


Abstract
Splice-switching antisense oligonucleotides (ASOs), which bind specific RNA-target sequences and modulate pre-mRNA splicing by sterically blocking the binding of splicing factors to the pre-mRNA, are a promising therapeutic modality to treat a range of genetic diseases. ASOs are typically 15–25 nt long and considered to be highly specific towards their intended target sequence, typically elements that control exon definition and/or splice-site recognition. However, whether or not splice-modulating ASOs also induce hybridization-dependent mis-splicing of unintended targets has not been systematically studied. Here, we tested the in vitro effects of splice-modulating ASOs on 108 potential off-targets predicted on the basis of sequence complementarity, and identified 17 mis-splicing events for one of the ASOs tested. Based on analysis of data from two overlapping ASO sequences, we conclude that off-target effects are difficult to predict, and the choice of ASO chemistry influences the extent of off-target activity. The off-target events caused by the uniformly modified ASOs tested in this study were significantly reduced with mixed-chemistry ASOs of the same sequence. Furthermore, using shorter ASOs, combining two ASOs, and delivering ASOs by free uptake also reduced off-target activity. Finally, ASOs with strategically placed mismatches can be used to reduce unwanted off-target splicing events.
INTRODUCTION
Antisense oligonucleotides (ASOs) are a powerful research and therapeutic tool for a diverse range of applications, including target knock-down, splice correction, reading-frame restoration, removal of missense mutations, translational inhibition and modulating nonsense-mediated mRNA decay (NMD) (1,2). The mechanism of action of therapeutic ASOs depends on the type and position of chemical modifications in the phosphate backbone, ribose sugar and bases (3). Given that ASOs bind to RNA target sites by Watson–Crick base pairing, their specificity is primarily determined by the sequence itself. The sequence specificity of unmodified DNA oligos injected into Xenopus oocytes was investigated early on (4). Mismatches to the target sequence were shown to be tolerated, and the authors concluded that nonspecific RNA targets are likely partially degraded, which may be tolerated during oocyte development in most cases (4). However, with improvements in ASO chemistries with higher affinity for RNA, such as second- and third-generation 2'-O-methoxyethyl (MOE), 2', 4'-constrained 2'-O-ethyl (cEt) and locked nucleic acid (LNA) modifications, extensive sequence complementarity may not be required for efficient target engagement. This property is especially relevant for RNase-H-mediated knockdown with ‘gapmer’ ASOs. Gapmer sequences typically consist of 8–10 DNA nucleotides flanked by 3–5 modified high-affinity nucleotide ‘wings’ (5). Upon binding to RNA, the DNA/RNA duplex is recognized by RNase H, resulting in cleavage and degradation of the RNA strand. Importantly, cleavage requires as few as five continuous base pairs (6), which could result in knock-down of partially hybridized targets. Even though mismatch-induced changes in the shape of the RNA/DNA duplex negatively affect RNase-H-mediated cleavage (7), hybridization-dependent knock-down of unintended targets with mismatches was demonstrated in vitro (8), and is associated with hepatotoxicity in vivo (9). Assessment of off-targets, both in silico and experimentally, has therefore been recommended as an important part of the antisense drug-discovery process (10,11).
For splice-modulating ASOs, even fully complementary binding to the target sequence is not always sufficient to elicit an effect. ASOs typically target short regulatory elements in introns and exons (splicing enhancers and silencers) or the splice sites directly, to sterically interfere with the binding of proteins or small RNAs involved in splicing (12). ASOs that bind just one or a few nucleotides upstream or downstream of these regulatory sites sometimes completely lack splice-switching activity, as is often evident in ASO walks along the target sequence of interest (13–15). Steric-blocking ASOs are typically uniformly modified to prevent RNase-H-mediated cleavage, and as a result, they usually have high affinity for RNA. However, even if ASOs bind to partially complementary sequences in unintended targets, aberrant splicing as a result of this interaction is likely rare, and has not been studied extensively. In a study in which three potential splicing-specific off-targets were considered, none were experimentally identified (16). One reason could be that the BLAST algorithm commonly used for identifying ASO sequence complementarity heavily penalizes gaps and bulges, and does not consider G:U wobble base-pairing, typically found in RNA structures (17). The extent of ASO-mediated mis-splicing of unintended targets could therefore be more common than previously expected, warranting a more detailed investigation.
In this study, we investigate sequence-specific off-target effects of splice-switching ASOs in vitro and in vivo. The two overlapping ASO sequences used here were previously shown to target an exonic splicing enhancer (ESE) in exon 10 of the PKM gene, which results in an isoform switch from PKM2 to PKM1 (14,18). We adapted RNAhybrid, a microRNA target-prediction tool, to identify sequence complementarities between the ASO and the human transcriptome. We then tested 108 potential ASO targets by RT-PCR, and identified 17 mis-splicing events for one of the ASOs tested, some of which were unexpectedly stronger than the on-target activity on PKM splicing. We also found that the choice of ASO chemistry has a strong influence in the extent of off-target activity, with cEt/DNA mixed-chemistry ASOs having greater specificity than uniformly modified MOE ASOs. Furthermore, shortening the ASO sequence, using two ASOs in combination at lower concentration, introducing targeted mismatches, and delivering ASOs by free uptake rather than transfection, also reduced off-target activity. In summary, ASOs targeting specific RNA motifs to modulate splicing can also cause detectable levels of off-target mis-splicing, which theoretically could contribute to potential ASO toxicities in vitro or in vivo.
MATERIALS AND METHODS
Identifying potential ASO target sites in the transcriptome
We generated an in silico library of exons, including ±200 nt of flanking sequence, for all principal and alternative isoforms (APPRIS database (19)) of protein-coding transcripts in the GRCh38/hg38 human genome assembly, and assigned unique identifiers (Unique ID) to each entry. We removed duplicate exon coordinates (arising from overlapping transcripts) to speed up the alignment process. The resulting in silico exon library served as a reference for RNAhybrid sequence alignment (20), which we used to identify the top two ASO-binding sites in each exon/intron, based on binding free energy. Importantly, we allowed gaps, loops, and G:U base-pairing, which were part of the minimum free energy (MFE) calculation for each ASO-target pair. We selected the following settings: maximum internal loop length and bulge size were set to 5 nt each, and the free energy threshold was variable depending on the ASO, but was at least 10 kcal/mol lower than the on-target binding free energy. We did not set helix constraints, and chose the compact output to help with data formatting. We then annotated target sequences for each exon with gene names, exon position (terminal, internal), strand information, MFE, number of mismatches, number of G:U base pairs, binding site (exon, intron, splice site), distance to the closest splice site, and whether the exon is part of the coding sequence or UTR. We also analyzed ASO target sequences for potential binding sites of RNA-binding proteins using the RBPmap online tool (rbpmap.technion.ac.il (21)). RNA-binding proteins included in this analysis were HNRNP-A1, -A2B1, -C, -F, -H1, -H2, -M, SNRNP70, SNRPA, SRSF1, SRSF2, SRSF3, SRSF5, SRSF6, SRSF7, SRSF9, SRSF10, TRA2A, TRA2B and U2AF2. We set the stringency level to ‘high’, and the conservation filter to ‘on’. Lastly, we obtained RNA expression levels (TPM) of the predicted targets for common human cell lines and tissues from The Protein Atlas (proteinatlas.org (22)), and added them to the results table.
Antisense oligonucleotides
All ASOs used in this study are listed in Table 1. 2'-O-methoxyethyl (MOE) synthesis, purification, and quantification were performed as described (23). Mixed-chemistry oligonucleotides were a mixture of constrained ethyl (cEt) and DNA, as shown in Table 1, and were synthesized as described (24). All ASOs had uniform phosphorothioate (PS) backbones and 5-methyl C. We dissolved the ASOs in water and stored them at −20°C. Oligonucleotide concentration was determined with a Nanodrop spectrophotometer. RNA-oligos used for Tm measurements were purchased from IDT.
Table 1.
