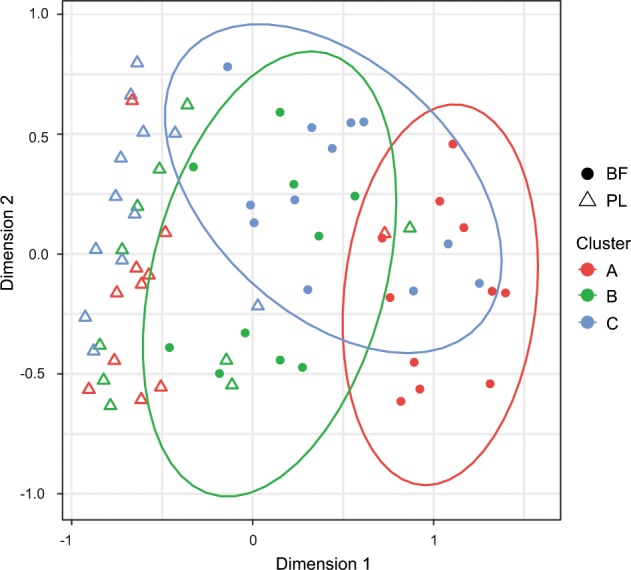
Biofilm (BF) but not planktonic (PL) transcriptional profiles show a grouping according to the biofilm structure. Ninety-five percent confidence intervals are displayed by ellipses. Each datapoint represents the transcriptional profile of one clinical isolate in a certain growth condition (BF: filled circles; PL: triangles). The color code represents the affiliation to a certain biofilm cluster: red—cluster A; green—cluster B; blue—cluster C.
Genetic background rather than the maturation status of the biofilms impacts the biofilm transcriptional profile
The analysis of dynamic transcriptional profiles of a selection of six isolates (two from each of the three biofilm clusters) over time (12, 24, 36, and 48 h of biofilm growth) revealed robust gene expression patterns for each strain and biofilm phenotype. Clustering of the transcriptional profiles was impacted by the strain background and its affiliation to a specific biofilm cluster, rather than by the time at which the transcriptional profiles were recorded (Supplementary Fig. 4). Thus, the observed convergence of transcriptional biofilm signatures of the individual isolates within the clusters is shaped by the genomic make-up of the clinical isolates rather than by the maturation status of the biofilm.
Cluster-specific biofilm transcriptional signatures
Figure 4 and Supplementary Fig. 5 depict the number of differentially expressed genes (DEGs) under biofilm versus planktonic growth conditions (Fig. 4a and Supplementary Fig. 5a) and the enrichment of functional gene categories (Fig. 4b and Supplementary Fig. 5b) of regulated genes among the structural groups. In line with the finding that iron acquisition systems are a central part of the upregulated core biofilm transcriptome genes, all three biofilm clusters showed higher expression of genes involved in pyoverdine biosynthesis; an enhanced production of pyoverdine was measured in all isolates under biofilm growth conditions (Fig. 4c). Our results imply that the production of pyoverdine is a general biofilm trait that is independent of the biofilm structure (Fig. 4d). Additionally, we identified genes that were not part of the biofilm core, but are upregulated in all three biofilm clusters (Supplementary Fig. 6a), such as genes encoding the multicomponent anthranilate dioxygenase (antABC) (Fig. 4a).
Fig. 4. Upregulated genes in biofilm growth in comparison to planktonic growth.

a The Venn diagram depicts commonly upregulated genes (70 genes) among all three biofilm clusters as well as cluster-specific regulated genes (A: 388; B: 33; C: 85 genes). b The GO term enrichment analysis of upregulated genes shows biological functions that are exclusively regulated in a certain biofilm cluster or shared by two or all three structural groups. The red color gradient represents the value of the enrichment factor. c A significant higher pyoverdine production was observed in biofilms (48 h) compared to planktonic cultures (24 h), as shown exemplarily for 33 clinical isolates. d Pyoverdine production in biofilm cultures was enhanced in all three biofilm clusters, independent of structural characteristics. Statistical significance was calculated with the Student’s t test and is displayed as *p < 0.05. Each datapoint represents one individual clinical isolate. Error bars represent the standard deviation.
We also identified cluster-specific biofilm gene expression patterns, indicating that there is a convergent evolution of distinct P. aeruginosa biofilm structures and transcriptional signatures. In the 11 cluster A isolates, which produced flat unstructured biofilms, 577 genes showed a specific expression profile in biofilms as compared to planktonic cultures (189 genes were downregulated, 388 genes were upregulated in biofilms) (Supplementary Data 1). We found a cluster A biofilm-specific upregulation of genes required for the production of extracellular polymeric substances. The functional analysis showed a significant enrichment for genes involved in the alginic acid biosynthetic process (algAGJKLX) (Fig. 4b). Further alg genes (algE, algI, alg44, and alg8) were upregulated in cluster A isolates; however, they were also found to be upregulated in cluster C isolates, which appear to share phenotypes consistent with both clusters A and B. Genes encoding the exopolysaccharide Pel (pelADEFG) and cup fimbrial genes (cupA2 and cupB2) encoding adhesin-like proteins that are both involved in biofilm formation45–47 were found to be specifically upregulated in cluster A biofilms. Further, cup genes (cupA3-5, cupB2-4, cupC2) were upregulated in both cluster A and cluster C biofilms. Of note, under planktonic growth conditions, the expression of alginate, pel and cup fimbrial genes exhibited no significant difference between the isolates of the three different clusters (Supplementary Data 2).
Genes encoding the type IV pilus genes (e.g., pilAOP) showed significantly lower expression in cluster A during biofilm growth in comparison to planktonic growth. Furthermore, we found genes encoding components of the type III secretion system (T3SS; e.g., pscD, exsC, pcrH, popB) (Supplementary Fig. 5b). Interestingly, cluster A showed a specific downregulation of transcriptional regulators important for other virulence- and biofilm-related functions. For instance, the global regulator mvaT involved in controlling the expression of virulence factors, such as pyocyanin, swarming motility and protease,48 and biofilm formation,49 was lower expressed in cluster A biofilms. Moreover, the alternative sigma factor sigX, involved in the regulation of the T3SS, swarming motility, biofilm formation, and c-di-GMP signaling50 was expressed at lower levels. The master regulator fleQ, which not only regulates flagellar gene expression51,52 but also Pel biosynthesis in a c-di-GMP-dependent manner,53 was under-expressed in biofilm conditions in comparison to planktonic conditions.
