


Abstract
Recently, newly developed ribosome profiling methods based on high-throughput sequencing of ribosome-protected mRNA footprints allow to study genome-wide translational changes in detail. However, computational analysis of the sequencing data still represents a bottleneck for many laboratories. Further, specific pipelines for quality control and statistical analysis of ribosome profiling data, providing high levels of both accuracy and confidence, are currently lacking. In this study, we describe automated bioinformatic and statistical diagnoses to perform robust quality control of ribosome profiling data (RiboQC), to efficiently visualize ribosome positions and to estimate ribosome speed (RiboMine) in an unbiased way. We present an R pipeline to setup and undertake the analyses that offers the user an HTML page to scan own data regarding the following aspects: periodicity, ligation and digestion of footprints; reproducibility and batch effects of replicates; drug-related artifacts; unbiased codon enrichment including variability between mRNAs, for A, P and E sites; mining of some causal or confounding factors. We expect our pipeline to allow an optimal use of the wealth of information provided by ribosome profiling experiments.
INTRODUCTION
The translation of genetic information into polypeptide sequences is a cellular process common to all kingdoms of life, involving a multitude of orchestrated interactions between mRNAs, translation factors, ribosomes and tRNAs. Translation is a highly regulated and fine-tuned process, which enables a fast response to metabolic and environmental changes and its regulation balances the pool of proteins actively translated from mRNAs (1). While mRNAs and proteins can be measured by RNA-seq and mass spectrometry, respectively, ribosome profiling allows to directly measure protein synthesis by detecting the position of ribosomes on mRNAs (2,3). As a result, Ribo-seq provides a quantitative profile of the translatome at high resolution, i.e. the set of mRNA species under active translation. More specifically, Ribo-Seq is based on the isolation and retrieval of mRNA fragments (footprints) when they are protected by a ribosome, followed by deep sequencing-based identification of ribosome footprints. Adequate alignment of these footprints allows to determine the position of translating ribosomes on mRNAs at single-codon resolution (3,4). This method has quickly been adopted by many laboratories, but at present, data analysis requires computational expertise (5), and the analysis so far has used visualization methods but few dedicated statistical estimates or quality diagnostics. Bioinformatics tools like Rqc (6) centered on quality assessment of reads (data structure, contaminants, etc.) can be used to assess the quality of sequencing but are not informative on the artifacts and batch effects detected on ribosome profiling datasets (Table 1). In this study we compared the performance of RiboVIEW with other existing tools dedicated to ribosome profiling analysis, like Gwips-viz (7), RiboProfiling (8) and riboSeqR (9). This comparison is presented in Table 2 and also includes tools with some quality control capabilities like RiboViz (10), mQC (11), RiboTools (12) and Ribo-TISH, (13) though none of these methods provides the full array of controls and visualization that we propose.
Table 1.
Artifacts and batch effects in ribosome profiling experiments
| Category | Impact | Ref |
|---|---|---|
| Replicate concordance | Single codon-level analysis (ORF, etc.) | (23) |
| Drugs | Leakage, ribosome run-off, biased codon occupancy | (15,29–31) |
| Experimental conditions | Ligation-digestion: end base bias, ambiguous FP location, loss of periodicity | (21,30) |
| Hybridization-subtraction: bias on codon enrichment | (9) | |
| EDTA: loss of information from large RPFs | ||
| RiboZero: loss of rRNA-like mRNA segments | ||
| Drop-off due to unwanted amino acid starvation, monitoring with metagene | (32,33) | |
| RNAse digestion | (34) | |
| Organismal specificities | Footprint loss through size selection in s.cerevisiae | (28) |
| CHX leakage in S. cerevisiae and S. pombe | (15,29) | |
| PCR, seq., post-treatment | Sequence preference, Biased counts due to inefficient alignment | (21) |
| Information loss | (35) | |
| Other batch effects | Non-relevant variability for one or all estimates | (21) |
Table 2.
Comparison of ribosome profiling tools
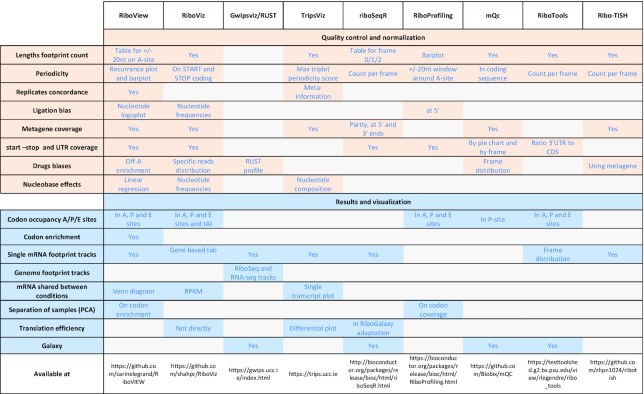
|
RiboVIEW visualizes translation elongation at codon level and provides relevant quality properties. Furthermore RiboVIEW provides unbiased estimates of codon enrichment, detects some (causal or confounding) covariates (Table 1, Supplementary Figure S1).
MATERIALS AND METHODS
Data preparation
Sequenced reads are submitted to adapter removal, using cutadapt (v1.8.1) with options ‘-a AGATCGGAAGAGCACACGTCT–error-rate=0.1 –times=2 –overlap=1’. Resulting reads are trimmed using Trimmomatic (v0.36) with 30 as minimum quality score, minimum length 11nt and maximum length 36nt (options -phred33 LEADING:30 TRAILING:30 MINLEN:11 CROP:36). Remaining reads are depleted of rRNA and other non-nuclear mRNA by aligning using Bowtie on a depletion reference (rRNA, tRNA and mitochondrial RNA sequences), with options –seedmms 2 –seedlen 11 –maqerr 70 –tryhard -k 1. Finally, reads that do not align to the depletion references are aligned to the transcriptome, using Bowtie with options –seedmms 2 –seedlen 11 –maqerr 70 -m 1.
