

Abstract
This study provide an insight that the panel genes methylation status in different clinical stage tended to reflect a different prognosis even in matched normal tissues, to clinical recommendation. We enrolled 153 colorectal cancer patients from a medical center in Taiwan and used the candidate gene approach to select five genes involved in carcinogenesis pathways. We analyzed the relationship between DNA methylation with different cancer stages and the prognostic outcome. There were significant trends of increasing risk of 5-year time to progression and event-free survival of subjects with raising number of hypermethylation genes both in normal tissue and tumor tissue. The group with two or more genes with aberrant methylation in the advanced cancer stages (Me/advanced) had lower 5-year event-free survival among patients with colorectal cancer in either normal or tumor tissue. The adjusted hazard ratios in the group with two or more genes with aberrant methylation with advanced cancer stages (Me/advanced) were 8.04 (95% CI, 2.80–23.1; P for trend <0.01) and 8.01 (95% CI, 1.92–33.4; P for trend <0.01) in normal and tumor tissue, respectively. DNA methylation status was significantly associated with poor prognosis outcome. This finding in the matched normal tissues of colorectal cancer patients could be an alternative source of prognostic markers to assist clinical decision making.
Subject terms: Prognostic markers, Cancer epidemiology
Introduction
Colorectal cancer (CRC) is the second most frequently diagnosed cancer in women (614,000 cases, 9.2% of the total female population) and the third most frequently diagnosed cancer in men (746,000 cases, 10% of the total male population) worldwide1–3. The prognosis of and options of therapy for CRC rely on pathological stages, the staging system used (Dukes system or the TNM system), 5-year survival rate, and disease-free rate. It is fabulous for early-stage disease, ranging between 91% and 80% for histological stages I and II, in patients who can benefit from curative treatment, but it is poor in patients with isolated liver or lung metastases (5-year survival ≤20%)4–7. Over the past two decades, various treatment strategies have resulted in significant improvement in survival among patients with CRC8. Extensive studies have been executed to recognize novel prognostic and predictive biomarkers for CRC, including both genetic and epigenetic abnormalities. Previous studies have demonstrated that aberrant DNA methylation, the most frequent aberrant epigenetic modification in cancer, is an important early biomarker in CRC that is related to transcriptional silencing of tumor suppressor genes and a marker for field cancerization9–11.
With these biomarkers, patients in the same tumor stage could be stratified by different individual molecular factors. It’s useful for prognosis prediction and individualized treatment12,13. Previous studies have identified that methylation of cyclin-dependent kinase inhibitor 2A (CDKN2A), O-6-methylguanine DNA methyltransferase (MGMT) and human mutL homolog 1 (hMLH1), correlated with carcinogenesis pathways through gene silencing, could serve as diagnostic prognostic markers for CRC14–17. We selected two other candidate genes, colony stimulating factor 2 (CSF2) and DIS3 mitotic control homolog (S. cerevisiae)-like 2 (DIS3L2), from a previous study database18 that involved inhibitory effects on tumor growth19–22. To understand the effect of specific gene methylation on the relationship between CRC prognosis including progression and mortality and histological stage, we explored DNA methylation status in tumor and adjacent normal tissues (matched normal) from subjects who received surgical resection for CRC. In this study, we provide insight that the selected genes methylation status in different clinical stages tended to reflect a different prognosis, even in matched normal tissues, to clinical recommendations.
Methods
Patients and specimen collections
We designed a hospital-based retrospective cohort study to estimate the 5-year prognosis of patients with CRC in Taiwan and have been described elsewhere17,23. Patients with a diagnosis of invasive CRC between 2006 and 2010 who underwent surgical resection were eligible for recruitment. Tumor stage was defined according to the American Joint Committee on Cancer TNM staging system24. Informed consent was obtained from all enrollees to evaluate prognosis (including recurrence, metastasis, and survival). This study was approved by the Institutional Review Board (IRB) of Tri-Service General Hospital (TSGH) (certificated number 098-05-292 and 2-105-05-129). The method of obtaining follow-up data for registered patients, including patients’ information on the prognosis including recurrence, metastasis and the cause of death, relied on medical records linked to data in a cancer registration database. In this study, according to the clinical practice guideline of the TSGH Division of Colon and Rectum, the enrollees should return for a checkup once every 3 months in the first year after receiving surgical resection and once every 3–6 months subsequently. All methods were organized under, and operated per International Conference on Harmonization (ICH) / WHO GCP and the applicable laws and regulations. In addition, we also confirmed that all methods were performed in accordance with the relevant guidelines and regulations. Progression was defined as local recurrence or metastases. Time to progression (TTP) has been described as the time interval from the date of receiving surgical resection to the date of disease progression. Overall survival (OS) was calculated from the date of receiving surgical resection to the date of death of any cause. Event-free survival (EFS) was defined as the interval from the date of receiving surgical resection to the date of disease progression or death as a result of any cause. Otherwise, the enrollees without progression and who were survivors were followed up until the latest date of checkup as the study endpoint.
Data on sex, age at surgery, cancer stage, adjuvant chemotherapy, histological grade, and tumor location were obtained from the patients’ medical files. On the basis of the inclusion criteria, we identified 153 tumor tissues and matched normal tissues (306 samples) from enrollees in TSGH. The mean values for TTP, OS, and EFS were 2.67 years, 3.53 years, and 3.53 years, respectively. The fresh tissue samples of participants were obtained in the operating room while tumor tissues were resected. From each patient, adjacent normal mucosa tissue samples were collected from resected, unaffected parts of the colon located at least 10 cm from the tumor site. Samples were immediately frozen in liquid nitrogen and stored at −80 °C for subsequent DNA extraction and methylation assays. Figure 1 depicts the flowchart of the study design.
Figure 1.
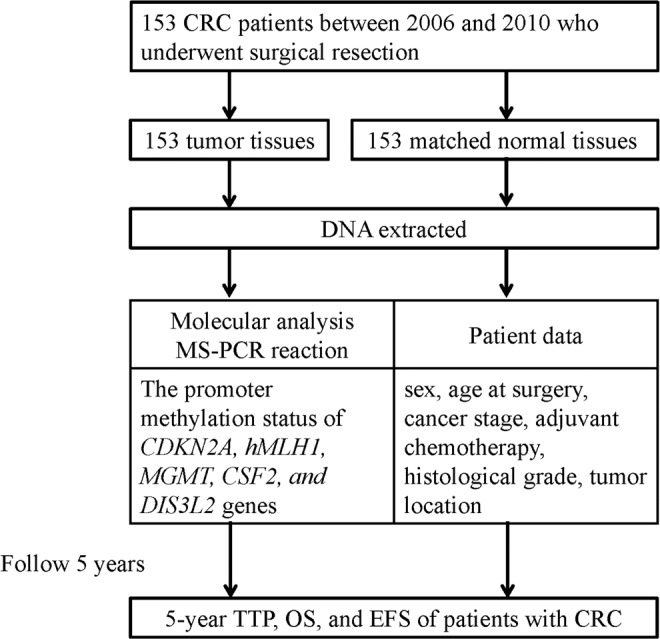
Flowchart of study design.
DNA extracted and MS-PCR reaction
We used the Genomic DNA Tissue Kit (catalogue no. 69504; Qiagen, Taipei, Taiwan) to extract the genomic DNA from the tissues according to the manufacturer’s protocol and the cellulose-coated magnetic beads with the MagCore Compact Automated Nucleic Acid Extractor (catalogue no. MCA0801; RBC Bioscience, Taipei, Taiwan). The isolated DNA was treated with sodium bisulfite using the EZ DNA Methylation Kit (Zymo Research Corporation, Orange, CA, USA).