ASOs used in this study
| ASO name | IONIS # | Length | Sequence 5'-3' | Chemistry |
|---|---|---|---|---|
| ASO1-MOE | 5644676 | 18 nt | CGGCGGAGTTCCTCAAAT | Uniform MOE |
| ASO1-cEt/DNA | 742114 | 18 nt | CGGCGGAGTTCCTCAAAT | kkddkddkddkddkddkk |
| ASO1-MOE/DNA | 759514 | 18 nt | CGGCGGAGTTCCTCAAAT | eeddeddeddeddeddee |
| ASO2-MOE | 564472 | 18 nt | AGGCGGCGGAGTTCCTCA | Uniform MOE |
| ASO2-cEt/DNA | 872245 | 18 nt | AGGCGGCGGAGTTCCTCA | kkddkddkddkddkddkk |
| GN3-ASO2-MOE | 1057288 | 18 nt | AGGCGGCGGAGTTCCTCA | 5'-GalNAc3-Uniform MOE |
| GN3-ASO2-cEt/DNA | 950663 | 18 nt | AGGCGGCGGAGTTCCTCA | 5'-GalNAc3-kkddkddkddkddkddkk |
| GN3-SMN-MOE | 699819 | 20 nt | ATTCACTTTCATAATGCTGG | 5'-GalNAc3-Uniform MOE |
| Ctrl. ASO-MOE (M4) | 759518 | 18 nt | CGGATGAGTGCCTGCAAT | Uniform MOE |
| Ctrl. ASO-cEt/DNA (M4) | 872243 | 18 nt | CGGATGAGTGCCTGCAAT | kkddkddkddkddkddkk |
| Scr. Ctrl ASO | 742116 | 18 nt | GCTGATCCGAGTAAGCTC | Uniform MOE |
| 12mer MOE | 759513 | 12 nt | CGGCGGAGTTCC | Uniform MOE |
| I9-MOE | 461378 | 15 nt | TACCTGCCCTTAGGG | Uniform MOE |
e: 2'-O-methoxyethyl (MOE) base modification; k: constrained ethyl (cEt) base modification; d: DNA base; GN3: tri-antennary N-acetyl galactosamine; all ASOs have a uniform PS backbone and 5-methyl-C modifications.
Cell culture and ASO delivery
U87 MG glioblastoma cells and HT-1080 fibrosarcoma cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin. HepG2 cells were cultured in EMEM supplemented with 10% FBS and 1% penicillin and streptomycin. We delivered ASOs to U87 and HepG2 cells using Lipofectamine 2000 (Invitrogen) at a final concentration of 30 nM. Alternatively, we added ASOs directly to the culture medium without transfection agent (free uptake), at the indicated concentrations, for up to 5 days.
Primer design and PCR
Total RNA was extracted using Trizol reagent, and reverse-transcribed with ImProm-II reverse transcriptase (Promega), using oligo-dT primers. PKM cDNA was amplified with AmpliTaq DNA polymerase (Thermo Fisher) using exon 8 Fwd 5'-AGAAACAGCCAAAGGGGACT-3' and exon 11 Rev 5'-CATTCATGGCAAAGTTCACC-3' primers. The PCR product was digested with PstI for 2 h at 37°C to distinguish PKM1 (undigested) from PKM2 (2 bands) (14). Primers used to test the effect of ASOs on potential off-targets are listed in Supplementary Table S1, and were designed using NCBI Primer-BLAST (25). Forward and reverse primers were positioned at least 1 exon upstream and downstream, respectively, of the exon predicted to be bound by the ASO. The primer Tm, GC content, and product length were similar for all tested off-targets, and a standard PCR program (55°C annealing; 25 cycles) was used for all amplicons. If no band was detected, the PCR was repeated using 30 cycles. Primers that did not yield a product or yielded non-specific bands (based on size) were excluded from the analysis. The [α-32P]-dCTP radiolabeled PCR products were separated on a 5% native polyacrylamide gel, visualized by autoradiography on a Typhoon 9410 phosphoimager (GE Healthcare), and quantified using Multi Gauge v2.3 (Fujifilm, Tokyo, Japan). The radioactive signal of each band was normalized to the G/C content to calculate relative changes in splice isoforms.
Tm measurements
Melting temperatures of RNA/ASO duplexes were determined by high-resolution melting curves performed on a QuantStudio 6 Flex Real-Time PCR System (26). Single-stranded fragments (1–2 μM each) were mixed with 6.25 μM Eva green (Biotium, Fremont, CA) and annealing buffer (final concentration 10 mM Tris pH 7.4, 1 mM EDTA, 50 mM NaCl). 10 μL of the sample was transferred onto 386-well plates for melting analysis. The samples were heated to 98°C for 2 min, cooled to 25°C for 1 min at a rate of 0.05°C/s for annealing, and heated to 98°C at a rate of 0.05°C/s for melting analysis (continuous measurement). The Tm of each duplex was measured a total of six times on two separate plates.
Animals and tumor model
Non-obese diabetic-severe combined immunodeficiency (NOD-SCID)-gamma (NSG) immunocompromised mice (strain 005557; The Jackson Laboratory) were housed in vented cages and bred in-house. ASGP-R1(H1a)-expressing U87 cells used to generate xenografts are described in Scharner et al. (27). Tumors were established by injecting 5 × 106 U87-H1a cells (50 000 cells/μl in Hank's Balanced Salt Solution) subcutaneously into the flanks of adult NSG mice. Once tumors were palpable (7–10 days post-transplantation), animals were treated with ASOs delivered by subcutaneous (s.c.) injection at 250 mg/kg/week, 5 times per week for two weeks, before collecting tumor tissue for splicing analysis. All animal protocols were performed in accordance with Cold Spring Harbor Laboratory's Institutional Animal Care and Use Committee (IACUC) guidelines.
Statistics and graph representation
Graphs were generated in Microsoft Excel and GraphPad Prism 6. Figures were compiled using Adobe Illustrator. Paired Student's t-test was used to test for statistical difference between off-target scores; unpaired Student's t-test and Fisher's exact test were used to test for statistical significance of feature characteristics of off-target sites. P-values <0.05 were considered to be statistically significant. For multiple hypotheses testing, P-values less than the Bonferroni critical value (α/n; specified in the figure legends) were considered to be statistically significant.
RESULTS
Identification of ASO sequence complementarities in the human transcriptome
To investigate whether ASO-mediated off-target splicing events are detectable in ASO-treated cells, we utilized the miRNA prediction tool RNAhybrid (20), to detect ASO sequence complementarities in the transcriptome, while allowing for G:U base pairing, loops, and gaps. We aimed to identify potential ASO-binding sites on or near exons, and then test the splicing outcome by RT-PCR. We therefore created a database of exon sequences, including up to 200 nt of flanking intronic sequences, in which to search for putative ASO-binding sites. After aligning the ASO to the exon database using RNAhybrid, we formatted, annotated, and exported the data for further analysis (Figure 1A). To characterize each ASO-binding site in the transcriptome, we also listed several descriptive features, including the number of mismatches, the number of G:U base pairs, the distance to the nearest splice site, etc. (Figure 1B, Supplementary Table S2). We then selected a wide range of predicted targets and tested the ASO’s effect on splicing of the nearest exon in an initial RT-PCR screen. When we detected an aberrant splice product, we further characterized the splicing event by RT-PCR (Figure 1C). The ASOs used for the initial screen were two uniformly modified, MOE-PS PKM splice-switching ASOs targeting exon 10 (ASO1 and ASO2), as well as a control ASO with five mismatches to the PKM target (Figure 1D, Table 1). The ASO modifications used increase RNA affinity and do not support RNase-H-mediated cleavage (12). Upon binding to PKM exon 10, these ASOs induce a splice switch between two mutually exclusive exons, promoting exon 10 skipping and exon 9 inclusion (14,18). We aligned each of the ASOs to the exon database using RNAhybrid, and ranked the hits by minimum free energy (MFE). The calculated MFE values of the top 50 and top 1000 targets shown in Figure 1E confirm PKM as the top hit, as well as the only hit without mismatches or G:U wobble base-pairing. For ASO2 we found a second predicted target with full complementarity (THSD7B); however, 6/18 base pairs are wobble base pairs, and this gene is not expressed in U87 cells. We also analyzed the overlap of predicted targets between different ASOs, and found that a 3-nt shift along the target sequence changed >70% of the predicted targets, whereas introducing five mismatches resulted in an almost completely new set of predicted binding targets (Figure 1F).
