

Abstract
Background
The dearth of relevant tumor models reflecting the heterogeneity of human central nervous system metastasis (CM) has hindered development of novel therapies.
Methods
We established 39 CM patient-derived xenograft (PDX) models representing the histological spectrum, and performed phenotypic and multi-omic characterization of PDXs and their original patient tumors. PDX clonal evolution was also reconstructed using allele-specific copy number and somatic variants.
Results
PDXs retained their metastatic potential, with flank-implanted PDXs forming spontaneous metastases in multiple organs, including brain, and CM subsequent to intracardiac injection. PDXs also retained the histological and molecular profiles of the original patient tumors, including retention of genomic aberrations and signaling pathways. Novel modes of clonal evolution involving rapid expansion by a minor clone were identified in 2 PDXs, including CM13, which was highly aggressive in vivo forming multiple spontaneous metastases, including to brain. These PDXs had little molecular resemblance to the patient donor tumor, including reversion to a copy number neutral genome, no shared nonsynonymous mutations, and no correlation by gene expression.
Conclusions
We generated a diverse and novel repertoire of PDXs that provides a new set of tools to enhance our knowledge of CM biology and improve preclinical testing. Furthermore, our study suggests that minor clone succession may confer tumor aggressiveness and potentiate brain metastasis.
Keywords: animal model, central nervous system metastasis, clonal evolution, patient-derived xenograft
Key Point.
1. Generation of novel CM PDX models identified a mode of PDX clonal evolution by minor clone expansion.
Importance of the Study.
A lack of tumor models reflecting the heterogeneity and complexity of human CM has presented a barrier to understanding the underlying biology and developing better therapies. The 39 novel CNS metastasis PDX models generated and comprehensively characterized in this study provide a new set of tools that will help further our understanding of the mechanisms of CNS metastasis and enable novel and relevant preclinical testing to help enable clinical trials. Our data also identify novel modes of clonal evolution in PDXs, involving explosive or rapid minor clone takeover, which could be related to aggressive tumor behaviors in both PDX models and the human cancers from which they derive.
Despite progress made in the treatment of many cancers, metastasis to the central nervous system (CNS) has remained a major source of mortality. CNS metastasis (CM) has become more common due to greater systemic control of the primary cancer.1–5 Improved surgical and radiation treatment techniques have led to modest improvements in survival. However, treatment for CM remains largely palliative and rarely curative, highlighting the fact that novel therapies and associated predictive biomarkers remain a critically unmet need for CM.1,3
Progress in treating CM has been hampered by a lack of translationally relevant models, access and availability to human tissue samples, and comprehensive molecular profiles reflecting the underlying heterogeneous biology. Current animal models of metastasis are principally derived from cell lines and do not represent the biological underpinnings of the human disease.6,7 Studies suggest that patient-derived xenograft (PDX) models more faithfully preserve the genetic features of patient tumors and are more predictive of clinical outcomes than are their predecessor cell line xenograft models.8–10
Herein we report the successful establishment and characterization of 39 PDXs derived from 8 different CM histological subtypes. Multi-omic analysis of the PDX and original patient tumor showed that most models accurately recapitulated the histological and molecular properties of their parent tumors. However, some PDX tumors diverged through the expansion of minor tumor clones, which were associated with spontaneous CM after flank implantation. Overall, this study has led to the identification of novel molecular profiles of CM and illustrated unique features of clonal evolution when using PDX models.
Materials and Methods
Sample Acquisition
Forty-five patients with brain metastases were consented for tissue collection under an institutional review board–approved protocol at Geisinger Health System, the University Health Network, the Mayo Clinic, or US Oncology between 2013 and 2016 (Supplementary Tables 1–3). Tumors excised during craniotomy were used for PDX generation, snap frozen for genomic profiling, and formalin fixed for histology where available.
Animal Studies
All animal experiments were conducted in accordance with Institutional Animal Care and Use Committee guidelines. Tumors for engraftment were minced and mixed with 1 part of Matrigel (Corning, #356231). Injected into the right flank of 4- to 6-week-old female NOG (Taconic) or nude (Narlan) mice were 0.3 mL tumor slurry. Tumors were measured using a digital caliper, and tumor volume was determined by the following formula: Length × Width × Width × 0.5. Tumors reaching 1.5 cm3 were harvested and passaged into new mice using the above protocol. Tumors were also snap frozen for sequencing, formalin fixed for histology, and cryopreserved according to standard protocol for future use.
Intracranial injections were performed using a stereotaxic frame (Stoelting). A 5–7 mm skin incision was made and a burr hole drilled 1–2 mm lateral to the midline and 2–3 mm vertical from the bregma suture. The automated injection system was used to slowly inject 2–3 µL of 104 cells into the brain parenchyma. For intracardiac injections, 1 × 105 cells in 50 µL of cells were injected into the left cardiac ventricle of anesthetized mice.
Histological Assessment
Tumor and/or organ tissues obtained at necropsy were formalin fixed and paraffin embedded (FFPE), and 5 µm sections were stained with hematoxylin and eosin (H&E). Metastatic tumors were identified by study pathologists at Mayo Clinic and TGEN/USC. Immunohistochemistry and immunofluorescence were performed using standard protocols. See Supplementary Material for details.
Next-Generation Sequencing and Microarrays
Whole exome sequencing and RNA sequencing (RNA-seq) were performed as previously described.11 Briefly, tissues were homogenized by the Bullet Blender (Next Advance) and DNA/RNA extracted with the AllPrep DNA/RNA Mini Kit (Qiagen). Whole blood was extracted with the QIAamp DNA Blood Maxi kit (Qiagen), and FFPE tissues were macrodissected and extracted with the QIAamp DNA FFPE Tissue Kit (Qiagen).
Libraries for whole-exome sequencing were prepared using the SureSelectXT Target Enrichment System (Agilent), and hybridization was accomplished with Human All Exon v5 (Agilent) baits. RNA-seq libraries were prepared using the TruSeq RNA Sample Preparation v2 (Illumina). For all libraries, paired end sequencing was performed on the Illumina HiSeq 2500 platform using TruSeq SBS v3 kits. DNA Methylation analysis was performed using the Infinium HumanMethylation450 BeadChip Kit (Illumina) as described previously.12 Reverse phase protein array (RPPA) was performed as previously described13 on a custom array (Supplementary Table 4). Sequencing and array data were analyzed as previously described.11 For further details on next-generation sequencing preparation, and analysis, see Supplementary Methods.