Cluster B isolates, which produced tall, highly structured biofilms had a smaller number of regulated genes. Overall, 49 genes were specifically differentially regulated (16 downregulated and 33 upregulated) under biofilm growth conditions in cluster B. Genes that were exclusively upregulated in cluster B include those involved in pyochelin biosynthesis (pchBCHI). Further, pyochelin genes (pchADEFG) were upregulated in biofilms of clusters B and C. Moreover, quorum sensing-related genes (rhlA, rhlB, rhlC, and lasA) as part of global regulatory networks involved in environmental adaptation54 were upregulated in biofilms of clusters B and C.
Biofilms of clusters A and C had higher expression levels of hasD, hasE, and hasR, all encoding for components of the heme uptake system,55 in comparison to those found in cluster B. In both clusters B and C, phz genes (phzA1-G1, phzA2-G2, phzM, and phzS), required for the biosynthesis of pyocyanin,56 were upregulated during biofilm growth (Fig. 4b). Interestingly, analysis of transcriptional profiles under planktonic growth conditions revealed that cluster A already exhibited higher phz gene expression levels compared to clusters B and C (Supplementary Table 2).
106 genes were found to be exclusively regulated in cluster C biofilms (21 genes were downregulated, 85 genes were upregulated in biofilms). Phage-related genes (gpFI, gpFII, gpI, gpW) were specifically downregulated, while the pqqCDE operon required for the biosynthesis of pyrroloquinoline quinone, a redox cofactor for ethanol oxidation,57 was upregulated (Fig. 4b). Beaudoin et al.58 found that both exaA and pqqC are upregulated in P. aeruginosa PA14 biofilms and are linked to biofilm-specific tolerance. Furthermore, we observed a specific upregulation of pqsABCDE encoding 4-quinolone signal molecules in cluster C biofilms. Along this line, the expression of phnAB, encoding an anthranilate synthase, which is an important precursor of 4-quinolone production, was also found to be upregulated in cluster C isolates.
Notably, T3SS genes were found to have the highest expression in cluster C biofilms, whereas cluster A biofilms showed intermediate and cluster B biofilms the lowest expression levels. Under planktonic growth conditions, no significant differences in the expression of T3SS genes could be observed between the three clusters (Supplementary Table 3).
Biofilm clustering correlates with other virulence-related P. aeruginosa phenotypes
We screened the 33 aforementioned clinical isolates, as part of the three distinct biofilm clusters, for other phenotypes such as colony morphology, motility, and pathogenicity (Supplementary Table 4). We found that cluster A isolates produced colonies on blood agar plates with a strong hemolytic zone, while hemolysis was not as pronounced in colonies of P. aeruginosa isolates of the other two clusters (images available in the database bactome; https://bactome.helmholtz-hzi.de). Figure 5a depicts the in vivo virulence phenotype of cluster A, B, and C isolates as determined in the Galleria mellonella assay.59 In stark contrast to isolates of cluster A and C, isolates of cluster B were almost avirulent. An in vitro infection assay using human epithelial A549 cells showed a significant reduced cytotoxicity of cluster B isolates (Fig. 5b). Moreover, while cluster A isolates exhibited the highest motility, cluster B isolates showed the lowest motility in swimming, twitching, and swarming assays (Fig. 5d–f). Remarkably, the avirulent cluster B isolates, with an overall reduced motility, produced significantly higher levels of the intracellular second messenger c-di-GMP (Fig. 5c). Moreover, cluster A isolates secreted significantly higher levels of pyocyanin than the other two clusters (Fig. 5g), which is in agreement with the higher expression of phz genes (Supplementary Table 2). Cluster A strains also showed an enhanced protease secretion, as was found for proteolytic activity in general (Fig. 5i), and particularly elastase secretion (Fig. 5h).
Fig. 5. Biofilm cluster-specific isolates differ in their production of various virulence factors.

a In vivo virulence using the Galleria mellonella model, b in vitro cytotoxicity on A549 epithelial cells, c intracellular second messenger c-di-GMP levels, d swimming motility, e twitching motility, f swarming motility, g pyocyanin production, h elastase secretion, and i protease production of selected isolates belonging to the three different biofilm clusters are depicted. Each dot represents one individual clinical isolate. Levels of statistical significance were calculated using Tukey’s HSD (honest significant difference) following analysis of variance (ANOVA) and are displayed as ****p < 0.0001, **p < 0.01, or *p < 0.05.
Discussion
The enormous success of environmental bacteria as human opportunistic pathogens is founded on their capability to efficiently adapt to the changing and challenging conditions within the human host.1 In general, there are two mechanisms that allow adaptation of a bacterial population to a changing environment: genetic diversification, which increases the ability of populations to adapt to new environmental conditions, and environment-driven transcriptional responses, which increase the fitness of the individual bacterium. It has become clear from many studies focusing on the various aspects of either of these two mechanisms that P. aeruginosa uses both strategies to adapt to host-specific niches.25,26,32,34,35 The complex interrelationships between genetic and environmental factors in determining bacterial traits, however, are largely unexplored and much remains to be learned about how genetic diversity is translated into differentially adapted phenotypes.
Similar to comparative genomics, comparative transcriptomics has the potential to uncover shared and unique features among multiple strains within the same species or among isolates that produce a specific organismal trait despite variation in environmental conditions.60 The detection of common responses of bacterial strains to a biofilm growth environment has been a focus of previous research.61–64 Uncovering core regulatory pathways that drive biofilm-associated tolerance towards environmental stressors promise to give clues to host and environmental interactions and could provide useful targets for new clinical interventions.
Previous studies described that different environmental conditions promote different biofilm-specific transcriptional profiles, and only a small number of common biofilm-specific transcripts could be detected.65,66 We found that a diverse array of clinical isolates shares a restricted core biofilm transcriptional profile, even if the isolates were cultivated under the same biofilm-promoting conditions. Interestingly, expression of previously well-described biofilm-associated genes, such as those involved in surface attachment and the production of biofilm matrix components,67–69 were not among the strict core biofilm genes. Instead, the core biofilm transcriptional profile was dominated by the expression of genes involved in stress responses and adaptation to oxygen and iron-limiting growth conditions.
