

Abstract
Autophagy is a highly conserved biological process essential to protein, cellular and organismal homeostasis. As autophagy plays a critical role in cellular responses to various external and internal stimuli, it is important to understand the mechanism underlying autophagy regulation. Here, we monitor the stability of 17 key autophagy factors in the yeast S. cerevisiae and show that Atg9 and Atg14 are degraded under normal growth conditions. Whereas Atg14 is regulated by both the proteasome and autophagy, Atg9 turnover is normally mediated by the proteasome but impeded upon starvation or rapamycin treatment. Interestingly, distinct segments of Atg9 confer instability, suggesting that multiple pathways are involved in Atg9 degradation. Our results provide the foundation to further elucidate the physiological significance of Atg9 turnover and also the interplay between two major proteolytic systems (i.e., autophagy and the proteasome).
Keywords: autophagy, proteasome, Atg9, protein degradation, stress response
Introduction:
The lysosome and 26S proteasome represent the major degradation machines in eukaryotic cells [1–4]. While the lysosome deals mainly with the removal of bulky contents (e.g., protein aggregates, organelles, intracellular pathogens), the 26S proteasome handles the majority of regulated protein turnover [3, 4]. Proper cell functioning requires delicate and efficient coordination of these degradation apparatus. Dysregulation of the lysosome and proteasome can lead to diseases ranging from cancers to neurologic disorders [1, 2, 4].
Macroautophagy (hereafter termed autophagy), an important branch of lysosome-mediated degradation system, is essential for cellular homeostasis, growth, differentiation and development [1, 5]. Autophagy delivers unwanted cytoplasmic components to the lysosome for destruction through an elaborated process that engulfs cellular materials by a double-membrane structure autophagosome, which is then transported and fused with the lysosome to degrade the sequestrated cargo. Autophagy requires more than 30 proteins to work in highly coordinated steps of autophagy initiation, phagophore elongation, autophagosome maturation, autophagosome-lysosome fusion, and cargo degradation [5, 6]. Most of the core autophagy factors were first identified in the yeast S. cerevisiae [1, 5], which provides powerful genetic and biochemical tools and found to be conserved in higher eukaryotes.
In a time of stress, autophagy plays a major role in helping cells face challenges and adapt to the environment as it can quickly remove harmful components and supply nutrients and energy [5, 7]. Whereas the mechanism underlying autophagy induction in response to stress is better understood, how autophagy is kept in check under normal conditions remains enigmatic [6, 8, 9]. We reasoned that some key autophagy factors may be limited due to normal constitutive degradation but become stabilized upon stress. As autophagy induction is highly regulated [6, 10], we therefore have systematically monitored the stability of 17 yeast proteins involved in earlier steps of autophagy. Under normal growth conditions, Atg9 and Atg14 are found to be degraded. Since Atg14 turnover has been previously dissected, we have mainly focused on Atg9 regulation and demonstrated that Atg9 degradation is proteasome-dependent and stress-responsive. Among the core autophagy regulators essential for autophagosome formation, Atg9 is the only integral membrane protein [11–14]. Our results suggest Atg9 may contain multiple degradation signals for various ubiquitin ligases. As uncontrolled autophagy activation can lead to dire consequences [1, 8], understanding Atg9 regulation would bring light to the control of autophagy activity and also the cooperation between the proteasome and autophagy.
Material and methods:
Yeast strains and plasmids.
Yeast S. cerevisiae strains lacking various ubiquitylation components or autophagy regulators in a BY4741 background were obtained from Dr. Mark Hochstrasser (Yale U.) and Open Systems (Huntsville, AL), respectively. The yeast strains bearing ATG9 wild-type or phosphorylation mutant allele appended with the tandem affinity purification (TAP) tag were provided by Dr. Claudine Kraft (University of Vienna). SM5925 strain bearing uba1-204 temperature sensitive mutation and isogenic wild-type strain were obtained from Dr. Susan Michaelis (Johns Hopkins University).