Results
Generation of a CM PDX Bank Representing Multiple Histological Subtypes
Forty-six fresh CM tumors were collected in the Salhia (n = 18) and Sarkaria (n = 28) labs immediately following craniotomy and transplanted into the right flanks of mice. Two independent metastatic lesions (T1 and T2) were resected from patient CM13, and both were collected and implanted. Thirty-nine tumor lines, between the 2 labs, were successfully maintained through multiple rounds of serial transplantation (Supplementary Tables 1–3). Tumor latency ranged from 7 to 183 days in the models from the Salhia lab and from 120 to 180 days in the models from the Sarkaria lab (Supplementary Fig. 1A, Supplementary Table 5). Tumor latency correlated significantly with patient survival (Supplementary Fig. 1B), demonstrating potential prognostic value of CM-derived PDXs. Lung CM tumors had a shorter average latency (43 days) compared with breast tumors (79 days). Tumors that did not grow included 4 CM tumors (CM03, CM05, CM10, CM13-T2) from the Salhia lab, and 3 CMs from the Sarkaria lab. All CM tumors from the Salhia lab were from patients without therapy prior to their CM surgical resection, while tumors from the Sarkaria lab came from patients with or without prior therapy for their CM tumors.
Histological assessment of patient tumors and corresponding PDXs by H&E staining demonstrated conservation in morphology (Fig. 1A, B). In both the patient and the PDX mouse model, the lung cancer squamous cell carcinoma shows areas with tumor cells infiltrating the dense fibrotic stroma in strings (Fig. 1A). In other parts the tumor is more solid and keratinization can be seen (Fig. 1A inset). However, the PDX model showed significantly less keratinization compared with the patient’s tumor, which might be an indication of it being less differentiated, but a sampling bias cannot be completely excluded. Both tumors are pleomorphic with nuclei showing sometimes serval nucleoli. The adenocarcinoma in the patient and in the PDX tumors are highly pleomorphic, with nuclei showing prominent nucleoli and tumor cells with abundant eosinophilic cytoplasm and distinct cell borders. Some gland-like growth patterns can also be observed in both tumors. The small cell carcinoma in the patient as well as in the PDX model shows a classic small cell morphology with dense packed tumor cells with scant cytoplasm, fine nuclear chromatin, and absence of nucleoli. The morphology can be especially well appreciated in the PDX models, since fixation is optimal. All breast tumors (CM1, CM13, and CM16) show tumor cell nest formation of variable size and tumor cells are pleomorphic. Interestingly, one breast tumor (CM01) shows morphologically 2 distinctive cell populations. In the background of eosinophilic tumor cells some tumor cells with a more clear cytoplasm could be appreciated (data not shown). The corresponding PDX model, however, shows only the eosinophilic cell population. The breast cancer cells from the flank tumor of CM07 grow in small cell nests that are intersected by thin fibrous stroma, while the tumor cells in the brain show a solid growth pattern. Perhaps this is due to the different tumor microenvironments. Still, both tumors are very pleomorphic; mitotic figures can be easily found; the cells have eosinophilic cytoplasm; and the tumors show large necrotic areas. The lung cancers (CM04) grown in the flank and in the brain both show classic small cell carcinoma morphology as described above.
Fig. 1.

Histological assessment by H&E staining of CM PDXs. (A–B) Representative images of PDX flank tumors with their patient-matched tumors from lung (A) or breast (B) CM. Inset in (A) show areas of keratinization patterns: clear visible eosinophilic keratinization and single cell keratinization (black arrow). (C–D) Evidence of brain tumor formation after intracranial injections (C) or intracardiac injections, which shows multifocal lesions (D). (E) Examples of extracranial macrometastasis formation from flank-implanted CM13 PDX in lung and liver. (F) Brain micrometastases were identified in flank-implanted CM13 PDX as evidenced by immunohistochemical staining for human-specific human leukocyte antigen A. Scale bars represent 200 μm (A–C) or 100 μm (E–F).
The ability of CM tumors to grow in the brain of immunocompromised mice was tested by intracranial injection. Intracranial growth was successful in 5/6 PDX lines from the Salhia lab, and 7/7 PDX lines tested from the Sarkaria lab. The one tumor that did not grow successfully in the brain was CM02; it was derived from a spinal cord metastasis, and not a brain metastasis. Intracranial tumors retained the morphology of the PDX flank (Fig. 1C), indicating that the site of implantation does not impact tumor morphology.
The metastatic and brain-tropic potential of PDX tumors was examined first by left ventricle cardiac injections. Five of 12 PDXs resulted in multifocal brain metastases, along with metastasis to the lung, liver, and other organs (Fig. 1D). Interestingly, necropsies revealed that spontaneous distant metastasis also occurred in flank-implanted PDXs. Three PDXs had evidence of metastasis to multiple tissues, including lung and liver (Fig. 1E, Supplementary Table 6). CM13-T1 was one of the most aggressive PDXs with a short latency period (40.5 days), and metastasized to 7 different locations: lung, liver, spleen, kidney, gastrointestinal (GI) tract, forearm and brain (Fig. 1F).
Maintenance of Molecular Profiles Between Patient Samples and Tumor Grafts
In order to compare the molecular profile of PDX tissue with its patient donor CM tissue, whole exome and RNA-seq was performed on 14 PDXs from the Salhia lab, which had patient matched tumor and germline tissue. The 3 CM tumors without corresponding PDXs were also sequenced. The F0 generation was analyzed for all PDXs, while additional passages (up to F4) were sequenced for 7 of the PDXs: CM04, CM06, CM09 (F0, F1); CM02 (F0, F1, F3); CM16 (F0-F2); CM07 (F0-F3); and CM01 (F0–F4). Both patient lesions in CM13 were sequenced, although only T1 formed a PDX. DNA methylation profiling used the 450K Infinium array, and RPPA was also performed on a subset of patient tumors and PDXs.