Closer inspection revealed that clinical P. aeruginosa isolates can be stratified based on the production of specific biofilm structures in vitro. The individual biofilm phenotypes evolved via parallel or convergent evolution and were produced repeatedly in several clinical isolates. Interestingly, the individual groups of biofilm phenotypes produced biofilm transcriptional profiles that included the expression of surface adhesive structures and extracellular matrix components, indicating that there is more than one way to make a biofilm70,71 even under identical environmental conditions.
The independent evolution of specific biofilm phenotypic traits in non-related clinical isolates indicates adaptation to similar environments. Of note, we found that the biofilm-specific grouping of the clinical isolates was also reflected in convergent motility and virulence phenotypes. Our data thus show that the groups of clinical isolates have undergone different paths of convergent evolution to produce a complex phenotype that goes beyond the production of a distinct biofilm phenotype. In this context, our observation that the variation of the transcriptional profiles of our clinical isolates was larger under biofilm growth conditions than under planktonic conditions is interesting. The conserved lower variation width of the transcriptional profile of the evolved strains under planktonic growth conditions could promote maintenance of genetic diversity within the population, as the pressure to change decreases.
Methods
Strains, media, and growth conditions
To determine biofilm structures in a high content screen, we used our strain collection of 414 well-characterized clinical P. aeruginosa isolates, for which in vivo virulence data in the Galleria mellonella infection model59 and images of the colony morphology (including hemolysis on Columbia agar)72 were available. Furthermore, transcriptional profiles under planktonic conditions have been recorded for all 414 strains in the frame of a previous study.42 In this study, we performed transcriptional profiling under biofilm growth conditions for a subset of 77 strains (Supplementary Table 1). All clinical strains used in this study were collected in clinical microbiology laboratories, in private practice laboratories, or were provided by strain collection curators. Strains originate from sampling sites across Germany, Spain, Hungary, and Romania. Unless otherwise stated, all experiments were performed at 37 °C in standard lysogeny broth (LB) in order to accommodate growth and to enable comparable growth rates for clinical strains originating from diverse infection sites. Planktonic cultures were incubated in an orbital shaker (180 r.p.m.); biofilms were cultivated under static conditions in a humid atmosphere.
Biofilm phenotype assay
A collection of 414 clinical P. aeruginosa isolates, which have previously been sampled from several clinical microbiology laboratories across Europe,42,59,72 was screened for biofilm phenotypes through the use of a high content static microtiter plate assay combined with automated CLSM.73 In brief, bacteria were grown overnight in LB at 37 °C and 100 µl of bacterial suspension with an adjusted OD600 of 0.002 were added to the wells of a sterile half-area 96-well µClear microtiter plate (Greiner Bio-One, Austria). The microtiter plate was sealed with an air-permeable BREATHseal cover foil (Greiner Bio-One) and incubated for 48 h in humid atmosphere at 37 °C. After 24 h, bacteria were stained by carefully adding 60 µl of a solution containing the fluorescent dyes Syto9 and propidium iodide (final concentrations of 2.1 and 12.5 µM, respectively) from the LIVE/DEAD® BacLight™ Bacterial Viability Kit (Molecular Probes, Life Technologies, CA, USA). Z-stacks of 48-h-old biofilms with a total height of 60 µm (20 focal planes; z-step size 3 µm) in the center of each well were acquired by using an automated confocal laser scanning microscope (SP8 System, Leica, Germany), including the matrix screener tool and equipped with an HC PL APO ×40/1.10 W motCORR CS2 water immersion objective. Imaris 7.6 (Bitplane, UK) was used for 3D reconstructions of biofilm structures. The biofilm phenotype was recorded for all clinical strains in four replicates. Major biofilm clusters with distinct structural characteristics were identified on the basis of microscopy images and clinical isolates were manually categorized by visual inspection. Additional minor biofilm clusters were not further described. For an accurate grouping, only strains that clearly reflected the respective structural properties of a certain biofilm cluster were considered for further investigation. Strains that did not fit the described selection criteria remained unassigned.
Crystal violet biofilm formation assay
Selected clinical isolates were subjected to a crystal violet biofilm formation assay with slight modifications according to a previously published method.74 Briefly, overnight cultures were adjusted to an OD600 of 0.02 and 100 µl of the inoculum were added to each well of a flexible, non-treated U-bottom PVC 96-well plate (Corning Inc., NY, USA). Plates were sealed with an air-permeable membrane and incubated at 37 °C in humid atmosphere for 24 h. Planktonic bacteria were removed and wells were washed with water prior to the addition of 150 µl of the crystal violet staining solution (0.1% w/v in water). After 30 min incubation, the staining solution was removed and wells were again washed and air-dried. For de-staining, 200 µl of 96% ethanol was added to each well and the plate was incubated for 30 min at room temperature (RT). 125 μl of the solution was transferred to a fresh 96-well flat bottom plate before absorbance was measured at 540 nm. Each clinical isolate was tested in two independent experiments with eight technical replicates each time.
RNA sequencing and bioinformatic analyses
We selected a subset of 77 clinical P. aeruginosa strains for transcriptional profiling of bacteria grown under both biofilm and planktonic conditions. Transcriptional profiles under planktonic conditions have been recorded in a previous study.42 Briefly, planktonic bacteria were cultivated to early stationary phase (OD600 = 2) in 10 ml LB under shaking conditions (37 °C, 180 r.p.m.). Three independent cultures were pooled and an equal volume of RNAprotect (Qiagen, The Netherlands) was added prior to cell harvest. Biofilm cultures were grown as described above. Dynamic transcriptional profiles were recorded over time. For this purpose, biofilm-grown cells were not only harvested at 48 h but also at 12, 24, and 36 h. To examine structural characteristics, all biofilms were evaluated by CLSM prior to harvest. At least ten wells of two biological replicates were pooled to obtain one transcriptional profile per strain and time point. Biofilm suspensions were mixed with an equal volume of RNAlater (Qiagen) and centrifuged. For RNA extraction, bacterial pellets were stored overnight at −80 °C and the RNeasy© Mini Kit (Qiagen) following QIAshredder™ columns (Qiagen) was used according to the manufacturer’s instruction. DNase treatment (DNA-free™ Kit DNase Treatment & Removal, Ambion, Life Technologies) was applied to remove remaining DNA. RNA samples were quality-checked by the use of the RNA Nano Kit with an Agilent Bioanalyzer 2100 (Agilent Technologies, CA, USA). To remove ribosomal RNA, the Ribo-Zero Bacteria Kit (Illumina, CA, USA) was used and cDNA libraries were generated using ScriptSeq™ v2 Kit (Illumina). Samples were sequenced in single-end mode on an Illumina HiSeq 2500 device (1 × 50 bp reads) or paired-end mode on a Novaseq (2 × 50 bp).