Figure 1.

Prediction of off-targets and validation by RT-PCR. (A) ASO sequences were aligned to an exon database, including 200 nt of each flanking intron. Sequence alignment was performed using RNAhybrid, allowing G:U wobble base pairing, internal loops, and bulges. The data were annotated and formatted for further analysis. (B) Features used to characterize the interaction between the ASO and the RNA target. (C) Potential off-targets were tested by radioactive RT-PCR, using primer pairs located at least 1 exon upstream and downstream of the binding site, respectively. Positive hits from the initial screen were analyzed further. (D) ASOs used in the initial off-target screen included two PKM splice-switching ASOs shifted by three nucleotides (ASO1-MOE and ASO2-MOE), and a control ASO with five mismatches to the target (Ctrl ASO-MOE). Nucleotides highlighted in red indicate mismatch positions. (E) Distribution of the minimum free energy (MFE; kcal/mol) of the top 1000 and top 50 hits from the RNAhybrid alignment prediction. PKM was the top hit for both ASO1 (−38.2 kcal/mol) and ASO2 (−44.4 kcal/mol). (F) Venn diagrams of overlapping targets among the top 1000 hits. ASO1 and the Ctrl ASO have 13 hits in common. ASO1 and ASO2 have 294 targets in common.
PKM splice-switching ASOs induce mis-splicing of some unintended targets
To validate predicted off-targets identified by RNAhybrid, we delivered 30 nM ASO to U87 glioblastoma cells by lipid transfection, and analyzed RNA splice products by radioactive RT-PCR 2 days post-transfection. This condition resulted in 31% and 32% PKM1 splice switching for MOE-PS modified ASO1 and ASO2, respectively (Figure 2). Importantly, the selected concentration did not saturate the effect on PKM splice-switching, or result in notable cell toxicity. (Supplementary Figure S1). We also tested a pair of cEt and DNA mixed-chemistry ASOs (cEt/DNA) with the same sequences (Table 1). Mixed-chemistry ASO1 and ASO2 resulted in 29% and 34% PKM1 splice switching, respectively, at 30 nM concentration. The control ASO with five mismatches to the intended target did not result in increased PKM1 splicing, compared to the untreated control, and was identified in an ASO mutation screen (Supplementary Figure S2).
Figure 2.

Validation of predicted off-targets by RT-PCR. (A) Representative autoradiographs and quantification of splice changes of exons in transcripts to which ASOs are predicted to bind. Bar charts represent mean ± SE; N = 3; radiolabeled MspI digest of pBR322 served as size markers. PKMds (double skipping) is an aberrant splice product that lacks both exons 9 and 10. The identities of PCR fragments labeled with an asterisk (*) were confirmed by Sanger sequencing. (B) Quantification of the fold-difference of off-target effects, relative to PKM on-target effects, normalized to the Lipofectamine control (OT score). An OT-score <1 indicates that the off-target effect is smaller than the on-target effect; values >1 indicate off-target effects larger than on-target effects. The OT-score takes variable on-target activity into consideration, and thus it can be used to directly compare the off-target activity of different ASOs. N = the number of validated off-targets for each ASO (effect > 10%); ** P < 0.01.
To test whether PKM splice-switching ASOs can induce unintended splicing changes of exons with substantial sequence complementarity, we selected 140 predicted targets and designed primers upstream and downstream of the exon closest to the ASO-binding site for an initial RT-PCR screen (Supplementary Table S1). Out of 140 primer pairs tested, 32 resulted in non-specific or no amplification, and we excluded them from further analysis. The remaining 108 primer pairs yielded correct amplicons, based on size. 22 of them gave aberrant splice products, which we investigated further (Supplementary Table S1). Figure 2A and Supplementary Figure S3 show radioactive RT-PCR gels and isoform quantification of all 22 tested off-targets. Out of those that resulted in a splicing change of >10%, 12 were common for both ASO1 and ASO2. We also detected an additional two unique off-targets for ASO1 and five unique off-targets for ASO2. The majority of the validated off-target splicing events resulted in skipping of constitutive exons. In three cases (CSNK1G3, ATP5SL, PKD1), we detected a change in relative expression of naturally occurring splice isoforms (Figure 2A and Supplementary Figure S3). Surprisingly, the mismatch control ASO (Ctrl. ASO) also induced exon 12 skipping in CLIP4, as well as exon 12 inclusion in CSNK1G3. Closer inspection of the sequences revealed the presence of Ctrl. ASO-binding sites distinct from the ASO1 binding site, consistent with a direct correlation between Ctrl. ASO binding and a splicing change (Supplementary Figure S4). Importantly, a different control ASO (scrambled) used in Figure 5 did not induce exon 12 skipping in CLIP4, or exon 12 inclusion in CSNK1G3, supporting a hybridization-dependent effect.
Figure 5.

Alternative strategies to reduce off-target effects in vitro. (A) ASOs used in this figure include a short 12mer ASO that overlaps both ASO1 and ASO2, another PKM splice-switching ASO that targets intron 9 (I9), and a scrambled-sequence control ASO. (B) The effect of alternative ASOs on PKM splice-switching. The on-target activity of MOE/DNA ASO is worse than the uniformly MOE-modified ASO1 (3.5% and 29.9% PKM1, respectively; see Figure 2). Both the 12mer MOE (45.5% PKM1) and the combination treatment (52.3% PKM1) performed better than either ASO1 or ASO2 alone. Individual ASOs were transfected at a final concentration of 30 nM. For the combination treatment, ASOs were transfected together, at a final concentration of 15 nM each. (C) Radioactive RT-PCR results of seven validated off-targets. Bar charts show mean ± SE from three independent experiments. (D) Average OT-scores for each ASO tested, for 7 different off-targets (AK2, PROM1, RER1, CLIP4, UCK2, PTOV1, CSNK1G3), compared to the ASO1 and ASO2 results. OT-scores for ASO1-MOE and ASO1-cEt/DNA serve as a point of comparison, and are the same as in Figure 2. Individual OT-scores for each off-target are listed in Table 2. The OT-score takes variable on-target activity into consideration, and thus it can be used to directly compare the off-target activity of different ASOs. * P < 0.05; ** P < 0.01;
Next, we sought to compare the propensity of each ASO for off-target effects. Given that each of the ASOs used in this study has slightly different effects on PKM splicing, we quantified the off-target effect relative to PKM, and calculated an Off-target score (OT-score = ΔPSI(Off-target)/ΔPSI(On-target), Table 2). Similar to on-target activity, off-target splicing events increase with ASO concentration, confirming that the observed effects are within the dynamic range of the assay at the 30-nM concentration used to compare on- and off-target effects (Supplementary Figure S1). However, potentially different EC50/Emax values between on- and off-target activity may result in different OT scores at higher ASO concentrations. OT-scores >1 represent splicing changes that are greater than the on-target effect, whereas OT-scores <1 represent splicing changes that are smaller than the on-target effect. MOE-modified ASOs 1 and 2 had average OT-scores of 1.08 and 1.15, respectively. In contrast, the OT-scores of cEt/DNA ASOs 1 and 2 were statistically significantly lower (0.47 and 0.33, respectively), showing that the mixed-chemistry design is beneficial in avoiding off-target effects in vitro (Figure 2B). The OT scores of six off-targets measured in HepG2 and HT1080 cells showed some variability (Supplementary Figure S4). However, consistent with the results we obtained in U87 cells, the cEt/DNA mixed-chemistry ASOs had reduced off-target activity, compared to the uniform-MOE ASOs, in HepG2 and HT1080 cells (Supplementary Figure S4).
Table 2.