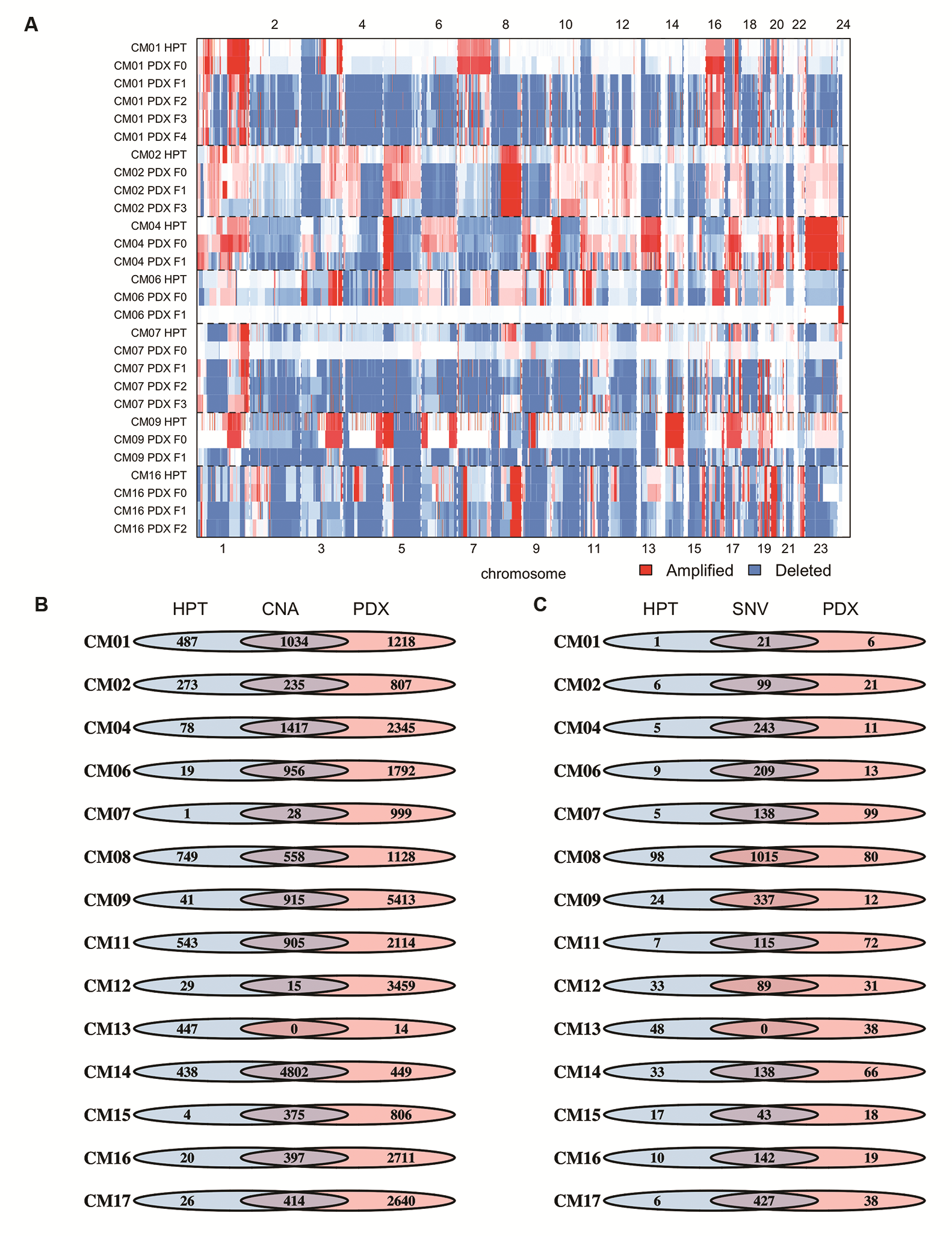
Copy number
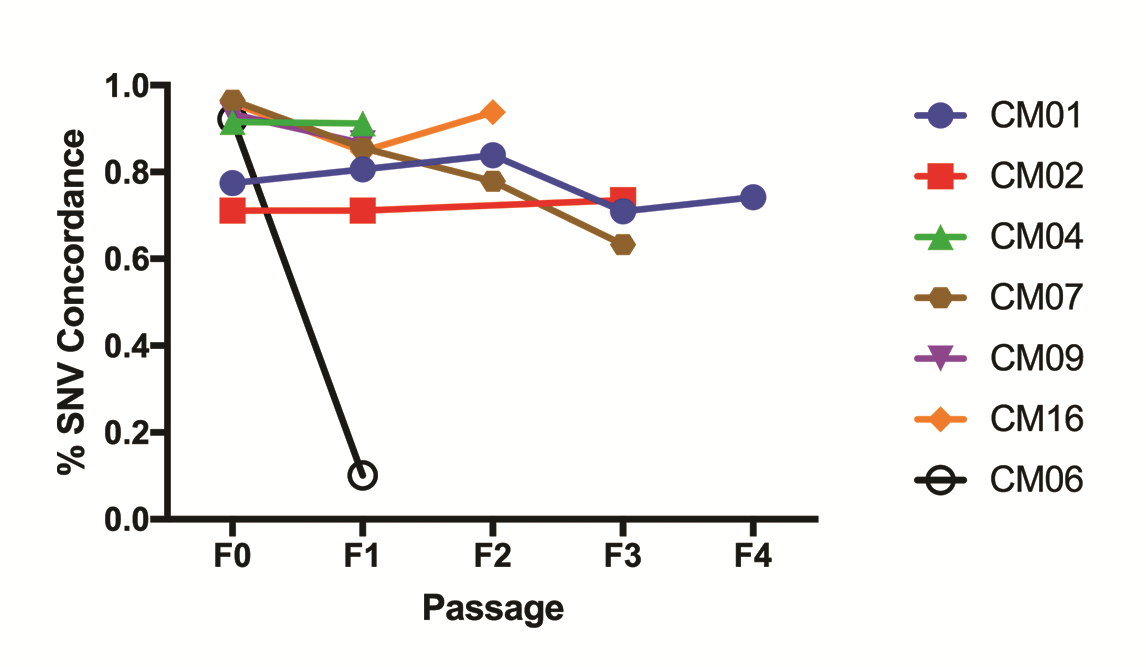
The patterns of DNA copy number variations (CNVs), determined by whole exome sequencing, in the original tumors were typically maintained in PDXs (Fig. 2A). On average, PDXs retained 73% of CNVs from patients, with similar retention rates in breast (75%) and lung CMs (73%). The only PDX to deviate dramatically from this trend was CM13, where the PDX appeared copy number neutral, unlike its patient tumor, which had numerous CNVs. This was confirmed in a second independent PDX tumor from CM13-T1. Most CNVs were also preserved over multiple generations in 6/7 PDXs sequenced (Supplementary Fig. 2A). The only PDX to deviate from this was CM06 in the F1 generation, which also reverted to a copy number neutral genome.
Fig. 2.

Comparison of genomic landscape between PDXs and their corresponding human patient tumors (HPTs). (A) Heatmap displaying identified in HPTs and the F0 generation of PDXs. The percentage of retained (%Ret) copy number alterations was high for most PDX–tumor pairs. (B) IVA analysis from PDXs and HPTs displaying the most significant pathways (left) and gene complexes (right) in HPTs and PDXs. The lines on the graph demarcate P-value in log scale and the bars denote the number of models with significant mutations in pathways or gene complexes. (C) Oncoprint map of common driver mutations and most frequent mutations identified in HPTs (top) and their corresponding status in PDXs (bottom).
Numerous homozygous deletions in tumor suppressor genes found in PDX tumors were either absent or present as single copy losses in the patient tumor (Supplementary Fig. 2B), suggesting expansion of a prominent clone after PDX establishment. Examples of this include loss of NF1, an inhibitor of RAS signaling, in CM01 and PTEN and FAS in CM06. The average number of CNVs increased from 1086 in the patient tumor to 2710 in the PDXs, a 2.5-fold enrichment. CM07 had the largest CNV enrichment, from 29 to 1027, representing a 35-fold increase. However, CNVs in the patient tumor of CM07 were largely preserved, including the retention of aberrations in phenotypically important loci, such as the 17q amplification bearing human epidermal growth factor receptor 2 (HER2) amplicon, which originated from a HER2+ breast cancer tumor.
