

Abstract
Sphingosine-1-phosphate is now emerging as an important player in cancer, inflammation, autoimmune, neurological and cardiovascular disorders. Abundance evidence in animal and humans cancer models has shown that SphK1 is linked to cancer. Thus, there is a great interest in the development new SphK1 inhibitors as a potential new treatment for cancer. In a search for new SphK1 inhibitors we selected the well-known SKI-II inhibitor as the starting structure and we synthesized a new inhibitor structurally related to SKI-II with a significant but moderate inhibitory effect. In a second approach, based on our molecular modeling results, we designed new structures based on the structure of PF-543, the most potent known SphK1 inhibitor. Using this approach, we report the design, synthesis and biological evaluation of a new series of compounds with inhibitory activity against both SphK1 and SphK2. These new inhibitors were obtained incorporating new connecting chains between their polar heads and hydrophobic tails.
On the other hand, the combined techniques of molecular dynamics simulations and QTAIM calculations provided complete and detailed information about the molecular interactions that stabilize the different complexes of these new inhibitors with the active sites of the SphK1. This information will be useful in the design of new SphK inhibitors.
Keywords: Sphingosine kinase 1 inhibitors, Quinolin-2-one-pyrimidine hybrids, Synthesis, Bioassays, Molecular modelling
1. Introduction
Sphingosine-1-phosphate (S1P) is a pleiotropic sphingolipid metabolite that regulates numerous physiological processes including cell growth, survival, differentiation, migration, lymphocyte trafficking, activation and immune response [1–3]. Due to its multiple roles, S1P is now emerging as an molecular target of interest in cancer, inflammation, autoimmune, neurological, and cardiovascular disorders [4–6]. S1P is formed intracellularly by phosphorylation of sphingosine, the backbone of all sphingolipids, catalyzed by two sphingosine kinases, SphK1 and SphK2 [7–9]. These kinases have distinct cellular localization and functions [10]. The two enzymes display partial redundancy since SphK1−/− or SphK2−/− mice are phenotypically normal whereas elimination of both genes is embryonic lethal due to neurological and vascular defects [11]. Also, SphK1 and SphK2 contain five conserved domains (C1-C5), with the ATP binding site placed in C2 region and the catalytic domain formed within C1-C3 [12].
Abundance evidence in animal cancer models and in humans has shown that SphK1 is linked to cancer. SphK1 expression is increased in many cancers and has been correlated with poor prognosis [13–15]. Thus, there is a great interest in development of potent SphK inhibitors as a potential new treatment avenue for cancer. Some compounds have recently been utilized in clinical trials, like Sonepcizumab (Asonep) (), Safingol (SphK1 inhibitor) in combination with Cisplatin (), ABC294640 (SphK2 inhibitor) () and Fingolimod (SphK2 inhibitor) (), among others.
We have recently reported two new structural scaffolds that are useful for the development of new inhibitors of SphK1. These compounds were obtained by virtual screening and molecular modeling based on the crystal structure of SphK1 which enable to identify structural changes that could increase affinity of SphK1 for these new inhibitors [16]. More recently, we expanded this study and specific inhibitors of SphK2 with potential anti-inflammatory properties were arisen [17]. In addition to the useful information about the structural characteristics of SphK1, we also found excellent correlations between theoretical calculations and experimental data [18]. In the search of more potent SphK1 inhibitors, in this study we have used a different strategy. As a first approach, we have selected the well-known SKI-II inhibitor as the starting structure and synthesized diverse derivatives [19–21]. In the second approach, based on our molecular modeling results, we designed new structures based on the structure of PF-543, the most potent SphK1 inhibitor known [22,23]. We are now reporting the design, synthesis and biological evaluation of a new series of compounds with improved inhibitory activity against SphK1.
2. Results and discussion
2.1. Obtaining analogues of SKI-II
The quinolones and their oxo-derivatives are an important class of heterocyclic compounds that are considered as being pharmacologically privileged for the structural design of new drugs [24]. We have recently designed new hybrids containing the quinoline pharmacophore in their structures with the aim of increasing their biological potency. Some examples of quinolone derivatives we have prepared coupled with benzimidazolo (I), diazepinone (II) and curcumin mimic (III) with biological activities are displayed in Fig. 1 [25–27].
Fig. 1.

Quinolone hybrids with biological activity.
Based on our experience in the design and synthesis of heterocyclic hybrids, we designed a new type of SphK inhibitors that mimic the shape and functionality of SKI-II (IC50 0.5 μM) [19,20], in which the heterocyclic core thiazole has been replaced by a pyrimidine unit, and a quinolone moiety is used as the polar head group and aryl groups, pchlorophenyl and naphthyl, as hydrophobic tails (see Fig. 2).
Fig. 2.

Designed strategy to achieve title compounds.
The first compound we synthesized had only a small modification, replacing the thiazole ring of SKI-II with a pyrimidine ring, thereby obtaining compound 4a (Fig. 3).
Fig. 3.

Chemical structure of the inhibitors of SphK1: PF-543, SKI-II and compound 4a obtained by replacing the thiazole ring of SKI-II with a pyrimidine ring (yellow circle) are shown with their respective IC50 values. The IC50 value for the compound marked with an asterisk (*) was obtained from the literature. It is expected that these values would be higher using the methods described in the current work.
The IC50 values for the inhibition of SphK1 for 4a was 43.9 μM (Fig. 4). In comparison, the IC50 for PF-543 was 0.016 μM [28]. Because the SKI-II compound was not evaluated in the current study, PF-543 was used as the reference compound to compare with the compounds synthesized in this study. Although 4a show inhibitory activity toward SphK1, the IC50 value was higher than those for SKI-II and PF-543.
Fig. 4.

SphK1 inhibition rate for compound 4a.
In order to obtain compounds with stronger inhibitory effects, a series of structural modifications was made to compound 4a in both the polar portion of the molecule (5a, 6a, 7a and 8a) and in the part intended to give hydrophobic interactions (4b, 5b, 6b, 7b and 8b) (Table 1). It should be noted that according to some recent results reported by Gustin et al. [29] compounds in which a naphthyl residue is in the hydrophobic portion result in an increase of potency against SphK1 showing IC50 values between 1 and 2 μM. Therefore, we have examined whether similar modifications also increased the potency in our pyrimidinic core derivatives.
Table 1.
Structure of the new synthesized compounds. The polar head and hydrophobic tail are shown in blue and orange, respectively.
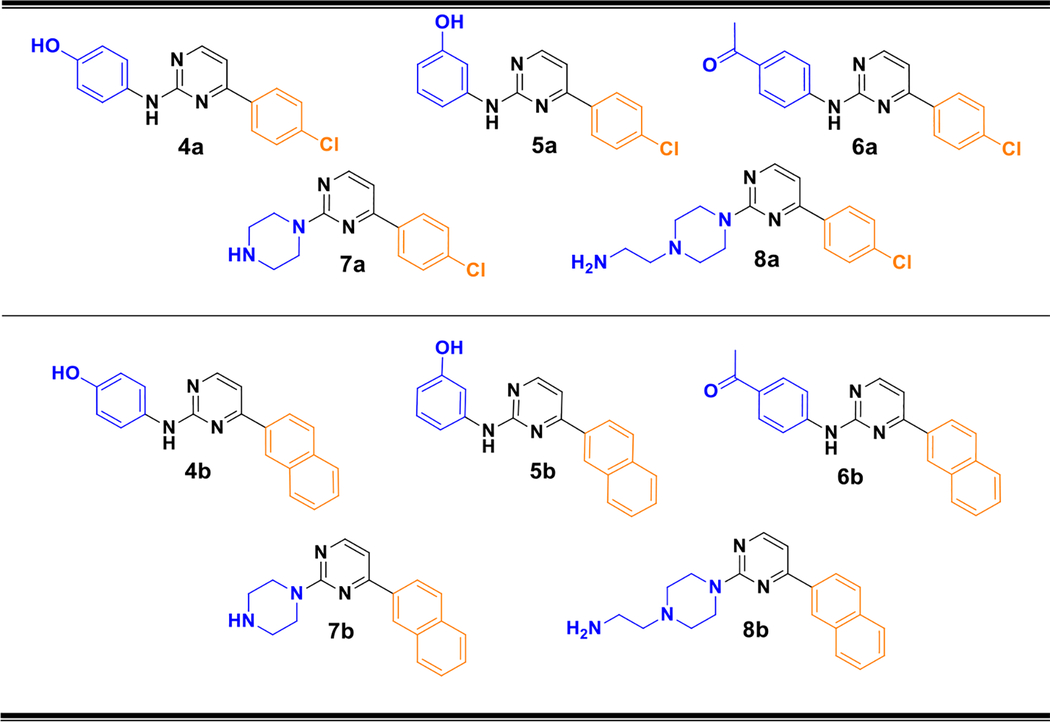
|
Unfortunately, none of these new compounds showed significant inhibitory activity against SphK1 and were even worse inhibitors than compound 4a (all compounds analyzed showed IC50 > 650 μM and therefore they were considered as inactive ones). While these results were disappointing, they showed that these structural changes have deleterious effects on inhibitory activity. In addition it is evident that it is not realistic to discover new compounds with inhibitory effects on SphK1 through structural changes made without a rational basis.
2.2. Structure based design of new SphK1 inhibitors
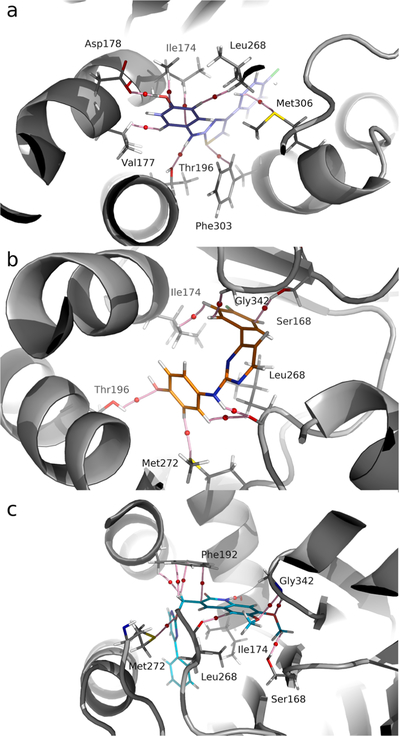
These results led us to try a different approach using structural based design and molecular docking. Through molecular docking studies, we observed that whereas SKI-II only adopts a highly preferred conformation corresponding to a shape similar to that of SKI-II in the crystal structure of SphK1 (PDB ID: 4VZD) (Fig. 5a), compound 4a can adopt two different conformations. In one of these conformations, although the polar and hydrophobic portions of the compounds are well oriented, their spatial arrangements are slightly shifted towards the center of the active site, compared to the shape adopted by SKI-II (see Fig. 5b). In contrast, the other conformation accommodates the polar and hydrophobic portion of 4a in inverted forms with respect to SKI-II (Fig. 5c). These explain, at least in part, why compound 4a have lower affinities for SphK1 than SKI-II. However, since it is well known that the results of docking should be considered as exploratory and preliminary and not definitive data; therefore we next carried out a more rigorous molecular modeling analysis. This molecular modeling study used molecular dynamics simulations (analysis by residue using MM-GBSA calculations) and also QTAIM (Quantum Theory of Atoms in Molecules) calculations for a more exhaustive evaluation of the different molecular complexes. The well-known inhibitor of SphK1, PF-543, which is the most potent inhibitor known to date, was used as a reference. In addition to the docking studies, simulations of molecular dynamics and analysis by residue obtained by MM-GBSA calculations were also performed.
Fig. 5.

(a) Crystal structure of SKI-II (pink) bound to SphK1. (b) Crystal structure of SKI-II (pink) bound to SphK1 superimposed with structure of 4a (cyan) in the conformation similar to SKI-II. (c) Crystal structure of SKI-II (pink) bound to SphK1 superimposed with 4a (cyan) in the inverted conformation respect to SKI-II. (d) Crystal structure of SKI-II (pink) bound to SphK1 together with 4a (cyan) superimposed on PF-543 (orange).
The docking study clearly shows that PF-543 is arranged along the pocket of the binding site of SphK1 in a conformation very similar to that found in the crystal structure (Fig. 5d). When the structures of 4a and SKI-II are superimposed, they occupy approximately the same site on SphK1 as PF-543 (Fig. 5d). Based on these results, we speculated that increasing the length of 4a might also increase its affinity with SphK1. Thus, we carried out molecular docking studies of compound 9, an elongated version of 4a, which has a larger polar portion (see Fig. 6). An isosteric replacement was also introduced into 9, changing the NH group to O in the linker chain to provide more flexibility. Before synthesizing compound 9, a docking study, molecular dynamics simulations and QTAIM calculations were carried out to evaluate its SphK1 binding behavior compared to the compounds previously evaluated (Figs. 7 and 8). In the docking studies, spatial superposition of the PF-543 with 9 showed that both were spatially arranged in a similar manner (Fig. 7). Strong hydrogen bond interactions were observed with Ile174 and Gly342 that stabilize their binding within the active site pocket of SphK1. This is consistent with Fig. 8, which clearly shows the importance of these two interactions with energies close to −8 and −4 Kcal/mol., respectively.
Fig. 6.

Chemical structure of compound 9.
Fig. 7.

Spatial view of PF-543 (orange) and compound 9 (violet) superimposed on the binding pocket of SphK1. The most important amino acids within the site are highlighted. The red lines represent the hydrogen bridge bonds that 9 establishes with different residues.
Fig. 8.

Superimposed histograms showing the interaction energies of: (a) 9 (cyan) versus SKI-II (magenta) and (b) 9 (cyan) versus PF-543 (orange) with the main SphK1 amino acids involved in inhibitor-SphK1 complex formation.
Fig. 8a and b show the analysis per residue obtained for 9, SKI-II and PF-543 from the molecular dynamics simulations. Compound 9 and SKI-II have similar interactions and in some cases the interactions obtained for 9 are stronger, for example with Ile174, Phe192, Leu259, Leu268, Met306 and Gly342. On the other hand, comparing 9 with PF-543, the lack of interaction with the Asp178 residue becomes clear, which is an important interaction that anchors the inhibitors within the active site [30]. However, in some cases, 9 has stronger interactions with Ile174 and Gly342 and also interactions of similar energy to those of PF-543, as is the case of Phe192, Leu268 and Met306. From a comparison of binding of these three inhibitors to different amino acids on SphK1, it can be predicted that the inhibitory potency of 9 against SphK1 should be intermediate between that of SKI-II and PF-543. Next, QTAIM calculations were carried out to analyze in more detail the molecular interactions with SphK1 of PF-543, SKI-II, 4a and 9.
For the QTAIM analysis, the molecules were divided into two portions (Fig. 9). It was considered as the non-polar part of the compounds (orange color, Fig. 9) all those atoms that are in the pyrimidine ring and in the p-chlorophenyl or naphthyl ring (structures “a” or “b”, respectively). The rest of the atoms of the different compounds were considered part of the polar zone (blue color, Fig. 9).
Fig. 9.

(a) General structure of the compounds showing the portions that were considered polar (blue) and non-polar (orange). (b) and (c) structures of PF-543 and compound 4a, respectively, highlighting the polar and non-polar regions of each one.
The QTAIM results are summarized in Fig. 10 which shows that rho total (Σρ(r)) value obtained for PF-543 is the highest of all of these compounds and that the greatest contribution to the interactions is provided by the portion of the molecule that corresponds to its cationic head (blue bar).
Fig. 10.