Off-target score of various ASO chemistries, lengths and combinations
| AK2 | PROM1 | RER1 | CLIP4 | UCK2 | PTOV1 | CSNK1G3 | |
|---|---|---|---|---|---|---|---|
| ASO1-MOE | 1.8 (0.14) | 1.5 (0.02) | 1.5 (0.21) | 1.6 (0.28) | 0.7 (0.08) | 0.0 (0.06) | 1.1 (0.10) |
| ASO1-cEt/DNA | 0.2 (0.04) | 1.5 (0.20) | 0.7 (0.16) | 1.0 (0.13) | 0.1 (0.0) | 0.1 (0.01) | 0.7 (0.10) |
| ASO2-MOE | 2.2 (0.15) | 2.2 (0.13) | 1.4 (0.23) | 1.5 (0.21) | 1.3 (0.20) | 1.2 (0.09) | 1.2 (0.11) |
| ASO2-cEt/DNA | 0.3 (0.06) | 0.0 (0.0) | 1.1 (0.20) | 0.3 (0.06) | 0.9 (0.09) | 0.0 (0.01) | 0.5 (0.12) |
| ASO1-MOE/DNA | 0.0 (0.0) | 1.0 (0.56) | 0.3 (0.06) | 1.5 (0.18) | 0.0 (0.0) | 0.2 (0.04) | 1.0 (0.37) |
| 12mer MOE | 1.3 (0.16) | 0.3 (0.01) | 1.1 (0.14) | 1.4 (0.13) | 0.1 (0.01) | 1.0 (0.09) | 0.0 (0.02) |
| MOE combination | 1.0 (0.06) | 0.7 (0.18) | 1.0 (0.09) | 0.8 (0.05) | 0.7 (0.06) | 0.4 (0.02) | 0.5 (0.05) |
Data represent mean (SE) relative to PKM.
Base-pairing of modified nucleotides in mixed-chemistry ASOs correlates with the off-target effects
In some cases, the MOE ASO had a more robust effect on splicing of the off-target gene than the cET/DNA (e.g. AK2 and PTOV1). In other cases (e.g., ASO1 PROM1, ASO2 UCK2), the outcome was very similar, irrespective of ASO chemistry. To gain insight into the reason for these differences in off-target effects, we analyzed the ASO/target binding structures predicted by RNAhybrid, and specifically looked at base-pairing of the modified bases (Figure 3A). Constrained-ethyl-modified nucleotides bind more tightly to RNA than MOE-modified nucleotides, which would result in excessively high Tm's if present uniformly. To balance the strong interactions of cEt, while maintaining a melting temperature comparable to that of uniformly modified MOE ASOs, cEt/DNA mixed-chemistry ASOs only comprise eight cEt-modified bases (Figure 3A, highlighted in red). Depending on the predicted duplex structure, some predicted target binding sites have more cEt residues engaged in base-pairing, whereas in other targets, some cEt residues are located at mismatch positions. Binding sites with 7/8 (UCK2, ASO2) or 8/8 (PROM1, ASO1) cEt residues engaged in base-pairing corresponded to moderately lower exon skipping (UCK2 −37%) or even slightly higher exon skipping (PROM1 +2.8%), compared to MOE-modified ASOs of the same sequence. In contrast, with six or fewer cEt residues engaged in base-pairing, the effects on off-target splicing were markedly reduced, compared to the effects of MOE ASOs, likely because the overall Tm of the ASO is affected a lot more by a cEt mismatch than by an MOE mismatch.
Figure 3.

Structures of individual ASO-binding sites. (A) Structures show the target sequence at the top (5'-3') and the ASO sequence at the bottom (3'-5'), as predicted (for RNA:RNA duplexes) by RNAhybrid. cEt-modified bases are highlighted in red, and the number of paired cEt bases is shown at the top right of each field. The numbers below each structure indicate the splicing change relative to the MOE-modified ASO of the same sequence. (B) Melting temperatures of RNA/DNA and RNA/ASO duplexes determined by high-resolution melting analysis. RNA (green), DNA (black), MOE (blue), cEt (red). Data points represent mean ± SE; N = 6.
To test the impact of cEt mismatches on the overall Tm of the RNA/ASO duplex, we performed high-resolution melting analysis (Figure 3B). First, we measured the Tm of ASO2-MOE and ASO2-cEt/DNA, as well as the equivalent DNA sequence, to a fully complementary RNA strand. As expected, the Tm's of the modified ASOs were significantly higher (MOE 85.7°C; cEt/DNA 92°C) compared to DNA (70.6°C). To assess the impact of MOE versus cEt residues on the Tm, we used the same RNA substrate and annealed ASO1, which is shifted by three nucleotides and thus has three fewer hybridizing nucleotides (AAT) at the 3'-end. The overall Tm of ASO1-MOE (15-bp match) was 5.3°C lower, compared to the fully complementary ASO2-MOE. However, the Tm of ASO1-cEt/DNA (15-bp match) was 6.9°C lower, compared to ASO2-cEt/DNA (18-bp match), confirming that non-hybridizing cEt-modified residues have a stronger impact on the Tm than non-hybridizing MOE residues; the difference was statistically significant (P = 0.0018) (Figure 3B).
Validated ASO off-targets lack shared characteristic features
One aim of this study was to identify key features that characterize off-targets, so as to facilitate the prediction of off-targets of new ASOs. A primary feature we looked at was the ASO binding free energy (MFE), as predicted by RNAhybrid. However, when we ranked all off-targets tested in this study by their MFEs, only 3/10 top targets for ASO1 and 1/10 top targets for ASO2 resulted in a measurable splicing change, indicating that MFE is a poor predictor of off-target activity. One caveat is that the program uses RNA-RNA hybrids for the calculations, rather than modified nucleotide-RNA. Furthermore, there was no correlation between MFE and splicing among validated off-targets for either ASO1 or ASO2 (Figure 4A). To see whether the MFE obtained by RNAhybrid is a good approximation of the actual Tm between an ASO and its RNA target, we measured the Tm of several RNA/ASO duplexes with various mismatches and G:U wobble base pairs (Figure 4B, Supplementary Table S3). Overall, the Tm of the RNA/ASO duplex correlated well with the predicted MFE (R2 = 0.78; Pearson's r = −0.89). Two of the data points (AK2 and CLIP4) slightly deviated from the trend line. Interestingly, both AK2 and CLIP4 RNA templates have significant predicted secondary structures (not observed in other RNA templates used in this study), which may influence the Tm measurements of the RNA/ASO duplex. The overall correlation between MFE and measured Tm may therefore be underestimated.
Figure 4.

Characterization of ASO-binding sites. (A) The splicing change for each of the validated off-targets does not correlate with the binding energy (MFE) (ASO1 R2 = 0.0) or is negatively correlated, with weaker interactions eliciting stronger splicing changes (ASO2 R2 = 0.16). (B) The empirically measured Tm of ASO2 and RNA templates corresponding to off-target binding sites correlates well with the predicted MFE (R2 = 0.88, N = 6). Data points show mean ± SEM Tm from 6 measurements. (C, G) Comparison of ASO-binding sites on validated off-targets (splicing change > 10%) with a control group, comprising the top 20 predicted binding sites based on MFE that did not result in a splice change. Both ASO1 and ASO2 are predicted to bind significantly more strongly to target sites in the control group, compared to sites on validated off-targets (ASO1 −30.17 kcal/mol and -31.95 kcal/mol, respectively; ASO2 −31.85 kcal/mol and −36.25 kcal/mol, respectively) Each data point indicates a unique ASO-binding site. (D, H) The relative distribution of ASO-binding sites (exon, intron, or splice site) is not statistically different between the off-target and control groups for ASO1 and ASO2. (E, I) Graph showing the proportion of ASO-binding sites with at least one RBP motif, as determined by RBPmap. (F, J) Chart indicating the distance of each ASO-binding site to the nearest splice site. Negative values indicate binding sites that span exon/intron junctions. n.s., not significant; P < 0.0056 (Bonferroni critical value) was considered to be statistically significant.