Mutations
A broad range of nonsynonymous somatic mutations, including missense, nonsense, insertion/deletion, as well as splice site variants, were identified from whole exome sequencing results. Similar to the CNVs, the mutational landscape of PDXs resembled their original tumors, retaining on average 84% of mutations (Supplementary Fig. 2C). Lung CMs had higher mutation retention (92%) than breast CMs (73%, or 88% if outlier CM13 was excluded). This was despite the higher frequency of mutations in lung patient tumors, which averaged 371 per patient, compared with 99 in breast (Supplementary Fig. 2C). Mutation profiles in PDXs were largely similar, with an average of 375 and 121 mutations per patient in lung and breast PDXs, respectively. Of the 7 PDXs tracked over multiple generations, excluding outlier CM06-F1, 83% of the variants were preserved (Supplementary Fig. 3). Unlike CNVs, however, there was no dramatic enrichment in mutations in PDXs, though CM07, which saw a large enrichment in CNVs, also had a 1.7-fold enrichment in mutations. CM13 and CM06-F1 were also outliers, retaining none of the called mutations of the patient tumor.
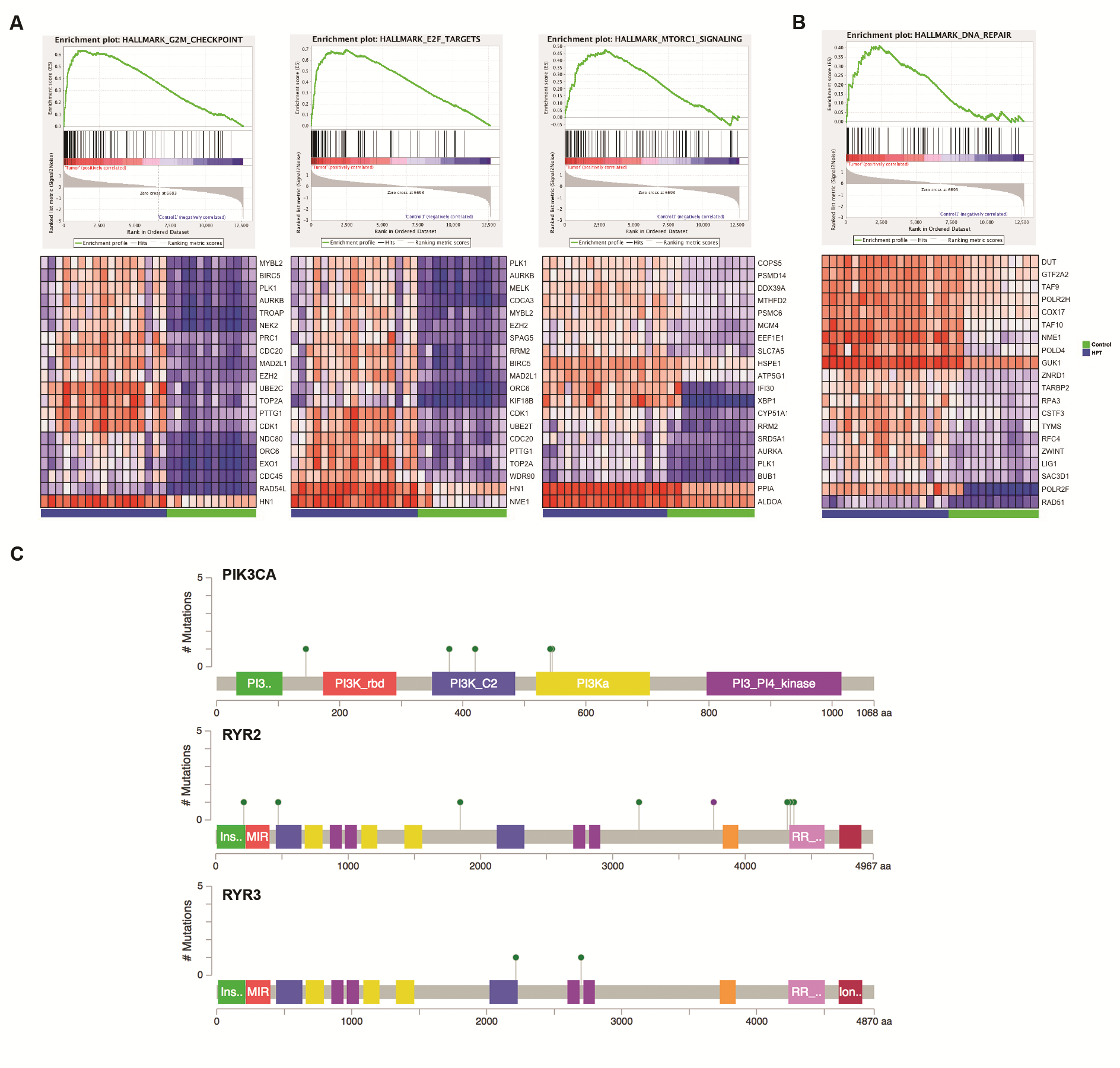
We next performed Ingenuity Variant Analysis (IVA) to determine if the overall mutational landscape was consistent between matched patient tumors and PDX (Fig. 2B). The top mutated pathways and gene groups identified in the patient tumors were also present in PDXs, including pathways related to cancer and neuronal signaling. In almost all cases, the important driver mutations were maintained in the PDXs (Fig. 2C). The most frequently occurring mutations were found in TP53, PIK3CA, RYR2, RYR3, ABCA8, DISP1, SYNE1, ZFHX4, and FSIP2.
Gene expression
Gene expressions from RNA-seq of patient tumors and PDXs were compared using a Pearson correlation matrix (Fig. 3A). Most tumors and PDX pairs were strongly correlated, with a mean of 0.86. Even CM07, with enrichments for CNV, had a high correlation (0.89). Notable exceptions included the PDXs of CM13 and CM06-F1, which bore no resemblance to their parent tumors (Fig. 3A).
Fig. 3.

Molecular landscape of PDXs and their corresponding human patient tumors (HPTs). (A) Pearson correlation matrix of RNA-seq data between PDXs and HPTs. Two independent PDX tumors of CM13 were sequenced to confirm the low correlation with their parent tumor. GSEA (B) and IPA (C) enriched pathways were similar in PDXs and HPTs. The numbers of PDX tumors and HPTs with statistically significant changes in the most highly differentially expressed IPA pathways are shown in (C). (D) Unsupervised hierarchical clustering of the top 100 differentially expressed genes between PDXs and HPTs. (E) Differentially expressed genes identified in (D) were then analyzed by IPA. The lines on the graph demarcate P-value in log scale and the bars denote the number of differentially expressed genes identified in each pathway. (F) Multi-omic correlation matrix. Each cell represents the degree of correlation between PDX and HPT for each listed technology. Gray cells indicate data not available.