Fasta-format mRNA and ncRNA reference, as well as GTF-format annotation, were downloaded from Ensembl FTP-download page https://www.ensembl.org/info/data/ftp/index.html. A template for data preparation under UNIX/Linux systems is provided in the Supplemental Information.
Workflow
Once the aligned reads are generated in a bam format, the next step is done using R command line. A template workflow is provided in the Supplementary Information. In this template, the user can define the addresses of the input files and the experimental conditions. Then, a set of commands generates results including the two HTML pages Results-QC.html and Results-MINE.html where the results can be viewed. All is coded as custom R and Python scripts. As a general rule, replicates of a same condition are either integrated or all shown. In some cases, this would have been impractical; the resulting plots are then saved to the output folder and only one replicate is shown in the HTML pages.
Calculating periodicity
The number of footprints which align close to the start of the annotated sequence of an mRNA are counted (regardless of codon identity). Tables of this coverage, stratified by footprint length and by position in a window of 20nt 5′ of the A in AUG-codon to 20nt on the 3′ side, are generated. Those tables are used to display periodicity using a recurrence plot (14), R function recurr from R package tseriesChaos, adapted for RiboVIEW). Recurrence plot usually represents time autocorrelation in dynamical systems, while it is used here to represent spatial autocorrelation. The recurrence plot is generated for each footprint length, alongside with a barplot, which shows the coverage achieved for each footprint length.
Selecting adequate footprint lengths
Recurrence plots per footprint length are displayed one at a time (function selectFPlen), after which an interactive dialog prompts the user to select a minimum and maximum footprint length which comply with recurrence every 3nt, starting at −12nt.
Metagene
A metagene is generated using the coverage in A site for each available position along each mRNA in one sample, which is calculated in enrichment.py and stored in output files *.metagene. Positions are normalized to the following metagene coordinates: [−1; 0] for the 5′ UTR, [0; 1] for the CDS and [1; 2] for the 3′ UTR. Coverage counts are normalized so as to add up to unity, and binned at 0.1 resolution (option res1 in function ‘metagene.all’). These normalized and binned values constitute the metagene profile. The percentage of reads in the UTRs relative to the CDS is calculated and informs possible selection artifact (indicative cutoffs of 1% and 10% are used). The percentage of reads in the first 15 codons stretch at CDS start, including the AUG codon, is calculated and compared to an indicative threshold of 1% for possible inflation around AUG. Leakage is examined at AUG and STOP codon. For AUG, a robust linear fit is applied to the metagene profile at and after AUG (metagene coordinates [−0.1; +0.3]). If the slope from this fit is positive and has a significant P-value at 0.05 level (respectively, 0.1), this yields an indication of strong (respectively, mild) leakage after AUG. Similarly, leakage at STOP is calculated as the percentage of metagene coverage after the STOP codon relative to shortly before (segments [1; 1.3] and [0.9; 1] in metagene coordinates). A percentage larger than 5% (respectively, 1%) triggers strong (mild) indication of STOP leakage.
Ligation biases if any are highlighted in logoplots at the nucleotide and codon level. These plots are automated from coverage counts at nucleotide and codon level as derived from enrichment.py and from adapted scripts from the R package ggseqlogo. A significant nucleotide or codon sequence bias is here indicated not by a P-value, but by the information content, measured in bits. Cutoffs of 0.2 (or 0.4) are used to indicate possible (or strong) bias on a sequence.
Correlation between replicates
Counts per mRNA per replicate are displayed in an RPKM plot for each set of replicates of the same condition, along with the Spearman correlation at gene level. Additionally, stretches of 3–100 codons are scanned for Spearman correlation higher than 0.4, or better, 0.6 between replicates. The relevant stretch, or 100 if none fulfills this criterion, is used to display a codon-level RPKM plot.
A heatmap with hierarchical clustering is produced for the full set of samples. Hierarchical clustering is compared to the actual experimental conditions and replicates using a Spearman correlation.
Codon enrichment, relative codon enrichment and codon occupancy
As a rationale for unbiased codon enrichment calculation, we considered the pool of mRNA actively translated, from which footprints derived. Focusing on a codon with identity c, we look for footprints where this codon appears at a certain offset i from the A-site (Supplementary Figure S2). For example, this offset i could be four codons away, downstream (5′ side) of the A-site. If there is no specific pausing or acceleration of this codon c at offset i, then one would expect codon c at offset i to appear in ribosome footprints at a frequency, which simply reflects its codon usage. Based on this rationale, unbiased codon enrichment is calculated as the observed codon usage relative to the expected codon usage. In practice, in the Python script dedicated to enrichment calculation sums first the observed codon usage at mRNA level and second over mRNAs, using weights. Weights by mRNA are defined as the number of reads per mRNA. Furthermore, we make the assumption that the expected codon usage is independent of the position, except in domains near AUG and STOP codons, which are excluded (15 codons near AUG, 5 codons near STOP codons). This yields equation (1), where in particular weights are simplified out when one sums over all mRNAs:
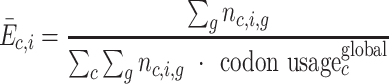 |
(1) |
where Ēc,i is the codon enrichment for codon identity c at position i (with, especially, i = 0 at A-site, i = 1 at P-site), averaged over mRNAs, nc,i,g is the number of codons c observed at position i in mRNA g, and global codon usage is defined by equation (2):
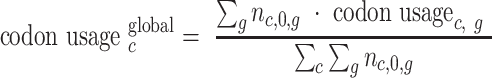 |
(2) |
Under the assumptions mentioned, enrichment Ēc,i is unbiased at unity (Supplementary Figure S2). This relies on the fact that, if codon c is nether paused nor accelerated and if the assumption that the expected codon usage is independent of the position holds, then the observed codon usage converges to the expected codon usage, as the coverage in the experiment becomes large enough, as in Equation (3):
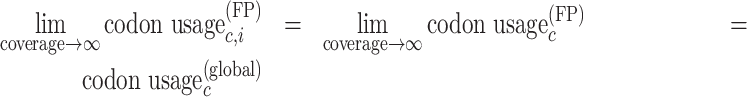 |
(3) |
As a consequence, still in the case when codon c is neither paused nor accelerated, enrichment Ēc,i, the ratio of observed to expected codon usage, should converge to 1, as coverage becomes large enough.