QTAIM analysis performed with PF-543, SKI-II, 4a and 9. The green bar indicates the rho total value (Σρ(r)) obtained for each compound. The blue and orange bars correspond to the rho value (ρ(r)) obtained for the polar and non-polar portions, respectively. The asterisk (*) represent the structure in which the compound is located “inverted” with respect to the crystal structure of SKI-II.
Compared with PF-543, the values obtained for SKI-II and 4a are substantially lower. Also there is a significant difference between SKI-II and 4a showing the first one better inhibitory activity. These results agree with our experimental data. In this analysis, the two conformations obtained for compound 4a were evaluated as two different molecules; the asterisk in Fig. 10 indicates the inverted structure, where the polar portion of the molecule is lodged in the hydrophobic region of the binding pocket of SphK1. The “correct” conformation (the one that locates its polar part in the polar region of the active site) has a greater interaction than the “inverted” form. The lower inhibitory activity of compound 4a compared to SKI-II could be explained due to the existence of these two conformations, since based on our simulations, it is not possible to determine what proportion of the compound is in each conformation.
The results obtained for 9 are particularly interesting, since the values of ρ(r) obtained for this compound indicate that it has stronger interactions with SphK1 than SKI-II and 4a. The increase in ρ(r) total is mainly due to the increases in interactions through its cationic head (almost double that for compound 4a). These interactions can be seen in more detail in the molecular structures of these compounds (Fig. 11).
Fig. 11.
Molecular graph of charge density obtained for the complexes of SphK1with: (a) SKI-II (blue), (b) compound 4a (orange) and (c) compound 9 (cyan). The pink lines connecting the nuclei are the binding routes, and the small red spheres on them are the binding critical points (BCP).
Fig. 11a shows the main interactions that stabilize SKI-II in its complex with SphK1. These interactions are with the following residues: Ile174, Val177, Asp178, Thr196, Leu268, Phe303 and Met306. The Bond Critical Point (BCP) and the corresponding binding pathways connecting the hydroxyl group of SKI-II with the carboxylic group of residue Asp178 is shown in Fig. 11a. The local charge density value (ρ(r)) in this BCP is 0.0631 atomic units (a.u.), which correspond to a strong hydrogen bond interaction [31].
SKI-II also interacts with Thr196 residue at the amino group level forming a moderate hydrogen bond with the OH group of this residue (ρ(r) = 0.0227 a.u.). In addition, SKI-II forms several hydrophobic intermolecular interactions with residues Ile174, Val177 and Leu268 in the region corresponding to the phenolic ring. Altogether, the interactions with Ile174, Val177 and Leu268 contribute 0.0277, 0.0114 and 0.0162 a.u. of charge density, respectively. On the other hand, the sulfur atom of the thiazide ring exhibits weak interactions with Phe303 (ρ(r) = 0.0143 a.u.). This interaction, although relatively weak, seems to be important for the adequate positioning of the inhibitor in the SphK1 cavity. Residue Met306 establishes weak interactions in the non-polar region of the ligand (ρ(r) = 0.0157 a.u.).
The structure of compound 4a was obtained replacing the thiazole ring of SKI-II with a pyrimidine ring. This produces changes at the level of the interactions stabilizing the complex in the active site of SphK1 as well as at the positioning within it (Fig. 11b). The hydroxyl group of the phenolic ring, instead of establishing interactions with Asp178, forms a weak hydrogen bond with the OH group of Thr196 (ρ(r) = 0.0055 a.u.). The main interaction in this complex corresponds to a moderate hydrogen bond N–H•••O=CO (ρ(r) = 0.0287 a.u.) with Leu268. In turn, the non-polar region of the molecule is stabilized by interactions with Ile174, Ser168 and Gly342 that contribute 0.0543, 0.0225 and 0.0088 a.u., respectively. These results are consistent with the experimental data since compound 4a has a weaker inhibitory activity than SKI-II.
Compound 9 (Fig. 11c), which has a quinolinic ring in the polar region of the molecule, has a greater number of interactions and significant strength compared to 4a. Interactions of the non-polar region contribute 0.0797 a.u., including both the p-chlorophenyl ring and the pyrimidine ring. This value is not very different to that obtained for compound 4a (Σρ(r) = 0.0693 a.u.). This indicates that the main interactions responsible for the affinity of 9 for SphK1 are located in the polar region of the molecule.
The data indicates that the methyl group substituent of the quinolinic ring forms hydrogen bonds (CH•••O) with Ser168, which are stabilizers within the structure of SphK1. These interactions are maintained throughout the dynamics of the protein and participate in the correlated movement (Σρ(r) = 0.0224 a.u.) [32]. Residue Gly342 establishes moderate interactions with the methoxy group of the quinolinic ring with a value of ρ(r) = 0.0245 a.u.
An important interaction in this complex is established between a hydrogen of the quinolinic ring and an oxygen of residue Ile174 (N–H•••O), with a strong hydrogen bond interaction corresponding to ρ(r) = 0.0528 a.u. A large number of stabilizing interactions of hydrophobic type with Phe192 are also observed, which contribute with Σρ(r) = 0.0235 a.u. In addition, Met272 and Leu268 contribute to a lesser extent to the anchoring of 9 with ρ(r) = 0.0033 and 0.0245 a.u. respectively.
These results led us to synthesize compound 9. The details of this synthesis are shown in Section 2.4. This compound showed IC50 values of 10.7 μM and 18.2 μM for SphK1 and SphK2, respectively, being more potent than 4a. While the inhibitory activity of compound 9 with SphK1 is weaker than that of PF-543, this result was very encouraging as it was in line with the molecular modeling results.
2.3. A new series of inhibitors of SphK1
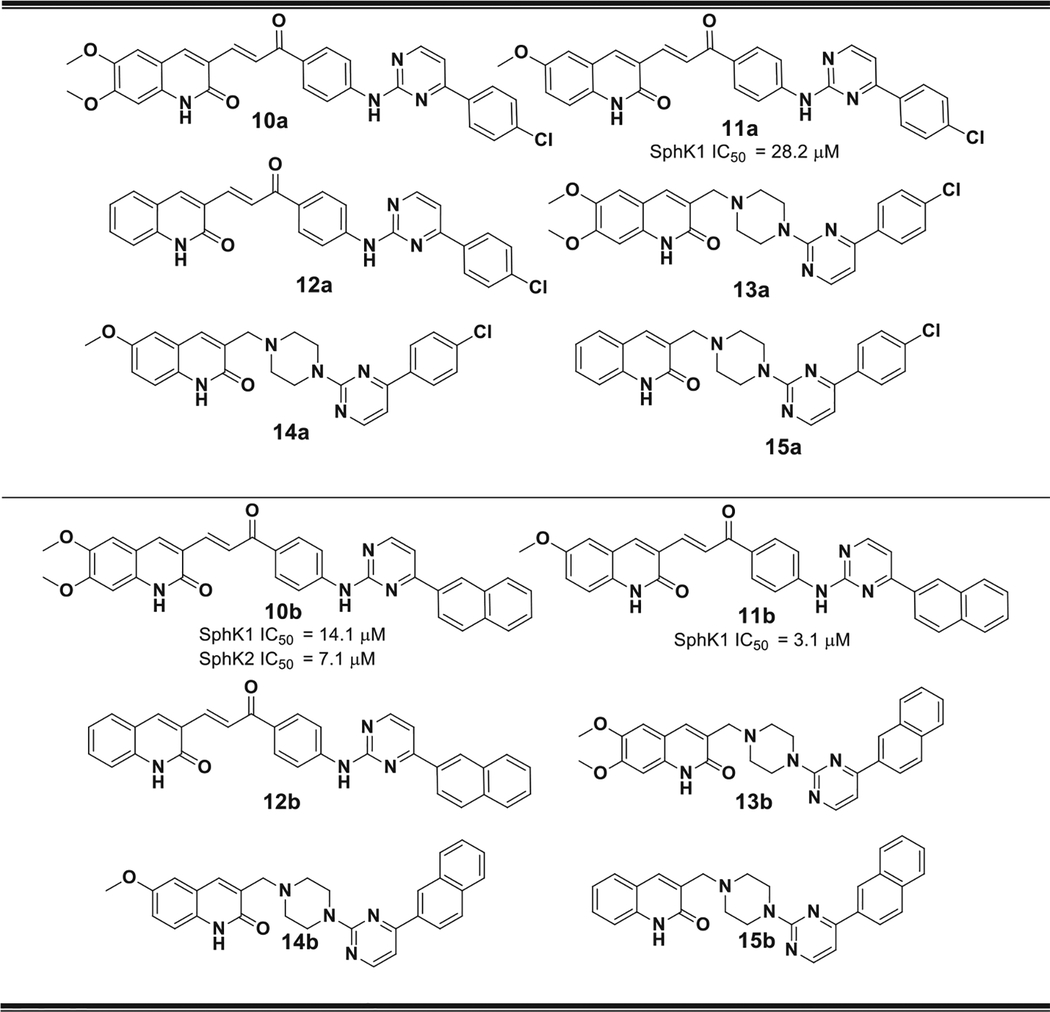
Analyzing in detail the binding site of SphK1, it can be seen that there is enough space to extend the structure of these compounds even further. Therefore, our next step was to construct a longer linker chain that joined the polar head with the hydrophobic portion. Three types of spacers were selected: a 1-phenylpropenone; a piperazine ring and aminophenyl fragments, a, b and c, respectively (shown in Fig. 12). This lead to the design and synthesis of compounds 10–19 (Tables 2 and 3).
Fig. 12.

Structure of the three types of linkers used: (a) 1-phenylpropenone fragment: 10–12, (b) a piperazine ring: 13–15 and (c) aminophenyl fragment: 16–19, respectively.
Table 2.
Structure of compounds 10–15. The IC50 values obtained for those compounds with IC50 < 650 μM are shown. All compounds with IC50 > 650 μM were considered inactive.
|
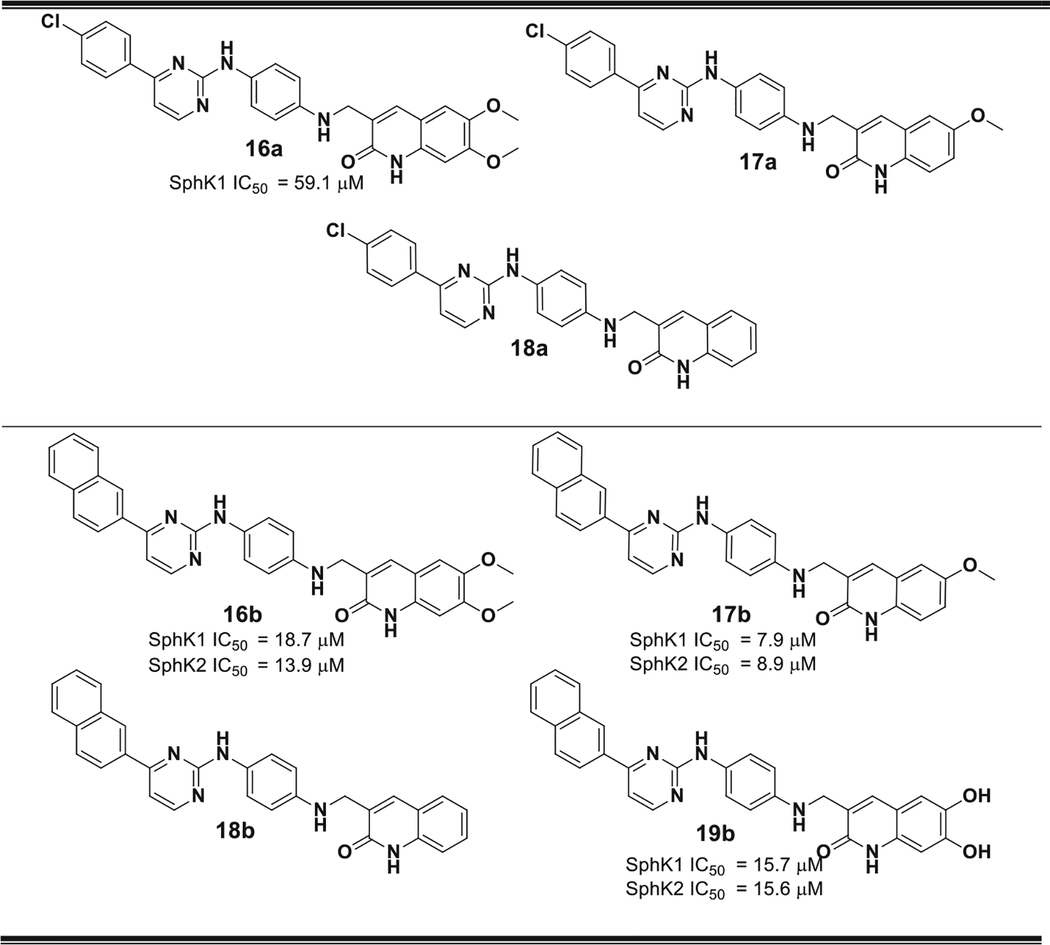
Table 3.
Structure of compounds 16–19. The IC50 values obtained for those compounds with IC50 < 650 μM are shown. All compounds with IC50 > 650 μM were considered inactive.
|
Fig. 13 summarizes the main results obtained for this new series of inhibitors of SphK1 and SphK2.
Fig. 13.

(a) SphK1 and (b) SphK2 inhibition rates for the indicated compounds. Only the IC50 values obtained for those compounds with IC50 < 650 μM are shown. All compounds with IC50 > 650 μM were considered inactive.
Some of these new compounds showed significantly stronger SphK inhibitory activities than compound 4a, including: 10b, 11a, 11b, 16a, 16b, 17b and 19b with IC50 values ranging 3.1–59.1 μM (Fig. 13). Although most of these compounds have inhibitory effects on both SphK1 and SphK2, the most active compound in the whole series, compound 11b, showed a selective inhibitory effect on SphK1 (IC50 = 3.1 μM).
With respect to the activity on SphK2, it is important to note that the compounds reported here have a strong inhibitory activity on this enzyme. It should be noted that the inhibitory effect reported for the well-known selective inhibitor of SphK2, compound K145 is 4.3 μM [33]. However, we reported previously that K145 displayed an inhibitory activity of 33.7 μM when it was evaluated using the technique used in this work [28]. On the basis of these results it is evident that the inhibitory activity on SphK2 of these new compounds is very strong, although not selective.
Considering, the structural characteristics of these compounds it is possible to make some correlations between structure and activity. For example, no compound with the “b” linker chain (13–15) showed significant inhibitory effects, displaying values of IC50 > 650 μM. Apparently this type of connecting chain gives rise to a spatial ordering that is not favorable. On the other hand, the substituent with naphthalene ring in the hydrophobic portion of the molecule has advantages over the benzene ring with Cl substituent in the para position. This can be seen by comparing the inhibitory activities of compounds 11a (IC50 = 28.2 μM) and 11b (IC50 = 3.1 μM), as well as between 16a (IC50 = 59.1 μM) and 16b (IC50 = 18.7 μM). On the other hand, there is no substantial improvement in the activity when the methoxyl group is replaced by a hydroxyl group as shown by the similar activities of 16b and 19b (IC50 = 15.7 μM).
In order to better understand the behavior of these compounds at the sub-molecular level, we carried out molecular simulation studies and QTAIM calculations for this new series.
Fig. 14 shows the results obtained from the analysis per residue for compounds 11b and 16b compared with PF-543. The data show that both (11b and 16b) have several similar interactions to those of PF-543, such as those with Leu259, Leu268, Phe303 and Leu319 for compound 11b and Ile174, Phe192, Thr196, Leu259 and Met306 for 16b. However, an important difference was evident when comparing the interaction with residue Asp81 in compounds 11b and 16b with that of PF-543, since this interaction is very weak (almost marginal) for PF-543 and quite significant for 11b and 16b, with almost −4 Kcal/mol in both cases (Fig. 14a–b).
Fig. 14.

Superimposed histograms showing the interaction energies of compounds: (a) 11b (green) and (b) 16b (blue) both with respect to the values obtained for the inhibitor PF-543 (orange).
In contrast, none of the new compounds shows significant interactions with Asp178, which is a very important residue for the anchoring of the ligands within the SphK active site [30].
QTAIM analyses of these compounds provided a fairly clear picture regarding the molecular interactions that are stabilizing the formation of the different molecular complexes.
Fig. 15 shows the values of rho (ρ(r)) obtained for compounds PF-543, 10b, 11a, 11b, 16a and 16b. As can be seen, these new compounds have weaker molecular interactions than PF-543, but very similar interactions to those of compound 9 and these results are in agreement with the experimental results obtained regarding their inhibitory activities. Comparison of PF-543 with respect the rest of the series shows that none of them have a strong interactions. In contrast, comparison of compounds 11a and 11b shows very similar values in the blue bars that correspond to the polar portion of the molecule. This is logical, as they have the same structure in that part of the molecule. However, there is a lower interaction in the hydrophobic region of compound 11a compared to compound 11b. This indicates that the naphthalene ring is the preferred hydrophobic portion. Similar conclusions are reached from the orange bars of compounds 16a and 16b.
Fig. 15.