Pathway analyses by gene set enrichment analysis (GSEA) and Ingenuity Pathway Analysis (IPA) resulted in similar pathways identified between the tumors and PDXs (Fig. 3B, C). Additionally, intrinsic molecular subtype analysis by the PAM50 classifier14 demonstrated that PDXs retained the molecular subtype of the parent tumor even in the case of CM13 (Supplementary Fig. 4A). These data further indicate that PDXs, even ones established in the flank, generally retained human tumor characteristics at the gene expression level. Enrichment of genomic aberrations in PDXs, which has been previously reported,15,16 had minimal effects on observed expression profiles.
However, to better understand the differences between the parent tissue and the PDX, we performed differential gene expression analysis. This analysis revealed significant downregulation of immunity related genes in the PDX (Fig. 3D, E). Among the top downregulated genes were immune cell markers, such as the CD4 T-cell marker, CD33 myeloid cell marker, and CSF1R (CD115) monocyte marker. This suggests that the biggest difference in gene expression between PDX and patient tumor is the lack of tumor-infiltrating immune cells in the immunocompromised PDX host, rather than those pertaining to the non-immune component of the tumor microenvironment.
DNA methylation and RPPA
Unsupervised hierarchical clustering for DNA methylation and RPPA data also showed that the methylome and proteome of patient and PDX tumors were maintained (Supplementary Fig. 4B, C). In both analyses the lung tumors (CM02, CM04, CM06) clustered separately from the breast tumors (CM01, CM07).
Taken together, the DNA copy number, somatic mutation, gene expression, DNA methylation, and RPPA provide evidence that tumor grafts generally maintain the prominent molecular characteristics of the original tumors, even when implanted in the flank (Fig. 3F). However, in some instances, unique clonal expansion events can occur and lead to differing molecular phenotypes.
Integrated Analysis of Patient CM Tumors with Relevance to PDX Models
Multiple analyses revealed that pathways related to tumorigenesis were significantly altered in both the patient tumors and PDXs (Figs. 2B, 3B). GSEA revealed enrichment of gene sets related to enhanced proliferative potential, including G2/M, E2F, and mammalian target of rapamycin complex 1 (Fig. 3B, Supplementary Fig. 5A). The Aurora kinases (AURKA, AURKB), which we have previously shown to be associated with CM3, were also overexpressed. An upregulation of DNA damage repair genes was also observed, including DNA damage sensing components (RAD51, ATM, ATR) and DNA repair enzymes (BRCA1, BRCA2, POLD1, POLD4) (Supplementary Fig. 5B). Increased DNA repair activity is often associated with an increase in replication stress caused by increased proliferation of tumor cells.17
In the top 10 enriched pathways, 4 IVA and 5 IPA pathways were related to neuronal signaling. Multiple studies have reported a tendency of brain metastasis tumors to adapt neuronal features.18,19 These features were shown to assist the metastasizing tumor cells in adapting to the CNS microenvironment. For example, we found increased expression of gamma-aminobutyric acid (GABA) receptors, such as GABRA1 and GABRA2, and associated proteins, such as GAD1, which were shown to enable brain metastasis tumors to utilize GABA present in the brain environment to promote cell proliferation.18
Calcium signaling was also altered in CM tumors. There were 20 genetic variants in 12 calcium channel genes in 56% of cases. Of these, the ryanodine receptors (RYR2, RYR3), both intracellular calcium channels, were found to be most frequently mutated (33% and 28%, respectively) (Supplementary Fig. 5C). Furthermore, mutation of RYR2 or RYR3 was associated with poorer patient survival, albeit not statistically significant due to small sample size (Supplementary Fig. 6A). Other intracellular calcium channels (RYR1, ITPR1) were also mutated at a lower frequency. Deregulation of calcium signaling is known to promote proliferation and migration of tumor cells,20 suggesting that deregulation of calcium signaling may potentiate CM. Interestingly, mutation of RYR3 appeared to be mutually exclusive with PIK3CA (Fig. 2C). Two samples had PIK3CA C2 domain mutations, which is known to facilitate phosphatidylinositol-3 kinase targeting to membrane surfaces as a consequence of calcium-binding, again implicating the importance of calcium regulation in CM.
Changes in calcium signaling were also apparent at the gene expression level, where calcium signaling was predicted to be downregulated by both IPA and GSEA (Supplementary Fig. 6B). Top downregulated genes point to a downregulation of phospholipase C (PLC)–mediated calcium signaling, where upstream signaling mediators GNAQ and PLCD1, along with calcium channels such as ITPR, were all downregulated. PLCD1 has been reported to be a tumor suppressor that is frequently silenced by promoter methylation in breast cancer.21 Its ability to induce cell cycle arrest could suggest that the shutdown of calcium signaling may be important for CM tumor survival.
Downregulation of inflammation response was observed in tumors through changes in the acute phase response signaling in IPA, and inflammatory response and tumor necrosis factor alpha (TNF-α) signaling in GSEA (Supplementary Fig. 6C). The brain environment is thought to have limited capacity for inflammation. The downregulation of inflammation response could be a reflection of this vastly different immune landscape of the CNS microenvironment compared with peripheral tissue where the tumor originated. However, high expression of TNF-α was associated with poor patient survival (Supplementary Fig. 6D). This suggests that the presence of pro-inflammatory cytokines such as TNF-α could be an indicator of increased tumor aggressiveness.
Subtypes of CM PDX Revealed Discrete Gene Expression Differences
Given our rich resource of CM PDXs representing a wide spectrum of histological subtypes, we performed multidimensional scaling (MDS) of the RNA-seq data from all 39 PDXs to study how CMs differ across the 8 histological subtypes represented. MDS identified 6 distinct subgroups of PDXs (Fig. 4A), based in large part on the histological subtype of the PDX. Intermixing of samples illustrated no bias between PDX samples generated in different labs (Supplementary Fig. 7). Subgroups 1 and 4 were most homogeneous and consisted of melanoma and upper GI PDXs, respectively. Subgroups 2, 3, and 5 were heterogeneous and consisted of a mix of breast and lung PDXs. Still, most non–small cell lung cancer (NSCLC) PDXs clustered in subgroup 3, while small cell lung cancer tumor PDXs were found in subgroups 2 and 5. HER2+/estrogen receptor and triple-negative basal-like breast PDXs were found in subgroup 3, while luminal B tumors were the most diverse of all the PDXs. Group 6 consisted solely of CM13.
Fig. 4.

Molecular signatures of histological subtypes in CM. (A) Multidimensional scaling (MDS) and k-means classification of the gene expression data for F0 generation of 39 PDX tumors resulted in 6 distinct tumor clusters, each indicated by a subgroup number and unique color on the plot. Cluster 6 represents CM13. (B) Differential gene expression analysis between the clusters and IPA analysis of gene lists was performed. Unsupervised clustering analysis of the union of the top 100 genes across the clusters was performed and is shown along with associated enriched pathways in the heatmap.