Standard deviation of codon enrichment is calculated similarly, using the number of reads as weights. This simplifies into equation (4):
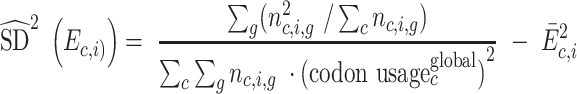 |
(4) |
Standard error of the average codon enrichment across mRNAs is taken as the standard deviation of codon enrichment divided by the square root of the number of mRNAs.
Relative codon enrichment is calculated as described previously by Hussmann (15). Relative codon enrichment for arginine codons are produced for each replicate and displayed in RiboQC. The full table of values for all codons is provided in the results folder.
A comparison of main differences in the calculation of enrichment in this study and in Hussmann et al. (2015) is given in Supplementary Figure S3).
Bulk codon occupancy corresponds to the counts of footprints stratified by codon identity. We provide the codons present in A-site, P-site and E-site, as well as three positions downstream and upstream of the ribosome. The rationale to assign a specific position for each codon was described previously (16). Briefly, the A-site is assigned at the footprint 5′ start +15nt, relaxed by ±1nt to match the closest codon in the main reading frame.
Enrichment per experimental condition
Enrichment in one condition is calculated as the weighted mean and standard deviation over replicates, where the weight associated to one replicate is 1/SE²(Ēc,i). Standard error across replicates is taken as the standard deviation across replicates divided by the square root of the number of replicates. Enrichment per condition is displayed in Results-Mine.html for each codon, along with an error bar corresponding to ±standard deviation.
Enrichment between experimental conditions
Enrichment in condition (a) relative to condition (b) is calculated by bootstrapping possible quotients from replicates in condition (a) relative to replicates from condition (b). This procedure yields the mean and standard error of the quotient of enrichments. This quotient is shown in Rresults-Mine.html, with error bars corresponding to the standard error.
mRNA tracks
Coverage in the A site is displayed in a barplot along the coding sequence of an mRNA, in every sample. By default, an mRNA with sufficient coverage is picked at random. Tracks for a specific mRNA can be requested using the option ‘mRNA=’ in RiboVIEW function visu.tracks.
Venn diagram
RiboView automatically retrieves the number of footprints per mRNA and per replicate for one condition and creates a Venn plot using R package VennDiagram. This is restricted to up to five replicates per condition (limitation of VennDiagram package).
Group effects
Group effects are evaluated using a principal component analysis on codon enrichment. A P-value for significant principal component is derived by bootstrapping 10000 times the elements of the matrix of occupancies for all samples. The PCA plot is displayed along with this P-value for interpretation as a batch effect (separation of replicates), or as a functional role (separation of conditions) by the user. Additionally, a tSNE plot is generated, using the average number of replicates to set the parameter ‘perplexity’.
Second, a linear regression is applied to codon occupancy with nucleobases a, c, g or u as explanatory variables. The slope, standard deviation and P-value are retrieved to produce a barplot for display in Results-Mine.html. Error bars signal the standard deviation, while a significant P-value is indicated in the text associated to this plot, for the user to identify, if either a batch effect between different replicates or a functional effect between different conditions is present.
RiboQC HTML page
Text and plot files are retrieved from R data files corresponding to each theme ‘Periodicity’, ‘Replicates’, ‘Selection’ and ‘Drugs’, and to each category within these themes. This hierarchy of themes and categories is specified via a nested list. The output page Results-Qc.html is generated in three phases: (i) HTML (Hyper Text Markup Language) commands for page initiation, definition and header are written to Results-Qc.html. This includes a style sheet ‘output-style’ written in CSS (Cascading Style Sheets) language. (ii) A loop for each theme generates one rounded box-frame per theme. Inside this frame, a nested loop generates one tab per category, containing one plot and corresponding text with relevant values. Plots are included as a character string using Python package ‘base64’. (iii) Footer and closing HTML commands are written to Results-Qc.html.
RiboMine HTML page
The procedure and structure is the same as for RiboQC HTML page. The different content is entirely defined by the hierarchy of themes and categories and by the corresponding R-data files loaded.
Tests
Using a Python script, synthetic mRNAs were generated and annotated. Following different relevant scenarii (footprint periodicity present or absent, enrichment or not at a specific codon, ribosome leakage or not), footprint reads were sampled from the pool of synthetic mRNAs and written to BAM files. These files were used as input into RiboVIEW in order to check its different functionalities. A checklist of functions and expected outputs was established.
RESULTS
Preparatory work and input
RiboVIEW is meant to be easily integrated into a general ribosome profiling workflow (Figure 1A). Cells could be treated with cycloheximide, or with different drugs or simply flash-frozen to arrest translating ribosomes. Cytoplasmic extracts from these cells are then treated with RNase to digest regions of mRNAs not protected by ribosomes. 80S monosomes, that mainly protect a ∼30-nucleotide footprint, are purified using a sucrose gradient or alternatively with a sucrose cushion. Nucleotide footprints are then size-selected and processed for Illumina high-throughput sequencing (Figure 1A).