The plasmids expressing the MORF-tagged genes ATG1, ATG3, ATG4, ATG5, ATG6, ATG7, ATG8, ATG9, ATG10, ATG11, ATG12, ATG13, ATG14, ATG17, ATG38, TRS85, VPS34 were obtained from Open Biosystems. The plasmid expressing GFP-tagged Atg8 was provided by Dr. Klionsky (U. of Michigan). Using the plasmid bearing MORF-ATG9 as the template, ATG9 deletion mutants were generated via Q5 site-directed mutagenesis kit (New England Biolab, MA).
Yeast cells were grown in rich YPD or synthetic medium containing 2% raffinose or 2% galactose. For nutrient starvation, actively growing yeast cells were grown in the medium without amino acids. Rapamycin and MG132 were purchased from Sigma-Aldrich (MO).
Antibodies.
Antibodies against Rpt5 and HA were obtained from Enzo Life Sciences (Farmingdale, NY) and BioLegend Inc. (San Diego, CA), respectively. Pgk1 antibody was obtained from Thermo Fisher Scientific (Waltham, MA). GFP antibody was purchased from Abeam (Cambridge, MA).
Expression shut-off assay.
Yeast cells bearing MORF-tagged autophagy regulators under the control of the galactose-inducible GAL1-promoter were grown to log phase at 30°C in synthetic SR medium, and 2% galactose was added to induce protein expression for 3 h. Protein synthesis was then shut off by the addition of cycloheximide. Samples were withdrawn at the indicated intervals. Proteins were extracted and processed for western blotting with anti-HA to detect MORF-tagged autophagy regulators. The stable protein Rpt5 and Pgk1 were employed as a loading control.
Results:
Atg9 and Atg14 are degraded under normal growth conditions.
The regulation of autophagy activity likely occurs mainly at early steps [6, 10]. To evaluate whether core autophagy machinery is subject to degradation control, we monitored the protein stability of 17 conserved autophagy factors that are involved in autophagy initiation and autophagosome formation [5, 6, 10]. These proteins are linked to MORF or GFP tags for easy detection. To assess the alteration of protein levels, wild-type yeast cells were grown in nutrient-rich media and later treated with cycloheximide to turn off protein expression. Only Atg9 and Atg14 were found to be degraded under normal growth conditions (Fig. 1).
Fig. 1.
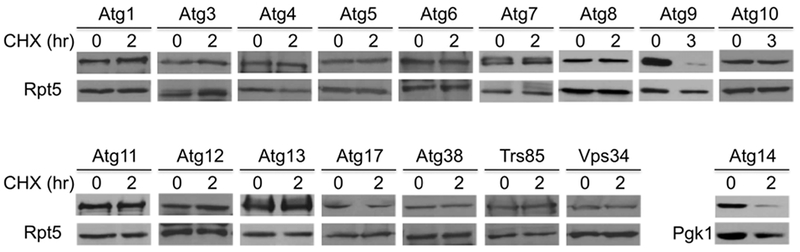
Two autophagy regulators are degraded under normal growth conditions. The protein stability of 17 autophagy regulators was examined in wild-type yeast cells by a protein expression shut-off assay. Yeast cells bearing GFP-Atg8 or MORF tagged autophagy proteins were grown to log phase, and samples were collected at indicated time points after cycloheximide (CHX) turned off protein synthesis. Extracts were analyzed by western blotting with anti-GFP for Atg8 or an HA antibody to detect MORF-tagged proteins. Equal loading was ascertained by immunoblotting with Rpt5 antibody (lower panels). For Atg14, we used Pgk1 as a loading control as the sizes of Atg14 and Rpt5 are similar. The identities of autophagy regulators are shown at the top.
ATG14 encodes a subunit of phosphatidylinositol 3-kinase (PI3K) complex I, which synthesizes phosphatidylinositol-3-phosphate and is essential for autophagosome membrane biogenesis [5, 10]. Atg14 brings the PI3K complex to the phagophore initiation sites and promotes phagophore expansion. Besides its key role in the early step of the autophagy, Atg14 also facilitates autophagosome-lysosome fusion [15]. Atg14 has been demonstrated previously to be degraded by the ubiquitin-proteasome system as a means to modulate autophagy activity [16].