Unsupervised clustering followed by IPA was used to understand underlying molecular differences between the subgroups (Fig. 4B). Subgroup 1 was associated with loss of cell-cell adhesion, such as claudins 3 and 7, which have been linked to epithelial mesenchymal transition22 and increased invasiveness. Subgroups 2, 3, and 5 were highly similar, and were associated with gene expression signatures related to p53 loss, consistent with its high mutation rate. Subgroup 3 has a more secretory phenotype, possibly implicating aggressive modulation of its tumor microenvironment. Subgroup 4 was associated with hallmarks of GI tumors, including deregulation of the hepatocyte nuclear factor (HNF) gene family, most notably HNF4A and FOXA2, which are important in proliferation and metastasis in GI cancers.23,24
Clonal Evolution from Patient to PDX and Minor Clone Expansion
Although most PDXs retained molecular features of their parent tumor, CM13-F0 and CM06-F1 PDXs emerged as outliers. They reverted to a copy number neutral profile, had no shared nonsynonymous mutations or driver mutations, and were not correlated by gene expression (Figs. 2A, 3A). We ruled out erroneous results by sequencing a second PDX from CM13-F0 (Fig. 3A), and confirmed that >95% of reads aligned to the human genome (Supplementary Fig. 8). The most prevalent mouse reads, when analyzed by IPA, were related to inflammation and immune cell motility. The mouse models, though immunocompromised, still have functional monocytes and neutrophils, and other nonfunction immune cells. Indeed, many mouse immune cell markers were found to be enriched in PDX tumors (Supplementary Fig. 9A). CM13-F0 PDX, a HER2+ inflammatory breast cancer CM, also had a short tumor latency (40.5 days) and was highly metastatic when grown in the flank (Fig. 1E). CM13-F0 flank tumors displayed spontaneous metastasis to lung, liver, spleen, kidney, GI tract, and forearm but most notably to brain (Fig. 1F). CM06-F1, which originated from a squamous cell carcinoma of the lung, bore little resemblance to its previous generation CM06-F0. Instead, it appeared more like CM13-F0 (Supplementary Fig. 9B), albeit with no evidence of spontaneous brain metastasis (which could be due to timing of when mice were euthanized).
These data suggest that a clonal expansion by a minor clone may have occurred in CM13-F0 and CM06-F1 which led to the apparent deviation from its donor tumor. To assess tumor clonality and evolution, we traced the phylogeny of PDX tumors from their originating patient tumor. Allele-specific copy number and somatic variants were used to reconstruct the phylogenetic history of each PDX lineage (Fig. 5, Supplementary Fig. 10). We found that all PDX and originating tumors displayed evidence of tumor clonal heterogeneity. Twelve of the 14 PDXs examined evolved neutrally or by expansion of a major clone (defined by an increase in the prevalence of a clone that was already present at a high [>10%] frequency in the tumor) (Fig. 5A–C, Supplementary Fig. 10). That is, at least one major clone was conserved in F0 and subsequent passages where tested.
Fig. 5.

Phylogeny tracing of PDXs. (A–F) Phylogenic reconstruction from allele-specific copy number and somatic variants. Timescape plots with associated phylogenetic trees are shown for selected PDXs. Each color represents a different clone within the tumor, with the height of the bar representing the prevalence of the clone within the entire tumor, and thicker bars indicating more prevalent clones. Three PDX tumors had neutral clones, where clonal prevalence of the major clones remained relatively unchanged (A and C) or expanded (B). However, CM06-F1 (D) and CM13-F0 (E) demonstrated rapid and explosive clonal succession, respectively, where expansion of one or multiple minor clones present at low frequencies in either the patient tumor (E) or the first passage (D) was seen.
On the other hand, CM13-F0 and CM06-F1 displayed strong evidence of clonal succession by a minor clone (CM06) or several minor clones (CM13), where a clone that was previously present at low frequencies in the original patient tumor—1% and 7%, respectively—is now the dominant clone in the tumor (Fig. 5D, E). Clonal succession was explosive in the case of CM13, because it occurred in the F0 generation and rapid in the case of CM06-F1, as it occurred after the first passage. This was certainly unlike all the other tumors that had either neutral clones or major clone expansion (Fig. 5). Three distinct modes of evolution in PDXs were observed: rapid (CM06) and explosive (CM13) clonal succession and neutral evolution (all other PDXs). These data suggest that minor clone expansion might confer tumor aggressiveness and potentiate metastasis in some patients.
Next, we wanted to determine the set of genetic alterations that differentiate CM13-F0 and CM06-F1 from other PDXs. The dramatic genetic changes in the clonally expanded CM13-F0 and CM06-F1 were reflected in their gene expression signatures, which did not resemble that of other CM PDXs (Supplementary Fig. 11A). As previously described, pathways such as E2F signaling and G2M checkpoint were upregulated in PDXs. However, these were downregulated in CM13-F0 and CM06-F1. Other pathways downregulated in most PDXs were instead upregulated in CM13-F0 and CM06-F1, including the immunity-related pathways (Supplementary Fig. 11B). IPA analysis of significantly differentially expressed genes revealed an enrichment of pathways related to immunity and neuronal signaling (Fig. 6A). Differentially expressed genes in these pathways included glia maturation factor gamma (GMFG), a growth factor important for both neuronal and immune cell development,25,26 and interferon regulatory factor 4 (IRF4), a transcriptional activator important in microglia activation27 (Fig. 6B). Immunofluorescent staining confirmed that GMFG and IRF4 were highly expressed in flank tumors and brain metastases of CM13-F0 and CM06-F1 (Fig. 6C, D) compared with the other PDXs, suggesting that the upregulation of immunity- and neuronal-related pathways may be associated with minor clone expansion and metastatic behavior.
Fig. 6.

Unique gene expression features of CM with minor clone expansion (CM13-F0, CM06-F1). (A) IPA analysis of differentially expressed genes between CM13/CM06-F1 versus the other PDXs. The lines on the graph indicate the log P-value and the bar denotes the number of differentially expressed genes identified in each pathway. Immune-related (red) and neuronal-related (blue) pathways are highlighted. (B) Hierarchical clustering of the top 100 differentially upregulated genes in CM13 and CM06-F1 have distinct expression profiles compared with the other models. Selected genes related to immunity (red), neuronal signaling (blue), or both (purple) are highlighted. (C–D) Immunofluorescence staining of 2 differentially expressed genes, (C) GMFG and (D) IRF4 (red staining), in the flank tumors of CM13, CM06-F1, and CM01 (non-minor clone tumor), and brain micrometastases from flank-implanted CM13, along with human leukocyte antigen A (green staining), a human-specific marker, and counterstained with 4′,6′-diamidino-2-phenylindole (blue staining). Scale bars represent 50 μm.