We evaluated the promoter methylation status of CDKN2A, hMLH1, MGMT, CSF2, and DIS3L2 genes through methylation-specific polymerase chain reaction (MS-PCR), as described in our previous research17,23. The reaction solution (25 μL) contained 1.2-μL aliquots of forward and reverse primers, 12.5 μL HotStart Taq Premix (RBC Bioscience) and bisulfite-converted DNA. The sequences, annealing temperature of each primer used for amplification, and PCR product sizes are described in Table 1. PCR cycling conditions were as follows: first, 10 min at 95 °C; then, 35 cycles of 30-s denaturation at 95 °C, 30-s annealing, and 30-s extension at 72 °C; finally, 4-min extension at 72 °C. After the amplification, PCR products were mixed with a loading buffer, electrophoresed on 2% agarose gel by using 0.2-μL gel-stained dye for 25 min, and visualized using an ultraviolet transilluminator.
Table 1.
Primer sequences, annealing temperature and product size for MSP of target genes.
| Genes | Forward primer (5′ → 3′) | Annealing temperature(oC) | Product size (bp) | |
|---|---|---|---|---|
| CDKN2A | M | F:TTATTAGAGGGTGGGGCGGATCGC | 62 | 150 |
| R:GACCCCGAACCGCGACCGTAA | ||||
| U | F:TTATTAGAGGGTGGGGTGGATTGT | 62 | 151 | |
| R:CAACCCCAAACCACAACCATAA | ||||
| hMLH1 | M | F:ACGTAGACGTTTTATTAGGGTCGC | 60 | 118 |
| R:CCTCATCGTAACTACCCGCG | ||||
| U | F:TTTTGATGTAGATGTTTTATTAGGGTTGT | 60 | 124 | |
| R:ACCACCTCATCATAACTACCCACA | ||||
| MGMT | M | F:TTTCGACGTTCGTAGGTTTTCGC | 53 | 81 |
| R:GCACTCTTCCGAAAACGAAACG | ||||
| U | F:TTTGTGTTTTGATGTTTGTAGGTTTTTGT | 53 | 93 | |
| R:AACTCCACACTCTTCCAAAAACAAAACA | ||||
| CSF2 | M | F: TGATTATTTAGGGAAAAGGTTTATC | 56 | 105 |
| R: ATAACCACAAAATACCAAAAAAACG | ||||
| U | F: ATTATTTAGGGAAAAGGTTTATTGT | 60 | 104 | |
| R: AATAACCACAAAATACCAAAAAAACA | ||||
| DIS3L2 | M | F: GTCGTAGTTGAATCGTCGATTAC | 54 | 134 |
| R: TTACTAAAAAAAATACTCTTCCGAA | ||||
| U | F: GTTGTAGTTGAATTGTTGATTATGA | 55 | 134 | |
| R: TTACTAAAAAAAATACTCTTCCAAA | ||||
MSP, methylation-specific polymerase chain reaction; CDKN2A, cyclin-dependent kinase inhibitor 2A; MLH1, mutL homolog 1; MGMT, O-6-methylguanine DNA methyltransferase; CSF2, colony stimulating factor 2; DIS3L2, DIS3 mitotic control homolog (S. cerevisiae)-like 2; M, methylation; U, unmethylation.
Statistical analysis
To determine the association of methylation in CDKN2A, hMLH1, MGMT, CSF2, and DIS3L2 with 5-year TTP, OS, and EFS of patients with CRC in different clinical stages, we separately evaluated the various stages and divided them into two subgroups (local and advanced stages) on the basis of the different pathological types of tissue: tumor and adjacent normal tissues (matched normal).
Patients have been split into two groups based on the methylation status of the five evaluated genes: a group with aberrant methylation at two or more genes and a group with aberrant methylation at less than two genes. We used the Kaplan–Meier survival analysis to estimate 5-year TTP, OS, and EFS. Log-rank tests were used to assess the significance of differences in the groups. We performed multivariate analyses with a Cox proportional hazards model adjusted for baseline characteristics, which included sex, age at surgery (continuous), cancer stage (1, 2, 3, or 4), adjuvant chemotherapy, lymphovascular invasion, histological grade, tumor location, and methylation status of candidate genes based on previous studies23 to evaluate hazard ratios (HRs) with 95% confidence intervals (CIs). All statistical tests were two-sided, and P values < 0.05 were considered statistically significant. Statistical analyses for the clinical study were conducted using IBM SPSS Statistics version 22 (IBM® SPSS® Statistics 22).
Results
During the study period, we identified 153 tumor samples of patients with CRC and matched normal samples from the TSGH tumor bank. The demographic features of the study population are shown in Table 2. Among the study patients, 51.0% were men, and the mean age was 64.1 years (standard deviation, 14.7 years). The patients were classified into four clinical subgroups: stage 1 (15.0%), stage 2 (35.3%), stage 3 (32.0%), and stage 4 (17.6%). The progression in 5 years indicated that 41.2% of the enrollees had cancer recurrence or metastasis and 38.5% died. In addition, on the basis of the tumor and matched normal tissues of the patients, we classified the patient characteristics according to five individual gene groups (CDKN2A, hMLH1, MGMT, CSF2, and DIS3L2) and according to those with two or more genes with aberrant methylation, stratified by different variables (sex, age at surgery, cancer stage, progression, all-cause death, progression including death, adjuvant chemotherapy, histological grade, and tumor location).
Table 2.
Characteristics and distribution of methylation status in patients with CRC (n = 153).