Reads were quality controlled and adapter clipped using fastq-mcf from the ea-utils package75 and mapped to the genome of the reference genome UCBPP-PA14 (NC_008463.1, available for download from the Pseudomonas Genome database: http://v2.pseudomonas.com) with bowtie276 using default parameters for single- or paired-end reads. The resulting sam-files were converted to indexed binary format and pile-up format using SAMtools.77 The program featureCounts78 was used to count the reads mapping to genes.
Read counts were used as the basis for further analyses. Differential gene expression analysis between biofilm and planktonic transcriptional profiles was performed with the R package edgeR (v.3.20.1).79 Normalization factors to scale the raw library sizes were calculated using the weighted trimmed mean of M values method. Multidimensional scaling plots were generated using the plotMDS function of edgeR taking all genes into account. Normalized read counts were used to calculate the Euclidean and Pearson’s distance (1 − Pearson’s correlation) between samples. Therefore, basic R functions dist and cor were used, respectively.
Differential gene expression (biofilm vs. planktonic growth) was calculated for all 77 clinical isolates to identify the biofilm core transcriptome. For this purpose, the 77 strains were regarded as replicates. For the analysis of DEGs in biofilm clusters, 10 to 12 strains per each structural group were regarded as replicates. A fold change of 4 (log 2 fold change [log 2 FC] 2) was used as a threshold with a false discovery rate (FDR) of <0.05. The identified genes were used as the input for a Venn diagram to identify the overlap of regulated genes between the three biofilm groups. Area-proportional Venn diagrams were adapted from the online tool BioVenn.80 Overlapping DEGs and the output of the Venn diagram were used for an enrichment analysis. Functional enrichment of Gene Ontology terms81,82 was performed using the hypergeometric test (R function phyper, adjusted p value (FDR) < 0.05). For the analysis of DEGs between structure-related isolates (cluster-wise pairwise comparison), the threshold was set to an FC of 2.83 (log 2 FC 1.5) with an FDR < 0.05.
Correlation between biofilm clusters and phenotypes of the isolates was assessed by analysis of variance, followed by Tukey’s post hoc test. Both were performed using basic functions provided by the stats package in the R statistical environment.83
A phylogenetic tree was created based on whole-genome sequencing data, which are available for all 414 of our clinical P. aeruginosa isolates42 and five additional reference strains (UCBPP-PA14, PAO1, PA7, LESB58, PACS2). Sequences from 3524 genes that were present in all strains were used. Gene sequences for the isolates were generated from DNA sequencing reads using the mpileup option of the SAMtools package.84 Phylogenetic distances were calculated based on 17-mers to create an alignment-free k-mer tree.85 These distances were then used to visualize the phylogeny using the neighbor-joining tree estimation method from the R package ape and the package ggtree.86,87
Quantification of cyclic diguanylate
Intracellular cyclic diguanylate (c-di-GMP) was extracted, and quantified by high-performance liquid chromatography-coupled tandem mass spectrometry according to a published protocol.50,88 In brief, bacterial cultures were grown for 24 h shaking at 37 °C and 5 ml of the bacterial suspensions were harvested. Isotope-labeled [13C15N]-c-di-GMP was used as an internal standard during methanol:acetonitrile:water (2:2:1) extraction. Intracellular c-di-GMP was determined for each clinical isolate in three independent experiments and the resulting concentration is given as pmol c-di-GMP/mg protein. Roti Nanoquant solution (Carl Roth, Germany) was used according to the manufacturer’s instructions to determine protein concentrations in a Bradford assay.
In vitro cytotoxicity
In vitro infection assays were performed in accordance with published protocols.50,89,90 Briefly, A549 cells from a human alveolar adenocarcinoma cell line (ACC 107) were maintained in Dulbecco’s modified Eagle medium (DMEM) (Gibco, Life Technologies) supplemented with 2 mM l-glutamine (Thermo Fisher Scientific, MA, USA), 1× non-essential amino acids (Gibco), 100 U/ml penicillin–streptomycin (Gibco), and 10% fetal calf serum (Sigma-Aldrich, MO, USA). For the infection assay, A549 cells were grown in 24-well plates at 37 °C and 5% CO2 to 90% confluence. Prior to bacterial infection, epithelial cells were washed once with PBS. Bacterial inocula in DMEM were prepared from planktonic cultures in early stationary phase (3 to 3.5 h of growth) with an adjusted bacterial cell number according to a multiplicity of infection (MOI) of 10. To facilitate contact between bacteria and eukaryotic cells, plates were centrifuged for 5 min at 500 × g and incubated for 24 h at 37 °C. Supernatants were collected and centrifuged to pellet out cell debris and bacterial cells. The supernatant was used for the Pierce lactate dehydrogenase assay according to the manufacturer’s instructions (Thermo Fisher Scientific). As a positive control, one well was treated with lysis buffer 45 min prior to the collection of the supernatant, resulting in maximum cytotoxicity (cytomax). Cytotoxicity for clinical strains is given as percentage of cytomax.