QTAIM calculations obtained for compounds PF-543, 10b, 11a, 11b, 16a and 16b. The blue and orange bars correspond to the rho value (Σρ(r)) for the polar and non-polar portions of each ligand, respectively.
In order to describe these molecular interactions in more detail, the molecular structures obtained for compounds 11b and 16b are shown in Fig. 16. These compounds are the most active of their respective series. Similar to the data in the graph obtained from decomposition per residue, the molecular structure for compound 11b (Fig. 16a) shows the hydrogen bond formed between the amino group of the quinolinic ring and the oxygen of Asp81 (NH•••OC=O) with a value of ρ(r) = 0.0346 a.u. Although this interaction is only moderate, it seems to be important for the correct anchoring of the 11b within the SphK active site. Compound 16b also has a similar interaction, but is slightly weaker with a ρ(r) = 0.0262 a.u. (Fig. 16b).
Fig. 16.

Molecular structures showing the charge density obtained for the complex of SphK1 with (a) 11b (green) and (b) 16b (yellow). The pink lines connecting the nuclei are the binding routes, and the small red spheres on them are the bind critical points (BCP).
Compound 11b also has three interactions of the stabilizing type with residue Asp178, which add up to a total of Σρ(r) = 0.0201 a.u. As mentioned above, such interactions are not present in compound 16b, and therefore, due to their importance, could be responsible for the lower inhibitory activity of this compound compared to 11b. In addition, compound 11b, establishes an interaction of the π stacking type with Phe192 that is visible in Fig. 16a, with a total rho value equal to 0.0274 a.u. The pyrimidine ring region in 11b has important interactions with Ile174, Thr196 and Phe303 with ρ(r) = 0.0079; 0.0380 and 0.0100 a.u., respectively. Moreover, the naphthalene ring is stabilized through an interaction with Leu302 (ρ(r) = 0.0128 a.u.). Similar interactions were obtained for compound 16b.
2.4. Synthesis
For the preparation of the target hybrids, we have designed a convergent synthetic pathway in which the quinolone fragment is coupled to the linker, that was previously inserted in the pyrimidine core, either by condensation reaction from 3-formylquinolone derivatives 1a-c or by nucleophilic substitution from the chloromethylene derivative 1d-f, which is achieved from 1a-c by reduction to give an alcohol intermediate and followed by chlorination reaction (Scheme 1). These quinolone precursors can be easily prepared following a previously reported methodology that involves a Meth-Cohn type reaction [34], which consists in a three steps sequence starting from the corresponding aniline derivative Ia-c. An acetylation, followed by cyclization via a chloro-formylation under Vilsmeier conditions, and finally a hydrolysis of the chlorine residue were performed in order to get the lactam functionality finally achieve the synthetic intermediate 3-formylquinolone derivatives 1a-c.
Scheme 1.

Synthesis pathway to afford quinolone-fragment precursors 1a-i. The commercial starting material and known intermediates to quinolones 1 are numbered in roman letters I-III.
To prepare the pyrimidine moiety, a synthetic pathway starting from the commercially available 2,4-chloropyrimidine (IV) has been designed. The first step consists in a Suzuki type coupling reaction catalyzed by palladium (0) in order to perform the reaction with the corresponding boronic aryl acid derivatives, in a selective fashion towards C4 at the pyrimidine nucleus yielding precursors 2a-b [35,36]. The next step involves a nucleophilic aromatic substitution with the selected amino-linker at the remaining chloro in C-2 of the pyrimidine 2a-b to afford 4-arylpyrimidine intermediates (3–7ab) (Table 4).
Table 4.
Synthesis of 4-arylpyrimidine intermediates 3–8ab.
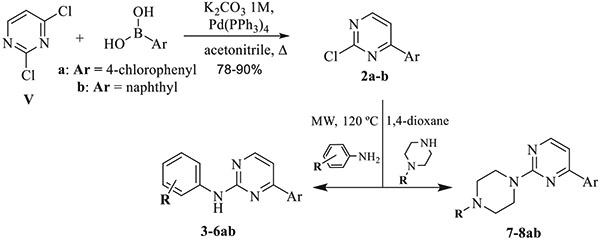 | |||
|---|---|---|---|
| Compound | R | Ar | Yield (%) |
| 3a | 4′-NH2 | p-Cl-Ph- | 71 |
| 3b | 4′-NH2 | Naphthyl | 65 |
| 4a | 4′-OH | p-Cl-Ph- | 85 |
| 4b | 4′-OH | naphthyl | 75 |
| 5a | 3′-OH | p-Cl-Ph- | 83 |
| 5b | 3′-OH | naphthyl | 64 |
| 6a | 4′-COCH3 | p-Cl-Ph- | 81 |
| 6b | 4′-COCH3 | naphthyl | 79 |
| 7a | H | p-Cl-Ph- | 76 |
| 7b | H | naphthyl | 68 |
| 8a | CH2-CH2NH2 | p-Cl-Ph- | 72 |
| 8b | CH2-CH2NH2 | naphthyl | 59 |
We firstly tried to introduce the quinolinone residue to the intermediates 8, for that, a reductive amination of quinolone-3-carbalde-hyde precursors 1a-c and 8a was performed, in a similar way to that below described with intermediates 4, but in this case instead of the expected coupling reaction, the linker was cleavage in the reaction conditions leading to compound 9. In turn, this resulted to be interesting to test the influence of the linker in the activity, so we have prepared directly from compound 1a, in a similar fashion to above reaction we performed the introduction quinolone fragment at pyrimidine by reaction of the 2-chloropyrimidine 1a, with the 3-hydro-xymethyl 1d yielding compound 9 in 53% yield (Scheme 2).
Scheme 2.