| Variables | Total | Methylation status | |||||
|---|---|---|---|---|---|---|---|
| CSF2 | DIS3L2 | ≥2 of genes | |||||
| Normal | Tumors | Normal | Tumors | Normal | Tumors | ||
| Sex, n (%) | |||||||
| Male | 78 (51.0) | 21 (26.9) | 49 (62.8) | 18 (23.1) | 19 (24.4) | 17 (21.8) | 47 (60.3) |
| Female | 75 (49.0) | 28 (37.3) | 43 (57.3) | 17 (22.7) | 20 (26.7) | 10 (13.3) | 39 (52.0) |
| Age at surgery | |||||||
| Mean (SD) | 64.1 (14.7) | 63.1 (15.0) | 62.6 (16.3) | 66.7 (12.6) | 68.1 (13.1) | 61.3 (16.0) | 63.3 (14.8) |
| <65, n (%) | 25 (33.3) | 51 (68.0) | 59 (77.6) | 15 (20.0) | 16 (21.3) | 16 (21.3) | 45 (60.0) |
| ≥65, n (%) | 24 (30.8) | 41 (52.6) | 58 (74.4) | 20 (25.6) | 23 (29.5) | 11 (14.1) | 41 (52.6) |
| Stage, n (%) | |||||||
| I | 23 (15.0) | 9 (39.1) | 11 (47.8) | 3 (13.0) | 6 (26.1) | 1 (4.3) | 8 (34.8) |
| II | 54 (35.3) | 19 (35.2) | 34 (63.0) | 12 (22.2) | 10 (18.5) | 9 (16.7) | 30 (55.6) |
| III | 49 (32.0) | 15 (30.6) | 31 (63.3) | 16 (32.7) | 15 (30.6) | 8 (16.3) | 33 (67.3) |
| IV | 27 (17.6) | 6 (22.2) | 16 (59.3) | 4 (14.8) | 8 (29.6) | 9 (33.3) | 15 (55.6) |
| Progression in 5 yr, n (%) | |||||||
| No | 90 (58.8) | 26 (28.9) | 52 (57.8) | 17 (18.9) | 20 (22.2) | 10 (11.1) | 43 (47.8) |
| Yes | 63 (41.2) | 23 (36.5) | 40 (63.5) | 18 (28.6) | 19 (30.2) | 17 (27.0) | 43 (68.3) |
| All-cause death in 5 yr, n (%) | |||||||
| No | 122 (79.7) | 39 (32.0) | 76 (62.3) | 30 (24.6) | 33 (27.0) | 20 (16.4) | 71 (58.2) |
| Yes | 31 (20.3) | 10 (32.3) | 16 (51.6) | 5 (16.1) | 6 (193.4) | 7 (22.6) | 15 (48.4) |
| Progression including death, in 5 yr, n (%) | |||||||
| No | 78 (51.0) | 22 (28.2) | 44 (56.4) | 15 (19.2) | 17 (21.8) | 7 (9.0) | 37 (47.4) |
| Yes | 75 (49.0) | 27 (36.0) | 48 (64.0) | 20 (26.7) | 22 (29.3) | 20 (26.7) | 49 (65.3) |
| Adjuvant chemotherapy, n (%) | |||||||
| No | 37 (24.2) | 20 (54.1) | 28 (75.7) | 16 (43.2) | 16 (43.2) | 6 (16.2) | 21 (56.8) |
| Yes | 97 (63.4) | 26 (26.8) | 57 (58.8) | 17 (17.5) | 20 (20.6) | 20 (20.6) | 57 (58.8) |
| Histological grade, n (%)* | |||||||
| Well or Moderately | 114 (74.6) | 39 (34.2) | 69 (60.5) | 29 (25.4) | 31 (27.2) | 23 (20.2) | 62 (54.4) |
| Poor or undifferentiated | 13 (8.5) | 2 (15.4) | 9 (69.2) | 1 (7.7) | 3 (23.1) | 3 (23.1) | 10 (76.9) |
| Tumor location, n (%)* | |||||||
| Colon | 104 (68.0) | 32 (30.8) | 63 (60.6) | 25 (24.0) | 25 (24.0) | 20 (19.2) | 58 (55.8) |
| Rectum | 30 (19.6) | 14 (46.7) | 22 (73.3) | 8 (26.7) | 11 (36.7) | 6 (20.0) | 20 (66.7) |
| Variables | Total | Methylation status | |||||
| CSF2 | DIS3L2 | ≥2 of genes | |||||
| Normal | Normal | Normal | Tumors | Normal | Tumors | ||
| Sex, n (%) | |||||||
| Male | 78 (51.0) | 51 (65.4) | 57 (73.1) | 21 (26.9) | 26 (33.3) | 41 (52.6) | 62 (79.5) |
| Female | 75 (49.0) | 61 (81.3) | 60 (80.0) | 24 (32.0) | 28 (37.3) | 45 (60.0) | 63 (84.0) |
| Age at surgery | |||||||
| Mean (SD) | 64.1 (14.7) | 62.4 (14.9) | 63.3 (14.9) | 59.7 (14.7) | 61.8 (15.0) | 61.3 (14.6) | 63.3 (15.3) |
| <65, n (%) | 25 (33.3) | 61 (81.3) | 59 (78.7) | 28 (37.3) | 33 (44.0) | 49 (65.3) | 65 (86.7) |
| ≥65, n (%) | 24 (30.8) | 51 (65.4) | 58 (74.4) | 17 (21.8) | 21 (26.9) | 37 (47.4) | 60 (76.9) |
| Stage, n (%) | |||||||
| I | 23 (15.0) | 17 (73.9) | 19 (82.6) | 9 (39.1) | 7 (30.4) | 13 (56.5) | 15 (65.2) |
| II | 54 (35.3) | 39 (72.2) | 39 (72.2) | 15 (27.8) | 19 (35.2) | 28 (51.9) | 42 (77.8) |
| III | 49 (32.0) | 35 (71.4) | 37 (75.5) | 11 (22.4) | 14 (28.6) | 28 (57.1) | 45 (91.8) |
| IV | 27 (17.6) | 21 (77.8) | 22 (81.5) | 10 (37.0) | 14 (51.9) | 17 (63.0) | 23 (85.2) |
| Progression in 5 yr, n (%) | |||||||
| No | 90 (58.8) | 65 (72.2) | 67 (74.4) | 27 (30.0) | 32 (35.6) | 45 (50.0) | 69 (76.7) |
| Yes | 63 (41.2) | 47 (74.6) | 50 (79.4) | 18 (28.6) | 22 (34.9) | 41 (65.1) | 56 (88.9) |
| All-cause death in 5 yr, n (%) | |||||||
| No | 122 (79.7) | 86 (70.5) | 89 (73.0) | 37 (30.3) | 41 (33.6) | 69 (56.6) | 100 (82.0) |
| Yes | 31 (20.3) | 26 (83.9) | 28 (90.3) | 8 (25.8) | 13 (41.9) | 17 (54.8) | 25 (80.6) |
| Progression including death, in 5 yr, n (%) | |||||||
| No | 78 (51.0) | 53 (67.9) | 55 (70.5) | 25 (32.1) | 28 (35.9) | 38 (48.7) | 58 (74.4) |
| Yes | 75 (49.0) | 59 (78.7) | 82 (82.7) | 20 (26.7) | 26 (34.7) | 48 (64.0) | 67 (89.3) |
| Adjuvant chemotherapy, n (%) | |||||||
| No | 37 (24.2) | 30 (81.1) | 34 (91.9) | 14 (37.8) | 14 (37.8) | 29 (78.4) | 34 (91.9) |
| Yes | 97 (63.4) | 71 (73.2) | 72 (74.2) | 29 (29.9) | 39 (40.2) | 51 (52.6) | 81 (83.5) |
| Histological grade, n (%)* | |||||||
| Well or Moderately | 114 (74.6) | 83 (72.8) | 87 (76.3) | 36 (31.6) | 45 (39.5) | 67 (58.8) | 95 (83.3) |
| Poor or undifferentiated | 13 (8.5) | 12 (92.3) | 13 (100) | 4 (30.8) | 6 (46.2) | 8 (61.5) | 13 (100) |
| Tumor location, n (%)* | |||||||
| Colon | 104 (68.0) | 77 (74.0) | 78 (75.0) | 35 (33.7) | 41 (39.4) | 59 (56.7) | 86 (82.7) |
| Rectum | 30 (19.6) | 24 (80.0) | 28 (93.3) | 8 (26.7) | 12 (40.0) | 21 (70.0) | 29 (96.7) |
CRC, colorectal cancer; CDKN2A, cyclin-dependent kinase inhibitor 2A; MGMT, O-6-methylguanine DNA methyltransferase; MLH1, mutL homolog 1.
*The total number of patients with CRC does not correspond because of missing data.
CRC, colorectal cancer; CSF2, colony stimulating factor 2; DIS3L2, DIS3 mitotic control homolog (S. cerevisiae)-like 2.