Motility assays
Motility experiments were carried out according to previously published protocols91 with slight modifications. For a screening attempt, swimming assays were carried out in 12-well plates (Nunc, Denmark) with 2.5 ml of BM2 glucose agar (62 mM potassium phosphate buffer [pH = 7], 7 mM (NH4)2SO4, 2 mM MgSO4, 10 µM FeSO4, 0.4% [w/v] glucose; containing 0.3% agar) per well. For swarming, BM2 medium without (NH4)2SO4 was supplemented with 0.1% casamino acids and 0.5% agar. Liquid cultures inoculated from overnight cultures were incubated until an OD600 of ~1.5 to 2.5 (log phase) was reached. Subsequently, all cultures were adjusted to an OD600 of 1. To evaluate swimming ability, 1 µl of the adjusted suspension was carefully added on top of the agar; for swarming 0.5 µl of the cellular suspension was pipetted into the agar. Plates were incubated for 15 h at 30 °C in a humid atmosphere prior to evaluation. For twitching assays, a hole was punched into LB agar plates solidified with 1.5% agar and pelleted bacteria from 1 ml of culture were carefully added onto the bottom of the petri dish. After 48 h of static incubation at 37 °C (humid atmosphere), the agar was removed and the twitching zone on the plastic surface of the petri dish was measured. For all motility experiments, two independent experiments were performed, including the lab strain PA14 as a reference. For evaluation, pictures were taken and the motility area for each clinical isolate was measured with ImageJ and adjusted to the respective PA14 control to account for inter-experimental variations.
Pyocyanin production
Pyocyanin production was analyzed as described elsewhere.92,93 Briefly, cultures were grown for 24 h shaking before harvesting 5 ml bacterial suspension. After centrifugation, pyocyanin was extracted by the addition of an equal volume of chloroform. Three milliliters of the organic phase was mixed with 1 ml 0.2 M hydrochloric acid (HCl) and absorbance was determined at 520 nm for the aqueous phase. Pyocyanin concentrations [µg/ml supernatant] were calculated by multiplication with the correction factor 17.072.
Pyoverdine production
To investigate pyoverdine production, clinical isolates were cultivated in iron-restricted succinate medium (6 g K2HPO4, 3 g KH2PO4, 4.2 g (NH4)2HPO4, 0.2 g MgSO4, 4 g succinic acid, 1.1 g NaOH; filled up to 1 l; pH = 7) as previously described.94,95 Clinical isolates were cultivated under both planktonic (16 h shaking) and biofilm conditions (48 h statically). Pyoverdine-specific absorbance was measured at 400 nm and normalized to bacterial growth (OD600).
Proteolytic activity
To assess proteolytic activity of clinical strains, a protocol modified from Casilag et al.90 was used. The assay was performed with cation-adjusted Müller–Hinton (MH) broth (Fluka analytical, Thermo Fisher Scientific) containing 10% (v/v) of commercially available homogenized, ultra-heated milk (fat content < 0.3%), solidified with 1.5% agar. Overnight cultures were adjusted to an OD600 of 0.025 and 5 µl of the bacterial suspensions were spotted on top of the agar plates. Plates were incubated for 24 h at 37 °C and the clearing zones, which indicate proteolytic activity, were measured in axial and horizontal direction. Relative protease activity was calculated as follows:
| 1 |
Elastase secretion
The elastolytic activity of proteases secreted by the clinical isolates was tested in an Elastin Congo Red (ECR) assay.96 Briefly, bacteria were cultivated in either planktonic or static conditions to promote biofilm formation. The standard biofilm assay was scaled up (15 times the volume) to 24-well plates (Nunc). Cells were harvested after 24 h (PL) or 48 h (BF), respectively, and 100 µl of the supernatant was mixed with 900 µl ECR buffer (100 mM Tris [pH 7.5], 1 mM CaCl2, supplemented with 22.5 mg/ml ECR [Sigma-Aldrich]). The suspension was incubated for 3 h at 37 °C and 900 r.p.m. After centrifugation, the absorbance of the supernatant was determined at 495 nm. Elastase secretion is indicated relative to the maximum absorbance.
Oxidative stress response
To investigate the tolerance of clinical strains to H2O2, a disk diffusion assay was applied.97,98 In brief, bacteria were grown overnight and adjusted to an OD600 of 3. 150 µl of the bacterial suspension were mixed with 3 ml of cooled down LB soft agar (0.6% w/v) and poured onto the surface of LB solidified with 1.5% agar. Sterile filter paper disks were placed on the overlaying soft agar and 8 µl of 30% H2O2 (Carl Roth) was added. Plates were incubated for 16 h at 37 °C, and the diameter of the inhibition zone was measured in both the vertical and horizontal direction. H2O2 sensitivity is depicted as the mean of three technical triplicates.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We gratefully thank Agnes Nielsen, Astrid Dröge, Annette Garbe, and Anna-Lena Hagemann for technical assistance and support with RNA-seq sample preparation and LC-MS/MS analysis. S.H. was funded by the EU (ERC Consolidator Grant COMBAT 724290) and received funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC 2155 “RESIST” – Project ID 39087428. S.H. and V.K. received additional funding from the Deutsche Forschungsgemeinschaft (DFG SPP 1879).
Author contributions
J.G.T., M.K. and N.G. conducted the experiments; J.G.T., J.T., M.P., S.P. and S.D.W. processed and analyzed data; V.K. was responsible for mass spectrometry; S.H., J.G.T. and M.M. designed the experiments; S.H. and J.G.T. wrote the paper; J.T. and M.M. critically reviewed the manuscript.
Data availability
Transcriptome data have been deposited to NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession numbers GSE134231 (biofilm transcriptomes) and GSE123544 (planktonic transcriptomes). DNA-seq data are available in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under the reference number PRJNA526797.
Code availability
Scripts for RNA-seq data processing and analysis as well as for phylogenetic analysis are available from the authors upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41522-019-0113-6.