Discussion
A lack of tumor models reflecting the heterogeneity and complexity of human CM has presented a barrier to understanding the underlying biology and developing better therapies. Even though CM PDXs have been developed in the past, they have been developed from a limited number of cases and tumor types originating only from NSCLC,10 HER2+ breast cancer,28 or melanoma29,30 and were not extensively characterized and/or focused mostly on PDXs from the primary tumor. In the current study, we developed 39 CM PDXs from 8 different histological subtypes, and performed comprehensive multi-omic characterization. Our study shows that in the CM tumors analyzed, melanoma and GI CM were genomically distinct histological subtypes, while breast and lung cancer–derived CM were relatively heterogeneous. Commonalities across CM include alterations leading to upregulation of neuronal signaling and downregulation of calcium signaling.
An important advantage of PDXs is their ability to preserve the genomic and molecular properties of the original patient tumor. Here we show that ectopic engraftment of CM tumors in mice resulted in the retention of the molecular genetic landscape of the original tumor in a process of neutral evolution. In addition, tracing of 7 PDXs over multiple generations (F0–F4) demonstrated that genomic and molecular changes were stable even after multiple passages in 6/7 PDX models.
Multiple studies have also shown that, although PDXs are genetically stable, they can undergo clonal evolution, resulting in deviation from their original tumor over time.16,31 Genomic drifting has been the mode of clonal evolution reported and involves gradual accumulation of genetic changes over many passages.15,32 This has been attributed to observed selection pressures in xenograft models.16 In this study, we identified a different mode of clonal evolution in both CM13-F0 and CM06-F1, which were characterized by explosive (F0) or rapid (F1) clonal succession where a minor clone preferentially expands in the mouse and becomes the major clone. While explosive minor clonal succession in orthotopically implanted PDXs has been shown previously,33 we found in our study that clonal succession can result in dramatic genomic and functional changes, including reversion to a diploid genome and loss of driver mutations, as well as aggressive tumor behavior. In addition, we demonstrated that clonal succession could occur in subsequent passages, as seen in CM06-F1. Additionally, the observation that the PDX with the highest metastatic potential (CM13) evolved by explosive clonal succession suggests a potentially highly clinically relevant mode of tumor evolution whereby minor clones in primary tumors give rise to aggressive metastases.
It was notable that the PDXs with rapid or explosive minor clone succession did not harbor classical driver gene mutations such as TP53. It can therefore be argued that such classical driver genes could be required for tumor initiation and virulence but not tumor spread. Hence, genes upregulated in minor clones, including GMFG and IRF4 are known for their role in tumor metastasis. GMFG, a chemotaxic factor generally involved in immune cell migration, is associated with increased metastatic potential of both colorectal and ovarian cancer.34,35 IRF4, a transcription factor involved in interferon regulation, has been associated with poor prognosis of multiple hematologic cancers36 and was found to be tumor promoting in NSCLC by the activation of Notch signaling, independently of the immune system.37 Upregulation of IRF4 has also been shown to increase the proliferation and metastasis of cholangiocarcinoma.38 Further studies will be required to confirm whether these genes are indeed responsible for enhancing CM potential.
Studying metastasis in tumor xenograft models has often been a challenge. Injection of tumor cells directly into the circulation or orthotopically fails to recapitulate many processes of metastasis such as intravasation and extravasation.39 While subcutaneous xenografts in the flank have been viewed as less relevant compared with orthotopic xenografts due to the lack of anatomic microenvironment, we showed that in addition to recapitulating the histological and molecular features of the original patient tumor, 3 out of 14 (21%) of Salhia lab models are capable of developing spontaneous metastasis to extracranial sites when implanted into the flank, despite being derived from a CM tumor. This suggests that the concept of organ tropism may need to be revisited and introduces the possibility that CM may be the result of tertiary metastases, especially in breast cancer, where CM is a late event. Interestingly, CM13 PDX formed numerous extracranial metastases and was the only PDX that also formed spontaneous metastases in the brain. While spontaneous distant metastasis from the flank to brain has been observed in CM cell lines,40,41 this is the first report demonstrating the ability of a CM PDX model to traverse multiple barriers and form spontaneous distant metastases, an event that would have been missed had we implanted the tissue directly into the brain.
Many studies have performed histological and genomic analyses of tumor xenografts. However, to our knowledge, there are no studies that have characterized the patient germline, the patient tumor and corresponding PDX tumor, including tumors from multiple PDX passages in CM. Furthermore, our samples were characterized by multiple orthogonal approaches, where PDX/patient tumor and germline were analyzed by histology and whole exome sequencing, patient/PDX tumor pairs were analyzed by RNA-seq DNA methylation analysis, and proteomics comprehensively assessed the degree to which PDXs recapitulate the histological and molecular makeup of the originating patient tumor.
In summary, the models reported in this study alongside the accompanying dataset provide a new set of tools that will help further our understanding of the mechanics of disease spread, enable novel and relevant preclinical testing, and provide a population-scale reference model for the deadliest of metastatic cancers. Finally, our data suggest that inherent plasticity may favor accelerated clonal evolution and predict aggressive tumor behavior in both PDX models and the human cancers from which they derive.
Supplementary Material
Supplementary data are available at Neuro-Oncology online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by the Flinn Foundation.
Conflict of interest statement. All authors report no conflicts of interest.
Authorship statement. Study conception and design: B. Salhia, J. Sarkaria. Data collection: B. Salhia, B. Y. Tew, G. Gooden, M. Schroeder, D. Ma, K. Johnson, R. A. Martinez, J. Sarkaria, S. Toms, M. Pierobon, E. Petricoin, J. O’Shaughnessy, C. Osborone, J. Glen, M. Bernstein. Data analysis and interpretation: B. Salhia, B. Y. Tew, C. Legendre, T. Triche Jr., G. Gooden, Y. Huang, C. Tapia, D. N. Buckley, S. Toms. Manuscript preparation: B. Salhia, B. Y. Tew, C. Legendre, G. Gooden, C. Tapia, D. N. Buckley, S. Toms, J. Sarkaria.
Acknowledgments
We would like to thank Mario Sepulveda at Translational Drug Development for his early assistance in the generation of PDX models.