Atg9 is stabilized upon starvation or rapamycin treatment.
Atg9 is the only transmembrane protein among core autophagy effectors and has not been previously shown to be degraded [11, 12]. We evaluated whether Atg9 turnover is altered under various stress conditions (Fig. 2). Upon nutrient depletion, which triggers autophagy induction, Atg9 was markedly stabilized (Fig. 2a). We also treated yeast cells with rapamycin, which triggers a starvation-like response and activates autophagy and found that Atg9 turnover was significantly impaired in the presence of rapamycin (Fig. 2b).
Fig. 2.
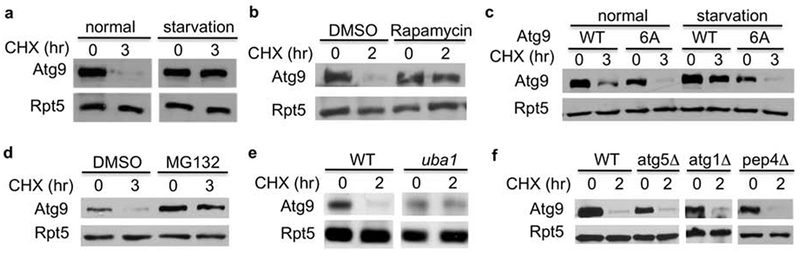
Atg9 turnover is hindered under cellular stress conditions that activate autophagy and regulated by the proteasome. (a,b) Immunoblot analysis of MORF-tagged Atg9 degradation in the absence or presence of rapamycin or nutrient depletion. (c) The degradation of Atg9 mutant defective for Atg1-phosphorylation is insensitive to starvation. The Atg9 mutant lacking Atg1 phosphorylation sites was previously dissected and the chromosomal copy of ATG9 is appended with a TAP tag at its C-terminus. The degradation of TAP-tagged wild-type and mutant Atg9 under the regulation of its endogenous promoter was analyzed with or without nutrient starvation. (d) Atg9 stability was evaluated in the presence and absence of the proteasome inhibitor MG132. (e) Atg9 degradation was assessed in wild-type and mutant cells bearing a temperature sensitive UBA1 mutation. The experiments were done as described above, except that cells were shifted to 37°C for I hr before the addition of cycloheximide. (f) Atg9 degradation was assessed in yeast cells lacking key autophagy regulators (i.e., ATG1, ATG5) or Pep4 protease.
Upon starvation, Atg9 is known to be phosphorylated directly by Atg1, a Ser/Thr kinase essential for autophagy initiation, which in turn is required for autophagosome formation and expansion [17]. Wild-type or phosphorylation-defective mutant Atg9 was appended with tandem affinity purification (TAP) tag at its C-terminus and their expression was under the regulation of its endogenous promoter [17]. Interestingly, Atg9 mutant devoid of Atg1 phosphorylation sites [17] appeared to have a faster turnover rate than wild-type Atg9 (Fig. 2c). Moreover, unlike wild-type Atg9 during starvation, Atg9 phosphorylation mutant remained to be degraded rapidly upon nutritional stress (Fig. 2c), suggesting that Atg1-mediated phosphorylation promotes Atg9 stabilization and in turn enhances autophagy activity. The results indicate that Atg9 degradation is responsive to stress conditions and likely modulated by phosphorylation.
The proteasome is required for Atg9 degradation.
Since two major intracellular degradation systems are the proteasome and autophagy [3], we then examined whether Atg9 turnover is carried out by the proteasome or autophagy. Atg9 was significantly stabilized upon the treatment of proteasome inhibitor MG132 (Fig. 2d), suggesting that Atg9 is degraded by the proteasome. Consistent with the requirement of ubiquitin in proteasome-mediated proteolysis, Atg9 turnover was impaired in yeast cells bearing uba1-204 mutant (Fig. 2e) that contains a temperature sensitive mutation in the E1 ubiquitin activating enzyme, which is essential for ubiquitin modification [18]. Atg9 degradation was not compromised in cells lacking ATG1 or ATG5, two essential autophagy effectors, or PEP4, a lysosomal protease (Fig. 2f).