Effective splice-modulating ASOs not only need to bind to the target RNA, but they also need to block the binding of splicing regulators. We therefore also looked at other features of ASO-binding sites, as well as the exons to which the ASO is predicted to bind (Figure 4 and Supplementary Figure S5). To identify relevant features for eliciting a splicing change upon ASO binding, we statistically compared all ASO-binding sites of validated off-targets (splicing change > 10%) with a control group, comprising the top 20 predicted ASO-binding sites, based on MFE, that did not result in a splice change upon ASO treatment. Both ASO1 and ASO2 were predicted to bind significantly more strongly to the target sites in the control group (no detected splicing change), compared to sites on validated off-targets (average ΔMFE 1.18 kcal/mol and 4.40 kcal/mol for ASO1 and ASO2 target sites, respectively (Figure 4C and D).
Given that our exon database includes exons and part of the flanking introns, we then asked whether ASOs that bind to exonic sequences are more likely to induce a splicing change. We observed a trend towards exonic binding sites, but the relative distribution of ASO-binding sites (exon, intron or splice site) was not statistically different between the off-target and control groups, both for ASO1 and ASO2 (Figure 4C and D).
Next, we asked whether the presence of RNA-binding protein (RBP) motifs is a predictive factor in off-target activity, considering that ASOs that interfere with exonic splicing enhancers or silencers should be more likely to result in aberrant splicing. We used RBPmap to predict binding sites for common RBPs involved in RNA splicing regulation, within the ASO-target site. This tool includes hnRNPs, TRA proteins, members of the SR-protein family, as well as others (the full list of RBPs included in this analysis is in the Material and methods section). Interestingly, RBP motifs were found in 85%, or 12 of 14, of predicted ASO1-binding sites that resulted in a splicing change, compared to 45% (9 of 20) of predicted ASO1-binding sites on exons in the control group, for which no splicing change was detected (Figure 4C). RBP motifs were also found in 76% or 13 of 17 ASO2-binding sites on mis-spliced exons, versus 60% for the control group, but this difference was not statistically significant (Figure 4D).
We also asked whether the distance between the ASO-binding site and the nearest splice site is a predictive feature of off-target activity. On average, ASO-binding sites that resulted in off-target splicing tended to be closer to one of the splice sites than those that did not result in detectable splice changes. However, as above, this trend was not significant, due to multiple hypotheses testing.
Lastly, we did not uncover any predictive features when looking at the larger context of mis-spliced exons, including the length of the targeted exon, the length of the flanking introns, whether the target exon is in-frame or out-of-frame, and the total number of exons in the gene (Supplementary Figure S5). Taken together, we observed positive trends for several parameters we investigated (although none of them was statistically significant), suggesting that the overall splicing outcome upon ASO binding depends on combinations of several features.
ASO combination enhances on-target and reduces off-target effects in vitro
Given that cEt/DNA mixed-chemistry ASOs reduced off-target effects for some transcripts in vitro, compared to their uniformly MOE modified counterparts, we investigated additional strategies to increase the specificity of splice-switching ASOs targeting PKM. We therefore designed additional ASOs, including an MOE/DNA mixed-chemistry ASO and a short 12mer ASO. We also found that ASOs targeting splicing-regulator binding sites in PKM intron 9 induce splice-switching, albeit to a lesser extent than exon 10 ASOs (manuscript in preparation), and so we tested a uniformly modified MOE ASO targeting PKM intron 9, in combination with ASO2 (Figure 5A).
The MOE/DNA ASO had a significantly lower on-target activity than the equivalent fully modified MOE ASO (4.1% and 30.6% PKM1, respectively, Figure 5B). We also observed a reduced OT-score, which is comparable to the effect seen with cEt/DNA mixed chemistry ASOs. However, given the low efficacy of the MOE/DNA ASO, it is difficult to attribute the apparent increase in specificity to ASO chemistry alone (Figure 5C).
Short 8–13mer ASOs, although less potent, have been used successfully in vitro for splice modulation (28,29). We hypothesized that short ASOs may be more sensitive to mismatches than 18mer ASOs, which show some resistance that depends on the mismatch position (Supplementary Figure S2). We therefore designed a uniformly MOE-modified PKM-targeting 12mer ASO that overlaps with both ASO1 and ASO2 (Figure 5A) and tested its on- and off-target activities. Interestingly, we observed a strong reduction of the OT-score of the shorter ASO with a subset of off-targets (PROM1, RER1 and CSNK1G3; Figure 5). Sequence alignment of the 12mer ASO with our exon/intron database revealed one perfect match (EXD3; not expressed in U87 cells), as well 34 additional matches containing a single G:U base pair. However, given that the 12mer sequence is contained in both the ASO1 and ASO2 sequences, the parent ASOs would presumably bind to a similar extent, resulting in largely overlapping off-target activity.
The last strategy we tested to increase on-target specificity was to use a combination of ASOs targeting distinct regulatory elements of the same splicing event. The complex regulation of PKM alternative splicing allows targeting different sites from exon 9 to exon 10 to efficiently induce PKM2 to PKM1 splice-switching (14,18). For PKM, these sites include a secondary regulatory region in exon 10, as well as a region in intron 9 (manuscript in preparation). We reasoned that the use of a second ASO targeting the same splicing event may allow lowering the concentration of each individual ASO, and could perhaps show synergy, potentiating the effects of single ASOs. We used an intron 9 MOE ASO in combination with ASO2-MOE, and transfected cells at a final concentration of 15 nM of each ASO. In combination, these ASOs induced stronger PKM splice-switching (52.9%) than 30 nM intron ASO2-MOE alone (32.1%). The intron 9 ASO is less potent than ASO1 or ASO2-MOE (manuscript in preparation). Importantly, off-target effects were significantly reduced when the ASOs were used in combination (Figure 5).
Strategically placed mismatches can be used to reduce specific off-target activity
To investigate whether mismatches can be used to reduce the activity on specific off-targets, we tested a sequence variant of ASO1, with 5 mismatches to PKM at the 3'-end of the ASO (Figure 6A and Supplementary Figure S2). The effect on PKM splice switching in transfected U87 cells was comparable to that of the fully complementary parent ASO (Supplementary Figure S2). The effect on off-target activity, however, depended on the target and location of mismatches (Figure 6B). We tested four different mis-splicing events induced by ASO1 (PTOV1, RER1, UCK2 and PROM1). The parent ASO is predicted to bind with mismatches to PTOV1 and RER1 targets; Therefore, changing the ASO sequence at the 3'-end did not significantly reduce off-target activity for these two targets (Figure 6A). In contrast, targets predicted to hybridize with the ASO at the 3'-end (UCK2 and PROM1) were negatively affected by the mutant ASO. As a result, we observed a statistically significant reduction in off-target mis-splicing (Figure 6B). In conclusion, strategically placed mismatches could therefore be used to prevent a particular off-target event, while maintaining on-target activity.
Figure 6.