We evaluated the relationship between the gene hypermethylation status and 5-year TTP, OS, and EFS of patients with CRC. In the multivariable analysis, compared with all genes in the unmethylated group, four hypermethylation genes in normal tissue were more highly associated with 5-year TTP of patients with CRC (HR 6.23; 95% CI 1.16–33.5). Moreover, a significant increasing trend of HR for 5-year TTP of patients with CRC was observed with increasing number of hypermethylation genes, both in normal tissue (P < 0.01) and in tumor tissue (P = 0.01) (Table 3). The 5-year TTP survival curves in normal tissue showed a significant difference between the group with 2 or more genes with aberrant methylation and the comparison group (P = 0.01). In tumor tissue, the group with two or more genes with aberrant methylation was borderline significantly associated with 5-year TTP of patients with CRC (P = 0.08) (Fig. 2). After multivariable adjustment for confounders, we observed a significant association between the group with two or more genes with aberrant methylation in normal tissue and 5-year TTP of patients with CRC (adjusted HR, 2.28; 95% CI, 1.17–4.44). A nonsignificant association was observed between the number of genes with hypermethylation status in both normal and tumor tissue and 5-year OS of patients with CRC (data not shown).
Table 3.
The relationship between the number of gene hypermethylation status and 5-year TTP of CRC patients.
| Normal tissues | Tumor tissues | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of subjects | No. of cases (%) | Crude | Adjusted | No. of subjects | No. of cases (%) | Crude | Adjusted | |||||
| HR | 95% CI | HR | 95% CI | HR | 95% CI | HR | 95% CI | |||||
| NO. of hypermethylation gene | ||||||||||||
| 0 | 18 | 5 (27.8) | 1.00 | Referent | 1.00 | Referent | 8 | 2 (25.0) | 1.00 | Referent | 1.00 | Referent |
| 1 | 49 | 17 (34.7) | 1.22 | (0.39 to 3.78) | 1.51 | (0.33 to 6.93) | 20 | 5 (25.0) | 0.85 | (0.16 to 4.64) | 1.24 | (0.13 to 12.0) |
| 2 | 49 | 23 (46.9) | 2.24 | (0.77 to 6.52) | 3.05 | (0.69 to 13.5) | 47 | 19 (40.4) | 1.27 | (0.28 to 5.67) | 1.84 | (0.23 to 14.5) |
| 3 | 27 | 12 (44.4) | 2.12 | (0.66 to 6.79) | 2.81 | (0.57 to 13.8) | 44 | 19 (43.2) | 1.76 | (0.40 to 7.66) | 3.00 | (0.38 to 23.6) |
| 4 | 10 | 6 (60.0) | 2.88 | (0.77 to 10.8) | 6.23 | (1.16 to 33.5) | 28 | 16 (57.1) | 2.99 | (0.69 to 13.1) | 4.12 | (0.53 to 32.5) |
| 5 | 0 | 0 (0) | N/A | N/A | N/A | N/A | 6 | 2 (33.3) | 1.47 | (0.21 to 10.4) | 3.12 | (0.26 to 36.9) |
| p for trend | 0.03 | <0.01 | 0.02 | 0.01 | ||||||||
| ≥2 of genes | 86 | 41 (47.7) | 1.97 | (1.09 to 3.56) | 2.28 | (1.17 to 4.44) | 125 | 56 (44.8) | 2.03 | (0.87 to 4.75) | 2.34 | (0.82 to 6.71) |
Abbreviations: TTP, time to progression; CRC, colorectal cancer; HR, hazard ratio; CI, confidence interval; N/A, not applicable.
Adjusted for gender, age at surgery (continuous), adjuvant chemotherapy, histological grade and tumor location.
Not applicable due to limited numbers of cases.
Figure 2.

Kaplan–Meier survival curves depicting the effect of the ≥ 2 aberrancy group on 5-year TTP of CRC patients in (A) normal tissue and (B) tumor tissue. Vertical tick marks indicate censored events. TTP, time to progression; CRC, colorectal cancer.
The significance of associations between four hypermethylation genes in normal tissue and 5-year EFS of patients with CRC was evaluated using Cox regression (HR, 5.85; 95% CI, 1.40–24.6). Furthermore, significant trends for increasing risk of 5-year EFS in patients with CRC were observed with increasing number of hypermethylated genes in both normal tissue (P < 0.01) and tumor tissue (P < 0.01) (Table 4). The Kaplan–Meier curves of 5-year EFS in patients with CRC among the group with two or more genes with aberrant methylation and the comparison group are shown in Fig. 3. The log-rank test revealed significant differences in both normal (P = 0.04) and tumor tissue (P = 0.01) over the entire Kaplan–Meier curve.
Table 4.
The relationship between the number of gene hypermethylation status and 5-year EFS of CRC patients.
| Normal tissues | Tumor tissues | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of subjects | No. of cases (%) | Crude | Adjusted | No. of subjects | No. of cases (%) | Crude | Adjusted | |||||
| HR | 95% CI | HR | 95% CI | HR | 95% CI | HR | 95% CI | |||||
| NO. of gene methylation | ||||||||||||
| 0 | 18 | 5 (27.8) | 1.00 | Referent | 1.00 | Referent | 8 | 2 (25.0) | 1.00 | Referent | 1.00 | Referent |
| 1 | 49 | 22 (44.9) | 1.51 | (0.57 to 3.98) | 1.69 | (0.50 to 5.75) | 20 | 6 (30.0) | 1.12 | (0.23 to 5.53) | 1.39 | (0.15 to 12.5) |
| 2 | 49 | 26 (53.1) | 191 | (0.74 to 4.99) | 1.70 | (0.48 to 5.98) | 47 | 23 (48.9) | 2.15 | (0.51 to 9.13) | 2.78 | (0.37 to 21.1) |
| 3 | 27 | 16 (59.3) | 2.83 | (1.04 to 7.72) | 3.09 | (0.85 to 11.2) | 44 | 23 (52.3) | 2.42 | (0.57 to 10.3) | 3.60 | (0.47 to 27.5) |
| 4 | 10 | 6 (60.0) | 2.72 | (0.83 to 8.91) | 5.85 | (1.40 to 24.6) | 28 | 19 (67.9) | 4.58 | (1.07 to 19.7) | 6.35 | (0.83 to 48.5) |
| 5 | 0 | 0 (0) | N/A | N/A | N/A | N/A | 6 | 2 (33.3) | 1.44 | (0.20 to 10.2) | 2.39 | (0.21 to 27.7) |
| p for trend | 0.01 | <0.01 | <0.01 | <0.01 | ||||||||
| ≥2 of genes | 86 | 48 (55.8) | 1.63 | (1.01 to 2.60) | 1.49 | (0.87 to 2.56) | 125 | 67 (53.6) | 2.39 | (1.15 to 4.99) | 2.84 | (1.12 to 7.22) |
Abbreviations: EFS, event-free survival; CRC, colorectal cancer; HR, hazard ratio; CI, confidence interval; N/A, not applicable.
Adjusted for gender, age at surgery (continuous), adjuvant chemotherapy, histological grade and tumor location.
Not applicable due to limited numbers of cases.
Figure 3.

Kaplan–Meier survival curves depicting the effect of the ≥ 2 aberrancy group on 5-year EFS of CRC patients in (A) normal tissue and (B) tumor tissue. Vertical tick marks indicate censored events. EFS, event-free survival; CRC, colorectal cancer.