Synthesis of compound 9.
Finally, the coupling between linker-pyrimidine intermediate 6a-b and quinolones 1a-c yielded the final pyrimidine-quinolone chalcone analogues 10–12ab via Claisen-Schmidt condensation (Table 5).
Table 5.
Synthesis of pyrimidine-quinolone chalcones analogues 10–12ab.
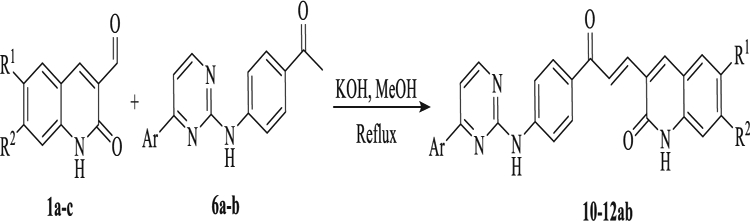 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | Ar | Yield (%) |
| 10a | CH3O− | CH3O− | p-Cl-Ph- | 43 |
| 10b | CH3O− | CH3O− | naphthyl | 50 |
| 11a | CH3O− | H | p-Cl-Ph- | 40 |
| 11b | CH3O− | H | naphthyl | 54 |
| 12a | H | H | p-Cl-Ph- | 39 |
| 12b | H | H | naphthyl | 65 |
For the coupling of quinolone residue to the amino functionality linker nucleophilic substitution reactions was needed to yield the desired quinolone-pyperazin-pyrimidine hybrids (13–15ab) (Table 6).
Table 6.
Synthesis of quinolone-pyperazin-pyrimidine hybrids 13–15ab.
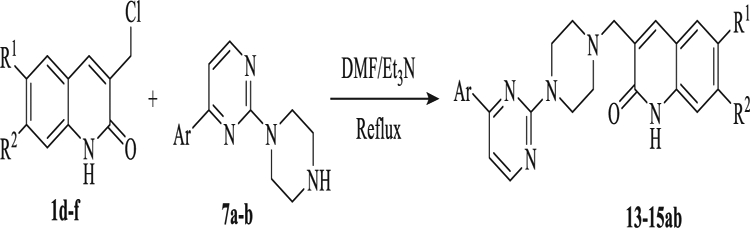 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | Ar | Yield (%) |
| 13a | CH3O− | CH3O− | p-Cl-Ph- | 50 |
| 13b | CH3O− | CH3O− | naphthyl | 57 |
| 14a | CH3O− | H | p-Cl-Ph- | 60 |
| 14b | CH3O− | H | naphthyl | 58 |
| 15a | H | H | p-Cl-Ph- | 55 |
| 15b | H | H | naphthyl | 52 |
In contrast to those reactions with a secondary amino group at linker like in 7a-b, when the linker has a primary amino as the reactive functionality like in 3a-b, this coupling reaction can proceed successfully by a reductive amination in a two-steps one pot reaction between the 3-formylquinolone 1a-c and pyrimidine-linker intermediates 3a-b to give the desired target products in good to excellent yields (Table 7).
Table 7.
Synthesis of quinolone-pyrimidine-pyrimidine hybrids 13–15ab.
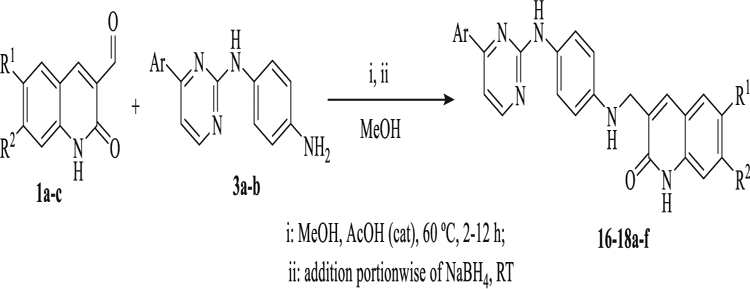 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | Ar | Yield (%) |
| 16a | CH3O− | CH3O− | p-Cl-Ph- | 86 |
| 16b | CH3O− | CH3O− | naphthyl | 80 |
| 17a | CH3O− | H | p-Cl-Ph- | 90 |
| 17b | CH3O− | H | naphthyl | 85 |
| 18a | H | H | p-Cl-Ph- | 79 |
| 18b | H | H | naphthyl | 82 |
Finally, to test the influence in the inhibition activity of the methoxy group vs hydroxyl at the quinolone residue, demethylation was carried out to one of those compound by treatment with borontribromide in dichloromethane at −78 °C; and so from 16b the corresponding demethylated derivative 19b was afforded in 38% yield.
3. Conclusions
In this study, we developed a new series of inhibitors of SphK1 and SphK2 by a rational study based on structural data, in which various molecular modeling techniques were used. This involved the design, synthesis and testing of a new series of inhibitors, some of them with strong inhibitory activity.
The molecular modeling we carried out using the potent SphK1 inhibitor PF-543 as a reference structure enabled us to design and synthesize another series of active compounds incorporating new connecting chains between the their polar heads and hydrophobic tails. Among the compounds reported here, molecules 9, 10b, 11b, 16b and 17b showed the best inhibitory activities. It is interesting that compound 11b which had the strongest inhibitory was a selective inhibitor of SphK1.
The combined techniques of molecular dynamics simulations and QTAIM calculations provided complete and detailed information about the molecular interactions that stabilize the different complexes of the new inhibitors with the active sites of the SphKs. This information will be useful in defining modifications and structural changes that could be made to increase the affinity for these types of compounds with SphK1. Furthermore, it will also help in the design of new SphK inhibitors that are structurally different.
4. Experimental section
4.1. Chemistry
Melting points were collected using a Brastead Electrothermal 9100 melting point apparatus, and the acquired data are uncorrected. IR spectra were recorded on a Fourier Bruker Tensor 27 Spectrophotometer using the ATR dura SamplIR accessory. NMR spectra were recorded in Bruker Avance 400 spectrometer at 400 MHz (1H) and 100 MHz (13C) at 298 K, using as solvents CDCl3 and DMSO‑d6 and as the internal reference tetramethylsilane (0 ppm), or the residual hydrogen and carbon solvent signals for 1H / 13C respectively, that is, 7.26 / 77.16 ppm and 2.50 / 39.52 ppm for CDCl3 and DMSO‑d6 respectively. DEPT and 2D-MNR (COSY, HSQC and HMBC) experiments were used for the assignment of carbon and hydrogen signals. Chemical shifts (δ) are given in ppm and coupling constants (J) are given in Hz. The following abbreviations are used for multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. The mass spectra were recorded on a Thermo model DSQ II spectrometer (equipped with a direct inlet probe) and operating at 70 eV. HPLCHRMS data were obtained on an Agilent Technologies Q-TOF 6520 spectrometer via an electrospray ionization (ESI, 4000 V). The reactions were monitored by TLC on a 0.2 mm pre–coated aluminium plates of silica gel (Merck 60 F254) and spots were visualized by UV irradiation (254 nm). Flash chromatography was performed on silica gel (40–63 μm). All reagents were purchased from commercial sources and used without further purification, unless otherwise noted. All starting materials were weighed and handled in air at room temperature. Precursors quinolone derivatives and 4-ary-2-chloropyrimidine were prepared according to reported procedures.
4.1.1. Synthesis of 2-oxoquinoline-3-carbaldehydes (1a-c) and 3-(hydroxymethyl)quinolinones (IVa-c) [34]
These compounds were prepared by following the Meth-Cohn method. N,N-dimethylformamide (9.6 mL, 0.125 mol) was cooled to 0 °C and phosphoryl chloride (32.2 mL, 0.35 mol) was added dropwise with stirring. To this solution, the corresponding acetanilides IIa-c (0.05 mol) was added and the temperature of the reaction mixture was raised to 80 °C during 8–19 h. The cooled reaction mixture was poured into ice-water (300 mL) and stirred for 10 min at 0–10 °C. The precipitate formed corresponding to 2-chloroquinoline-3-carbaldehydes IIIa-c was filtered off, washed with water (100 mL), dried, and recrystallized from acetonitrile. A suspension of these aldehydes III (1 mmol) in 70% aqueous acetic acid (50 mL) was heated under reflux for 24–72 h. Completion of the reaction was checked by TLC. After cooling the solid formed was filtered, washed well with water, dried and purified by recrystallization from DMF to obtain 1a-c. To a stirred solution of 2-oxo-1,2-dihydroquinoline-3-carbaldehydes 1a-c (1 mmol) in 15 mL methanol sodium borohydride (3 mmol) was slowly added at room temperature. The progress reaction was monitored by TLC (20:1 DCM:MeOH). After completion of the reaction (18 h), the solvent was removed under reduced pressure. To the residue formed, ice cold water was added and the obtained solid was filtered off and recrystallized from MeOH to obtain 1d-f. To a stirred solution of 1d-f (1 mmol) in DCM (10 mL), a solution of SOCl2 (2 mL) in DCM (5 mL) was added dropwise. After the addition was complete, four drops of DMF was added and the mixture was stirred for 2 h at reflux. The reaction progress was monitored by the TLC (20:1 DCM:MeOH). After the solvent was removed under reduced pressure the compound 3-(chloromethyl)-quinolin-2(1H)-ones 1g-i were obtained as a syrup and further used without purification.
4.1.2. General procedure for the synthesis of 2-chloro-4-(4-aryl) pyrimidines (2a-b) [35,36]
An aqueous solution of K2CO3 (3.86 mL, 1 M) was added to a solution of 2,4-dichloropyrimidine (575 mg, 3.86 mmol), the corresponding 4-chlorophenyl or naphtthylboronic acid (600 mg, 3.86 mmol) and Pd (PPh3)4 (45 mg, 0.039 mmol) in acetonitrile (13 mL). The mixture was heated to 90 °C for 7 h (for Ar = naphthyl) and 4 h (for Ar = 4-chlorophenyl) and monitored by TLC. The reaction mixture was reduced to half volume and the left to room temperature to appear a solid that was filtered, washed with water and dried. If the solid does not appear, the mixture was poured into water (30 mL), and extracted with EtOAc (50 mL × 2). The combined organic layers-phases were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. After removal of the solvent, the residue was purified by chromatography (ethylacetate/n-hexane 1:10) on silica gel to give 2-chloro-4-(4-aryl)pyrimidines 2a-b.
4.1.3. General procedure for the synthesis 1-(4-((4-phenylpyrimidin-2-yl) amino)phenyl)ethan-1-one and N1-(4-arylpyrimidin-2-yl)benzene-1,4-diamine (3–6ab) and 2-(4-(4-arylpyrimidin-2-yl)piperazin-1-yl)ethan-1-amines (7–8ab)
A mixture of pyrimidines 2a-b (1 mmol) and the corresponding substituted aniline (3 mmol) or piperazine (3 mmol) in 1,4-dioxane (1 mL) was subjected to microwave irradiation for 15 to 60 min at 120 °C, and 250 W of power. Then, the solvent was removed under reduced pressure and the crude was purified by recrystallization from EtOH and by column chromatography on silica gel using in a mixture CH2Cl2/MeOH (10:1) as eluent.
4.1.3.1. Synthesis of compound 3a-8b
4.1.3.1.1. 4-(4-(4-chlorophenyl)pyrimidin-2-yl)benzene-1,4-diamine (3a).
Yellow solid. 71% yield, mp, 161 °C, IR (ATR, cm−1): ν 3435 and 3357 (NH2), 3187 (NH); 1H NMR (400 MHz, DMSO‑d6) δ = 4.76 (s, 2H, NH2), 6.55 (d, J = 8.6 Hz, 2H, H-Ar), 7.23 (d, J = 5.1 Hz, 1H, CH), 7.37 (d, J = 8.6 Hz, 2H, H-Ar), 7.58 (d, J = 8.5 Hz, 2H, H-Ar), 8.12 (d, J = 8.5 Hz, 2H, H-Ar), 8.43 (d, J = 5.1 Hz, 1H, CH), 9.14 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 106.4 (CH) 113.9 (CH), 121.4 (CH), 128.5 (CH), 128.8 (CH), 129.4 (Cq), 135.4 (Cq), 135.7 (Cq), 143.8 (Cq), 159.1 (CH), 160.6 (Cq), 162.1 (Cq) ppm. EI MS (70 eV): m/z (%): 296/298 (M+, 100/32), 295 (94). ESI-QTOF (positive ionization) M + H calc. for C16H13ClN4 297.0906 found 297.0902.
4.1.3.1.2. N1-(4-(naphthalen-2-yl)pyrimidin-2-yl)benzene-1,4-diamine (3b).
Yellow solid. 65% yield, mp, 130–131 °C, IR (ATR, cm−1): ν 3387 and 3313 (NH2), 3191 (NH); 1H NMR (400 MHz, DMSO‑d6) δ = 4.76 (s, 2H, NH2), 6.58 (d, J = 8.8 Hz, 2H, H-Ar), 7.39 (d, J = 5.2 Hz, 1H, CH), 7.44 (d, J = 8.8 Hz, 2H, H-Ar), 7.68–7.52 (m, 2H), 8.00–7.93 (m, 1H), 8.09–8.00 (m, 2H), 8.24 (dd, J = 8.6, 1.8 Hz, 1H, CH), 8.47 (d, J = 5.2 Hz, 1H, CH), 8.70 (d, J = 1.2 Hz, 1H, CH), 9.18 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 106.9 (CH), 113.9 (CH), 121.4 (CH), 123.9 (CH), 126.6 (CH), 126.7 (CH), 127.3 (CH), 127.6 (CH), 128.3 (CH), 128.8 (CH), 129.6 (Cq), 132.7 (Cq), 134.0 (Cq), 134.3 (Cq), 143.7 (Cq), 158.9 (CH), 160.6 (Cq), 163.3 (Cq) ppm. EI MS (70 eV): m/z (%): 312 (M+, 100), 156 (15). ESI-QTOF (positive ionization) M + H calc. for C20H16N4 313,1453 found 313,1446.
4.1.3.1.3. 4-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenol (4a).
Beige solid. 85% yield, mp, 152 °C, IR (ATR, cm−1): ν 3272 (OH, NH), 1H NMR (400 MHz, CDCl3) δ = 6.02 (s, OH), 6.81 (d, J = 8.8 Hz, 2H, H-Ar), 7.06 (d, J = 5.3 Hz, 1H, CH), 7.15 (s, 1H, NH), 7.49–7.36 (m, 4H, H-Ar), 7.97 (d, J = 8.7 Hz, 2H, H-Ar), 8.41 (d, J = 5.3 Hz, 1H, CH). 13C NMR (101 MHz, CDCl3) δ = 107.6 (CH), 115.8 (CH), 122.5 (CH), 128.4 (CH), 129.0 (CH), 132.1 (Cq), 135.5 (Cq), 137.0 (Cq), 152.0 (Cq), 158.5 (CH), 160.6 (Cq), 164.0 (Cq) ppm. MS (70 eV): m/z (%): 297/299 (M+, 73/22), 296/298 (100/43), ESI-QTOF (positive ionization) M + H calc. for C16H12ClN3O 298.0747 found 298.0742.
4.1.3.1.4. 4-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenol (4b).
Beige solid. 75% yield, mp, 187 °C, IR (ATR, cm−1): ν 3388 (OH, NH) 1H NMR (400 MHz, DMSO‑d6) δ = 6.75 (d, J = 8.2 Hz, 2H, H-Ar), 7.45 (d, J = 5.2 Hz, 1H, CH), 7.67–7.52 (m, 4H), 8.01–7.91 (m, 1H, CH), 8.05 (d, J = 8.2 Hz, 2H, H-Ar), 8.25 (dd, J = 8.7, 1.8 Hz, 1H, CH), 8.50 (d, J = 5.2 Hz, 1H, CH), 8.71 (s, 1H, CH), 9.04 (s, 1H, NH), 9.35 (s, 1H, OH). 13C NMR (101 MHz, DMSO‑d6) δ = 107.3 (CH), 115.0 (CH), 121.2 (CH), 123.9 (CH), 126.7 (CH), 126.8 (CH), 127.4 (CH), 127.6 (CH), 128.3 (CH), 128.8 (CH), 132.1 (Cq), 132.7 (Cq), 134.0 (Cq), 134.24 (Cq), 152.3 (Cq), 158.9 (CH), 160.5 (Cq), 163.4 (Cq) ppm. MS (70 eV): m/z (%): 313 (M+, 79), 312 (100), 155 (14), 152 (16). ESI-QTOF (positive ionization) M + H calc. for C20H15N3O 314.1293 found 314.1288.
4.1.3.1.5. 3-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenol (5a).