Figure 1.

RiboQC: Periodicity, metagene and ligation analysis. (A) Ribosome profiling workflow including the major steps of the ribosomal profiling approach. (B) Periodicity and A site identification: (top) Anatomy of a ribosome footprint, with P-site offset for 30 mer reads indicated. (bottom) Sum of the footprints aliened at the start codons, used for identifying the A-site. Footprints were first stratified by length (26–32nt). The first nucleotide of each footprint at the 5′ terminus that mapped to the proximity of its start codon was summed for the annotated mouse genes. The 0 position is the first nucleotide of the start codon. The highest peak for this representative read length of 30nt is 12nt before the start (P site). Hence, the A site for reads of 30 nt in this sample was identified as +15 from the start of the footprint. (C) Recurrence plots for footprints of length 28nt (left) and 32nt (right) with blue arrow indicating presence and red arrow absence of initiation peak at −12nt. Further, blue triangles indicate 3nt recurrence and red triangles no recurrence at positions −9nt, −6nt, …, +18nt. (D) Representative metagene plot of 27–30 mer ribosome footprints at coding start and stop site. (E) Logo nucleotide analysis of 5′ and 3′ footprints ligation sites.
Besides the analysis pipeline, we also provide, in the Supplementary Methods, our in-house experimental protocol adapted from (17,18).
The next steps are computational: first the footprints are trimmed from adapter and low quality bases, and depleted if they align to rRNA (or further RNA sequences which could be ambiguously aligned to mRNA, like tRNA for example). Remaining reads are mapped to the transcriptome.
We routinely use Bowtie (19) with a seed region of 20 nucleotides and one mismatch allowed. We chose Bowtie because it is fast and dedicated to short read alignments up to 50 bp, which is compatible with the length of a ribosome footprint, for which it is faster and/or more sensitive than Bowtie2 that was developed for reads longer than 50 bp (http://bowtie-bio.sourceforge.net). STAR aligner (20) could be a valid alternative. Mapping should preferably be unique (21), which results in the loss of some coverage, but avoids skewed codon enrichment or artifacts in translation efficiency (#FP/#mRNA) results.
Afterwards, the resulting BAM files, reference mRNA sequences (fasta format), and annotation of their coding sequence (table format, generated from a gtf file) can be entered in RiboView. We show here example results of the analysis of our in-house samples (16), obtained from HeLa cells treated with the elongation inhibitor cycloheximide (Supplementary HTML files).
1.5 to 2.4 M reads for minus queuine medium samples (denoted L) and +queuine medium (denoted L+Q) samples (category) aligned to known CDS regions. This corresponds to 88.3–92.7% of remaining reads after depletion of rRNA, tRNA and mitochondrial RNAs (7.6–12.3M reads were depleted).
We further validated RiboVIEW using independent datasets. An example obtained from c. elegans samples (22) is provided in Supplementary Figures S4 and S5 and Supplementary HTML files.
RiboQC
Reproducibility and quality control are a concern for any experimental procedure. RiboQC offers a collection of tools to scan own data for the most relevant aspects of ribosome profiling quality control.
Periodicity
For any given mRNA, ribosome footprint sequences should mainly correspond to the protein coding portion of the transcript, extending from the start codon to the stop codon. Footprint-allocated position of the A-site should also show a strong preference for the first nucleotide position within each codon, in agreement with the reading frame. In order to monitor these aspects, we propose both the classical coverage representation (barplot, Figure 1B), which represents well the coverage obtained from different footprint lengths, and a recurrence plot, which is an unsupervised way to display recurring patterns in a series (14). Recurrence plot was previously developed for dynamical systems and can be adapted in a straightforward way to ribosome profiling data. A recurrence plot is robust to non-periodic coverage variations, and preserves the positional information, whereas a Fourier transform would yield a summary value over all positions, and methods based on coverage in 0, +1 or +2 frames lose positional information and could be biased for outlier peaks. Sufficient periodicity is attained if a peak at −12nt is present on the recurrence plot (−12nt corresponding to AUG initiation in P-site), and distinct recurrence patterns occur every 3nt, at −9nt, −6nt, etc. We call ‘distinct’ a recurrence black band centered around −12, −9, …, −18 nt and separated of the next band by grey to white bands. In our demonstration dataset, the peak at AUG initiation is clearly visible as a dark band at −12nt and recurrence starting at −12nt is shown by dark bands at −12nt, −9nt, …, +18nt, each well separated by a lighter band (Figure 1C left panel). This was the case for footprints of length 27–30nt. By contrast, footprints of length 32nt possess a diffuse peak at −12nt (encompassing positions −13nt and −12nt), and lack recurrence at positions −9, +3, +9, +12, +15, +18 nt (Figure 1C right panel).
Metagene
A metagene profile is an average of quantitative ribosome density of all mRNAs, along a normalized transcript. In our examples the metagene profile showed the restriction of footprints to the genes coding region (Figure 1D), indicating sufficient monosome selection, and absence of drop-off. Further, there were no indication of inflation around AUG or leakage either at AUG or STOP codon, as indicated by detailed zoom and diagnostic values around AUG and STOP (Figure 1D, Supplementary Figure S4B).