We examined whether the interaction of prognosis of CRC and different cancer stages (local and advanced) was reported for the methylation status of the five genes in tumor tissues and normal tissues. Table 5 shows a significant association between the group with two or more genes with aberrant methylation in normal tissue with advanced cancer stages (Me/advanced) and 5-year TTP of patients with CRC. The crude and adjusted HRs were 9.76 (95% CI, 3.78–25.3; P for trend < 0.01) and 15.0 (95% CI, 3.78–25.3; P for trend < 0.01), respectively. We found similar results in tumor tissue, for which the crude and adjusted HRs were 6.47 (95% CI, 2.00–21.0; P for trend < 0.01) and 11.5 (95% CI, 1.56–85.2; P for trend < 0.01), respectively. As regard to the association between gene promoter region methylation and different cancer stages for 5-year OS of patients with CRC, no significant relationship was observed between CDKN2A, hMLH1, and CSF2 methylation in the advanced cancer stages (Me/advanced) and cancer mortality in 5 years in both normal and tumor tissue (data not shown). Moreover, we estimated the association between the group with two or more genes with aberrant methylation with cancer stage and 5-year OS of patients with CRC. In normal and tumor tissue, the adjusted HRs of the group with two or more genes with aberrant methylation in advanced cancer stages (Me/advanced) were 3.97 (95% CI, 0.83–19.1; P for trend = 0.31) and 4.42 (95% CI, 0.57–34.5; P for trend = 0.10), respectively (Table 6). A significant relationship was observed between the group with two or more genes with aberrant methylation in the advanced cancer stages (Me/advanced) and 5-year EFS of patients with CRC in both normal and tumor tissue (Table 7). The group with two or more genes with aberrant methylation in the advanced cancer stages (Me/advanced) had lower 5-year EFS among patients with CRC in either normal or tumor tissue. The adjusted HRs in the group with two or more genes with aberrant methylation with advanced cancer stages (Me/advanced) were 8.04 (95% CI, 2.80–23.1; P for trend < 0.01) and 8.01 (95% CI, 1.92–33.4; P for trend < 0.01) in normal and tumor tissue, respectively.
Table 5.
The interaction between gene promoter region methylation and different cancer stages for 5-year TTP of CRC patients.
| Normal tissues | Tumor tissues | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of subjects | No. of cases (%) | Crude | Adjusted | No. of subjects | No. of cases (%) | Crude | Adjusted | |||||
| HR | 95% CI | HR | 95% CI | HR | 95% CI | HR | 95% CI | |||||
| ≧2 of genes | ||||||||||||
| UnMe/local (1&2) | 36 | 6 (16.7) | 1.00 | Referent | 1.00 | Referent | 20 | 4 (20.0) | 1.00 | Referent | 1.00 | Referent |
| UnMe/advanced (3&4) | 31 | 16 (51.6) | 3.49 | (1.21 to 10.1) | 6.30 | (1.39 to 28.6) | 8 | 3 (37.5) | 2.50 | (0.50 to 12.4) | 7.36 | (0.76 to 71.7) |
| Me/local (1&2) | 41 | 3 (7.3) | 0.19 | (0.02 to 1.60) | 0.32 | (0.03 to 3.74) | 57 | 5 (8.8) | 0.34 | (0.07 to 1.69) | 0.51 | (0.04 to 5.82) |
| Me/advanced (3&4) | 45 | 38 (84.4) | 9.76 | (3.78 to 25.3) | 15.0 | (3.52 to 63.6) | 68 | 51 (75.0) | 6.47 | (2.00 to 21.0) | 11.5 | (1.56 to 85.2) |
| p for trend | <0.01 | <0.01 | <0.01 | <0.01 | ||||||||
Abbreviations: TTP, time to progression; CRC, colorectal cancer; HR, hazard ratio; CI, confidence interval.
Adjusted for gender, age at surgery (continuous), adjuvant chemotherapy, histological grade and tumor location.
UnMe/loccal (1&2): gene promoter region unmethylated with cancer stage 1 or 2.
UnMe/advanced (3&4): gene promoter region unmethylated with cancer stage 3 or 4.
Me/local (1&2): gene promoter region methylated with cancer stage 1 or 2.
Me/advanced (3&4): gene promoter region methylated with cancer stage 3 or 4.
Table 6.
The interaction between gene promoter region methylation and different cancer stages for 5-year OS of CRC patients.
| Normal tissues | Tumor tissues | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of subjects | No. of cases (%) | Crude | Adjusted | No. of subjects | No. of cases (%) | Crude | Adjusted | |||||
| HR | 95% CI | HR | 95% CI | HR | 95% CI | HR | 95% CI | |||||
| ≧2 of genes | ||||||||||||
| UnMe/local (1&2) | 36 | 6 (16.7) | 1.00 | Referent | 1.00 | Referent | 20 | 4 (20.0) | 1.00 | Referent | 1.00 | Referent |
| UnMe/advanced (3&4) | 31 | 8 (25.8) | 1.73 | (0.60 to 5.00) | 4.69 | (0.98 to 22.5) | 8 | 2 (25.0) | 1.06 | (0.20 to 5.81) | 4.74 | (0.42 to 53.2) |
| Me/local (1&2) | 41 | 7 (17.1) | 1.15 | (0.39 to 3.41) | 1.23 | (0.22 to 6.91) | 57 | 9 (15.8) | 0.82 | (0.25 to 2.66) | 1.23 | (0.14 to 10.9) |
| Me/advanced (3&4) | 45 | 10 (22.2) | 1.74 | (0.63 to 4.79) | 3.97 | (0.83 to 19.1) | 68 | 16 (23.5) | 1.46 | (0.49 to 4.38) | 4.42 | (0.57 to 34.5) |
| p for trend | 0.44 | 0.31 | 0.40 | 0.10 | ||||||||
Abbreviations: OS, overall survival; CRC, colorectal cancer; HR, hazard ratio; CI, confidence interval.
Adjusted for gender, age at surgery (continuous), adjuvant chemotherapy, histological grade and tumor location.
UnMe/loccal (1&2): DNA promoter region unmethylated with cancer stage 1 or 2.
UnMe/advanced (3&4): DNA promoter region unmethylated with cancer stage 3 or 4.
Me/local (1&2): DNA promoter region methylated with cancer stage 1 or 2.
Me/advanced (3&4): DNA promoter region methylated with cancer stage 3 or 4.
Table 7.
The interaction between gene promoter region methylation and different cancer stages for 5-year EFS of CRC patients.
| Normal tissues | Tumor tissues | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of subjects | No. of cases (%) | Crude | Adjusted | No. of subjects | No. of cases (%) | Crude | Adjusted | |||||
| HR | 95% CI | HR | 95% CI | HR | 95% CI | HR | 95% CI | |||||
| ≧2 of genes | ||||||||||||
| UnMe/local (1&2) | 36 | 8 (22.2) | 1.00 | Referent | 1.00 | Referent | 20 | 5 (25.0) | 1.00 | Referent | 1.00 | Referent |
| UnMe/advanced (3&4) | 31 | 19 (61.3) | 3.36 | (1.47 to 7.68) | 5.44 | (1.83 to 16.1) | 8 | 3 (37.5) | 1.32 | (0.32 to 5.53) | 3.52 | (0.58 to 21.3) |
| Me/local (1&2) | 41 | 10 (24.4) | 2.26 | (0.50 to 3.18) | 0.95 | (0.26 to 3.43) | 57 | 13 (22.8) | 0.99 | (0.35 to 2.77) | 1.09 | (0.23 to 5.25) |
| Me/advanced (3&4) | 45 | 38 (84.4) | 5.48 | (2.55 to 11.8) | 8.04 | (2.80 to 23.1) | 68 | 54 (79.4) | 4.48 | (1.79 to 11.2) | 8.01 | (1.92 to 33.4) |
| p for trend | <0.01 | <0.01 | <0.01 | <0.01 | ||||||||
Abbreviations: EFS, event-free survival; CRC, colorectal cancer; HR, hazard ratio; CI, confidence interval.