Beige solid. 83% yield, mp, 162 °C, IR (ATR, cm−1): ν 3399 (OH, NH); 1H NMR (400 MHz, DMSO‑d6) δ = 7.06 (t, J = 8.1 Hz, 1H, CH), 7.21 (ddd, J = 8.1, 1.9, 0.8 Hz, 1H, CH), 7.36 (t, J = 2.2 Hz, 1H, CH), 7.38 (d, J = 5.2 Hz, 1H, CH), 7.60 (d, J = 8.7 Hz, 2H, H-Ar), 7.60 (d, J = 8.7 Hz, 2H, H-Ar), 8.54 (d, J = 5.2 Hz, 1H, CH), 9.25 (s, 1H, CH), 9.56 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 106.1 (CH), 107.7 (CH), 108.7 (CH), 109.9 (CH), 128.7 (CH), 128.9 (CH), 129.0 (CH), 135.5 (Cq), 135.6 (Cq), 141.5 (Cq), 157.5 (Cq), 159.1 (CH), 160.2 (Cq), 162.3 (Cq) ppm. MS (70 eV): m/z (%): 297/299 (M+, 52/17), 296 (100), ESI-QTOF (positive ionization) M + H calc. for C16H12ClN3O 298.0747 found 298.0743.
4.1.3.1.6. 3-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenol (5b).
Beige solid. 64% yield, mp, 158 °C, IR (ATR, cm−1): ν 3389 (OH), 3117 (NH); 1H NMR (400 MHz, DMSO‑d6) δ = 6.41 (dd, J = 7.9, 1.3 Hz, 1H, CH), 7.10 (t, J = 8.0 Hz, 1H, CH), 7.28 (d, J = 7.9 Hz, 1H, CH), 7.45 (s, 1H, CH), 7.53 (d, J = 5.2 Hz, 1H, CH), 7.70–7.55 (m, 2H), 8.02 – 7.92 (m, 1H, CH), 8.06 (d, J = 8.3 Hz, 2H), 8.29 (d, J = 8.5 Hz, 1H, CH), 8.57 (d, J = 5.2 Hz, 1H, CH), 8.76 (s, 1H, CH), 9.29 (s, 1H, NH), 9.59 (s, 1H, OH), 13C NMR (101 MHz, DMSO‑d6) δ = 106.1 (CH), 108.1 (CH), 108.7 (CH), 110.0 (CH), 124.0 (CH), 126.7 (CH), 127.0 (CH), 127.4 (CH), 127.6 (CH), 128.4 (CH), 128.8 (CH), 129.1 (CH), 132.7 (Cq), 134.1 (Cq x2), 141.6 (Cq), 157.5 (Cq), 158.9 (CH), 160.2 (Cq), 163.5 (Cq) ppm. MS (70 eV): m/z (%): 313 (M+, 56), 312 (100), 156 (18). ESI-QTOF (positive ionization) M + H calc. for C20H15N3O 314.1293 found 314.1287.
4.1.3.1.7. 1-(4-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl) ethan-1-one (6a).
Yellow solid. 81% yield, mp, 201–202 °C, IR (ATR, cm−1): ν 3310 (NH), 1663 (C=O); 1H NMR (400 MHz, DMSO‑d6) δ = 2.49 (s, 3H, CH3), 7.49 (d, J = 5.3 Hz, 1H, CH), 7.60 (d, J = 8.8 Hz, 2H, H-Ar), 7.91 (d, J = 9.1 Hz, 1H, H-Ar), 7.91 (d, J = 9.1 Hz, 1H, H-Ar), 8.18 (d, J = 8.8 Hz, 2H, H-Ar), 8.61 (d, J = 5.2 Hz, 1H, CH), 10.13 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 26.3 (CH3), 108.9 (CH), 117.6 (CH), 128.7 (CH), 129.0 (CH), 129.4 (CH), 129.9 (Cq), 135.2 (Cq), 135.8 (Cq), 145.1 (Cq), 159.3 (CH), 159.7 (Cq), 162.5 (Cq), 196.1 (C=O) ppm. EI MS (70 eV): m/z (%): 323/325 (M+, 68/22), 307 (100), 279 (37). ESI-QTOF (positive ionization) M + H calc. for C18H14ClN3O 324.0904 found 324.0897.
4.1.3.1.8. 1-(4-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl) ethenone (6b).
Yellow solid. 79% yield, mp, 130–131 °C, IR (ATR, cm−1): ν 3309 (NH), 1564 (C=O); 1H NMR (400 MHz, DMSO‑d6) δ = 2.53 (s, 3H, CH3), 7.62–7.56 (m, 2H), 7.65 (d, J = 5.2 Hz, 1H), 8.15–7.89 (m, 7H), 8.30 (dd, J = 8.6, 1.7 Hz, 1H), 8.66 (d, J = 5.2 Hz, 1H), 8.78 (s, 1H), 10.18 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 26.3 (CH3), 109.3 (CH), 117.6 (CH), 123.9 (CH), 126.7 (CH), 127.2 (CH), 127.5 (CH), 127.6 8 (Cq), 128.5 (CH), 128.9 (CH), 129.5 (CH), 129.8 8 (Cq), 132.7 8 (Cq), 133.8 8 (Cq), 134.1 8 (Cq), 145.2 8 (Cq), 159.1 (CH), 159.8 8 (Cq), 163.7 8 (Cq), 196.2 (C=O) ppm. EI MS (70 eV): EI MS (70 eV): m/z (%): 339 (M+, 100), 324 (87), 296 (45), 161 (34). ESI-QTOF (positive ionization) M + H calc. for C22H16N3O 340.1450 found 340.1442.
4.1.3.1.9. 4-(4-chlorophenyl)-2-(piperazin-1-yl)pyrimidine (7a).
White solid. 76% yield, mp, 89–90 °C, IR (ATR, cm−1): ν 3296 (NH); 1H NMR (400 MHz, CDCl3) δ = 3.28–2.74 (m, 4H), 4.17–3.73 (m, 4H), 6.88 (d, J = 5.1 Hz, 1H, CH), 7.42 (d, J = 8.7 Hz, 2H, H-Ar), 7.98 (d, J = 8.7 Hz, 2H, H-Ar), 8.37 (d, J = 5.1 Hz, 1H, CH), NH not detected. 13C NMR (101 MHz, CDCl3) δ = 45.0 (CH2) 46.1 (CH2), 105.2 (CH), 128.3 (CH), 128.8 (CH), 136.2 (Cq), 136.5 (Cq), 158.5(CH), 162.0 (Cq), 163.0 (Cq) ppm. EI MS (70 eV): m/z (%): 274/276 (M+, 13/4), 231 (78), 217 (42), 205 (100), 188 (21). ESI-QTOF (positive ionization) M + H calc. for C14H15ClN4 275.1063 found 275.1058.
4.1.3.1.10. 4-(naphthalen-2-yl)-2-(piperazin-1-yl)pyrimidine (7b).
White solid. 68% yield, mp, 112–113C, IR (ATR, cm−1): ν 3309 (NH); 1H NMR (400 MHz, CDCl3) δ = 3.05 (br, 4H, CH2), 4.00 (br, 4H, CH2), 7.11 (d, J = 5.1 Hz, 1H, CH), 7.71–7.45 (m, 2H), 8.06–7.86 (m, 3H), 8.19 (dd, J = 8.5, 1.4 Hz, 1H, CH), 8.44 (d, J = 5.0 Hz, 1H, CH), 8.56 (s, 1H, CH), NH not detected. 13C NMR (101 MHz, CDCl3) δ = 44.9 (CH2). 45.9 (CH2), 106.0 (CH), 124.2 (CH), 126.4 (CH), 127.1 (CH), 127.1 (CH), 127.7 (CH), 128.4 (CH), 129.0 (CH), 133.2 0 (Cq), 134.50 (Cq), 135.1 (Cq), 158.3 (CH), 162.1 0 (Cq), 164.3 0 (Cq) ppm. EI MS (70 eV): m/z (%): 290 (M+, 18), 248 (67), 234 (52), 222 (100), 204 (32). ESI-QTOF (positive ionization) M + H calc. for C18H18N4 291.1610 found 291.1604.
4.1.3.1.11. 2-(4-(4-(4-chlorophenyl)pyrimidin-2-yl)piperazin-1-yl) ethan-1-amine (8a).
White solid. 72% yield, mp, 93 °C, IR (ATR, cm−1): [3164, 3330,] (NH2). 1H NMR (400 MHz, CDCl3) δ = 1.68 (s, 2H, NH2), 2.49 (t, J = 6.1 Hz, 2H, CH2), 2.65 – 2.52 (m, 4H, CH2), 2.86 (t, J = 6.1 Hz, 2H, CH2), 4.09–3.82 (m, 4H, CH2), 6.88 (d, J = 5.1 Hz, 1H, CH), 7.43 (d, J = 8.6 Hz, 2H, H-Ar), 7.98 (d, J = 8.6 Hz, 2H, H-Ar), 8.37 (d, J = 5.1 Hz, 1H, CH). 13C NMR (101 MHz, CDCl3) δ = 38.8 (CH2), 43.8 (CH2), 53.2 (CH2), 61.1 (CH2), 105.3 (CH), 128.3 (CH), 128.8 (CH), 136.2 (Cq), 136.5 (Cq), 158.5 (CH), 161.9 (Cq), 163.0 (Cq) ppm. EI MS (70 eV): m/z (%): 317 (M+, 1), 287/289 (100/32, M−CH2NH2), 288 (18), 258 (49), 232 (54). ESI-QTOF (positive ionization) M + H calc. for C16H20ClN5 318,1485 found 318,1480.
4.1.3.1.12. 2-(4-(4-(naphthalen-2-yl)pyrimidin-2-yl)piperazin-1-yl) ethan-1-amine (8b).
White solid. 59% yield, mp, 107C, IR (ATR, cm−1): ν (3373, 3319 NH2); 1H NMR (400 MHz, CDCl3) δ = 1.63 (s, 2H, NH2), 2.50 (t, J = 6.1 Hz, 2H, CH2), 2.64–2.53 (m, 4H, CH2), 2.86 (t, J = 6.1 Hz, 2H, CH2), 4.06–3.88 (m, 4H, CH2), 7.07 (d, J = 5.2 Hz, 1H, CH), 7.57–7.47 (m, 2H), 7.89–7.84 (m, 1H, CH), 7.92 (d, J = 8.7 Hz, 1H, CH), 7.99–7.93 (m, 1H, CH), 8.16 (dd, J = 8.6, 1.8 Hz, 1H, CH), 8.41 (d, J = 5.2 Hz, 1H, CH), 8.52 (d, J = 1.2 Hz, 1H, CH) 13C NMR (101 MHz, CDCl3) δ = 38.8 (CH2), 43.9 (CH2), 53.3 (CH2), 61.2 (CH2), 105.9 (CH), 124.1(CH), 126.4 (CH), 127.0 (CH), 127.0 (CH), 127.7 (CH), 128.3 (CH), 128.9 (CH), 133.2, 134.5, 135.0, 158.3 (CH), 161.9, 164.2 ppm. EI MS (70 eV): m/z (%): 333 (M+, 2), 316 (7), 304 (21), 303 (100, M−CH2NH2), 274 (45), 248 (58), 204 (25). ESI-QTOF (positive ionization) M + H calc. for C20H23N5 334.2032 found 334.2025.
4.1.4. Synthesis of 3-((4-(4-chlorophenyl)pyrimidin-2-yl)-1-yl)oxymethyl) quinolin-2(1H)-ones (9)
A mixture of 2-chloro-4-(4-aryl)pyrimidine 2a (1 mmol) and 3-hydroxymethylquinolinein-2-one-3-carbaldehyde 1d-f in 1,4-dioxane (1 mL) was subjected to microwave irradiation for 15 min at 120 °C and 250 W of power. Then, the solvent was removed under reduced pressure and the crude was purified by recrystallization from EtOH.
The residue obtained was a beige solid. 53% yield, mp, 206 °C, IR (ATR, cm−1): ν 3199 (NH), 1645 (C=O), 1H NMR (400 MHz, DMSO‑d6) δ = 3.80 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 4.11 (s, 2H, OCH2), 6.95 (s, 1H, Quin-H), 7.18 (s, 1H, Quin-H), 7.27 (d, J = 3.5 Hz, 1H, CH), 7.57 (d, J = 8.6 Hz, 2H, Ar-H), 8.07 (s, 1H, Quin-H), 8.16 (d, J = 8.5 Hz, 2H, Ar-H), 8.48 (d, J = 4.9 Hz, 1H, CH), 12.07 (s, 1H, NH), 13C NMR (101 MHz, DMSO‑d6) δ = 46.2 (CH2), 55.6 (OCH3), 55.8 (OCH3), 97.6 (CH), 105.8 (CH), 108.7 (CH), 111.9 (Cq), 115.2 (CH), 119.9 (Cq), 128.6 (CH), 128.8 (CH), 134.6 (Cq), 135.5 (Cq), 135.6 (Cq), 140.8 (CH), 145.2 (Cq), 152.5 (Cq), 159.2 (CH), 161.4 (Cq), 162.1 (C=O) ppm. EI MS (70 eV): m/z (%): 423 (0.55), 287 (100), 258 (55), 232 (62), 217 (89) 128 (50). ESI-QTOF (positive ionization) M + H calc. for C22H18ClN3O4 424,1064 found 424.1058.
4.1.5. General procedure for the synthesis of (E)-3-(3-oxo-3-(4-((4-phenylpyrimidin-2-yl)amino)phenyl)prop-1-en-1-yl)quinolin-2(1H)-ones (10–12ab)
A mixture of the quinoline-3-carbaldehydes 1a-c (0.05 mmol), ethanones 6a-c (0.05 mmol) and solid KOH (3.2 mmol) in 30 mL MeOH/H2O (10/1) was heated under reflux for 3 days until the starting reagents were not detected by TLC. Then, water (2 mL) was added and the resulting precipitate was collected by filtration, washed with water and recrystallized from DMF.
4.1.5.1. Synthesis of compound 10a-12b
4.1.5.1.1. (E)-3-(3-(4-((4-(4-chlorophenyl)pyrimidin-2-yl)amino) phenyl)-3-oxoprop-1-en-1-yl)-6,7-dimethoxyquinolin-2(1H)-one (10a).
Yellow solid. 43% yield, mp, ˃300 °C, IR (ATR, cm−1): ν [3007, 3061] (NH), [1662, 1579] (C=O) 1537, 1415. 1H NMR (400 MHz, DMSO‑d6) δ = 4.02 (s, 3H, OCH3), 4.05 (s, 3H, OCH3), 6.94 (s, 1H, Quin-H), 7.21 (s, 1H, Quin-H), 7.55 (d, J = 4.8 Hz, 1H, CH), 7.65 (d, J = 7.9 Hz, 2H, Ar-H), 7.76 (d, J = 14.6 Hz, 1H, =CH), 8.02 (d, J = 8.5 Hz, 2H, Ar-H), 8.05 (d, J = 8.5 Hz, 2H, Ar-H), 8.20 (d, J = 7.9 Hz, 2H, Ar-H), 8.27 (d, J = 14.6 Hz, 1H, =CH), 8.39 (s, 1H, Quin-H), 8.60 (d, J = 4.8 Hz, 1H, CH), 10.05 (s, 1H, NH), 11.65 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6, 120 °C) δ = 55.4 (CH3), 56.0 (CH3), 98.2 (CH), 108.2 (CH), 110.5 (CH), 112.3 (Cq), 117.7 (CH), 123.1 (Cq), 123.3 (CH), 127.8 (CH), 128.1 (CH), 128.4 (CH), 131.0 (Cq), 135.0 (Cq), 135.0 (Cq), 135.1 (Cq), 137.6 (CH), 139.1 (CH), 144.1 (Cq), 145.2 (Cq), 153.4 (Cq), 158.2 (CH), 159.3 (Cq), 159.9 (Cq), 162.3 (C=O), 187.8 (C=O) ppm. EI MS (70 eV): m/z (%): 534 (M+,1), 323 (20), 307 (30), 229 (100). ESI-QTOF (positive ionization) M + H calc. for C30H23ClN4O4 539.1481 found 539.1477.
4.1.5.1.2. (E)-6,7-dimethoxy-3-(3-(4-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl)-3-oxoprop-1-en-1-yl)quinolin-2(1H)-one (10b).
Yellow solid. 50% yield, mp, ˃300 °C, IR (ATR, cm−1): ν [3261, 3165] (NH), [1660, 1570] (C=O) 1531, 1411. 1H NMR (400 MHz, DMSO‑d6) δ = 3.84 (s, 3H, OCH3). 3.87 (s, 3H, OCH3), 6.92 (s, 1H, Quin-H), 7.20 (s, 1H, Quin-H),7.69–7.59 (m, 2H, Ar-H), 7.72 (d, J = 5.2 Hz, 1H, CH), 7.81 (d, J = 15.4 Hz, 1H, =CH), 8.04 (m, 2H, Ar-H), 8.18–8.10 (m, 6H, Ar-H, Ar-H), 8.35 (d, J = 15.5 Hz, 1H, =CH), 8.36 (d, J = 8.8 Hz, 1H, Ar-H), 8.45 (s, 1H, Quin-H), 8.72 (d, J = 5.2 Hz, 1H, CH), 8.83 (s, 1H, NH), 10.26 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 56.1 (CH3), 56.2 (CH3), 109.0 (CH), 109.9 (CH), 113.4 (Cq), 118.4 (CH), 122.3 (CH), 122.4 (CH), 123.1 (Cq), 123.1 (Cq), 124.4 (CH), 127.3 (CH), 127.7 (CH), 128.1 (CH), 128.2 (CH), 129.1 (CH), 129.5 (CH), 130.1 (CH), 131.5 (Cq), 133.3 (Cq), 134.4 (Cq), 134.7 (Cq), 140.3 (CH), 140.6 (CH), 145.4 (Cq), 145.4 (Cq), 145.7 (Cq), 153.7 (Cq), 159.7 (CH), 160.3 (Cq), 164.3 (C=O), 188.2 (C=O) ppm. EI MS (70 eV): m/z (%): 554 (M+, 2), 339 (39), 324 (40), 296 (32), 244 (47), 230 (100), 200 (38). ESI-QTOF (positive ionization) M + H calc. for C34H26N4O4 555.2032 found 555.2018.
4.1.5.1.3. (E)-3-(3-(4-((4-(4-chlorophenyl)pyrimidin-2-yl)amino) phenyl)-3-oxoprop-1-en-1-yl)-6-methoxyquinolin-2(1H)-one (11a).
Yellow solid. 40% yield, mp, ˃300 °C, IR (ATR, cm−1): ν [3278, 3165] (NH), [1674, 1581] (C=O) 1546, 1415. 1H NMR (400 MHz, DMSO‑d6) δ = 3.84 (s, 3H, OCH3). 7.26–7.16 (m, 2H, Quin-H), 7.33 (d, J = 8.8 Hz, 1H, Quin-H), 7.43 (d, J = 5.2 Hz, 1H, CH), 7.59 (d, J = 8.5 Hz, 2H, Ar-H), 7.74 (d, J = 15.6 Hz, 1H, =CH), 8.09–7.99 (m, 4H, Ar-H), 8.17 (d, J = 8.5 Hz, 1H, Ar-H), 8.23 (d, J = 15.6 Hz, 1H, =CH), 8.35 (s, 1H, Quin-H), 8.61 (d, J = 5.2 Hz, 1H, CH), 9.65 (s, 1H, NH), 11.45 (s, 1H, NH), 13C NMR (101 MHz, DMSO‑d6) δ = 55.2 (CH3). 108.4 (CH), 109.4 (CH), 115.7 (CH), 117.7 (CH), 119.2 (Cq), 120.2 (CH), 124.6 (CH), 126.4 (Cq), 128.0 (CH), 128.2 (CH), 128.7 (CH), 130.7 (Cq), 133.2 (Cq), 135.0 (Cq), 135.2 (Cq), 137.1 (CH), 139.1 (CH), 144.4 (Cq), 154.1 (Cq), 158.4 (CH), 159.3 (Cq), 159.8 (Cq), 162.3 (C=O), 187.7 (C=O) ppm. EI MS (70 eV): m/z (%): 508 (M+, 1), 281 (14), 200 (100) 169 (40). ESI-QTOF (positive ionization) M + H calc. for C29H21ClN4O3 509,1380 found 509.1371.
4.1.5.1.4. (E)-6-methoxy-3-(3-(4-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl)-3-oxoprop-1-en-1-yl)quinolin-2(1H)-one (11b).