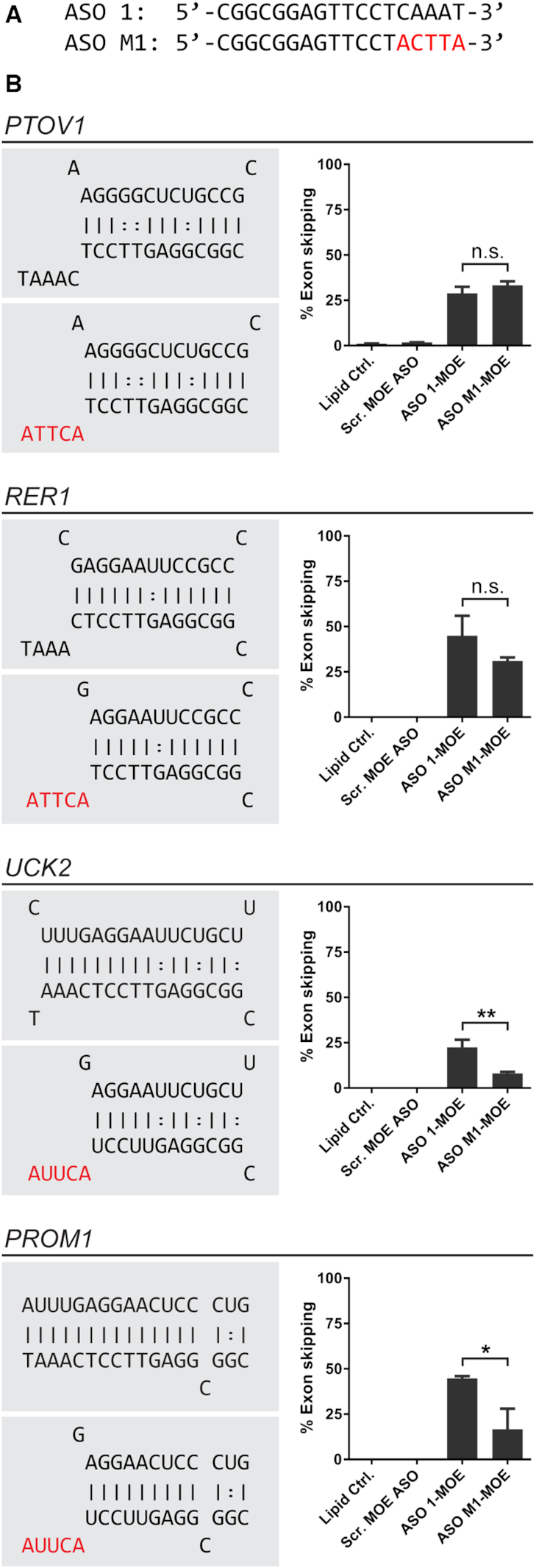
ASO mismatches differentially affect off-target splicing. (A) MOE-ASOs used to compare the effects of mismatches on off-target splicing include MOE-ASO1 (fully complementary to PKM) and an ASO with five mismatches (highlighted in red). Mismatches at the 3'-end of the ASO have a minimal effect on PKM splice switching (see Supplementary Figure S1). (B) ASO/RNA duplex structures (left) and quantification of the resulting splice change in U87 cells (right). ASOs were delivered by lipid transfection at a final concentration of 30 nM. Splicing changes were analyzed 48 hrs post-transfection. Bar graphs represent mean ± SE; N = 3; * P < 0.05; ** P < 0.01;
Free uptake reduces off-target effects of splice-switching ASOs
In vitro, ASOs are routinely delivered by lipid transfection directly into the cytoplasm, bypassing natural uptake and endosomal-release pathways. In addition, cationic lipids are known to enhance nucleic acid hybridization (30). Alternatively, in some cell lines ASOs can also be delivered by free uptake (31–33). However, only small quantities of ASOs delivered by free uptake escape the endosome and reach the target in the nucleus. Thus, higher ASO concentrations are required, and the effects on RNA splicing are usually observed a few days later, compared to lipid transfection. We tested whether the delivery method (lipid transfection vs. free-uptake) affects off-target splicing of six validated events (AK2, CLIP4, PROM1, PTOV1, RER1 and UCK2). U87 cells were exposed to ASOs in the medium at concentrations up to 30 μM for 5 days. PKM on-target splicing increased in a concentration-dependent manner, with ASO2-cEt/DNA performing better than ASO2-MOE (Figure 7). In contrast, mis-splicing of off-targets was markedly reduced when ASOs were delivered by free uptake, compared to lipid transfection. The average OT-score measured for ASO2-MOE delivered by free uptake was significantly lower than by lipid transfection (0.7 and 1.8, respectively; 2.6 fold; P = 0.002). Off-target activity of ASO2-cEt/DNA was not detectable with 4/6 targets. However, the 5-fold reduction of OT-score compared to lipid transfection (0.1 and 0.5, respectively), was not statistically significant (P = 0.068).
Figure 7.

Free uptake reduces off-target effects in vitro. (A) U87 cells were exposed to ASO2-MOE and ASO2-cEt/DNA (0–30 μM) in the culture medium for 5 days. Autoradiographs show representative results, and bar charts show mean ± SE from three independent experiments. (B) Average off-target scores of all 6 tested off-target splicing events (AK2, CLIP4, PROM1, PTOV1, RER1, and UCK2), relative to PKM, at 30 μM. Error bars represent mean ± SE OT-score of all tested off-targets. n.s. not significant; * P < 0.05; ** P < 0.01
To test off-target activity of PKM ASOs in vivo, we used a receptor/ligand ASO delivery system (27). Tri-antennary N-acetyl galactosamine (GN3)-conjugated MOE and cEt/DNA ASOs were delivered subcutaneously to animals bearing U87 xenografts expressing the GN3-receptor (ASGP-R1). We only observed a significant increase in PKM1 levels in the animals treated with the higher affinity cEt/DNA ASO (Supplementary Figure S6B). We then tested the same off-target splicing events as in our in vitro free-uptake experiment (AK2, CLIP4, PROM1, PTOV1, RER1 and UCK2) and found increased levels of RER1 exon 6 skipping, similar to the results in the free-uptake experiments (Supplementary Figure S6B). Aberrant splicing of UCK2 (observed by free uptake in vitro) was not detectable in vivo. Off-target scores comparing delivery methods in vitro and in vivo are listed in Table 3. This set of results, although limited to one ASO, suggests that off-target mis-splicing is of greater concern in in vitro experiments, compared to applications for which ASOs are typically delivered by natural uptake mechanisms, including in vivo.
Table 3.
Off-target score of ASO2-cEt/DNA delivered in vitro and in vivo
| % PKM1 | AK2 | PROM1 | RER1 | CLIP4 | UCK2 | PTOV1 | |
|---|---|---|---|---|---|---|---|
| Transfection in vitro | 34.0 (2.9) | 0.3 (0.06) | 0.0 (0.0) | 1.1 (0.20) | 0.3 (0.06) | 0.9 (0.09) | 0.0 (0.01) |
| Free uptake in vitro | 18.0 (1.8) | 0.0 | 0.0 | 0.1 (0.01) | 0.0 | 0.1 (0.0) | 0.0 |
| s.c. delivery in vivo* | 11.7 (1.3) | 0.0 | not expr. | 0.3 (0.07) | 0.0 | 0.0 | 0.0 |
Data represent mean (SE) relative to PKM; s.c. subcutaneous; *GN3-conjugated ASO2-cEt/DNA.
DISCUSSION
Splice-modulating ASOs can be highly specific, and depending on their length, can be designed to only match a single complementary site in the transcriptome. Even if there are additional ASO-binding sites present, it is unlikely that they overlap with a critical sequence element involved in splice regulation, such as a splicing enhancer or silencer. In this study, we investigated off-target mis-splicing events of splice-modulating PKM ASOs, and identified several splicing changes in constitutive exons, as well as alternative splice isoforms of ASO off-targets. All of the off-target splicing events appear to be sequence-specific, as in most cases neither a mismatch control ASO nor a scrambled ASO had an effect on these off-targets. In both cases in which the control ASO did affect the exon of interest, there were additional binding sites for the control ASO, supporting the notion that the observed effects are hybridization-dependent.
Closer inspection of ASO/RNA duplex structures revealed that in general, G:U wobble base pairs, terminal mismatches, and short internal loops and bulges are readily tolerated. For one of the off-targets (FRYL), we detected multiple exon-sipping events, even though only 12/18 nucleotides of ASO2 are predicted to base-pair with the target, two of which are wobble base pairs. This observation underscores the importance of choosing the right tool to identify sequence complementarities in the transcriptome. A BLAST search, even optimized for short sequences, only results in a limited number of hits, based on sequence alignment. However, this search algorithm ignores G:U base-pairing, and the results seldom have internal mismatches, which are commonly found in natural RNA structures, limiting its value for ASO target prediction.
We therefore used RNAhybrid, a program originally designed to predict miRNA target sites, based on RNA secondary structures. The advantage of RNAhybrid is that the number of hits and the cutoff can be manually set, resulting in extensive lists that can be sorted and annotated to fit the need of the user. However, the predicted structures and predicted minimum free energy of the interaction are based on RNA/RNA binding, which at best provides only an approximation of the binding energy of uniformly modified ASO interactions with RNA. For mixed-chemistry ASOs with high affinity cEt residues mixed with DNA residues, the resulting duplex structures, and by extension the binding energies, may be substantially different from what is predicted for RNA-RNA duplexes. Unfortunately, alignment algorithms that take into consideration ASO modifications in mixed-chemistry ASOs do not yet exist, and are likely crucial for accurately predicting ASO binding affinity to target sites. In addition, sequence-based affinity calculations ignore the larger context of secondary structures and RNA-binding proteins, both of which are known to influence the off-target activity of gapmer ASOs (34).