Atg9 degradation remains in various ubiquitylation mutants.
Proteasomal substrates are usually first recognized by a ubiquitin ligase (E3), which works with a ubiquitin-activating enzyme (E1) and a ubiquitin-conjugating enzyme (E2) to decorate the substrates with ubiquitin molecule [2, 18]. Ubiquitin modified substrates are then delivered to the proteasome for destruction. To understand the specific role of proteasome-mediated Atg9 turnover, it is pivotal to identify the specific pathway that selects Atg9 for degradation. There is one E1, ~12 E2s and over 60 E3s involved in ubiquitin-mediated degradation in the yeast S. cerevisiae [18]. As the ubiquitin ligase E3 is the rate limiting and substrate-recognition component of the ubiquitin-proteasome system, we evaluated Atg9 degradation kinetics in more than 17 mutant yeast cells lacking one of the non-essential ubiquitin ligases (Fig. 3a). However, Atg9 turnover was not significantly impaired in these mutants (Fig. 3a).
Fig. 3.
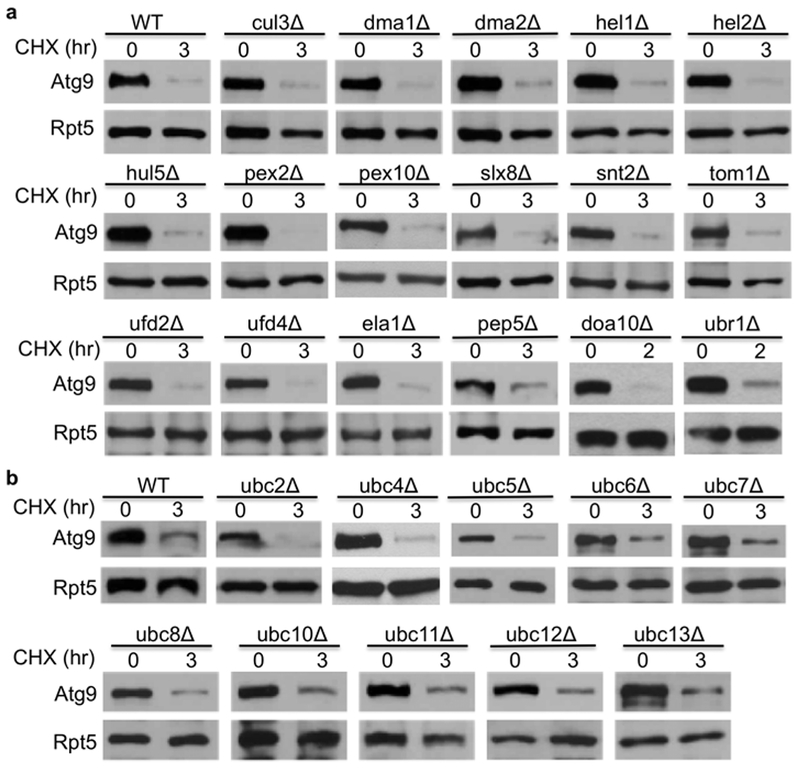
Atg9 turnover is not compromised in yeast cells lacking various ubiquitylation components. Atg9 degradation was evaluated in wild-type and E3-deficient (a) or E2-deficient cells (b). A plasmid bearing MORF-tagged Atg9 was transformed into indicated cells, and Atg9 degradation was determined as described above.
Since the number of ubiquitin-conjugating enzyme E2s is much smaller than E3s [18], we also examined Atg9 turnover in 10 non-essential E2 mutants (Fig. 3b). We found that Atg9 degradation was not significantly compromised in yeast cells lacking these E2 enzymes.
Multiple sequence elements contribute to Atg9 instability.