Yellow solid. 54% yield, mp, ˃300 °C, IR (ATR, cm−1): ν [3265, 3165] (NH), [1660, 1627] (C=O) 1579, 1411. 1H NMR (400 MHz, DMSO‑d6) δ = 3.84 (s, 3H, OCH3), 7.25 (s, 1H, Quin-H), 7.26 (d, J = 7.1 Hz, 1H, Quin-H), 7.33 (d, J = 9.7 Hz, 1H, Ar-H), 7.70–7.59 (m, 2H, Ar-H), 7.73 (d, J = 5.2 Hz, 1H, CH), 7.83 (d, J = 15.5 Hz, 1H, =CH), 8.08–8.00 (m, 1H, Ar-H), 8.21–8.11 (m, 6H, Ar-H), 8.36 (d, J = 7.0 Hz, 1H, Quin-H) 8.39 (d, J = 15.4 Hz, 1H, =CH), 8.58 (s, 1H, Ar-H), 8.73 (d, J = 5.2 Hz, 1H, CH), 8.84 (s, 1H, Quin-H), 10.30 (s, 1H, NH), 12.0 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 56.0 (CH3), 109.8 (Cq), 110.0 (CH), 117.01 (CH), 118.4 (CH), 120.19, 121.8 (CH), 124.4 (CH), 124.6 (CH), 125.7 (Cq), 127.3 (CH), 127.8 (CH), 128.1 (CH), 128.2 (CH), 129.1 (CH), 129.5 (CH), 130.3 (CH), 131.0 (Cq), 133.3 (Cq), 134.2 (Cq), 134.3 (Cq), 134.7 (Cq), 138.4 (CH), 140.7 (CH), 146.0 (Cq), 154.9 (Cq), 159.7 (CH), 160.3 (Cq), 161.1 (C=O), 164.3, 188.0 (C=O) ppm. EI MS (70 eV): m/z (%): 524 (M+, 4), 340 (17), 297 (38), 199 (100). ESI-QTOF (positive ionization) M + H calc. for C33H24N4O3 525.1927 found 525.1910.
4.1.5.1.5. (E)-3-(3-(4-((4-(4-chlorophenyl)pyrimidin-2-yl)amino) phenyl)-3-oxoprop-1-en-1-yl)quinolin-2(1H)-one (12a).
Yellow solid. 39% yield, mp, ˃300 °C, IR (ATR, cm−1): ν [3261, 3167] (NH), [1658, 1649] (C=O) 1583, 1421. 1H NMR (400 MHz, DMSO‑d6) δ = 7.21 (t, J = 7.5 Hz, 1H, Quin-H), 7.38 (d, J = 8.2 Hz, 1H, Quin-H), 7.43 (d, J = 5.2 Hz, 1H, CH), 7.53 (t, J = 7.7 Hz, 1H, Quin-H), 7.60 (d, J = 8.6 Hz, 2H, Ar-H), 7.71 (d, J = 7.9 Hz, 1H, Quin-H), 7.75 (d, J = 15.7 Hz, 1H, =CH), 8.09–7.97 (m, 4H, Ar-H), 8.17 (d, J = 8.6 Hz, 2H, Ar-H), 8.23 (d, J = 15.7 Hz, 1H, =CH), 8.40 (s, 1H, Quin-H), 8.62 (d, J = 5.2 Hz, 1H, CH), 9.66 (s, 1H, NH), 11.56 (s, 1H, NH), 13C NMR (101 MHz, DMSO‑d6) δ = 108.4 (CH), 113.8 (Cq), 114.4 (CH), 117.7 (CH), 118.6 (Cq), 121.4 (CH), 124.6 (CH), 126.1 (Cq), 127.8 (CH), 128.0 (CH), 128.2 (CH), 128.7 (CH), 130.5 (CH), 135.0 (Cq), 135.2 (Cq), 137.0 (CH), 138.4 (Cq), 139.5 (CH), 144.4 (Cq), 158.4 (CH), 159.3 (Cq), 160.2 (Cq), 162.3 (C=O), 187.7 (C=O) ppm. EI MS (70 eV): m/z (%): 478 (M+, 5), 281 (16), 267 (17), 170 (100). ESI-QTOF (positive ionization) M + H calc. for C28H19ClN4O2 479.1275 found 479.1262.
4.1.5.1.6. (E)-3-(3-(4-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino) phenyl)-3-oxoprop-1-en-1-yl)quinolin-2(1H)-one (12b).
Yellow solid. 65% yield, mp, ˃300 °C, IR (ATR, cm−1): ν [3267, 3170] (NH), [1660, 1579] (C=O) 1533, 1409. 1H NMR (400 MHz, DMSO‑d6) δ = 7.27 (t, J = 7.2 Hz, 1H, Quin-H). 7.39 (d, J = 8.1 Hz, 1H, Quin-H), 7.59 (t, J = 7.3 Hz, 1H, Quin-H), 7.68–7.62 (m, 2H), 7.72 (d, J = 5.1 Hz, 1H, CH), 7.75 (d, J = 7.8 Hz, 1H, Ar-H), 7.84 (d, J = 15.5 Hz, 1H, =CH), 8.03 (d, J = 4.9 Hz, 1H Ar-H), 8.0.08–8.21 (m, 6H, Ar-H), 8.36 (d, J = 7.1 Hz, 1H, Quin-H), 8.38 (d, J = 15.1 Hz, 1H, =CH), 8.64 (s, 1H, Quin-H), 8.72 (d, J = 4.9 Hz, 1H, NH), 8.83 (s, 1H, NH), 13C NMR (101 MHz, DMSO‑d6) δ = 108.9 (CH), 114.5 (CH), 117.6 (CH x2), 118.7 (Cq), 121.4 (CH), 123.3 (CH), 124.6 (CH), 125.9 (CH), 126.1 (Cq), 126.5 (CH), 126.7 (CH), 126.9 (CH), 127.7 (CH), 127.8 (CH), 128.2 (CH), 128.8 (CH x2), 130.5 (CH), 130.6 (Cq), 132.3 (Cq), 133.6 (Cq), 133.7 (Cq), 137.0 (CH), 138.5 (Cq), 139.5 (CH), 144.6 (Cq), 158.2 (CH), 159.4 (Cq), 160.2 (Cq), 163.5 (C=O), 187.7 (C=O) ppm. EI MS (70 eV): m/z (%): 494 (M+, 5), 340 (31), 324 (38), 297 (77), 170 (100). ESI-QTOF (positive ionization) M + H calc. for C32H22N4O2 495.1821 found 495.1814.
4.1.6. Synthesis of 3-((4-arylpyrimidin-2-yl)piperazin-1-yl)methyl) quinolin-2(1H)-ones (13–15ab)
To the crude products 1g-i dissolved in DMF (10 mL), 4-aryl-2-(piperazin-1-yl)pyrimidines 7a-b (1 mmol) and Et3N (three drops) were added. The stirring was continued for 8 h at reflux and after complete conversion (monitored by the TLC, DCM:MeOH 20:1), the cooled reaction mixture was poured into ice-water. The precipitate formed was filtered off, washed with water, dried and purified by column chromatography on silica gel using a mixture CH2Cl2/MeOH (20:1) as eluent.
4.1.6.1. Synthesis of compound 13a-15b
4.1.6.1.1. 3-((4-(4-(4-chlorophenyl)pyrimidin-2-yl)piperazin-1-yl) methyl)-6,7-dimethoxyquinolin-2(1H)-one (13a).
White solid. 50% yield, mp, 231–231 °C, IR (ATR, cm−1): ν 3309 (NH), 1645 (C=O), 1571, 1517. 1H NMR (400 MHz, CDCl3) δ = 2.71 (br, 4H, CH2). 3.68 (s, 2H, CH2), 6.87 (s, 1H), 394 (br, 4H, CH2), 3.98 (s, 6H, OCH3), 6.89 (d, J = 5.0 Hz, 1H, CH), 6.90 (s, 1H, CH, Quin-H), 6.98 (s, 1H, Quin-H), 7.42 (d, J = 8.2 Hz, 2H, H-Ar), 7.86 (s, 1H, Quin-H), 7.97 (d, J = 8.2 Hz, 2H, H-Ar), 8.37 (d, J = 5.0 Hz, 1H, CH), 12.00 (s, 1H, NH). 13C NMR (101 MHz, CDCl3) δ = 43.9 (CH2), 53.2 (CH2), 56.2 (OCH3), 56.2 (OCH3), 56.3 (CH2), 97.9 (CH), 105.3 (CH, Cq), 108.0 (CH), 113.5 (Cq), 126.1 (Cq), 128.3 (CH), 128.8 (CH), 133.5 (Cq), 136.1 (Cq), 136.5 (Cq), 138.1 (CH), 145.9 (Cq), 152.2 (Cq), 158.5 (CH, Cq), 161.8 (Cq), 163.1 (Cq), 163.9 (C=O), ppm. EI MS (70 eV): m/z (%): 491 (M+, 4), 273 (37) 217 (100). ESI-QTOF (positive ionization) M + H calc. for C26H26ClN5O3 492.1797 found 492.1793.
4.1.6.1.2. 6,7-dimethoxy-3-((4-(4-(naphthalen-2-yl)pyrimidin-2-yl) piperazin-1-yl)methyl)quinolin-2(1H)-one (13b).
White solid. 57% yield, mp, 262–263 °C, IR (ATR, cm−1): ν 3313 (NH), 1653 (C=O), 1581, 1555. 1H NMR (400 MHz, CDCl3) δ = 2.63–2.80 (m, 4H, CH2 × 2), 3.70 (s, 2H, CH2), 3.93 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 4.12–4.01 (m, 4H, CH2 × 2), 6.89 (s, 1H, CH), 6.98 (s, 1H, Quin-H), 7.08 (d, J = 5.2 Hz, 1H, Quin-H), 7.59–7.46 (m, 2H, CH), 8.00–7.81 (m, 4H), 8.15 (dd, J = 8.6, 1.7 Hz, 1H), 8.41 (d, J = 5.2 Hz, 1H, CH), 8.52 (s, 1H, Quin-H), 12.19 (s, 1H, NH). 13C NMR (101 MHz, CDCl3) δ = 43.9 (CH2). 53.3 (CH2), 56.2 (CH3), 56.2 (CH3), 56.2 (CH2), 97.9 (CH), 105.9 (CH), 107.9 (CH), 113.5 (Cq), 124.1 (CH), 126.4 (CH), 127.0 (CH), 127.0 (CH), 127.7 (CH), 128.2 (CH), 128.9 (CH), 133.2 (Cq), 133.5 (Cq), 134.5 (Cq), 135.0 (Cq), 145.2 (Cq), 152.18 (Cq), 158.3 (CH × 2), 161.9 (Cq), 164.0 (Cq), 164.2 (C=O) ppm. EI MS (70 eV): m/z (%): 507 (M+, 7), 289 (40) 234 (36). 217 (100), 204 (22). ESI-QTOF (positive ionization) M + H. calc. for C30H29N5O3 508.2343 found 508.2339.
4.1.6.1.3. 3-((4-(4-(4-chlorophenyl)pyrimidin-2-yl)piperazin-1-yl) methyl)-6-methoxyquinolin-2(1H)-one (14a).
White solid. 60% yield, mp, 262–263 °C, IR (ATR, cm−1): ν 3151 (NH), 1653 (C=O), 1620, 1562. 1H NMR (400 MHz, DMSO‑d6) δ = 2.58 (br. 4H, CH2 × 2), 3.48 (s, 2H, CH2), 3.80 (s, 3H, OCH3), 3.90 (br, 4H, CH2 × 2), 7.13 (d, J = 8.8 Hz, 1H, Quin-H), 7.22 (d, J = 5.0 Hz, 1H, CH), 7.26 (d, J = 8.8 Hz, 2H, Quin-H), 7.27 (s, 1H, Quin-H), 7.57 (d, J = 8.2 Hz, 2H, Ar-H), 7.90 (s, 1H Quin-H), 8.16 (d, J = 8.2 Hz, 2H, Ar-H), 8.46 (d, J = 5.0 Hz, 1H, CH), 11.70 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 44.0 (CH2), 53.2 (CH2), 55.9 (OCH3), 56.3 (CH2), 105.9 (CH), 109.7 (CH), 116.5 (CH), 119.3 (CH), 120.3 (Cq), 129.1 (CH), 129.3 (CH), 130.3 (Cq), 133.0 (Cq), 136.0 (Cq), 136.2 (Cq), 136.4 (Cq), 137.0 (CH), 154.6 (Cq × 2), 159.6 (CH), 161.9 (Cq), 161.9 (Cq), 162.5 (C=O) ppm. EI MS (70 eV): m/z (%): 461 (M+, 10), 273 (54), 256 (34), 231 (31), 217 (26), 188 (100). ESI-QTOF (positive ionization) M + H calc. for C25H24ClN5O2 462.1691 found 462.1687.
4.1.6.1.4. 6-methoxy-3-((4-(4-(naphthalen-2-yl)pyrimidin-2-yl) piperazin-1-yl)methyl)quinolin-2(1H)-one (14b).
White solid. 58% yield, mp, 247–248 °C, IR (ATR, cm−1): ν 3154 (NH), 1656 (C=O), 1622, 1568. 1H NMR (400 MHz, DMSO‑d6) δ = 2.61 (br, 4H, CH2 × 2). 3.50 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 3.96 (br, 4H, CH2 × 2), 7.13 (d, J = 8.9 Hz, 1H, Ar-H), 7.27 (d, J = 9.2 Hz, 1H, Ar-H), 7.29 (s, 1H, Quin-H), 7.38 (d, J = 5.1 Hz, 1H, CH), 7.64–7.54 (m, 2H, Ar-H), 7.92 (s, 1H, Quin-H), 8.01–7.96 (m, 1H, Ar-H), 8.04 (d, J = 8.6 Hz, 1H, Quin-H), 8.09 (d, J = 5.9 Hz, 1H, Ar-H), 8.28 (d, J = 8.6 Hz, 1H, Quin-H), 8.50 (d, J = 5.1 Hz, 1H, Ar-H), 8.75 (s, 1H, Ar-H), 11.71 (s, 1H, NH), 13C NMR (101 MHz, DMSO‑d6) δ = 44.1 (CH2), 53.3 (CH2), 55.9 (CH3), 56.3 (CH2), 106.3 (CH), 109.7 (CH), 116.5 (CH), 119.4 (CH), 120.3 (Cq), 124.4 (CH), 127.1 (CH), 127.3 (CH), 127.8 (CH), 128.1 (CH), 128.8 (CH), 129.4 (CH, Cq), 130.4 (Cq), 132.9 (Cq), 133.3 (Cq), 134.6(Cq), 134.8, 137.0 (CH), 154.6 (Cq × 2), 159.4 (Cq), 161.9 (Cq), 162.0 (Cq), 163.6 (C=O) ppm. EI MS (70 eV): m/z (%): 477 (M+, 16), 289 (84), 256 (54), 234 (71), 188 (100). 173 (23). ESI-QTOF (positive ionization) M + H calc. for C29H27N5O2 478.2238 found 478.2233.
4.1.6.1.5. 3-((4-(4-(4-chlorophenyl)pyrimidin-2-yl)piperazin-1-yl) methyl)quinolin-2(1H)-one (15a).
White solid. 55% yield, mp, 251–252 °C, IR (ATR, cm−1): ν 3165 (NH), 1652 (C=O), 1618, 1562. 1H NMR (400 MHz, DMSO‑d6) δ = 2.58 (br, 4H, CH2 × 2). 3.49 (s, 2H, CH2), 3.90 (br, 4H, CH2 × 2), 7.19 (t, J = 8.1 Hz, 1H, Quin-H), 7.23 (d, J = 5.2 Hz, 1H, CH), 7.33 (d, J = 8.1 Hz, 1H, Quin-H), 7.48 (t, J = 8.5 Hz, 1H, Quin-H), 7.58 (d, J = 8.6 Hz, 2H, Ar-H), 7.71 (d, J = 7.1 Hz, 1H, CH), 7.94 (s, 1H, Quin-H), 8.17 (d, J = 8.6 Hz, 2H, Ar-H), 8.46 (d, J = 5.1 Hz, 1H, CH), 11.81 (s, 1H, NH), 13C NMR (101 MHz, DMSO‑d6) δ = 44.0 (CH2), 53.2 (CH2), 56.2 (CH2), 105.9 (CH), 115.3 (CH), 119.7 (Cq), 122.2 (CH), 128.2 (CH), 129.1 (CH), 129.3 (CH), 129.9 (Cq), 130.2 (CH), 136.0 (Cq), 136.2 (Cq × 2), 137.5 (CH), 138.5 (Cq × 2), 159.6 (CH), 161.9 (Cq), 162.4 (Cq), 162.5 (C=O) ppm. EI MS (70 eV): EI MS (70 eV): m/z (%): 431 (M+, 9), 273 (49), 217 (36), 213 (44), 157 (100), 129 (28). ESI-QTOF (positive ionization) M +H calc. for C24H22ClN5O 432.1586 found 432.1585.
4.1.6.1.6. 3-((4-(4-(naphthalen-2-yl)pyrimidin-2-yl)piperazin-1-yl) methyl)quinolin-2(1H)-one (15b).
White solid. 52% yield, mp, 251–252 °C, IR (ATR, cm−1): ν 3158 (NH), 1656 (C=O), 1618, 1566. 1H NMR (400 MHz, DMSO‑d6) δ = 2.65 (t, J = 5.1 Hz, 4H, CH2 × 2), 3.55 (s, 2H, CH2), 3.94 (t, J = 5.1 Hz, 4H, CH2 × 2), 7.14 (t, J = 8.5 Hz, 1H, Quin-H), 7.25 (d, J = 5.1 Hz, 1H, CH), 7.34 (d, J = 8.4 Hz, 1H, Quin-H), 7.43 (t, J = 8.4 Hz, 1H, Ar-H), 7.59–7.50 (m, 2H, Ar-H), 7.63 (dd, J = 7.8, 1.0 Hz, 1H, Ar-H), 7.89 (s, 1H, Ar-H), 7.96–7.90 (m, 1H, Ar-H), 7.99 (d, J = 8.6 Hz, 1H, Quin-H), 8.19 (dd, J = 8.5, 1.8 Hz, 1H, Quin-H), 8.45 (d, J = 5.1 Hz, 1H, CH), 8.63 (s, 1H, Quin-H), 11.24 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 43.2 (CH2), 52.0 (CH2), 55.0 (CH2), 105.3 (CH), 114.1 (CH), 118.7 (Cq), 120.8 (CH), 123.3 (CH), 125.7 (CH), 126.0 (CH), 126.4 (CH), 126.8 (CH × 2), 127.5 (CH), 128.1 (CH), 128.7 (CH, Cq), 129.2 (Cq), 132.3 (Cq), 133.5 (Cq × 2), 134.2 (Cq), 136.4 (CH), 137.7 (Cq), 157.9 (CH), 161.2 (Cq × 2), 162.9 (C=O), EI MS (70 eV): m/z (%): 447 (M+, 16), 289 (78), 274 (30), 246 (49), 234 (100), 226 (59), 204 (50) 158 (48). ESI-QTOF (positive ionization) M + H calc. for C28H25N5O 448.2132 found 448.2129.
4.1.7. General procedure for the synthesis of the novel 3-(((4-((4-Arylpyrimidin-2-yl)amino)phenyl)amino)methyl)quinolin-2(1H)-ones (16–18ab)
A mixture of quinoline-3-carbaldehyde 1a-c (0.05 mmol), amines 4a-b (0.05 mmol) and catalytic amount of AcOH (a few drops) in MeOH (10 mL), was heated at 60 °C for 2 – 12 h. After complete disappearance of the starting materials (monitored by TLC), the mixture was allowed to cool to room temperature. Subsequently, solid NaBH4 (5.0 mmol) was added portion-wise and the stirring was continued at ambient temperature for 24 h. After the reaction was complete (monitored by TLC), the volume of the reaction mixture was concentrated to 1 mL under reduced pressure. Then, water (2 mL) was added and the resulting precipitate was collected by filtration, washed with water and recrystallized from EtOH.
4.1.7.1. Synthesis of compound 16a-18b
4.1.7.1.1. 3-(((4-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl) amino)methyl)-6,7-dimethoxyquinolin-2(1H)-one (16a).
Yellow solid. 86% yield, mp, 261–262 °C, IR (ATR, cm−1): ν [3384, 3271, 3268] (NH), 1648 (C=O), 1640, 1560. 1H NMR (400 MHz, DMSO‑d6) δ = 3.73 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 4.10 (d, J = 5.8 Hz, 2H, CH2), 5.84 (t, J = 5.8 Hz, 1H, CH2-NH), 6.56 (d, J = 8.9 Hz, 2H, Ar-H), 6.86 (s, 1H, Quin-H), 7.12 (s, 1H, Quin-H), 7.24 (d, J = 5.2 Hz, 1H, CH), 7.43 (d, J = 8.8 Hz, 2H, Ar-H), 7.56 (d, J = 8.6 Hz, 2H, Ar-H), 7.66 (s, 1H, Quin-H), 8.11 (d, J = 8.6 Hz, 2H, Ar-H), 8.42 (d, J = 5.2 Hz, 1H, CH), 9.19 (s, 1H, NH), 11.63 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 42.4 (CH2) 55.5 (OCH3), 55.7 (OCH3), 97.5 (CH), 106.5 (CH), 108.6 (CH), 112.2 (CH), 112.3 (CH), 121.5 (CH), 127.9 (Cq), 128.5 (CH), 128.8 (CH), 129.7 (Cq), 133.2 (Cq), 134.5 (CH), 135.4 (Cq), 135.7 (Cq), 144.0 (Cq), 144.8 (Cq), 151.2 (Cq), 159.1 (CH), 160.5 (Cq), 161.5 (C=O), 162.1(Cq) ppm. EI MS (70 eV): m/z (%): 513/515 (M+, 50/18), 296 (100), 217 (95). ESI-QTOF (positive ionization) M + H calc. for C28H24ClN5O3 514.1646 found 514.1618.