References
- 1.Brown, S. P., Cornforth, D. M. & Mideo, N. Evolution of virulence in opportunistic pathogens: generalism, plasticity, and control. Trends Microbiol. 20, 336–342 (2012). [DOI] [PMC free article] [PubMed]
- 2.Aujoulat F, et al. From environment to man: genome evolution and adaptation of human opportunistic bacterial pathogens. Genes (Basel) 2012;3:191–232. doi: 10.3390/genes3020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bervoets I, Charlier D. Diversity, versatility and complexity of bacterial gene regulation mechanisms: opportunities and drawbacks for applications in synthetic biology. FEMS Microbiol. Rev. 2019;43:304. doi: 10.1093/femsre/fuz001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pérez-Rueda E, Collado-Vides J. The repertoire of DNA-binding transcriptional regulators in Escherichia coli K-12. Nucleic Acids Res. 2000;28:1838–1847. doi: 10.1093/nar/28.8.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galagan James, Lyubetskaya Anna, Gomes Antonio. Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012. ChIP-Seq and the Complexity of Bacterial Transcriptional Regulation; pp. 43–68. [DOI] [PubMed] [Google Scholar]
- 6.Price TD, Qvarnström A, Irwin DE. The role of phenotypic plasticity in driving genetic evolution. Proc. Biol. Sci. 2003;270:1433–1440. doi: 10.1098/rspb.2003.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fusco G, Minelli A. Phenotypic plasticity in development and evolution: facts and concepts. Philos. Trans. R. Soc. Ser. B. 2010;365:547–556. doi: 10.1098/rstb.2009.0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly, S. A., Panhuis, T. M. & Stoehr, A. M. in Comprehensive Physiology Vol. 2, 1417–1439 (Wiley, 2012). [DOI] [PubMed]
- 9.Forsman A. Rethinking phenotypic plasticity and its consequences for individuals, populations and species. Heredity (Edinb.) 2015;115:276–284. doi: 10.1038/hdy.2014.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elena SF, Lenski RE. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat. Rev. Genet. 2003;4:457–469. doi: 10.1038/nrg1088. [DOI] [PubMed] [Google Scholar]
- 11.Silby MW, Winstanley C, Godfrey SAC, Levy SB, Jackson RW. Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev. 2011;35:652–680. doi: 10.1111/j.1574-6976.2011.00269.x. [DOI] [PubMed] [Google Scholar]
- 12.Herring CD, et al. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat. Genet. 2006;38:1406–1412. doi: 10.1038/ng1906. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg JB. Pseudomonas: global bacteria. Trends Microbiol. 2000;8:55–57. doi: 10.1016/S0966-842X(99)01671-6. [DOI] [PubMed] [Google Scholar]
- 14.Gellatly SL, Hancock REW. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog. Dis. 2013;67:159–173. doi: 10.1111/2049-632X.12033. [DOI] [PubMed] [Google Scholar]
- 15.Costerton JW. Bacterial biofilms: a common cause of persistent infections. Science (80−.) 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 16.Bjarnsholt T. The role of bacterial biofilms in chronic infections. APMIS. 2013;121:1–58. doi: 10.1111/apm.12099. [DOI] [PubMed] [Google Scholar]
- 17.Wagner VE, Iglewski BH. P. aeruginosa biofilms in CF infection. Clin. Rev. Allergy Immunol. 2008;35:124–134. doi: 10.1007/s12016-008-8079-9. [DOI] [PubMed] [Google Scholar]
- 18.Høiby N, et al. The clinical impact of bacterial biofilms. Int. J. Oral. Sci. 2011;3:55–65. doi: 10.4248/IJOS11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mah T-F, O’Toole GA. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001;9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 20.Stewart PS, Costerton JW. Antibiotic resistance of bacteria in biofilms. Lancet (Lond., Engl.) 2001;358:135–138. doi: 10.1016/S0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- 21.Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents. 2010;35:322–332. doi: 10.1016/j.ijantimicag.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 22.Stewart, P. S. Antimicrobial tolerance in biofilms. Microbiol. Spectr. 3, MB-0010-2014, 10.1128/microbiolspec.MB-0010-2014 (2015). [DOI] [PMC free article] [PubMed]
- 23.Liao J, Schurr MJ, Sauer K. The MerR-like regulator BrlR confers biofilm tolerance by activating multidrug efflux pumps in Pseudomonas aeruginosa biofilms. J. Bacteriol. 2013;195:3352–3363. doi: 10.1128/JB.00318-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall CW, Mah T-F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017;41:276–301. doi: 10.1093/femsre/fux010. [DOI] [PubMed] [Google Scholar]
- 25.Rossi E, Falcone M, Molin S, Johansen HK. High-resolution in situ transcriptomics of Pseudomonas aeruginosa unveils genotype independent patho-phenotypes in cystic fibrosis lungs. Nat. Commun. 2018;9:3459. doi: 10.1038/s41467-018-05944-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cornforth DM, et al. Pseudomonas aeruginosa transcriptome during human infection. Proc. Natl Acad. Sci. USA. 2018;115:E5125–E5134. doi: 10.1073/pnas.1717525115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kordes A, et al. Genetically diverse Pseudomonas aeruginosa populations display similar transcriptomic profiles in a cystic fibrosis explanted lung. Nat. Commun. 2019;10:3397. doi: 10.1038/s41467-019-11414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finnan S, Morrissey JP, O’Gara F, Boyd EF. Genome diversity of Pseudomonas aeruginosa isolates from cystic fibrosis patients and the hospital environment. J. Clin. Microbiol. 2004;42:5783–5792. doi: 10.1128/JCM.42.12.5783-5792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klockgether J, Cramer N, Wiehlmann L, Davenport CF, Tümmler B. Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2011;2:150. doi: 10.3389/fmicb.2011.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mowat E, et al. Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am. J. Respir. Crit. Care Med. 2011;183:1674–1679. doi: 10.1164/rccm.201009-1430OC. [DOI] [PubMed] [Google Scholar]
- 31.Marvig RL, Sommer LM, Molin S, Johansen HK. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 2015;47:57–64. doi: 10.1038/ng.3148. [DOI] [PubMed] [Google Scholar]
- 32.Folkesson A, et al. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat. Rev. Microbiol. 2012;10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 33.Ciofu O, Mandsberg LF, Wang H, Høiby N. Phenotypes selected during chronic lung infection in cystic fibrosis patients: implications for the treatment of Pseudomonas aeruginosa biofilm infections. FEMS Immunol. Med. Microbiol. 2012;65:215–225. doi: 10.1111/j.1574-695X.2012.00983.x. [DOI] [PubMed] [Google Scholar]