Ligation
We assess the frequency of specific nucleotide or codon sequences at the 3′ end and 5′ end of ribosome-protected fragments likely resulting from ligase specificity, or due to PCR amplification of cDNA that may selectively amplify certain sequences, thus distorting the relative abundance of reads. To this aim, we propose a logo nucleotide and codon analysis (Figure 1E, Supplementary Figure S4C). The reduction of last a and over representation of a, c, u, further signaled by an information content larger than 0.4, suggests a bias. However, this corresponds to adapter trimming which leads to the systematic removal of a terminal a in the footprint. This distortion is seemingly not functional, since codon logo shows no bias at footprint 3′ end (this includes a-terminated codons, whose identity is recovered through the alignment), as well as no codon bias both at 3′ and 5′ ends (Supplementary Figure S7).
Reproducibility
We assess reproducibility between replicates in several diagnostic tools, tailored for different experimental aims. Gene-level reproducibility is assessed by a classical RPKM plot. In our data we obtained a strong correlation of 0.953, in line with the best experiments (Figure 2A; (23)). Codon-level reproducibility, important for ORF finding or local analyses, is shown in a codon-level RPKM plot (Figure 2B). We also indicate the stretch length needed for codon-level reproducibility, corresponding to a strong Spearman correlation between replicates. In our examples, local analyses were not a primary aim, and the correlation we obtain is logically lower (rS = 0.6902 in -q and 0.7141 in +q on 100 codons, and stretches of at least 30 codons needed for local analyses, rS>0.4). Further, a heatmap with hierarchical clustering on codon enrichment provides an objective way to assess if replicates cluster together as should be expected (Figure 2C). In our −q and +q samples this is the case, indicated by a Spearman correlation larger than 0.8, between automatically-guessed and experimentally-defined clusters.
Figure 2.
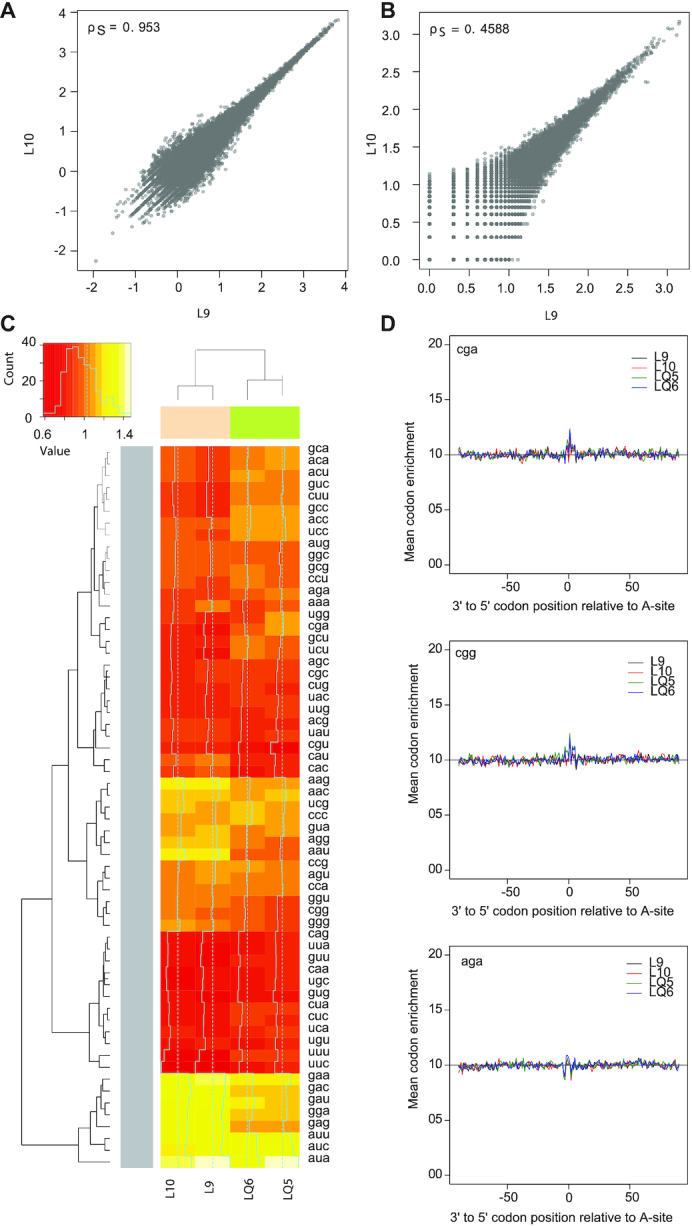
Replicates, batch effect and drug effect. Correlation between ribosome profiling replicates as measured by RPKM values (A) and codon-level RPKM values (B). Spearman correlations of 0.95 and 0.46 were computed for the two replicates. (C) Heatmap for codon occupancy replicates in conditions –q (pink) and +q (green), with hierarchical clustering tree for samples (top) and codons (left). (D) Relative codon enrichment for Arginine codons cga, cgg and aga from top to bottom.
Relative codon enrichments
We measure how frequently ribosomes are observed with their A-site positioned at a particular offset upstream or downstream of a given codon identity, as calculated and normalized by Hussmann ((15), Supplementary Figure S3), who showed that in yeast, pretreatment with CHX may produce artificial patterns in ribosome density downstream of arginine codons (Figure 2D, arginine codons cga, cgg and aga). However, enrichment estimates are perfectible: Hussmann estimates do not automatically converge to unity, which prompted us to design an improved estimation for codon enrichment (Figure 3, Supplementary Figure S2, S3 and S6).
Figure 3.

Codon enrichment. (A) Relative codon enrichment according to calculation and normalization steps described in Hussmann et al. (2015), for codons aan. (B) Unbiased codon enrichment for codons aan, (C) with magnification on ribosome-covered part of mRNAs, for unbiased codon enrichment.
RiboMine
Each ribosomal footprint read is related to a specific codon along the mRNA, since it was generated when that codon in one of the mRNA molecules was positioned in the A, P or E site of a ribosome. Based on the count or enrichment of footprints aligned to an mRNA, it is possible to infer translation elongation process roles. For example, slower codons can be detected based on the fact that they are covered by ribosomes for longer periods of time, resulting in a higher number of reads, as ribosomal densities inversely reflect ribosomal speed (24).