4.1.7.1.2. 6,7-dimethoxy-3-(((4-((4-(naphthalen-2-yl)pyrimidin-2-yl) amino)phenyl)amino)methyl)quinolin −2(1H)-one (16b).
Yellow solid. 80% yield, mp, 266–267 °C, IR (ATR, cm−1): ν [3394, 3290, 3286] (NH), 1645 (C=O), 1624, 1571. 1H NMR (400 MHz, DMSO‑d6) δ = 3.72 (s, 3H, OCH3). 3.79 (s, 3H, OCH3), 4.13 (d, J = 5.5 Hz, 2H, CH2), 5.84 (t, J = 6.1 Hz, 1H, CH2-NH), 6.59 (d, J = 8.8 Hz, 2H, Ar-H), 6.86 (s, 1H, Quin-H), 7.12 (s, 1H, Quin-H), 7.39 (d, J = 5.2 Hz, 1H, CH), 7.48 (d, J = 8.8 Hz, 2H, Ar-H), 7.62–7.52 (m, 2H, Ar-H), 7.68 (s, 1H, Quin-H), 8.10–7.88 (m, 3H, Ar-H), 8.23 (dd, J = 8.6, 1.7 Hz, 1H, Ar-H), 8.46 (d, J = 5.2 Hz, 1H, CH), 8.69 (s, 1H, CH, Ar-H), 9.21 (s, 1H, NH), 11.64 (s, 1H, NH), 13C NMR (101 MHz, DMSO‑d6) δ = 42.5 (CH2), 55.4 (CH3), 55.7 (CH3), 97.5 (CH), 106.9 (CH), 108.6 (CH), 112.2 (CH), 112.3 (Cq), 121.5 (CH), 123.9 (CH), 126.6 (CH), 126.7 (CH), 127.3 (CH), 127.6 (CH), 127.9 (Cq), 128.3 (CH), 128.8 (CH), 129.8 (Cq), 132.7 (Cq), 133.2 (Cq), 134.0 (Cq). 134.3 (Cq), 134.5 (CH), 144.0 (Cq), 144.8 (Cq), 151.2 (Cq), 158.9 (CH), 160.6 (Cq), 161.6 (C=O), 163.3 (Cq) ppm. EI MS (70 eV): m/z (%): 529 (M+, 16), 312 (100), 218 (22). ESI-QTOF (positive ionization) M + H calc. for C32H27N5O3 530.2187 found 530.2169.
4.1.7.1.3. 3-(((4-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl) amino)methyl)-6-methoxyquinolin-2(1H)-one (17a).
Yellow solid. 90% yield, mp, 255–256 °C, IR (ATR, cm−1): ν [3417, 3369, 3346] (NH), 1660 (C=O), 1620, 1514. 1H NMR (400 MHz, DMSO‑d6) δ = 3.75 (s, 3H, OCH3). 4.17 (d, J = 5.5 Hz, 2H, CH2), 5.92 (t, J = 5.5 Hz, 1H, CH2-NH), 6.60 (d, J = 8.6 Hz, 2H, Ar-H), 7.11 (dd, J = 8.9, 2.5 Hz, 1H, Quin-H), 7.16 (s, 1H, Quin-H), 7.34–7.24 (m, 2H, Quin-H, CH), 7.47 (d, J = 8.6 Hz, 2H, Ar-H), 7.59 (d, J = 8.3 Hz, 2H, Ar-H), 7.74 (s, 1H, Quin-H), 8.14 (d, J = 8.3 Hz, 2H, Ar-H), 8.45 (d, J = 5.1 Hz, 1H, CH), 9.23 (s, 1H, NH), 11.78 (s, 1H, NH), 13C NMR (101 MHz, DMSO‑d6) δ = 43.1 (CH2), 55.9 (OCH3), 107.1 (CH), 109.3 (CH), 112.7 (CH), 116.6 (CH), 119.3 (CH), 120.3 (Cq), 122.0 (CH), 129.0 (CH), 129.4 (CH), 130.3 (Cq), 132.1 (Cq), 132.7 (Cq), 134.9 (CH), 135.9 (Cq), 136.2 (Cq), 144.4 (Cq), 154.7 (Cq), 159.6 (CH), 161.0 (Cq), 161.8 (C=O), 162.7 (Cq) ppm. EI MS (70 eV): m/z (%): 483/485 (M+, 55/18), 294 (100), 279 (13). ESI-QTOF (positive ionization) M + H calc. for C27H22ClN5O2; 484.1535 found 484.1522.
4.1.7.1.4. 6-methoxy-3-(((4-((4-(naphthalen-2-yl)pyrimidin-2-yl) amino)phenyl) amino)methyl)quinolin-2(1H)-one (17b).
Yellow solid. 90% yield, mp, 294–295 °C, IR (ATR, cm−1): ν [3431, 3257, 3170] (NH), 1658 (C=O), 1610, 1560. 1H NMR (400 MHz, DMSO‑d6) δ 3.75 (s, 3H, OCH3). 4.20 (d, J = 5.4 Hz, 2H, CH2), 5.93 (t, J = 5.4 Hz, 1H, CH2-NH), 6.64 (d, J = 8.7 Hz, 2H, Ar-H), 7.11 (dd, J = 8.9, 2.3 Hz, 1H, Quin-H), 7.17 (s, 1H, Quin-H), 7.29 (d, J = 8.9 Hz, 1H, Quin-H), 7.42 (d, J = 5.1 Hz, 1H, CH), 7.53 (d, J = 8.5 Hz, 2H, Ar-H), 7.67–7.56 (m, 2H, Ar-H), 7.77 (s, 1H, Ar-H), 7.98 (d, J = 7.4 Hz, 1H, Ar-H), 8.04 (d, J = 8.7 Hz, 2H, Ar-H), 8.25 (d, J = 8.5 Hz, 1H, Ar-H), 8.49 (d, J = 5.1 Hz, 1H, CH), 8.72 (s, 1H Quin-H), 9.26 (s, 1H, NH), 11.80 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 43.2 (CH2), 55.9 (CH3), 107.5 (CH), 109.4 (CH), 112.8 (CH), 116.6 (CH), 119.3 (CH), 120.3 (Cq), 122.1 (CH), 124.4 (CH), 127.2 (CH), 127.3 (CH), 127.8 (CH), 128.1 (CH), 128.8 (CH), 129.3 (CH), 130.5 (Cq), 132.1 (Cq), 132.7 (Cq), 133.2 (Cq), 134.5 (Cq), 134.8 (CH), 134.9 (Cq), 154.7 (Cq × 2), 159.4 (CH), 16.1 (Cq), 161.8 (C=O), 163.8 (Cq) ppm. EI MS (70 eV): m/z (%): 499 (M+, 61), 311 (100). ESI-QTOF (positive ionization) M +H calc. for C31H25N5O2 500.2081 found 500.2065.
4.1.7.1.5. 3-(((4-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl) amino)methyl)quinolin-2(1H)-one (18a).
Yellow solid. 79% yield, mp, 261–262 °C, IR (ATR, cm−1): ν [3419, 3379, 3270] (NH), 1647 (C=O), 1571, 1533. 1H NMR (400 MHz, DMSO‑d6) δ = 4.21 (s, 2H, CH2), 5.37 (s, 1H, CH2-NH), 6.66 (d, J = 8.8 Hz, 2H, Ar-H), 7.12 (t, J = 8.0 Hz, 1H, Quin-H), 7.16 (d, J = 5.1 Hz, 1H, CH), 7.3 (d, J = 7.7 Hz, 1H, Quin-H), 7.41 (t, J = 8.3 Hz, 1H, Quin-H), 7.44 (d, J = 8.8 Hz, 2H, Ar-H), 7.51 (d, J = 8.6 Hz, 2H, Ar-H), 7.54 (d, J = 8.1 Hz, 1H, Quin-H), 7.80 (s, 1H, Quin-H), 8.07 (d, J = 8.6 Hz, 2H, Ar-H), 8.41 (d, J = 5.1 Hz, 1H, CH), 8.59 (s, 1H, NH), 11.34 (s, 1H, NH). 13C NMR (101 MHz DMSO‑d6) δ = 42.4 (CH2), 106.1 (CH), 112.2 (CH), 114.2 (CH), 118.7 (Cq), 120.9 (CH), 121.2 (CH), 126.6 (CH), 127.8 (CH), 128.0 (CH), 128.6 (CH), 129.6 (Cq), 130.8 (Cq), 134.6 (CH), 134.8 (Cq), 135.5 (Cq), 137.5 (Cq), 143.7 (Cq), 158.2 (CH), 160.3 (Cq), 161.1 (C=O), 161.9 (Cq) ppm. EI MS (70 eV): m/z (%): 453/455 (M+, 75/25), 294 (100), 249 (14). ESI-QTOF (positive ionization) M + H calc. for C26H20ClN5O 454.1429 found 454.1412.
4.1.7.1.6. 3-(((4-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl) amino) methyl)quinolin-2(1H)-one (18b).
Yellow solid. 82% yield, mp, 294–295 °C, IR (ATR, cm−1): ν [3402, 3287, 3175] (NH), 1651 (C=O), 1570, 1448. 1H NMR (400 MHz, DMSO‑d6) δ = 4.20 (d, J = 5.6 Hz, 2H, CH2), 5.90 (t, J = 5.9 Hz, 1H, CH2-NH), 6.65 (d, J = 8.6 Hz, 2H, Ar-H), 7.15 (t, J = 7.5 Hz, 1H, Quin-H), 7.35 (d, J = 8.2 Hz, 1H, Quin-H), 7.42 (d, J = 5.1 Hz, 1H, CH), 7.47 (t, J = 7.6 Hz, 1H, Quin-H), 7.53 (d, J = 8.6 Hz, 2H, Ar-H), 7.67–7.56 (m, 3H, Ar-H), 7.81 (s, 1H, Ar-H), 7.99 (d, J = 8.3 Hz, 1H, Ar-H), 8.05 (d, J = 8.7 Hz, 2H, Ar-H), 8.26 (d, J = 8.5 Hz, 1H, Quin-H), 8.49 (d, J = 5.1 Hz, 1H, CH), 8.72 (s, 1H, Quin-H), 9.25 (s, 1H, NH), 11.89 (s, 1H, NH). 13C NMR (101 MHz, DMSO‑d6) δ = 43.1 (CH2), 107.5 (CH), 112.8 (CH), 115.4 (CH), 119.7 (Cq), 122.3 (CH), 122.0 (CH), 124.4 (CH), 127.2 (CH), 127.3 (CH), 127.8 (CH), 128.0 (CH), 128.1 (CH), 128.8 (CH), 129.3 (CH), 130.0 (CH), 131.7 (Cq), 130.5 (Cq), 133.2 (Cq), 134.5 (Cq), 134.8 (Cq), 135.4 (CH), 138.3 (Cq), 144.4 (Cq), 159.4 (CH), 161.1 (Cq), 162.3 (C=O), 163.8 (Cq) ppm. EI MS (70 eV): m/z (%): 469 (M+, 64), 311 (100). ESI-QTOF (positive ionization) M + H calc. for C30H23N5O 470.1975 found 470.1968.
4.1.8. Procedure for the synthesis of the novel 6,7-dihydroxy-3-(((4-((4-(naphthalene-2-yl)pyrimidin-2-yl)amino)phenyl)amino)methyl)quinolin-2(1H)-one (19b)
To a stirred solution of 6,7-dimethoxy-3-(((4-((4-(naphthalen-2-yl) pyrimidin-2-yl)amino)phenyl)amino)methyl)quinolin-2(1H)-one 16b (0.5 mmol) in dry CH2Cl2 (15 mL) cooled at − 78 °C was slowly added a solution of BBr3 (7 equiv) over a period of 30 min at −78 °C. After the reaction mixture was warmed to RT and stirred for further 24 h when no starting material was detected by TLC. Thereafter, the solvent was removed under reduced pressure and cold water was added. The obtained crude solid was filtered under vacuum and purified by column chromatography on silica gel with DCM/MeOH.
The residue obtained was a red solid. 38% yield, mp, ˃300 °C, IR (ATR, cm−1): ν 3267 (OH), [3267, 3120, 3055] (NH), 1645 (C=O), 1570, 1448. 1H NMR (400 MHz, DMSO -d6) δ = 4.08 (s, 2H, CH2). 5.75 (s, 1H, NH), 6.60 (d, J = 8.7 Hz, 2H, H-Ar), 6.77 (s, 1H), 6.87 (s, 1H), 7.40 (d, J = 5.2 Hz, 1H, CH), 7.48 (d, J = 8.6 Hz, 2H, H-Ar), 7.57 (s, 3H), 8.06–7.89 (m, 4H), 8.22 (d, J = 9.0 Hz, 1H, CH), 8.46 (d, J = 5.1 Hz, 1H, CH), 8.69 (s, 1H), 9.19 (s, 1H, CH), 11.47 (s, 1H, NH), 13C NMR (101 MHz, DMSO‑d6) δ = 42.5 (CH2). 100.6 (CH), 106.9 (CH), 111.5 (CH), 112.1 (Cq), 112.3 (CH), 121.5 (CH), 123.9 (CH), 126.6 (Cq), 126.6 (CH), 126.7 (Cq), 127.3 (CH), 127.6 (CH), 128.3 (CH), 128.8 (CH), 129.8 (Cq), 132.6 (Cq), 132.7 (Cq), 134.0 (Cq), 134.3 (Cq), 134.8 (CH), 141.7 (Cq), 144.1 (Cq), 148.9 (Cq), 158.9 (CH), 160.6 (Cq), 161.6 (Cq), 163.3 (C=O) ppm. EI MS (70 eV): m/z (%): 484 (M-OH, 100), 296 (12), 280 (19), 242 (25). ESI-QTOF (positive ionization) M +H calc. for C30H23N5O3 502,1874 found 502,1866.
4.2. SphK inhibition assays
SphK1 and SphK2 activity was determined fluorometrically with NBD-sphingosine as substrate as previously described [28]. Briefly, compounds were dissolved in DMSO and inhibitory activity determined at a concentration of 650 μM. Compounds showing significant inhibition were further examined at different concentrations to obtain IC50 values. The assays contained 100 nM recombinant SphK1 or SphK2, 30 mM Tris-HCl [pH 7.4], 0.05% Triton X-100, 150 mM NaCl, 10% glycerol, 0.05% triton X-100, and 1% DMSO. Formation of NBD-sphingosine-1-phosphate was measured in a TECAN Infinite M1000 fluorescence plate reader (Männedorf, Switzerland) at 37 °C with excitation at 550 nm and emission at 584 nm. Data were analyzed with Prism (GraphPad, La Jolla, USA).
4.3. Molecular modelling
4.3.1. Molecular docking
AutoDock4 [37] was used to perform the docking of each compound in the active SphK1 site. A Lamarckian genetic algorithm with local search of pseudo-Solis and Wets was used [38]. Initial population of trial ligands was established by 150 individuals and the maximum number of generations was set to 2.7 × 104. The maximum number of energy evaluations was 25.0 × 106. 200 conformations were generated for each docking job. All other run parameters were maintained at their default setting. The resulting docked conformations were clustered into families by considering the backbone rmsd. The lowest and most populated docking-energy conformation was considered the most favorable orientation [39].
4.3.2. MD simulations
The protein–ligand complex obtained from the docking process were soaked in octahedral boxes of explicit water using the TIP3P model [40] and subjected to MD simulation. MD simulations were accomplished using the Amber software package [41] with periodic boundary conditions. Other parameters were fixed as follows: particle mesh Ewald method (PME) [42] was applied using a grid spacing of 1.2 Å, a spline interpolation order of 4 and a real space direct sum cutoff of 10 Å. The SHAKE algorithm was employed with an integration time step of 2 fs. MD simulations were carried out at 310 K temperature. The NPT ensemble was employed using Berendsen coupling to a baro/thermostat (target pressure 1 atm, relaxation time 0.1 ps). MD simulations of 50 ns were conducted in triplicate for each system under different starting velocity distribution functions; thus, in total 150 ns were simulated for each complex. Post MD analysis was carried out with program ptraj [43].
4.3.3. Quantum calculations setup
From the MD simulation, the reduced 3D model systems were constructed, which include the tested compound and the interacting residues from the Sphk1 binding pocket. In this work, we identified the binding site residues of the receptors by using the free energy decomposition method (MM/GBSA) [44]. The side chains of the binding site residues that contributed with a |ΔG| higher than 1.0 kcal/mol in the per residue energy decomposition together with each inhibitor were included in the reduced model.
4.3.4. Atoms in molecules theory
The reduced models were used as input for quantum theory of atoms in molecules (QTAIM) analysis [45], which was performed with the help of Multiwfn software [46]. The wave function used as input for these calculations were computed with the Gaussian 09 package [47] by employing the B3LYP functional with dispersion correction (B3LYPD) and 6–31G(d) as basis set. The empirical dispersion correction for the B3LYP functional was applied by invoking the IOp 3/124 = 3 keyword in Gaussian 09. This type of calculations have been used in recent works because it ensures a reasonable compromise between the wave function quality required to obtain reliable values of the derivatives of ρ(r) and the computer power available, due to the extension of the system in study [48,49].
4.4. Additional materials
Spectra of the synthetized compounds can be found in the supporting information.
Supplementary Material
Acknowledgements
Grants from Universidad Nacional de San Luis (UNSL-Argentina) partially supported this work. Marcela Vettorazzi thanks a doctoral fellowship of CONICET-Argentina. This work was supported in part by the National Institute of Health grant R01GM043880 (to S.S), the Spanish Ministerio de Ciencia, Innovación y Universidades, project #RTI2018-098560-B-C22, Universidad de Jáen, Consejería de Innovación, Ciencia y Empresa (Junta de Andalucía, Spain), COLCIENCIAS and Universidad del Valle (Colombia). Authors would like to thank “Centro de Instrumentación Científico-técnico de la Universidad de Jaén (UJAEN)” and the staff for data collection.