The difficulty in uncovering a specific pathway (e.g., E2, E3) responsible for Atg9 degradation could be due to multiple E2s and E3s are involved. We then turned to delineate cis-elements critical for Atg9 destruction through deletion analysis (Fig. 4). Atg9 contains cytoplasmic domains at its N- and C-termini, and the conserved multi-transmembrane region in the middle (Fig. 4a). We constructed five deletion mutants missing various sections of Atg9 and then assessed their degradation in vivo (Fig. 4). Interestingly, both cytosolic fragments at N- and C-termini are rapidly degraded (Fig. 4b), and the membrane segment with either N- or C-terminal portion is degraded as well, indicating that not a single region is essential for Atg9 degradation. Our results suggest that Atg9 contains separate domains that promote its degradation, which likely requires multiple degradation pathways (e.g., E2s and E3s).
Fig. 4.
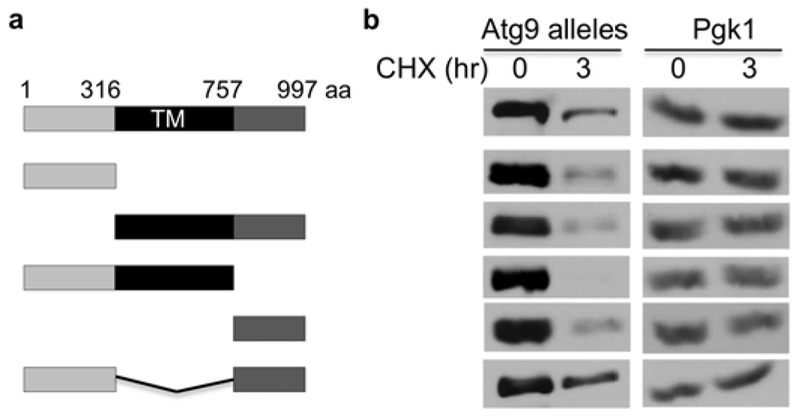
Cis-elements crucial for Atg9 degradation. (a) Domain structure of Atg9 and various mutants constructed. Atg9 contains two cytosolic regions on N- and C-end, and is membrane-anchored through six membrane-spanning domains (TM). (b) Degradation kinetics of various Atg9 deletion mutants. To ensure equal loading, we employed Pgk1 as a loading control. PGK1 encodes 3-phosphoglycerate kinase that is abundantly present in the cytosol.
Discussion:
Autophagy is a fundamental biological process critical for cellular homeostasis as it can quickly carry out mass destruction of various cellular components to meet the demand for cell growth and survival [1, 5]. Autophagy normally occurs at basal levels to maintain diverse cellular needs, and is activated by various stresses and catabolizes harmful contents to provide nutrients. Too much or too little autophagy can be detrimental to cell growth and development [8, 9]. We have systematically probed the degradation of 17 autophagy proteins and identified specific proteasomal targets (i.e., Atg9 and Atg14), uncovering new links between the proteasome and autophagy.
How autophagy activity is modulated in response to various cellular and environmental challenges remains poorly understood. Core autophagy genes have been shown to be regulated by various transcription activators (e.g., TFEB, TFE3, FOXO, p53, Gln3 and Dal80) or repressors (e.g., BRD4, Pho23, Ume6) [6, 9, 19, 20]. Post-translational modifications (e.g., phosphorylation, acetylation, methylation, ubiquitylation and sumoylation) play pivotal roles in controlling autophagy activity [1, 8, 19, 21–23]. For example, whereas AMP-activated protein kinase (AMPK) promotes autophagy through the phosphorylation of the transcription factor TFEB and several key autophagy regulators (e.g., Atg1 kinase, Atg6 and Vps34), TORC1 and AKT1 kinases restrain autophagy through their phosphorylation of Atg13 and FOXO proteins, respectively [1, 8, 19, 23]. Furthermore, p300-mediated acetylation on substrates including Atg5-8 and Atg12 inhibits autophagy, which can be reversed by SIRT1 deacetylase [1, 8, 19, 22].