Bulk codon occupancy (BCO)
BCO reveals ribosome positions at single nucleotide resolution, and thus has the potential to identify translational defects affecting single codons. Ribosome dwell time at specific codons is determined as follows. The positions of the A, P and E site codons within ribosome footprints of various lengths (25–31nt) is determined by examining the 5′ ends of footprints mapping to start codons, where initiating ribosomes are expected to contain start codons in their P sites. A-site position was assigned according to an offset equal to +15nt for 26–30 read lengths offset from the 5′ end of the reads (17). Reads were assigned to a codon when mapped to −1, 0, +1 relative to the first nucleotide of the codon (17,18). Occupancy of A-site codons is normalized by the frequency of the same codon in the non-decoded +1, +2, +3 triplets relative to the A-site, the first 15 codons and last 5 codons of each ORF are excluded from this analysis (21,25). BCO is provided as a table named BCO-<sample-name>.
Unbiased codon enrichment
Codon enrichment provides the observed usage of a certain codon in comparison with the expected codon usage. If a codon is found more frequently in the ribosomal A site in comparison with the frequency of the same codon in the observable segment of the coding sequence, then there is enrichment higher than unity. The calculations aim at providing an improved estimate for ribosome acceleration or stalling. By comparison to BCO and relative codon enrichment, we applied a rigorous normalization, which yields unbiased estimates of codon stalling (Figure 3, Supplementary Figure S6). Similar to BCO, a high enrichment reveals ribosome pausing or acceleration at single codons. However, BCO only allows comparison between conditions, whereas codon enrichment additionally allows quantifications of codon translation speed and its variability, for one specific sample, or for a specific condition.
For instance in our example dataset, codon aaa has an enrichment of 0.8001 (SEM: 0.0134 - adimensional) in the -q condition (Figure 4A). This can be interpreted as 1/0.8 = 1.250 (SEM: 0.0209) times the average ribosome translation speed. Further, conditions are compared by dividing enrichment in a condition (-q) by its value in another condition (+q), providing also the corresponding standard error. In our examples, this shows slower translation for codons cau, aau, and so forth, while uuc for instance is accelerated (Figure 4B). Unbiased enrichments by sample, by condition and by comparison are provided as tables containing means and standard deviations, or standard errors where relevant.
Figure 4.

RiboMine: Codon enrichment and footprints. (A) Unbiased codon enrichment in condition –q and +q summarized over two independent biological replicates per condition. (B) Unbiased codon enrichment of condition –q compared to +q. (C) Footprints are plotted according to the codon in the ribosome A site on individual transcripts.
mRNA footprint tracks
Footprint coverage is plotted according to the position of its codon in the ribosome A site along the mRNA to which it aligns (Figure 4C).
Venn diagram
The count of mRNA shared between different conditions might indicate adaptation of the mRNA pool. This is displayed on a Venn diagram, however this is limited to 5 samples maximum due to limitation of the R package it relies on (Supplementary Figure S8).
Nucleobase effects
While mining for causal or confounding factors, we observed sometimes a dependence on one or the other nucleobase a, c, g or u. In Ribomine, the linear link between codon enrichment and each of these base is explored and shown on a barplot (Figure 5A). In our dataset, a significant decrease of c-contaning codon enrichment (Benjamini–Hochberg-adjusted P-values lower or equal to 0.00399, linear regression of enrichment against c content) is observed (Figure 5A). While effect on c is similar between samples (−0.123 with SEM = 0.0.0356 to −0.153 with SEM = 0.0412 per additional c in the codon), other effects might in principle correspond to an artifact or a batch effect. In order to rule this out we correct the enrichment from significant effects (cutoff: type I error α = 0.05) due to a, c, g or u content. As a result, queuine still has a significant effect on Q-codons translation speed (Supplementary Table S1).