- 34.Smith EE, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl Acad. Sci. USA. 2006;103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winstanley C, O’Brien S, Brockhurst MA. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 2016;24:327–337. doi: 10.1016/j.tim.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oliver A, Cantón R, Campo P, Baquero F, Blázquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 37.Lee DG, et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006;7:R90. doi: 10.1186/gb-2006-7-10-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dötsch A, et al. The Pseudomonas aeruginosa transcriptional landscape is shaped by environmental heterogeneity and genetic variation. MBio. 2015;6:e00749–15. doi: 10.1128/mBio.00749-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Guo T, Mu Q, Li X, Yu J. Genomic and environmental determinants and their interplay underlying phenotypic plasticity. Proc. Natl Acad. Sci. USA. 2018;115:6679–6684. doi: 10.1073/pnas.1718326115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müsken M, et al. Towards individualized diagnostics of biofilm-associated infections: a case study. npj Biofilms Microbiomes. 2017;3:22. doi: 10.1038/s41522-017-0030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Müsken M, Di Fiore S, Dötsch A, Fischer R, Häussler S. Genetic determinants of Pseudomonas aeruginosa biofilm establishment. Microbiology. 2010;156:431–441. doi: 10.1099/mic.0.033290-0. [DOI] [PubMed] [Google Scholar]
- 42.Khaledi, A. et al. Fighting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. Preprint at 10.1101/643676v1 (2019). [DOI] [PMC free article] [PubMed]
- 43.Ringel MT, Brüser T. The biosynthesis of pyoverdines. Microb. Cell. 2018;5:424–437. doi: 10.15698/mic2018.10.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Q, et al. 2,3-Butanediol catabolism in Pseudomonas aeruginosa PAO1. Environ. Microbiol. 2018;20:3927–3940. doi: 10.1111/1462-2920.14332. [DOI] [PubMed] [Google Scholar]
- 45.Vallet I, Olson JW, Lory S, Lazdunski A, Filloux A. The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc. Natl Acad. Sci. USA. 2001;98:6911–6916. doi: 10.1073/pnas.111551898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vasseur P, Vallet-Gely I, Soscia C, Genin S, Filloux A. The pel genes of the Pseudomonas aeruginosa PAK strain are involved at early and late stages of biofilm formation. Microbiology. 2005;151:985–997. doi: 10.1099/mic.0.27410-0. [DOI] [PubMed] [Google Scholar]
- 47.Jennings LK, et al. Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc. Natl Acad. Sci. USA. 2015;112:11353–11358. doi: 10.1073/pnas.1503058112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diggle SP, Winzer K, Lazdunski A, Williams P, Cámara M. Advancing the quorum in Pseudomonas aeruginosa: MvaT and the regulation of N-acylhomoserine lactone production and virulence gene expression. J. Bacteriol. 2002;184:2576–2586. doi: 10.1128/JB.184.10.2576-2586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vallet I, et al. Biofilm formation in Pseudomonas aeruginosa: fimbrial cup gene clusters are controlled by the transcriptional regulator MvaT. J. Bacteriol. 2004;186:2880–2890. doi: 10.1128/JB.186.9.2880-2890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blanka A, et al. Identification of the alternative sigma factor SigX regulon and its implications for Pseudomonas aeruginosa pathogenicity. J. Bacteriol. 2014;196:345–356. doi: 10.1128/JB.01034-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arora SK, Ritchings BW, Almira EC, Lory S, Ramphal R. A transcriptional activator, FleQ, regulates mucin adhesion and flagellar gene expression in Pseudomonas aeruginosa in a cascade manner. J. Bacteriol. 1997;179:5574–5581. doi: 10.1128/jb.179.17.5574-5581.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dasgupta N, et al. A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol. Microbiol. 2003;50:809–824. doi: 10.1046/j.1365-2958.2003.03740.x. [DOI] [PubMed] [Google Scholar]
- 53.Baraquet C, Murakami K, Parsek MR, Harwood CS. The FleQ protein from Pseudomonas aeruginosa functions as both a repressor and an activator to control gene expression from the pel operon promoter in response to c-di-GMP. Nucleic Acids Res. 2012;40:7207–7218. doi: 10.1093/nar/gks384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams P, Cámara M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 2009;12:182–191. doi: 10.1016/j.mib.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 55.Ochsner UA, Johnson Z, Vasil ML. Genetics and regulation of two distinct haem-uptake systems, phu and has, in Pseudomonas aeruginosa. Microbiology. 2000;146:185–198. doi: 10.1099/00221287-146-1-185. [DOI] [PubMed] [Google Scholar]
- 56.Recinos DA, et al. Redundant phenazine operons in Pseudomonas aeruginosa exhibit environment-dependent expression and differential roles in pathogenicity. Proc. Natl Acad. Sci. USA. 2012;109:19420–19425. doi: 10.1073/pnas.1213901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Görisch H. The ethanol oxidation system and its regulation in Pseudomonas aeruginosa. Biochim. Biophys. Acta. 2003;1647:98–102. doi: 10.1016/S1570-9639(03)00066-9. [DOI] [PubMed] [Google Scholar]
- 58.Beaudoin T, Zhang L, Hinz AJ, Parr CJ, Mah T-F. The biofilm-specific antibiotic resistance gene ndvB is important for expression of ethanol oxidation genes in Pseudomonas aeruginosa biofilms. J. Bacteriol. 2012;194:3128–3136. doi: 10.1128/JB.06178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kordes Adrian, Grahl Nora, Koska Michal, Preusse Matthias, Arce-Rodriguez Alejandro, Abraham Wolf-Rainer, Kaever Volkhard, Häussler Susanne. Establishment of an induced memory response in Pseudomonas aeruginosa during infection of a eukaryotic host. The ISME Journal. 2019;13(8):2018–2030. doi: 10.1038/s41396-019-0412-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Breschi A, Gingeras TR, Guigó R. Comparative transcriptomics in human and mouse. Nat. Rev. Genet. 2017;18:425–440. doi: 10.1038/nrg.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cromie GA, et al. Transcriptional profiling of biofilm regulators identified by an overexpression screen in Saccharomyces cerevisiae. G3 (Bethesda) 2017;7:2845–2854. doi: 10.1534/g3.117.042440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu Xia, Siehnel Richard J., Garudathri Jayanthi, Staudinger Benjamin J., Hisert Katherine B., Ozer Egon A., Hauser Alan R., Eng Jimmy K., Manoil Colin, Singh Pradeep K., Bruce James E. In Vivo Proteome of Pseudomonas aeruginosa in Airways of Cystic Fibrosis Patients. Journal of Proteome Research. 2019;18(6):2601–2612. doi: 10.1021/acs.jproteome.9b00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suriyanarayanan T, et al. Quantitative proteomics of strong and weak biofilm formers of Enterococcus faecalis reveals novel regulators of biofilm formation. Mol. Cell. Proteom. 2018;17:643–654. doi: 10.1074/mcp.RA117.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Erdmann J, et al. The core proteome of biofilm-grown clinical Pseudomonas aeruginosa isolates. Cells. 2019;8:1129. doi: 10.3390/cells8101129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patell S, et al. Comparative microarray analysis reveals that the core biofilm-associated transcriptome of Pseudomonas aeruginosa comprises relatively few genes. Environ. Microbiol. Rep. 2010;2:440–448. doi: 10.1111/j.1758-2229.2010.00158.x. [DOI] [PubMed] [Google Scholar]