Our understanding of Atg9 regulation remains rudimentary. As the only transmembrane protein among the core autophagy proteins, Atg9 is essential for autophagosome biogenesis and postulated to be a key player in supplying membranes to autophagosomes [11, 12, 14]. As Atg9 level was shown to be a critical determinant for the amount of autophagosomes [24], Atg9 expression and activity must be tightly regulated [6, 9]. Atg9 can be activated by Atg1 kinase, which in turn is required for pre-autophagosomal structure assembly and also efficient recruitment of additional autophagy proteins (e.g., Atg18, Atg2, Atg8) and subsequent phagophore elongation [11, 13, 17, 25]. Our data suggest that Atg9 is degraded by the proteasome under normal conditions, but stabilized upon starvation. Moreover, Atg1-mediated phosphorylation may affect Atg9 stability. The Atg9 phosphorylation mutant exhibits autophagy defects [17], some of which may be attributed to its instability. Given its unique membrane-spanning property among core autophagy proteins and its demonstrated effect on autophagosome biogenesis, Atg9 degradation may be a key factor in autophagy regulation. As multiple sequence elements and degradation pathways (e.g., ubiquitin ligase E3s) likely govern Atg9 instability (Fig. 4), we plan to delineate the proteolytic pathways associated with the individual cis-elements and further dissect their physiological significance in autophagy.
The systematic approach we employed in surveying substrate degradation can be easily extended to define the mechanism underlying the regulation of these autophagy factors under various conditions. Key autophagy proteins are subjected to diverse modification events [1, 22]. For example, Beclin1, an essential subunit of the class III phosphatidylinositol 3-kinase (PI3KC) complex, is regulated by multiple modules that control its stability, activity and interaction with relevant partners [19, 26, 27]. Beclin1 is regulated by more than 6 kinases, four of which (i.e., AMPK, DAPK, MAPKAP2, Atg1/ULK1) promote its activity and two of which (i.e., AKT and EGFR) play negative roles [22, 27]. Moreover, there are at least five ubiquitin ligases (e.g., Parkin, Traf6, NEDD4, the KLHL20-CUL3 complex and the Ambra1-Cul4-Rbx1 E3 complex) that can covalently decorate Beclin1 with distinct ubiquitin chains to influence its activity under various conditions [26, 28]. As similar strategies may be widely used in autophagy regulation, it’d be important to systematically probe various modifications of these key autophagy proteins before, during and after nutrient starvation. Ultimately we hope to gain a comprehensive understanding of the cellular cross talks involved in autophagy activation and recovery.
Our study brings novel insights into the mechanism underlying autophagy regulation and the functional interplay between two major cellular degradation machineries [1, 3]. Many diseases have been intimately tied to the dysregulation of autophagy or the proteasome [1, 4]. A better understanding of the regulatory pathways controlling autophagy would allow us to manipulate autophagy for clinic intervention, which could be invaluable in our battle against various autophagy related disorders and promote overall health.
Highlights.
Autophagy regulator Atg9 is degraded by the proteasome system
Atg9 turnover is affected by nutrient depletion and likely phosphorylation
Atg9 degradation is promoted by multiple proteolytic pathways
Acknowledgement:
We are grateful to Drs. D. Klionsky, C. Kraft, M. Hochstrasser, S. Lee, S. Michaelis, J. Frydman, P. Carvalho and D. Wolf for strains and plasmids. H.R. is supported by grants from the William & Ella Owens Medical Foundation, the Cancer Prevention Institute of Texas (RP170686, RP180769), the Mays Cancer Center, the National Institute of Health (GM118350) and the National Center for Advancing Translational Science (UL1TR002645).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest statement:
There is no potential conflict of interest.