Adjusted for gender, age at surgery (continuous), adjuvant chemotherapy, histological grade and tumor location.
UnMe/loccal (1&2): DNA promoter region unmethylated with cancer stage 1 or 2.
UnMe/advanced (3&4): DNA promoter region unmethylated with cancer stage 3 or 4.
Me/local (1&2): DNA promoter region methylated with cancer stage 1 or 2.
Me/advanced (3&4): DNA promoter region methylated with cancer stage 3 or 4.
Discussion
DNA methylation, an important epigenetic mechanism, regulates gene expression through reversible modifications of histone acetylation that influence chromatin structure and the accessibility of transcription factors to their binding sites. In this study, the methylation status of five selected gene promoters (CDKN2A, hMLH1, MGMT, CSF2, and DIS3L2) confirmed the presence of DNA methylation in tumor tissues and matched normal tissues. We analyzed and tested the prognostic outcome, including TTP, OS, and EFS, in patients with CRC and found that the presence of hypermethylated DNA in the normal tissues could be predictive of worse outcomes in terms of TTP, OS, and EFS. Furthermore, the group with two or more genes with aberrant methylation with advanced cancer stages (Me/advanced) in normal tissue were highly associated with 5-year EFS of patients with CRC. The studies of DNA methylation biomarker panels showed that the hypermethylation of multiple candidate genes was associated with greater susceptibility to CRC25,26.
After correcting for potential confounders for 5-year TTP of CRC through multivariate analysis, an increasing number of hypermethylation genes was associated with poorer TTP, even in normal tissue (P < 0.01). In terms of OS, the group with two or more genes with aberrant methylation conferred shorter survival without reaching statistical significance. This finding is in accordance with review articles in which researchers used different panel candidate genes to predict prognosis27,28. Kim et al. evaluated the methylation status of 10 genes in patients with metastatic or recurrent CRC and found there was an independent association between a higher number of methylated genes among the genes examined and poorer clinical outcome29. This result is consistent with our finding that the candidate genes selected in our studies were involved in multiple molecular events in tumorigenesis. Some studies have reported no relationship between the candidate gene methylation status and prognosis in CRC30,31. These inconsistent results can be explained at least in part by the panels studied.
In this study, we used the other rate, EFS, which can be used to measure the prognosis in CRC clinical trials32. With respect to the relationship between the methylation of multiple genes and clinicopathological variables, we observed a significant relationship of normal tissue and 5-year EFS in the group with two or more genes with aberrant methylation. The result is consistent with recent findings of a trend for increases in EFS and 5-year OS for patients with gastric cancer who had zero or one methylated gene in their tumors33. Another study showed that hypermethylation of multiple genes was significantly associated with lower EFS in patients with neuroblastoma34. However, global DNA hypomethylation, which has been recognized to contribute to oncogenesis through various mechanisms, including genomic instability, resulted in the re-expression of proto-oncogenes or imprinted genes and was significantly associated with worse prognosis35,36.
Although genetic and environmental factors have been proposed as independent predictors of CRC prognosis37, the TNM staging system, which has defined the extent of cancer based solely on anatomic pathology since the 1940s, has been considered as the most comprehensive tool for predicting CRC prognosis38,39. Nevertheless, several studies have proposed that tumors of the same stage can differ unpredictably in both prognosis and treatment response because of heterogeneity by molecular subclassification39,40. This is consistent with our study of using DNA methylation patterns to perform further stratification.
Our study indicated that there was a positive association between the group with two or more genes with aberrant methylation with advanced cancer stages and prognostic outcome, including TTP and EFS, especially in normal tissue. Recently, growing evidence has demonstrated that DNA methylation profiles are changed by carcinogenic factors at the early precancerous stages in various organs25,41–44. Different DNA methylation profiles were observed at the chronic hepatitis or liver cirrhosis stage as a precancerous condition for liver cancer41. Sato et al. revealed aberrant DNA methylation in several genes in noncancerous tissues obtained from patients with lung cancer. The association between carcinogenetic factors such as cigarette smoking and epigenetic clustering of lung cancer based on DNA methylation profiles in adjacent lung tissue has been examined43. With regard to the precancerous condition for stomach cancer, aberrant DNA methylation is reportedly induced by Helicobacter pylori infection44,45. The evidence demonstrated that alterations in tissues surrounding prostate adenocarcinomas might be the result of carcinogenic factors affecting a whole organ, called a cancer field effect.
The surrounding tissues, which have been named tumor indicating normal tissue (TINT), may indicate the essence and nature of tumors. According to this information, the diagnostics and prognostics of prostate cancer could be improved46,47. These results were in accordance with our findings that we could find abnormalities of DNA methylation in adjacent normal tissue. The finding of aberrant DNA methylation in normal tumour-adjacent colorectal tissues could indicate a worse prognosis after surgical resection. Therefore, the TINT of CRC could be an alternative source of prognostic markers to help in clinical decision making.
The most frequently studied biomarker was CDKN2A (p16)48, which is as a negative regulator in the G1/S phase of the cell cycle by disrupting the complexes of CDK442. Studies including subgroup evaluations have demonstrated that aberrant CDKN2A methylation was significantly correlated to a poor prognosis49,50, even though the finding of Sanz-Casla et al. demonstrated an inconsistent result51. The DNA mismatch-repair system is inactivated by MLH1 hypermethylation, which reduces the MLH1 protein expression and stops the formation of MLH1 protein and blocks the activation of mismatch repair (MMR) genes. Inactivation of DNA mismatch repair caused by promoter hypermethylation of MLH1 lead to cell proliferation and genomic instability to the point of CRC formation52. The significant finding by Iida et al. and Kuan et al. showed that MLH1 hypermethylation was associated with worse prognosis in TNM stages 1 to 423,53.
MGMT is in charge of repairing DNA damage produced by alkylating agents. Aberrant MGMT methylation may be involved in CRC tumorigenesis54. For MGMT promoter hypermethylation, Kuan et al. showed that MGMT methylation in TNM stages 3 to 4 indicates a poor prognosis. However, the study of Nilsson et al. showed conflicting results with a better prognosis28.
With regard to CSF2, Lee et al. clarified that CSF2 was the mainly upregulated gene of importance for carcinoma development and invasiveness among those involved in positive regulation of tyrosine phosphorylation of STAT5. CSF2 could play an as an important role of prognosticator and a future therapeutic target of urothelial carcinoma55. DIS3L2 inactivation has been connected to modified expression of mitotic checkpoint proteins and mitotic abnormalities. Knockdown of DIS3L2 enhanced the growth of human cancer cells, and overexpression prohibited these cells growth21. There are few studies assessed the relationship between CSF2 or DIS3L2 promoter methylation and CRC prognosis
The findings of the this study must be interpreted within the context of some limitations. An important consideration in assessing the relation between the five selected gene promoters methylation and the CRC prognostic outcome due to KRAS and BRAF mutation analysis was not conducted in this study. There is mutually exclusiveness between KRAS and BRAF, worse survival in patients with methylated p16 and BRAF mutations is not influenced by KRAS status23,27. Furthermore, the present study did not include subjects with colorectal benign adenoma and healthy individuals. The development of an acceptable protocol could help in the study of the methylation status of tumor suppressor genes; their distribution in promoter regions; their distribution in the proximal colon, distal colon, and rectum; and their time sequence dependence in healthy individuals, particularly in those who develop CRC. Wu et al. used an animal model to simulate the methylation status of the adenoma–carcinoma sequence, which is a precursor of animal cancer progression; but they did not study humans18. Finally, the results of the present study should be carefully interpreted because of the small number of patients who were analyzed. A larger prospective cohort study is warranted to validate these results.