Footnotes
Declaration of Competing Interest
The authors confirm that this article content has no conflict of interest.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2019.103414.
References
- [1].Blaho VA, Hla T, Regulation of mammalian physiology, development, and disease by the sphingosine 1-phosphate and lysophosphatidic acid receptors, Chem. Rev 111 (2011) 6299–6320, 10.1021/cr200273u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hannun YA, Obeid LM, Principles of bioactive lipid signalling: lessons from sphingolipids, Nat. Rev. Mol. Cell Biol 9 (2008) 139–150, 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- [3].Ponnusamy S, Meyers-Needham M, Senkal CE, Saddoughi SA, Sentelle D, Selvam SP, Salas A, Ogretmen B, Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance, Future Oncol. 6 (2010) 1603–1624, 10.2217/fon.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Spiegel S, Milstien S, The outs and the ins of sphingosine-1-phosphate in immunity, Nat. Rev. Immunol 11 (2011) 403–415, 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Maceyka M, Spiegel S, Sphingolipid metabolites in inflammatory disease, Nature 510 (2014) 58–67, 10.1038/nature13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pyne S, Pyne NJ, Translational aspects of sphingosine 1-phosphate biology, Trends Mol. Med 17 (2011) 463–472, 10.1016/j.molmed.2011.03.002. [DOI] [PubMed] [Google Scholar]
- [7].Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S, Molecular cloning and functional characterization of murine sphingosine kinase, J Biol Chem. 273 (1998) 23722–23728. [DOI] [PubMed] [Google Scholar]
- [8].Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S, Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform, J. Biol. Chem 275 (2000) 19513–19520, 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- [9].Pitson SM, Moretti PA, Zebol JR, Xia P, Gamble JR, Vadas MA, D’Andrea RJ, Wattenberg BW, Expression of a catalytically inactive sphingosine kinase mutant blocks agonist-induced sphingosine kinase activation. A dominant-negative sphingosine kinase, J. Biol. Chem 275 (2000) 33945–33950, 10.1074/jbc.M006176200. [DOI] [PubMed] [Google Scholar]
- [10].Alvarez SE, Milstien S, Spiegel S, Autocrine and paracrine roles of sphingosine-1-phosphate, Trends Endocrinol. Metab 18 (2007) 300–307, 10.1016/j.tem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- [11].Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL, Essential role for sphingosine kinases in neural and vascular development, Mol. Cell. Biol 25 (2005) 11113–11121, 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pitson SM, Moretti PAB, Zebol JR, Zareie R, Derian CK, Darrow AL, Qi J, D’Andrea RJ, Bagley CJ, Vadas MA, Wattenberg BW, The nucleotide-binding site of human sphingosine kinase 1, J. Biol. Chem 277 (2002) 49545–49553, 10.1074/jbc.M206687200. [DOI] [PubMed] [Google Scholar]
- [13].Chen M-H, Yen C-C, Cheng C-T, Wu R-C, Huang S-C, Yu C-S, Chung Y-H, Liu C-Y, Chang PM-H, Chao Y, Chen M-H, Chen Y-F, Chiang K-C, Yeh T-S, Chen TC, Huang C-YF, Yeh C-N, Identification of SPHK1 as a therapeutic target and marker of poor prognosis in cholangiocarcinoma, Oncotarget. 6 (2015) 23594–23608, 10.18632/oncotarget.4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang F, Wu Z, Sphingosine kinase 1 overexpression is associated with poor prognosis and oxaliplatin resistance in hepatocellular carcinoma, Exp. Ther. Med 15 (2018) 5371–5376, 10.3892/etm.2018.6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Meng X-D, Zhou Z-S, Qiu J-H, Shen W-H, Wu Q, Xiao J, Increased SPHK1 expression is associated with poor prognosis in bladder cancer, Tumour Biol. 35 (2014) 2075–2080, 10.1007/s13277-013-1275-0. [DOI] [PubMed] [Google Scholar]
- [16].Vettorazzi M, Angelina E, Lima S, Gonec T, Otevrel J, Marvanova P, Padrtova T, Mokry P, Bobal P, Acosta LM, Palma A, Cobo J, Bobalova J, Csollei J, Malik I, Alvarez S, Spiegel S, Jampilek J, Enriz RD, An integrative study to identify novel scaffolds for sphingosine kinase 1 inhibitors, Eur. J. Med. Chem 139 (2017) 461–481, 10.1016/j.ejmech.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vettorazzi M, Vila L, Lima S, Acosta L, Yépes F, Palma A, Cobo J, Tengler J, Malík I, Alvarez S, Marques P, Cabedo N, Sanz M-J, Jampilek J, Spiegel S, Enriz RD, Synthesis and biological evaluation of sphingosine kinase 2 inhibitors with anti-inflammatory activity, Arch. Pharm. (Weinheim) 352 (2019), 10.1002/ardp.201800298. [DOI] [PubMed] [Google Scholar]
- [18].Vettorazzi M, Menéndez C, Gutiérrez L, Andujar S, Appignanesi G, Enriz RD, Theoretical models to predict the inhibitory effect of ligands of sphingosine kinase 1 using QTAIM calculations and hydrogen bond dynamic propensity analysis, J. Comput. Aided. Mol. Des (2018), 10.1007/s10822-018-0129-7. [DOI] [PubMed] [Google Scholar]
- [19].French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD, Discovery and evaluation of inhibitors of human sphingosine kinase, Cancer Res. 63 (2003) 5962–5969. [PubMed] [Google Scholar]
- [20].French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD, Antitumor activity of sphingosine kinase inhibitors, J. Pharmacol. Exp. Ther 318 (2006) 596–603, 10.1124/jpet.106.101345. [DOI] [PubMed] [Google Scholar]
- [21].Wang Z, Min X, Xiao S-H, Johnstone S, Romanow W, Meininger D, Xu H, Liu J, Dai J, An S, Thibault S, Walker N, Molecular basis of sphingosine kinase 1 substrate recognition and catalysis, Structure 21 (2013) 798–809, 10.1016/j.str.2013.02.025. [DOI] [PubMed] [Google Scholar]
- [22].Wang J, Knapp S, Pyne NJ, Pyne S, Elkins JM, Crystal Structure of Sphingosine Kinase 1 with PF-543, ACS Med. Chem. Lett 5 (2014) 1329–1333, 10.1021/ml5004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Byun H-S, Pyne S, MacRitchie N, Pyne NJ, Bittman R, Novel sphingosine-containing analogues selectively inhibit sphingosine kinase (SK) isozymes, induce SK1 proteasomal degradation and reduce DNA synthesis in human pulmonary arterial smooth muscle cells, Medchemcomm. 4 (2013), 10.1039/C3MD00201B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bongarzone S, Bolognesi ML, The concept of privileged structures in rational drug design: focus on acridine and quinoline scaffolds in neurodegenerative and protozoan diseases, Expert Opin. Drug Discov 6 (2011) 251–268, 10.1517/17460441.2011.550914. [DOI] [PubMed] [Google Scholar]
- [25].Abonia R, Cortés E, Insuasty B, Quiroga J, Nogueras M, Cobo J, Synthesis of novel 1,2,5-trisubstituted benzimidazoles as potential antitumor agents, Eur. J. Med. Chem 46 (2011) 4062–4070, 10.1016/j.ejmech.2011.06.006. [DOI] [PubMed] [Google Scholar]
- [26].Insuasty D, Robledo SM, Velez ID, Cuervo P, Insuasty B, Quiroga J, Nogueras M, Cobo J, Abonia R, A Schmidt rearrangement-mediated synthesis of novel tetrahydro-benzo[1,4]diazepin-5-ones as potential anticancer and anti-protozoal agents, Eur. J. Med. Chem 141 (2017) 567–583, 10.1016/j.ejmech.2017.10.024. [DOI] [PubMed] [Google Scholar]
- [27].Abonia R, Insuasty D, Castillo J, Insuasty B, Quiroga J, Nogueras M, Cobo J, Synthesis of novel quinoline-2-one based chalcones of potential anti-tumor activity, Eur. J. Med. Chem 57 (2012) 29–40, 10.1016/j.ejmech.2012.08.039. [DOI] [PubMed] [Google Scholar]
- [28].Lima S, Milstien S, Spiegel S, A real-time high-throughput fluorescence assay for sphingosine kinases, J. Lipid Res 55 (2014) 1525–1530, 10.1194/jlr.D048132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gustin DJ, Li Y, Brown ML, Min X, Schmitt MJ, Wanska M, Wang X, Connors R, Johnstone S, Cardozo M, Cheng AC, Jeffries S, Franks B, Li S, Shen S, Wong M, Wesche H, Xu G, Carlson TJ, Plant M, Morgenstern K, Rex K, Schmitt J, Coxon A, Walker N, Kayser F, Wang Z, Structure guided design of a series of sphingosine kinase (SphK) inhibitors, Bioorg. Med. Chem. Lett 23 (2013) 4608–4616, 10.1016/j.bmcl.2013.06.030. [DOI] [PubMed] [Google Scholar]
- [30].Yokota S, Taniguchi Y, Kihara A, Mitsutake S, Igarashi Y, Asp177 in C4 domain of mouse sphingosine kinase 1a is important for the sphingosine recognition, FEBS Lett. 578 (2004) 106–110, 10.1016/j.febslet.2004.10.081. [DOI] [PubMed] [Google Scholar]
- [31].Parthasarathi R, Subramanian V, Sathyamurthy N, Hydrogen Bonding without Borders: An Atoms-in-Molecules Perspective, J. Phys. Chem. A 110 (2006) 3349–3351, 10.1021/jp060571z. [DOI] [PubMed] [Google Scholar]
- [32].Yesselman JD, Horowitz S, Brooks CL 3rd, Trievel RC, Frequent side chain methyl carbon-oxygen hydrogen bonding in proteins revealed by computational and stereochemical analysis of neutron structures, Proteins. 83 (2015) 403–410, 10.1002/prot.24724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu K, Guo TL, Hait NC, Allegood J, Parikh HI, Xu W, Kellogg GE, Grant S, Spiegel S, Zhang S, Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a selective sphingosine kinase-2 inhibitor and anticancer agent, PLoS One 8 (2013) e56471, 10.1371/journal.pone.0056471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Laali KK, Insuasty D, Abonia R, Insuasty B, Bunge SD, Novel quinoline-imidazolium adducts via the reaction of 2-oxoquinoline-3-carbaldehyde and quinoline-3-carbaldehydes with 1-butyl-3-methylimidazolium chloride [BMIM][Cl], Tetrahedron Lett. 55 (2014) 4395–4399, 10.1016/j.tetlet.2014.05.094. [DOI] [Google Scholar]
- [35].Ceide SC, Montalban AG, Microwave-assisted, efficient and regioselective Pdcatalyzed C-phenylation of halopyrimidines, Tetrahedron Lett. (2006), 10.1016/j.tetlet.2006.04.082. [DOI] [Google Scholar]
- [36].Mavunkel B, Jin Xu Y, Goyal B, Lim D, Lu Q, Chen Z, Wang DX, Higaki J, Chakraborty I, Liclican A, Sideris S, Laney M, Delling U, Catalano R, Higgins LS, Wang H, Wang J, Feng Y, Dugar S, Levy DE, Pyrimidine-based inhibitors of CaMKIIδ, Bioorganic. Med. Chem. Lett 18 (2008) 2404–2408, 10.1016/j.bmcl.2008.02.056. [DOI] [PubMed] [Google Scholar]
- [37].Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ, AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility, J. Comput. Chem 30 (2009) 2785–2791, 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ, Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem 19 (1998) 1639–1662, . [DOI] [Google Scholar]
- [39].Ewing TJA, Kuntz ID, Critical evaluation of search algorithms for automated molecular docking and database screening, J. Comput. Chem 18 (1997) 1175–1189, . [DOI] [Google Scholar]
- [40].Mark P, Nilsson L, Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K, J. Phys. Chem. A 105 (2001) 9954–9960, 10.1021/jp003020w. [DOI] [Google Scholar]
- [41].Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker, Wang J, Wolf RM, Wu X, Kollman PA, {Amber 14} OR - University of California, San Francisco, 2014. citeulike-article-id: 14028955. [Google Scholar]
- [42].Darden T, York D, Pedersen L, Particle mesh Ewald: An N•log(N) method for Ewald sums in large systems, J. Chem. Phys 98 (1993). [Google Scholar]
- [43].Roe DR, Cheatham TE, PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data, J. Chem. Theory Comput 9 (2013) 3084–3095, 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- [44].Genheden S, Ryde U, The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities, Expert Opin. Drug Discov 10 (2015) 449–461, 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bader RFW, Atoms in molecules, Acc. Chem. Res 18 (1985) 9–15, 10.1021/ar00109a003. [DOI] [Google Scholar]
- [46].Lu T, Chen F, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem 33 (2012) 580–592, 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- [47].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Had M, Fox DJ, Gaussian 09 package, Gaussian, Inc, Wallingford CT: (2009). [Google Scholar]
- [48].Barrera Guisasola EE, Gutierrez LJ, Andujar SA, Angelina E, Rodriguez AM, Enriz RD, Pentameric models as alternative molecular targets for the design of new antiaggregant agents, Curr. Protein Pept. Sci 17 (2016) 156–168. [DOI] [PubMed] [Google Scholar]
- [49].Luchi AM, Angelina EL, Andujar SA, Enriz RD, Peruchena NM, Halogen bonding in biological context: a computational study of D2 dopamine receptor, J. Phys. Org. Chem 29 (2016) 645–655, 10.1002/poc.3586. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.