Reference:
- [1].Levine B, Kroemer G, Biological Functions of Autophagy Genes: A Disease Perspective, Cell, 176 (2019) 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hu R, Hochstrasser M, Recent progress in ubiquitin and ubiquitin-like protein (Ubl) signaling, Cell Res, 26 (2016) 389–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dikic I, Proteasomal and Autophagic Degradation Systems, Annu Rev Biochem, 86 (2017) 193–224. [DOI] [PubMed] [Google Scholar]
- [4].Kwon YT, Ciechanover A, The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy, Trends Biochem Sci, 42 (2017) 873–886. [DOI] [PubMed] [Google Scholar]
- [5].Ohsumi Y, Historical landmarks of autophagy research, Cell Res, 24 (2014) 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yin Z, Pascual C, Klionsky DJ, Autophagy: machinery and regulation, Microb Cell, 3 (2016) 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dikic I, Elazar Z, Mechanism and medical implications of mammalian autophagy, Nat Rev Mol Cell Biol, 19 (2018) 349–364. [DOI] [PubMed] [Google Scholar]
- [8].Klionsky DJ, Why do we need to regulate autophagy (and how can we do it)? A cartoon depiction, Autophagy, 14 (2018) 1661–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jin M, Klionsky DJ, Regulation of autophagy: modulation of the size and number of autophagosomes, FEBS Lett, 588 (2014) 2457–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hurley JH, Young LN, Mechanisms of Autophagy Initiation, Annu Rev Biochem, 86 (2017) 225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Noda T, Autophagy in the context of the cellular membrane-trafficking system: the enigma of Atg9 vesicles, Biochem Soc Trans, 45 (2017) 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Feng Y, Klionsky DJ, Autophagic membrane delivery through ATG9, Cell Res, 27 (2017) 161–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Papinski D, Kraft C, Atg1 kinase organizes autophagosome formation by phosphorylating Atg9, Autophagy, 10 (2014) 1338–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhao YG, Zhang H, Autophagosome maturation: An epic journey from the ER to lysosomes, J Cell Biol, 218 (2019) 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, Pfuetzner RA, Brunger AT, Zhong Q, ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes, Nature, 520 (2015) 563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF, Chen RH, Cul3-KLHL20 Ubiquitin Ligase Governs the Turnover of ULK1 and VPS34 Complexes to Control Autophagy Termination, Mol Cell, 61 (2016) 84–97. [DOI] [PubMed] [Google Scholar]
- [17].Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, Lee SS, Brezovich A, Lou JH, Turk BE, Aebersold R, Ammerer G, Peter M, Kraft C, Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase, Mol Cell, 53 (2014) 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Finley D, Ulrich HD, Sommer T, Kaiser P, The ubiquitin-proteasome system of Saccharomyces cerevisiae, Genetics, 192 (2012) 319–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Corona Velazquez AF, Jackson WT, So Many Roads: the Multifaceted Regulation of Autophagy Induction, Mol Cell Biol, 38 (2018) e00303–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sakamaki JI, Long JS, New M, Van Acker T, Tooze SA, Ryan KM, Emerging roles of transcriptional programs in autophagy regulation, Transcription, 9 (2018) 131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Reidick C, El Magraoui F, Meyer HE, Stenmark H, Platta HW, Regulation of the Tumor-Suppressor Function of the Class III Phosphatidylinositol 3-Kinase Complex by Ubiquitin and SUMO, Cancers (Basel), 7 (2014) 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Popelka H, Klionsky DJ, Post-translationally-modified structures in the autophagy machinery: an integrative perspective, FEBS J, 282 (2015) 3474–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Noda T, Regulation of Autophagy through TORC1 and mTORC1, Biomolecules, 7 (2017) 52, doi: 10.3390/biom7030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jin M, He D, Backues SK, Freeberg MA, Liu X, Kim JK, Klionsky DJ, Transcriptional regulation by Pho23 modulates the frequency of autophagosome formation, Curr Biol, 24 (2014) 1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Feng Y, Backues SK, Baba M, Heo JM, Harper JW, Klionsky DJ, Phosphorylation of Atg9 regulates movement to the phagophore assembly site and the rate of autophagosome formation, Autophagy, 12 (2016) 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hill SM, Wrobel L, Rubinsztein DC, Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation, Cell Death Differ, 26 (2019) 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Menon MB, Dhamija S, Beclin 1 Phosphorylation - at the Center of Autophagy Regulation, Front Cell Dev Biol, 6 (2018) 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Boutouja F, Brinkmeier R, Mastalski T, El Magraoui F, Platta HW, Regulation of the Tumor-Suppressor BECLIN 1 by Distinct Ubiquitination Cascades, Int J Mol Sci, 18 (2017) 2541. [DOI] [PMC free article] [PubMed] [Google Scholar]