In summary, this study demonstrated that DNA methylation status was significantly associated with poor CRC prognosis, particularly in the matched normal tissues with advanced stage, because the molecular changes could not be examined on the basis of clinical pathology. To mark out the implication of DNA aberrant methylation in CRC scenario, future research addressing the relationship should be prospective and make attempts to include subjects with colorectal benign adenoma and healthy subjects. We suggest using these findings in the matched normal tissues of patients with CRC as an alternative source of prognostic markers to assist in clinical decision making.
Acknowledgements
This manuscript was edited by Wallace Academic Editing. This research was funded by the Ministry of Science and Technology, Taiwan (R.O.C.), grant number MOST 104-2314-B-016-010-MY2 and MOST 106-2320-B-016-018 and the Ministry of National Defense, Taiwan (R.O.C.), grant number MAB-107-075, MAB-108-057 and MAB-109-061.
Author contributions
C.H. Hsu: Contributed to the data research and writing of the manuscript. C.W. Hsiao: Conception and design of the study, data research and writing of the manuscript. C.A. Sun: The accuracy of the data analysis and full access to all the data. W.C. Wu: The accuracy of the data analysis and full access to all the data. T. Yang: Statistical analysis and full access to all the data. J.M. Hu: Data acquisition and full access to all the data. Y.C. Liao: Statistical analysis and full access to all the data. C.H. Huang: Statistical analysis and full access to all the data. C.Y. Chen: Data acquisition and full access to all the data. F.H. Lin: Statistical analysis and full access to all the data. Y.C. Chou: Resources and funding, study supervision, and the guarantor of the manuscript and takes responsibility for the integrity of the work as a whole.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pfütze K, et al. Methylation status at HYAL2 predicts overall and progression-free survival of colon cancer patients under 5-FU chemotherapy. Genomics. 2015;106:348–354. doi: 10.1016/j.ygeno.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay, J. et al. (2017).
- 3.Navarro M, Nicolas A, Ferrandez A, Lanas A. Colorectal cancer population screening programs worldwide in 2016: An update. World Journal of Gastroenterology. 2017;23:3632–3642. doi: 10.3748/wjg.v23.i20.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo Y, et al. Clinical outcomes after surgical resection of colorectal cancer in 1,294 patients. Hepatogastroenterology. 2012;59:1398–1402. doi: 10.5754/hge11676. [DOI] [PubMed] [Google Scholar]
- 5.Parente F, et al. Improved 5-year survival of patients with immunochemical faecal blood test-screen-detected colorectal cancer versus non-screening cancers in northern Italy. Dig Liver Dis. 2015;47:68–72. doi: 10.1016/j.dld.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 6.Kuipers EJ, et al. Colorectal cancer. Nature Reviews Disease Primers. 2015;1:15065. doi: 10.1038/nrdp.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Society, A. C. Colorectal Cancer Facts & Figures 2014–2016. (2016).
- 8.Graham JS, Cassidy J. Adjuvant therapy in colon cancer. Expert Review of Anticancer Therapy. 2012;12:99–109. doi: 10.1586/era.11.189. [DOI] [PubMed] [Google Scholar]
- 9.Luo, Y., Yu, M. & Grady, W. M. Field cancerization in the colon: a role for aberrant DNA methylation? Gastroenterology Report, got039 (2014). [DOI] [PMC free article] [PubMed]
- 10.Kim T-O, et al. DNA hypermethylation of a selective gene panel as a risk marker for colon cancer in patients with ulcerative colitis. International journal of molecular medicine. 2013;31:1255–1261. doi: 10.3892/ijmm.2013.1317. [DOI] [PubMed] [Google Scholar]
- 11.Lam K, Pan K, Linnekamp JF, Medema JP, Kandimalla R. DNA methylation based biomarkers in colorectal cancer: a systematic review. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2016;1866:106–120. doi: 10.1016/j.bbcan.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Yi JM, et al. Genomic and Epigenomic Integration Identifies a Prognostic Signature in Colon Cancer. Clinical Cancer Research. 2011;17:1535. doi: 10.1158/1078-0432.CCR-10-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walther A, et al. Genetic prognostic and predictive markers in colorectal cancer. Nature Reviews Cancer. 2009;9:489. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- 14.Lee BB, et al. Aberrant methylation of APC, MGMT, RASSF2A, and Wif-1 genes in plasma as a biomarker for early detection of colorectal cancer. Clinical Cancer Research. 2009;15:6185–6191. doi: 10.1158/1078-0432.CCR-09-0111. [DOI] [PubMed] [Google Scholar]
- 15.Oberwalder M, et al. SFRP2 methylation in fecal DNA—a marker for colorectal polyps. International journal of colorectal disease. 2008;23:15–19. doi: 10.1007/s00384-007-0355-2. [DOI] [PubMed] [Google Scholar]
- 16.Imperiale TF, et al. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014;2014:1287–1297. doi: 10.1056/NEJMoa1311194. [DOI] [PubMed] [Google Scholar]
- 17.Chang HF, et al. Clinical stage and risk of recurrence and mortality: interaction of DNA methylation factors in patients with colorectal cancer. J. Investig. Med. 2016;64:1200–1207. doi: 10.1136/jim-2016-000086. [DOI] [PubMed] [Google Scholar]
- 18.Wu W-C, et al. Predicting the progress of colon cancer by DNA methylation markers of the p16 gene in feces-Evidence from an animal model. Genetics and molecular biology. 2013;36:323–328. doi: 10.1590/S1415-47572013000300004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butterfield LH, et al. Immune correlates of GM-CSF and melanoma peptide vaccination in a randomized trial for the adjuvant therapy of resected high-risk melanoma (E4697) Clinical Cancer Research, clincanres. 2017;3016:2016. doi: 10.1158/1078-0432.CCR-16-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urdinguio RG, et al. Immune-dependent and independent antitumor activity of GM-CSF aberrantly expressed by mouse and human colorectal tumors. Cancer research. 2013;73:395–405. doi: 10.1158/0008-5472.CAN-12-0806. [DOI] [PubMed] [Google Scholar]
- 21.Astuti D, et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nature genetics. 2012;44:277. doi: 10.1038/ng.1071. [DOI] [PubMed] [Google Scholar]
- 22.Costa, P., Romão, L. & Gama-Carvalho, M. Transcriptomic screen for DIS3 and DIS3L1 exosome subunits associated functional networks in colorectal cancer. BioSys PhD Day, Faculdade de Ciências da Universidade de Lisboa, 11 dezembro2015 (2015).