Figure 5.
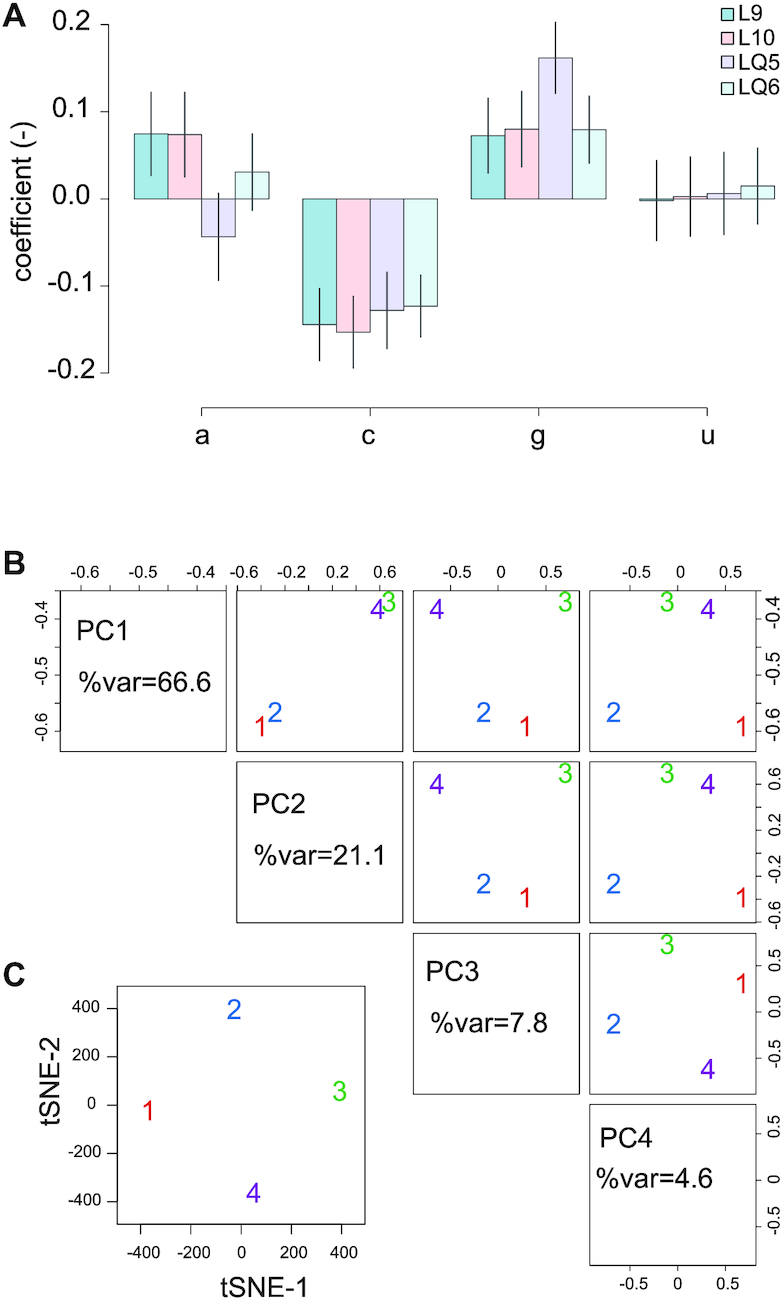
Nucleobase effects and separation of samples. (A) Regression coefficient for enrichment relative to base a, c, g or u. (B) Principal component analysis, axes 1 to 4 for samples –q (samples indicated 1, red and 2, blue) and samples +q (samples 3, green and 4, purple). (C) tSNE axes 1 and 2.
Separation of samples
Causal or confounding factors can sometimes be highlighted by unsupervised structure or clustering detection. To this aim, we include a PCA plot for unsupervised learning and a tSNE plot for cluster visualization (Figure 5B,C). –q and +q conditions were separated by principal components 1 and 2. tSNE plot didn’t show any clusters, however this plot is more likely to be useful for a higher number of samples.
Tests
All tests are listed in the supplementary file and were successful (Supplementary_software_RiboVIEW.zip).
DISCUSSION
We developed the pipeline RiboVIEW to automate calculations of key variables from ribosome profiling data, assess replicate concordance and other relevant quality parameters, providing easy data visualization. We display these parameters and views in HTML files, to quickly scan own data. We demonstrated the application using data from human cells (control and cells with queuosine deficiency (16)) and in c.elegans (control and tut1 mutant worms (22)). Monitoring of quality parameters using RiboQC and exploitation of unbiased codon estimates using RiboMINE will allow users to control and explore their own data in a comprehensive and handy manner.
The analysis highlights relevant aspects revealing the quality of ribosome profiling datasets. In particular, we suggest an unsupervised way to display periodicity using recurrence plots, which has the advantage relative to other methods based on a Fourier transform to provide offset information about −12nt for initiation in P site, and to determine if the main peaks are in the main reading frame, or offset by +1nt, +2nt. In addition, the relative codon enrichments described by Hussmann for S. Cerevisiae samples (15) as well as other artifacts should be easier to catch. Further, comparisons between several species should be facilitated, and mining might reveal additional ways to approach the data.
We also propose an unbiased estimate of codon enrichment as part of RiboVIEW. Codon enrichment provides the ratio of the observed frequency of a certain codon in comparison with its expected frequency. In contrast to traditional codon occupancy, unbiased enrichment provides direct quantification of codon-specific translation speed in each sample or across replicates. Together with mRNA footprint tracks, we expect that this improved analysis of codon pausing will open new interpretation possibilities of ribosome footprint data.
A limitation of RiboVIEW is that it focuses solely on ribosome footprints, not providing translation efficiency estimates, or ORF finding, however these aspects are covered elsewhere (26,27), and can be easily integrated into RiboVIEW outputs. Another limitation is a focus on footprint sizes 25–35nt, not considering shorter or longer footprints, which may be relevant in some application (28).
While computational and web tools for ribosome profiling exist (a summary is provided in (5) and Table 2, for example Riboviz or GWIPS-viz (7,10), few resources focus on checking quality measures or discovering authentic new information from ribosome profiling data. RiboVIEW specifically addresses these two points. The user can further select interesting aspects for adapting data processing, or to setup further analyses. The variety of new applications dedicated to ribosome profiling and the wide array of new tools developed for data analysis and quality control calls for a rigorous testing ensuring that claimed properties are effectively attained in various experimental conditions. Thus, a benchmark approach, separated from the developing of tools, would be useful to clarify the respective qualities of the different methods.
In conclusion, RiboVIEW permits to setup and verify ribosome profiling analyses, which can be further used for integration of transcriptomic and proteomic data.