As mentioned above, even if there is an adequate estimate of binding affinity, binding itself is not enough to induce a splice change, for the same reason that not every ASO designed against a target is effective in changing its splicing. In our study, we identified many unaffected target sites with predicted binding affinities that were significantly stronger than those of validated off-target sites. We tested various hypotheses to identify features with strong predictive value. Overall, our results did not point to any dominant individual feature; however, we observed several trends. For example, ASO-binding sites in exons harboring RBP motifs tended to be more likely to induce off-target mis-splicing. We also detected a slightly higher proportion of unaffected off-target sites located on out-of-frame exons, which result in premature termination codons and potentially in nonsense-mediated mRNA decay (NMD). Some NMD targets are rapidly degraded, and may not be detected as an alternative splice isoform by RT-PCR, potentially masking unidentified off-target splicing events. A comprehensive RNA-sequencing study, including protein-synthesis inhibitors to abrogate NMD, might help address whether predictive features do exist, and to what extent NMD hides some mis-splicing events.
The present study addresses off-target effects of a limited number of PKM ASOs. To derive universal rules for off-target prediction, it will be desirable to study additional ASOs to other targets, in multiple different cell lines. Testing additional ASOs would also address whether the potential secondary structure of the ASO itself, the base composition, or the GC-content, play a role in the extent of off-target activity. In addition to effects on RNA splicing, ASOs may also affect other steps of gene expression, including transcription, RNA processing, export and turnover, and translation. ASOs are very effective translation inhibitors when targeting the start codon or 5'UTR of a transcript, and are commonly used for targeted knockdown of genes in zebrafish (35,36). In contrast, Liang and colleagues showed that some ASOs targeting the 5'UTR of certain mRNAs can increase their translation (37). Splicing-independent off-target effects may be relevant for some splice-modulating ASOs, but we did not address them in this study. Potential effects on translation would require different detection methods. Finally, ASOs could potentially also bind to, and neutralize, miRNAs or their precursors, or interfere with lncRNA processing or function, which could in turn have global effects on gene expression.
Given that we are currently unable to robustly predict off-target effects on splicing, our focus shifted to identifying strategies to reduce off-target activity. Both on- and off-target splicing were concentration-dependent, prompting us to use two ASOs in combination, each at half the concentration. Alternative splicing events, the primary target of splice-modulating ASOs, are often regulated by multiple enhancer and silencer elements. PKM exon 10 splicing, for example, is regulated by multiple elements located in intron 9 and exon 10 (18,38) (manuscript in preparation). ASOs designed against intron 9 and exon 10, used in combination, increased PKM splice switching, while reducing off-target effects. These results reinforce the notion that the off-target effects observed in this study are hybridization-dependent, rather than a secondary effect caused by modulating PKM isoforms and the consequent alterations in cell physiology. Thus, ASO combinations can be an effective way to increase target specificity. In cases in which using ASO combinations is not feasible, we showed that introducing mismatches can also be used to avoid some off-target splicing effects, while maintaining on-target activity. However, in either scenario, every additional ASO may have new, non-overlapping sets of off-target sites, adding an extra layer of complexity.
Another strategy we explored to increase ASO specificity was to use a shorter 12mer MOE ASO, with a sequence that overlaps both 18mer ASOs used in this study. Compared to ASO2-MOE, the 12mer ASO resulted in a strong reduction of off-target activity for a subset of targets. However, we did not screen for 12mer ASO-binding sites specifically, and may well have missed off-target activity for targets we did not test by RT-PCR. Ultrashort ASOs have been used successfully, both in vitro and in vivo. For example, uniformly modified 8mer LNA ASOs have been used to block microRNA seed regions, without any obvious effects on off-target transcription or translation inhibition (39). In other studies, a 2'-O-methyl-modified 8mer ASO was used both in vitro (28) and in vivo (40) to induce SMN2 exon 7 inclusion. Considering that a short 8–12nt target site can be sufficient to elicit an efficient splicing change, using longer ASOs (although more potent) could theoretically decrease their specificity when ASOs are present in excess. Woolf et al. calculated the number of perfectly complementary 10mer sites in a 2 × 107 base RNA pool to be 19 for any given 10mer ASO, not considering G:U base pairing (4). For a 15mer containing 6 unique 10mer sequences, the number of complementary sites in the RNA pool increases to 210.
As expected, single-nucleotide mismatches appear to affect short ASOs more severely than longer ASOs: we showed that depending on the mismatch location, uniformly MOE-modified 18mer PKM ASOs with five mismatches can still induce efficient splice switching; in contrast, single-nucleotide mismatches of LNA nucleotides introduced at various positions of a 13mer LNA/DNA mixed-chemistry ASO designed to enhance SMN exon 7 inclusion—even at the 3' end of the ASO—significantly lowered inclusion efficiency (29). Even though longer ASOs may be more resistant to mismatches, and hybridize with off-target sequences in vitro, longer sequences might also provide an advantage at the lower, non-saturating concentrations typically obtainable in vivo. However, any such advantage with respect to their effect on off-target activity in vivo has yet to be determined, but is beyond the scope of this study.
Another important point raised in this study is that the mode of delivery of ASOs influences the extent of off-target splicing. ASOs are typically delivered in vitro by lipid transfection, and directly reach the cytoplasm. From there, they can readily reach the nucleus to engage the pre-mRNA target. In contrast, ASOs delivered in vivo and by free uptake in vitro, are internalized by endocytosis and to a small extent by micropinocytosis (41). Only a fraction of ASOs trapped in endosomes escapes, resulting in a much lower amount of ASO available to engage the target. Our results show that ASOs are more specific towards on-target splice-switching when delivered by free-uptake and in vivo, possibly because of more gradual and consistent internalization at lower concentrations, thus avoiding excess amounts of ASO in the nucleus, relative to the pre-mRNA target. Off-target mis-splicing may therefore be a greater issue in cell culture than in vivo applications. Given that lead ASOs are usually identified in cell-based screening assays, often by lipid transfection, care has to be taken not to misinterpret results that may stem from off-target activity. When feasible, mixed-chemistry ASOs, ASO combinations, or targeted mismatches can be used to minimize off-target effects. Furthermore, when appropriate, functional assays should include cDNA rescue experiments, to verify that the observed ASO activity is on-target.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Michael Wigler (CSHL) for providing HT-1080 cells.
Author contributions: Conceptualization, J.S. and A.R.K.; Investigation, J.S., K.W.M., Q.Z., K.T.L.; Writing – Original Draft J.S.; Writing – Review & Editing, J.S., A.R.K., F.R., C.F.B.; Resources, A.R.K., F.R., C.F.B.; Supervision, A.R.K.
Notes
Present address: Juergen Scharner, Stoke Therapeutics, Bedford, MA, USA.
Present address: The work was performed at Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NCI Program Project Grant [CA13106] and NIH grant [GM42699]. Funding for open access charge: NIH grant [GM42699]; PO number from Cold Spring Harbor Laboratory.
Conflict of interest statement. F.R. and C.F.B. are employees of Ionis Pharmaceuticals and they own stock options. J.S. is an employee and A.R.K. is a founder and director of Stoke Therapeutics, and they own stock options.