References
- 1. Cruz-Muñoz W, Kerbel RS. Preclinical approaches to study the biology and treatment of brain metastases. Semin Cancer Biol. 2011;21(2):123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Owonikoko TK, Arbiser J, Zelnak A, et al. Current approaches to the treatment of metastatic brain tumours. Nat Rev Clin Oncol. 2014;11(4):203–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salhia B, Kiefer J, Ross JT, et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS One. 2014;9(1):e85448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Svokos KA, Salhia B, Toms SA. Molecular biology of brain metastasis. Int J Mol Sci. 2014;15(6):9519–9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wong ET, Berkenblit A. The role of topotecan in the treatment of brain metastases. Oncologist. 2004;9(1):68–79. [DOI] [PubMed] [Google Scholar]
- 6. Tominaga N, Kosaka N, Ono M, et al. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat Commun. 2015;6:6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoneda T, Williams PJ, Hiraga T, Niewolna M, Nishimura R. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J Bone Miner Res. 2001;16(8):1486–1495. [DOI] [PubMed] [Google Scholar]
- 8. DeRose YS, Wang G, Lin YC, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17(11):1514–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klinghammer K, Raguse JD, Plath T, et al. A comprehensively characterized large panel of head and neck cancer patient-derived xenografts identifies the mTOR inhibitor everolimus as potential new treatment option. Int J Cancer. 2015;136(12):2940–2948. [DOI] [PubMed] [Google Scholar]
- 10. Lee HW, Lee JI, Lee SJ, et al. Patient-derived xenografts from non-small cell lung cancer brain metastases are valuable translational platforms for the development of personalized targeted therapy. Clin Cancer Res. 2015;21(5):1172–1182. [DOI] [PubMed] [Google Scholar]
- 11. Tew BY, Legendre C, Gooden GC, et al. Isolation and characterization of patient-derived CNS metastasis-associated stromal cell lines. Oncogene. 2019;38(21):4002–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Legendre CR, Demeure MJ, Whitsett TG, et al. Pathway implications of aberrant global methylation in adrenocortical cancer. PLoS One. 2016;11(3):e0150629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pin E, Federici G, Petricoin EF 3rd. Preparation and use of reverse protein microarrays. Curr Protoc Protein Sci. 2014;75:Unit 27 27. [DOI] [PubMed] [Google Scholar]
- 14. Chia SK, Bramwell VH, Tu D, et al. A 50-gene intrinsic subtype classifier for prognosis and prediction of benefit from adjuvant tamoxifen. Clin Cancer Res. 2012;18(16):4465–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eirew P, Steif A, Khattra J, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2015;518(7539):422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ben-David U, Ha G, Tseng YY, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017;49(11):1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Técher H, Koundrioukoff S, Nicolas A, Debatisse M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet. 2017;18(9):535–550. [DOI] [PubMed] [Google Scholar]
- 18. Neman J, Termini J, Wilczynski S, et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A. 2014;111(3):984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park ES, Kim SJ, Kim SW, et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc Natl Acad Sci U S A. 2011;108(42):17456–17461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11(8):609–618. [DOI] [PubMed] [Google Scholar]
- 21. Xiang T, Li L, Fan Y, et al. PLCD1 is a functional tumor suppressor inducing G(2)/M arrest and frequently methylated in breast cancer. Cancer Biol Ther. 2010;10(5):520–527. [DOI] [PubMed] [Google Scholar]
- 22. Bhat AA, Pope JL, Smith JJ, et al. Claudin-7 expression induces mesenchymal to epithelial transformation (MET) to inhibit colon tumorigenesis. Oncogene. 2015;34(35):4570–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lehner F, Kulik U, Klempnauer J, Borlak J. The hepatocyte nuclear factor 6 (HNF6) and FOXA2 are key regulators in colorectal liver metastases. FASEB J. 2007;21(7):1445–1462. [DOI] [PubMed] [Google Scholar]
- 24. Walesky C, Apte U. Role of hepatocyte nuclear factor 4α (HNF4α) in cell proliferation and cancer. Gene Expr. 2015;16(3):101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lim R, Miller JF, Zaheer A. Purification and characterization of glia maturation factor beta: a growth regulator for neurons and glia. Proc Natl Acad Sci U S A. 1989;86(10):3901–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi Y, Chen L, Liotta LA, Wan HH, Rodgers GP. Glia maturation factor gamma (GMFG): a cytokine-responsive protein during hematopoietic lineage development and its functional genomics analysis. Genomics Proteomics Bioinformatics. 2006;4(3):145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Al Mamun A, Chauhan A, Yu H, Xu Y, Sharmeen R, Liu F. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. Eur J Neurosci. 2018;47(2):140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ni J, Ramkissoon SH, Xie S, et al. Combination inhibition of PI3K and mTORC1 yields durable remissions in mice bearing orthotopic patient-derived xenografts of HER2-positive breast cancer brain metastases. Nat Med. 2016;22(7):723–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krepler C, Sproesser K, Brafford P, et al. A comprehensive patient-derived xenograft collection representing the heterogeneity of melanoma. Cell Rep. 2017;21(7):1953–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garman B, Anastopoulos IN, Krepler C, et al. Genetic and genomic characterization of 462 melanoma patient-derived xenografts, tumor biopsies, and cell lines. Cell Rep. 2017;21(7):1936–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruna A, Rueda OM, Greenwood W, et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell. 2016;167(1):260–274 e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davies NJ, Kwok M, Gould C, et al. Dynamic changes in clonal cytogenetic architecture during progression of chronic lymphocytic leukemia in patients and patient-derived murine xenografts. Oncotarget. 2017;8(27):44749–44760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stewart E, Federico SM, Chen X, et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature. 2017;549(7670):96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang H, Chen Z, Chang H, et al. Expression of glia maturation factor γ is associated with colorectal cancer metastasis and its downregulation suppresses colorectal cancer cell migration and invasion in vitro. Oncol Rep. 2017;37(2):929–936. [DOI] [PubMed] [Google Scholar]
- 35. Zuo P, Ma Y, Huang Y, et al. High GMFG expression correlates with poor prognosis and promotes cell migration and invasion in epithelial ovarian cancer. Gynecol Oncol. 2014;132(3):745–751. [DOI] [PubMed] [Google Scholar]
- 36. Tsuboi K, Iida S, Inagaki H, et al. MUM1/IRF4 expression as a frequent event in mature lymphoid malignancies. Leukemia. 2000;14(3):449–456. [DOI] [PubMed] [Google Scholar]
- 37. Qian Y, Du Z, Xing Y, Zhou T, Chen T, Shi M. Interferon regulatory factor 4 (IRF4) is overexpressed in human non‒small cell lung cancer (NSCLC) and activates the Notch signaling pathway. Mol Med Rep. 2017;16(5):6034–6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wei CX, Wong H, Xu F, Liu Z, Ran L, Jiang RD. IRF4-induced upregulation of lncRNA SOX2-OT promotes cell proliferation and metastasis in cholangiocarcinoma by regulating SOX2 and PI3K/AKT signaling. Eur Rev Med Pharmacol Sci. 2018;22(23):8169–8178. [DOI] [PubMed] [Google Scholar]
- 39. Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66(23):11271–11278. [DOI] [PubMed] [Google Scholar]
- 40. Klein A, Sagi-Assif O, Meshel T, et al. CCR4 is a determinant of melanoma brain metastasis. Oncotarget. 2017;8(19):31079–31091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schwartz H, Blacher E, Amer M, et al. Incipient melanoma brain metastases instigate astrogliosis and neuroinflammation. Cancer Res. 2016;76(15):4359–4371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.