DATA AVAILABILITY
SOFTWARE AVAILABILITY
RiboVIEW is available as a source R package at https://github.com/carinelegrand/RiboVIEW and as supplementary material (Supplementary_software_Riboview.zip).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Frank Lyko for his extensive support and Ann Ehrenhofer-Murray and Martin Müller for constructive discussions, which seeded some aspects of this project. FT was supported by the Institute of Genetics and Biophysics “A. Buzzati-Traverso, C.N.R.”, Naples (Italy).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) [TU5371-1 to F.T., SPP1784 to F.L.]; DKFZ NCT3.0 Integrative Project in Cancer Research [NCT3.0_2015.54 DysregPT to F.L.]; Funding for open access charge: DFG [DFG TU5371-1 to F.T.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Sonenberg N., Hinnebusch A.G.. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009; 136:731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ingolia N.T. Ribosome profiling: new views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014; 15:205–213. [DOI] [PubMed] [Google Scholar]
- 4. Zinshteyn B., Gilbert W.V.. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLos Genet. 2013; 9:e1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang H., Wang Y., Xie Z.. Computational resources for ribosome profiling: from database to Web server and software. Brief. Bioinform. 2019; 20:144–155. [DOI] [PubMed] [Google Scholar]
- 6. Lopes-Cendes I., de Sá Carvalho B., de Souza W.. Rqc: a bioconductor package for quality control of high-throughput sequencing data. J. Stat. Softw. Code Snippets. 2018; 87:1–14. [Google Scholar]
- 7. Michel A.M., Kiniry S.J., O’Connor P.B.F., Mullan J.P., Baranov P.V.. GWIPS-viz: 2018 update. Nucleic Acids Res. 2018; 46:D823–D830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Popa A., Lebrigand K., Paquet A., Nottet N., Robbe-Sermesant K., Waldmann R., Barbry P.. RiboProfiling: a Bioconductor package for standard Ribo-seq pipeline processing [version 1; peer review: 3 approved]. F1000Res. 2016; 5:1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung B.Y., Hardcastle T.J., Jones J.D., Irigoyen N., Firth A.E., Baulcombe D.C., Brierley I.. The use of duplex-specific nuclease in ribosome profiling and a user-friendly software package for Ribo-seq data analysis. RNA. 2015; 21:1731–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carja O., Xing T., Wallace E.W.J., Plotkin J.B., Shah P.. riboviz: analysis and visualization of ribosome profiling datasets. BMC Bioinformatics. 2017; 18:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verbruggen S., Menschaert G.. mQC: A post-mapping data exploration tool for ribosome profiling. Comput. Methods Programs Biomed. 2018; 181:104806. [DOI] [PubMed] [Google Scholar]
- 12. Legendre R., Baudin-Baillieu A., Hatin I., Namy O.. RiboTools: a Galaxy toolbox for qualitative ribosome profiling analysis. Bioinformatics. 2015; 31:2586–2588. [DOI] [PubMed] [Google Scholar]
- 13. Zhang P., He D., Xu Y., Hou J., Pan B.F., Wang Y., Liu T., Davis C.M., Ehli E.A., Tan L. et al.. Genome-wide identification and differential analysis of translational initiation. Nat. Commun. 2017; 8:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eckmann JP, Oliffson Kamphorst S., Ruell D.. Recurrence plots of dynamical systems. EPL (Europhys. Lett.). 1987; 4:973–977. [Google Scholar]
- 15. Hussmann J.A., Patchett S., Johnson A., Sawyer S., Press W.H.. Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLos Genet. 2015; 11:e1005732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tuorto F., Legrand C., Cirzi C., Federico G., Liebers R., Muller M., Ehrenhofer-Murray A.E., Dittmar G., Grone H.J., Lyko F.. Queuosine-modified tRNAs confer nutritional control of protein translation. EMBO J. 2018; 37:e99777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ingolia N.T. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 2010; 470:119–142. [DOI] [PubMed] [Google Scholar]
- 18. Tuorto F., Herbst F., Alerasool N., Bender S., Popp O., Federico G., Reitter S., Liebers R., Stoecklin G., Grone H.J. et al.. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 2015; 34:2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lecanda A., Nilges B.S., Sharma P., Nedialkova D.D., Schwarz J., Vaquerizas J.M., Leidel S.A.. Dual randomization of oligonucleotides to reduce the bias in ribosome-profiling libraries. Methods. 2016; 107:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nedialkova D.D., Leidel S.A.. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell. 2015; 161:1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diament A., Tuller T.. Estimation of ribosome profiling performance and reproducibility at various levels of resolution. Biol. Direct. 2016; 11:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tuller T., Waldman Y.Y., Kupiec M., Ruppin E.. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weinberg D.E., Shah P., Eichhorn S.W., Hussmann J.A., Plotkin J.B., Bartel D.P.. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 2016; 14:1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duc K.D., Song Y.S.. Correction: The impact of ribosomal interference, codon usage, and exit tunnel interactions on translation elongation rate variation. PLos Genet. 2018; 14:e1007620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gritsenko A.A., Hulsman M., Reinders M.J., de Ridder D.. Unbiased quantitative models of protein translation derived from ribosome profiling data. PLoS Comput. Biol. 2015; 11:e1004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lareau L.F., Hite D.H., Hogan G.J., Brown P.O.. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. Elife. 2014; 3:e01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duncan C.D.S., Mata J.. Effects of cycloheximide on the interpretation of ribosome profiling experiments in Schizosaccharomyces pombe. Sci. Rep. 2017; 7:10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gerashchenko M.V., Gladyshev V.N.. Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res. 2014; 42:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sharma P., Nilges B.S., Wu J., Leidel S.A.. The translation inhibitor cycloheximide affects ribosome profiling data in a species-specific manner. 2019; 10.1101/746255, 24 August 2019, preprint: not peer reviewed. [DOI]
- 32. Johnson G.E., Li G.W.. Genome-wide quantitation of protein synthesis rates in bacteria. Methods Enzymol. 2018; 612:225–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sin C., Chiarugi D., Valleriani A.. Quantitative assessment of ribosome drop-off in E. coli. Nucleic Acids Res. 2016; 44:2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gerashchenko M.V., Gladyshev V.N.. Ribonuclease selection for ribosome profiling. Nucleic Acids Res. 2017; 45:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chugunova A., Navalayeu T., Dontsova O., Sergiev P.. Mining for small translated ORFs. J. Proteome Res. 2018; 17:1–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.