REFERENCES
- 1. Nomakuchi T.T., Rigo F., Aznarez I., Krainer A.R.. Antisense oligonucleotide-directed inhibition of nonsense-mediated mRNA decay. Nat. Biotechnol. 2016; 34:164–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scharner J., Figeac N., Ellis J.A., Zammit P.S.. Ameliorating pathogenesis by removing an exon containing a missense mutation: a potential exon-skipping therapy for laminopathies. Gene Ther. 2015; 22:503–515. [DOI] [PubMed] [Google Scholar]
- 3. Bennett C.F., Swayze E.E.. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010; 50:259–293. [DOI] [PubMed] [Google Scholar]
- 4. Woolf T.M., Melton D.A., Jennings C.G.. Specificity of antisense oligonucleotides in vivo. PNAS. 1992; 89:7305–7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lima W.F., Rose J.B., Nichols J.G., Wu H., Migawa M.T., Wyrzykiewicz T.K., Siwkowski A.M., Crooke S.T.. Human RNase H1 discriminates between subtle variations in the structure of the heteroduplex substrate. Mol. Pharmacol. 2007; 71:83–91. [DOI] [PubMed] [Google Scholar]
- 6. Wu H., Lima W.F., Crooke S.T.. Properties of cloned and expressed human RNase H1. J. Biol. Chem. 1999; 274:28270–28278. [DOI] [PubMed] [Google Scholar]
- 7. Magner D., Biala E., Lisowiec-Wachnicka J., Kierzek R.. Influence of mismatched and bulged nucleotides on SNP-preferential RNase H cleavage of RNA-antisense gapmer heteroduplexes. Sci. Rep. 2017; 7:12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamola P.J., Kitson J.D., Turner G., Maratou K., Eriksson S., Panjwani A., Warnock L.C., Douillard Guilloux G.A., Moores K., Koppe E.L. et al.. In silico and in vitro evaluation of exonic and intronic off-target effects form a critical element of therapeutic ASO gapmer optimization. Nucleic Acids Res. 2015; 43:8638–8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burel S.A., Hart C.E., Cauntay P., Hsiao J., Machemer T., Katz M., Watt A., Bui H.H., Younis H., Sabripour M. et al.. Hepatotoxicity of high affinity gapmer antisense oligonucleotides is mediated by RNase H1 dependent promiscuous reduction of very long pre-mRNA transcripts. Nucleic Acids Res. 2016; 44:2093–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lindow M., Vornlocher H.P., Riley D., Kornbrust D.J., Burchard J., Whiteley L.O., Kamens J., Thompson J.D., Nochur S., Younis H. et al.. Assessing unintended hybridization-induced biological effects of oligonucleotides. Nat. Biotechnol. 2012; 30:920–923. [DOI] [PubMed] [Google Scholar]
- 11. Gagnon K.T., Corey D.R.. Guidelines for experiments using antisense oligonucleotides and double-stranded RNAs. Nucleic Acid Ther. 2019; 29:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kole R., Krainer A.R., Altman S.. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012; 11:125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hua Y., Vickers T.A., Baker B.F., Bennett C.F., Krainer A.R.. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007; 5:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Z., Jeon H.Y., Rigo F., Bennett C.F., Krainer A.R.. Manipulation of PK-M mutually exclusive alternative splicing by antisense oligonucleotides. Open Biol. 2012; 2:120133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sinha R., Kim Y.J., Nomakuchi T., Sahashi K., Hua Y., Rigo F., Bennett C.F., Krainer A.R.. Antisense oligonucleotides correct the familial dysautonomia splicing defect in IKBKAP transgenic mice. Nucleic Acids Res. 2018; 46:4833–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hinrich A.J., Jodelka F.M., Chang J.L., Brutman D., Bruno A.M., Briggs C.A., James B.D., Stutzmann G.E., Bennett D.A., Miller S.A. et al.. Therapeutic correction of ApoER2 splicing in Alzheimer's disease mice using antisense oligonucleotides. EMBO Mol. Med. 2016; 8:328–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Varani G., McClain W.H.. The G x U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000; 1:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Z., Chatterjee D., Jeon H.Y., Akerman M., Vander Heiden M.G., Cantley L.C., Krainer A.R.. Exon-centric regulation of pyruvate kinase M alternative splicing via mutually exclusive exons. J. Mol. Cell Biol. 2012; 4:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rodriguez J.M., Rodriguez-Rivas J., Di Domenico T., Vazquez J., Valencia A., Tress M.L.. APPRIS 2017: principal isoforms for multiple gene sets. Nucleic Acids Res. 2018; 46:D213–D217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R.. Fast and effective prediction of microRNA/target duplexes. RNA. 2004; 10:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paz I., Kosti I., Ares M. Jr., Cline M., Mandel-Gutfreund Y.. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014; 42:W361–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A. et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 23. Baker B.F., Lot S.S., Condon T.P., Cheng-Flournoy S., Lesnik E.A., Sasmor H.M., Bennett C.F.. 2'-O-(2-Methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J. Biol. Chem. 1997; 272:11994–12000. [DOI] [PubMed] [Google Scholar]
- 24. Seth P.P., Vasquez G., Allerson C.A., Berdeja A., Gaus H., Kinberger G.A., Prakash T.P., Migawa M.T., Bhat B., Swayze E.E.. Synthesis and biophysical evaluation of 2',4'-constrained 2'O-methoxyethyl and 2',4'-constrained 2'O-ethyl nucleic acid analogues. J. Org. Chem. 2010; 75:1569–1581. [DOI] [PubMed] [Google Scholar]
- 25. Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L.. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012; 13:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J., Pan X., Liang X.. Assessment for melting temperature measurement of nucleic acid by HRM. J. Anal. Methods Chem. 2016; 2016:5318935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scharner J., Qi S., Rigo F., Bennett C.F., Krainer A.R.. Delivery of GalNAc-conjugated splice-switching ASOs to non-hepatic cells through Ectopic expression of Asialoglycoprotein receptor. Mol. Ther. Nucleic Acids. 2019; 16:313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh N.N., Shishimorova M., Cao L.C., Gangwani L., Singh R.N.. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009; 6:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Touznik A., Maruyama R., Hosoki K., Echigoya Y., Yokota T.. LNA/DNA mixmer-based antisense oligonucleotides correct alternative splicing of the SMN2 gene and restore SMN protein expression in type 1 SMA fibroblasts. Sci. Rep. 2017; 7:3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pontius B.W., Berg P.. Rapid renaturation of complementary DNA strands mediated by cationic detergents: a role for high-probability binding domains in enhancing the kinetics of molecular assembly processes. PNAS. 1991; 88:8237–8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geary R.S., Norris D., Yu R., Bennett C.F.. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug. Deliv. Rev. 2015; 87:46–51. [DOI] [PubMed] [Google Scholar]
- 32. Gonzalez-Barriga A., Nillessen B., Kranzen J., van Kessel I.D.G., Croes H.J.E., Aguilera B., de Visser P.C., Datson N.A., Mulders S.A.M., van Deutekom J.C.T. et al.. Intracellular distribution and nuclear activity of antisense oligonucleotides after unassisted uptake in myoblasts and differentiated myotubes in vitro. Nucleic Acid Ther. 2017; 27:144–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stein C.A., Hansen J.B., Lai J., Wu S., Voskresenskiy A., Hog A., Worm J., Hedtjarn M., Souleimanian N., Miller P. et al.. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2010; 38:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lima W.F., Vickers T.A., Nichols J., Li C., Crooke S.T.. Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS One. 2014; 9:e101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nasevicius A., Ekker S.C.. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000; 26:216–220. [DOI] [PubMed] [Google Scholar]
- 36. Eisen J.S., Smith J.C.. Controlling morpholino experiments: don’t stop making antisense. Development. 2008; 135:1735–1743. [DOI] [PubMed] [Google Scholar]
- 37. Liang X.H., Sun H., Shen W., Wang S., Yao J., Migawa M.T., Bui H.H., Damle S.S., Riney S., Graham M.J. et al.. Antisense oligonucleotides targeting translation inhibitory elements in 5' UTRs can selectively increase protein levels. Nucleic Acids Res. 2017; 45:9528–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. David C.J., Chen M., Assanah M., Canoll P., Manley J.L.. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010; 463:364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Obad S., dos Santos C.O., Petri A., Heidenblad M., Broom O., Ruse C., Fu C., Lindow M., Stenvang J., Straarup E.M. et al.. Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet. 2011; 43:371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keil J.M., Seo J., Howell M.D., Hsu W.H., Singh R.N., DiDonato C.J.. A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Mol. Ther. Nucleic Acids. 2014; 3:e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H.. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:230–237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.