- 23.Kuan JC, et al. DNA methylation combinations in adjacent normal colon tissue predict cancer recurrence: evidence from a clinical cohort study. PLoS One. 2015;10:e0123396. doi: 10.1371/journal.pone.0123396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Annals of surgical oncology. 2010;17:1471–1474. doi: 10.1245/s10434-010-0985-4. [DOI] [PubMed] [Google Scholar]
- 25.Luo X, et al. Methylation of a panel of genes in peripheral blood leukocytes is associated with colorectal cancer. Scientific reports. 2016;6:29922. doi: 10.1038/srep29922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gyparaki M-T, Basdra EK, Papavassiliou AG. DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. Journal of molecular medicine. 2013;91:1249–1256. doi: 10.1007/s00109-013-1088-z. [DOI] [PubMed] [Google Scholar]
- 27.Kim SH, et al. p16 Hypermethylation and KRAS mutation are independent predictors of cetuximab plus FOLFIRI chemotherapy in patients with metastatic colorectal cancer. Cancer research and treatment: official journal of Korean Cancer Association. 2016;48:208. doi: 10.4143/crt.2014.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nilsson TK, Löf-Öhlin ZM, Sun X-F. DNA methylation of the p14ARF, RASSF1A and APC1A genes as an independent prognostic factor in colorectal cancer patients. International journal of oncology. 2013;42:127–133. doi: 10.3892/ijo.2012.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SH, et al. CpG Island Methylator Phenotype and Methylation of Wnt Pathway Genes Together Predict Survival in Patients with Colorectal Cancer. Yonsei medical journal. 2018;59:588–594. doi: 10.3349/ymj.2018.59.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia M, et al. No association of CpG island methylator phenotype and colorectal cancer survival: population-based study. British journal of cancer. 2016;115:1359. doi: 10.1038/bjc.2016.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallois C, Laurent-Puig P, Taieb J. Methylator phenotype in colorectal cancer: a prognostic factor or not? Critical reviews in oncology/hematology. 2016;99:74–80. doi: 10.1016/j.critrevonc.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Brody, T. In Clinical Trials (Second Edition) (ed. Tom Brody) 331-376 (Academic Press, 2016).
- 33.Napieralski R, et al. Methylation of tumor-related genes in neoadjuvant-treated gastric cancer: relation to therapy response and clinicopathologic and molecular features. Clinical Cancer Research. 2007;13:5095–5102. doi: 10.1158/1078-0432.CCR-07-0241. [DOI] [PubMed] [Google Scholar]
- 34.Hoebeeck J, et al. Aberrant methylation of candidate tumor suppressor genes in neuroblastoma. Cancer letters. 2009;273:336–346. doi: 10.1016/j.canlet.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 35.Fouad MA, et al. Impact of Global DNA Methylation in Treatment Outcome of Colorectal Cancer Patients. Frontiers in pharmacology. 2018;9:1173. doi: 10.3389/fphar.2018.01173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura K, et al. UHRF1 regulates global DNA hypomethylation and is associated with poor prognosis in esophageal squamous cell carcinoma. Oncotarget. 2016;7:57821–57831. doi: 10.18632/oncotarget.11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bishehsari F, Mahdavinia M, Vacca M, Malekzadeh R, Mariani-Costantini R. Epidemiological transition of colorectal cancer in developing countries: environmental factors, molecular pathways, and opportunities for prevention. World journal of gastroenterology: WJG. 2014;20:6055. doi: 10.3748/wjg.v20.i20.6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greene FL, Sobin LH. The staging of cancer: a retrospective and prospective appraisal. CA: a cancer journal for clinicians. 2008;58:180–190. doi: 10.3322/CA.2008.0001. [DOI] [PubMed] [Google Scholar]
- 39.Hari DM, et al. AJCC Cancer Staging Manual 7th edition criteria for colon cancer: do the complex modifications improve prognostic assessment? Journal of the American College of Surgeons. 2013;217:181–190. doi: 10.1016/j.jamcollsurg.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puppa G, Sonzogni A, Colombari R, Pelosi G. TNM staging system of colorectal carcinoma: a critical appraisal of challenging issues. Archives of pathology & laboratory medicine. 2010;134:837–852. doi: 10.5858/134.6.837. [DOI] [PubMed] [Google Scholar]
- 41.Nagashio R, et al. Carcinogenetic risk estimation based on quantification of DNA methylation levels in liver tissue at the precancerous stage. International journal of cancer. 2011;129:1170–1179. doi: 10.1002/ijc.26061. [DOI] [PubMed] [Google Scholar]
- 42.Saadallah-Kallel A, et al. Clinical and prognosis value of the CIMP status combined with MLH1 or p16INK4amethylation in colorectal cancer. Medical Oncology. 2017;34:147. doi: 10.1007/s12032-017-1007-1. [DOI] [PubMed] [Google Scholar]
- 43.Sato T, et al. Epigenetic clustering of lung adenocarcinomas based on DNA methylation profiles in adjacent lung tissue: Its correlation with smoking history and chronic obstructive pulmonary disease. International journal of cancer. 2014;135:319–334. doi: 10.1002/ijc.28684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ushijima T, Hattori N. Molecular pathways: Involvement of Helicobacter pylori–triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clinical Cancer Research. 2012;18:923–929. doi: 10.1158/1078-0432.CCR-11-2011. [DOI] [PubMed] [Google Scholar]
- 45.Wen X-Z, et al. Methylation of GATA-4 and GATA-5 and development of sporadic gastric carcinomas. World journal of gastroenterology: WJG. 2010;16:1201. doi: 10.3748/wjg.v16.i10.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nonn L, Ananthanarayanan V, Gann PH. Evidence for field cancerization of the prostate. The Prostate. 2009;69:1470–1479. doi: 10.1002/pros.20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halin S, Hammarsten P, Adamo H, Wikström P, Bergh A. Tumor indicating normal tissue could be a new source of diagnostic and prognostic markers for prostate cancer. Expert opinion on medical diagnostics. 2011;5:37–47. doi: 10.1517/17530059.2011.540009. [DOI] [PubMed] [Google Scholar]
- 48.Draht MX, et al. Prognostic DNA methylation markers for sporadic colorectal cancer: a systematic review. Clinical epigenetics. 2018;10:35. doi: 10.1186/s13148-018-0461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bihl MP, Foerster A, Lugli A, Zlobec I. Characterization of CDKN2A(p16) methylation and impact in colorectal cancer: systematic analysis using pyrosequencing. Journal of Translational Medicine. 2012;10:173. doi: 10.1186/1479-5876-10-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kohonen‐Corish MR, et al. KRAS mutations and CDKN2A promoter methylation show an interactive adverse effect on survival and predict recurrence of rectal cancer. International journal of cancer. 2014;134:2820–2828. doi: 10.1002/ijc.28619. [DOI] [PubMed] [Google Scholar]
- 51.Sanz-Casla M.T., Maestro M.L., Vidaurreta M., Maestro C., Arroyo M., Cerdán J. p16 Gene Methylation in Colorectal Tumors: Correlation with Clinicopathological Features and Prognostic Value. Digestive Diseases. 2005;23(2):151–155. doi: 10.1159/000088597. [DOI] [PubMed] [Google Scholar]
- 52.Niv Y. Microsatellite instability and MLH1 promoter hypermethylation in colorectal cancer. World journal of gastroenterology: WJG. 2007;13:1767. doi: 10.3748/wjg.v13.i12.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iida S, et al. PIK3CA mutation and methylation influences the outcome of colorectal cancer. Oncology letters. 2012;3:565–570. doi: 10.3892/ol.2011.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen S-P, et al. The association of methylation in the promoter of APC and MGMT and the prognosis of Taiwanese CRC patients. Genetic testing and molecular biomarkers. 2009;13:67–71. doi: 10.1089/gtmb.2008.0045. [DOI] [PubMed] [Google Scholar]
- 55.Lee Y-Y, et al. CSF2 overexpression is associated with STAT5 phosphorylation and poor prognosis in patients with urothelial carcinoma. Journal of Cancer. 2016;7:711. doi: 10.7150/jca.14281. [DOI] [PMC free article] [PubMed] [Google Scholar]