- 66.Tolker-Nielsen, T. Biofilm development. Microbiol. Spectr. 3, MB-0001-2014 (2015). [DOI] [PubMed]
- 67.Wei Q, Ma LZ. Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2013;14:20983–21005. doi: 10.3390/ijms141020983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fong, J. N. C. & Yildiz, F. H. Biofilm matrix proteins. Microbiol. Spectr. 3, 201–222 (2015). [DOI] [PMC free article] [PubMed]
- 69.Petrova OE, Sauer K. Sticky situations: key components that control bacterial surface attachment. J. Bacteriol. 2012;194:2413–2425. doi: 10.1128/JB.00003-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O’Toole GA. To build a biofilm. J. Bacteriol. 2003;185:2687–2689. doi: 10.1128/JB.185.9.2687-2689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Toole GA, Kolter R. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 1998;28:449–461. doi: 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- 72.Hornischer K, et al. BACTOME—a reference database to explore the sequence- and gene expression-variation landscape of Pseudomonas aeruginosa clinical isolates. Nucleic Acids Res. 2019;47:D716–D720. doi: 10.1093/nar/gky895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Müsken M, Di Fiore S, Römling U, Häussler S. A 96-well-plate-based optical method for the quantitative and qualitative evaluation of Pseudomonas aeruginosa biofilm formation and its application to susceptibility testing. Nat. Protoc. 2010;5:1460–1469. doi: 10.1038/nprot.2010.110. [DOI] [PubMed] [Google Scholar]
- 74.O’Toole GA. Microtiter dish biofilm formation assay. J. Vis. Exp. 2011 doi: 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aronesty, E. ea-utils: Command-Line Tools for Processing Biological Sequencing Data, http://code.google.com/p/ea-utils (Durham, NC, 2011).
- 76.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 79.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hulsen T, de Vlieg J, Alkema W. BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics. 2008;9:488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carbon S, et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47:D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.R Core Team. R: A Language and Environment for Statistical Computing (R Core Team, 2018).
- 84.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leekitcharoenphon P, Nielsen EM, Kaas RS, Lund O, Aarestrup FM. Evaluation of whole genome sequencing for outbreak detection of Salmonella enterica. PLoS ONE. 2014;9:e87991. doi: 10.1371/journal.pone.0087991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paradis, E., Claude, J. & Strimmer, K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics20, 289–290 (2004). [DOI] [PubMed]
- 87.Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017;8:28–36. doi: 10.1111/2041-210X.12628. [DOI] [Google Scholar]
- 88.Spangler C, Böhm A, Jenal U, Seifert R, Kaever V. A liquid chromatography-coupled tandem mass spectrometry method for quantitation of cyclic di-guanosine monophosphate. J. Microbiol. Methods. 2010;81:226–231. doi: 10.1016/j.mimet.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 89.Gödeke J, Pustelny C, Häussler S. Recycling of peptidyl-tRNAs by peptidyl-tRNA hydrolase counteracts azithromycin-mediated effects on Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013;57:1617–1624. doi: 10.1128/AAC.02582-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Casilag F, et al. The LasB elastase of Pseudomonas aeruginosa acts in concert with alkaline protease AprA to prevent flagellin-mediated immune recognition. Infect. Immun. 2016;84:162–171. doi: 10.1128/IAI.00939-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Overhage J, Bains M, Brazas MD, Hancock REW. Swarming of Pseudomonas aeruginosa is a complex adaptation leading to increased production of virulence factors and antibiotic resistance. J. Bacteriol. 2008;190:2671–2679. doi: 10.1128/JB.01659-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu H, et al. Influence of ptsP gene on pyocyanin production in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2005;253:103–109. doi: 10.1016/j.femsle.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 93.Pustelny C, et al. The peptide chain release factor methyltransferase PrmC is essential for pathogenicity and environmental adaptation of Pseudomonas aeruginosa PA14. Environ. Microbiol. 2013;15:597–609. doi: 10.1111/1462-2920.12040. [DOI] [PubMed] [Google Scholar]
- 94.Hoegy Françoise, Schalk Isabelle J. Methods in Molecular Biology. New York, NY: Springer New York; 2014. Monitoring Iron Uptake by Siderophores; pp. 337–346. [DOI] [PubMed] [Google Scholar]
- 95.Meyer JM, Abdallah MA. The fluorescent pigment of Pseudomonas fluorescens: biosynthesis, purification and physicochemical properties. J. Gen. Microbiol. 1978;107:319–328. doi: 10.1099/00221287-107-2-319. [DOI] [Google Scholar]
- 96.Rust L, Messing CR, Iglewski BH. Elastase assays. Methods Enzymol. 1994;235:554–562. doi: 10.1016/0076-6879(94)35170-8. [DOI] [PubMed] [Google Scholar]
- 97.Hassett DJ, et al. Quorum sensing in Pseudomonas aeruginosa controls expression of catalase and superoxide dismutase genes and mediates biofilm susceptibility to hydrogen peroxide. Mol. Microbiol. 1999;34:1082–1093. doi: 10.1046/j.1365-2958.1999.01672.x. [DOI] [PubMed] [Google Scholar]
- 98.Nalca Y, et al. Quorum-sensing antagonistic activities of azithromycin in Pseudomonas aeruginosa PAO1: a global approach. Antimicrob. Agents Chemother. 2006;50:1680–1688. doi: 10.1128/AAC.50.5.1680-1688.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptome data have been deposited to NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession numbers GSE134231 (biofilm transcriptomes) and GSE123544 (planktonic transcriptomes). DNA-seq data are available in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under the reference number PRJNA526797.
Scripts for RNA-seq data processing and analysis as well as for phylogenetic analysis are available from the authors upon request.