

Abstract
The intra-tumor heterogeneity is associated with cancer progression and therapeutic resistance, such as in breast cancer. While the existing methods for studying tumor heterogeneity only analyze variant allele frequency (VAF), the genotype of variant is also informative for inferring subclones, which can be detected by long reads or paired-end reads. We developed GenoClone to integrate VAF with the genotype of variant innovatively, so it showed superior performance of inferring the number of subclones, estimating the fractions of subclones and identifying somatic single-nucleotide variants composition of subclones. When GenoClone was applied to 389 TCGA breast cancer samples, it revealed extensive intra-tumor heterogeneity. We further found that a few somatic mutations were relevant to the late stage of tumor evolution, including the ones at the oncogene PIK3CA and the tumor suppress gene TP53. Moreover, 52 subclones that were identified from 167 samples shared high similarity of somatic mutations, which were clustered into three groups with the sizes of 24, 14 and 14. It is helpful for understanding the development of breast cancer in certain subgroups of people and the drug development for population level. Furthermore, GenoClone also identified the tumor heterogeneity in different aliquots of the same samples. The implementation of GenoClone is available at http://www.healthcare.uiowa.edu/labs/au/GenoClone/.
Keywords: tumor heterogeneity, subclone inference, VAF, somatic mutation, germline mutation
Introduction
Tumor evolution is a reiterative process of clonal expansion driven by sequential somatic mutation and Darwinian nature selection [1, 2]. Therefore, tumors are composed of remarkable distinct cell populations (referred as ‘subclones’), which is termed intra-tumor heterogeneity. The explicit studies of subclones within tumor samples can greatly improve the understanding of tumor evolution and thus benefit drug development and precision medicine [3, 4]. For example, intra-tumor heterogeneity has been shown in breast cancer [5–7] and causes target therapy may not function on the therapy-resistant subclones [8]. Moreover, the resistant subclones may reduce the successful rate of subsequent treatments and lead to tumor relapse and therapy failure [9]. Thus, it is important to correctly identify the subclones and estimate their fractions for intra-tumor study.
The development of new sequencing technologies allows us to perform genome-wide study of tumor heterogeneity [10–12]. Single-cell sequencing, which avoids the confounding factors of bulk sequencing of the whole tumor, is useful to address the heterogeneity problems [13, 14]. However, in addition to the technical problems of amplification bias and allele dropout [15, 16], single-cell sequencing is also limited by the high cost for sequencing a number of single cells, which is required to identify the subclones and estimate their fractions. Alternatively, identification of tumor subclones by bulk sequencing of a whole-tumor sample, followed by appropriate bioinformatics analysis, is more affordable and thus is of broader utility.
Variant allele frequency (VAF) of single-nucleotide mutations or variants has been used to infer the tumor subclones from bulk sequencing in recent studies [17, 18], because the fractions of subclones are linearly associated with VAFs. The observed VAFs depend on the stochastics process of selecting fragments from library construction, so it strongly correlates with sequencing depth. Deep sequencing is helpful to address the uncertainty of VAF. Roth et al. developed a statistical inference model, PyClone, to identify the tumor subclones [19]. PyClone identifies somatic mutations and copy number variants by the whole-genome or exome sequencing data, and next applies targeted deep sequencing to estimate VAFs. However, PyClone only infers cellular abundance of each variant, rather than the fractions of subclones. The other existing tools based on clustering approach overcome the substantial variability of observed VAFs measurement and further infer the fractions of subclones [20, 21]. For example, SciClone maximizes the posterior probability of VAFs based on a Dirichlet Process mixture model while the number of clusters is not fixed in advance. Subsequently, SciClone identifies the number of variant clusters by discarding clusters that does not contribute to the model and finally estimates the fractions of subclones. However, in addition to VAFs, the fractions of subclones also depend on genotype. For example, if the variant is adenine to guanine (A to G), then the genotype of the variant would be AG or GG. Lee et al. developed a Bayesian feature allocation model to identify the genotype of variants as well as the number of subclones and their fractions while it simultaneously increases the solution space and thus results in higher uncertainty of the solution [22]. Determination of the genotype of variants and reduction of the VAF uncertainty are the main problems in subclone inference.
Direct determination of the genotype of variants in each subclone is to identify whether variants are from paternal or maternal alleles [23–25]. However, it is difficult to obtain the entire paternal and maternal alleles (e.g. family trio data are required). Instead, if the linkage between somatic mutation (i.e. somatic single-nucleotide variant/sSNV) and germline mutation (single-nucleotide polymorphism/SNP) is known, we can determine the origins of sSNVs: maternal, paternal or both. Since the matched pair of normal and tumor samples shares the same SNPs, we can firstly distinguish sSNVs from SNPs from sequencing data. Then the sSNV–SNP linkage can be detected from long reads (e.g. PacBio, Oxford Nanopore Technologies and 454 sequencing) or paired-end reads covering sSNVs and the corresponding adjacent SNPs. This linkage information can reduce the dimension of solution space and thus a more accurate subclone inference can be obtained.
We develop a novel method, GenoClone (http://www.healthcare.uiowa.edu/labs/au/GenoClone/), to study tumor heterogeneity by innovatively integrating VAFs and genotype of sSNVs (Figure 1). Comparing to two existing methods, GenoClone showed superior performance of identifying the number of subclones, estimating their fractions and determining their sSNVs compositions in simulation data. By GenoClone, we analyzed 389 breast invasive carcinoma samples from The Cancer Genome Atlas (TCGA-BRCA) and revealed tumor heterogeneity in nearly all the samples. The results also showed that the mutations of the oncogene PIK3CA and the tumor suppressor gene TP53 may occur at the late stage of tumor evolution of breast cancer. Moreover, the similarity analysis among the subclones identified from 167 samples showed high similarity of 52 subclones, which were clustered into three groups with the sizes of 24, 14 and 14. Within these clusters of subclones, a few shared sSNVs, such as chr2_55679588_CA, chr3_179234297_AG and chr3_179218303_GA, provided informative foundation for drug design and treatment in certain subgroups of patients. In addition, we revealed the tumor heterogeneity in different aliquots of the same tumor samples and found the VAF of TP53 varied in different aliquots.
Figure 1.

Flowchart of GenoClone. VarScan2 is used to detect the somatic mutations (sSNVs) and germline mutations (SNPs) from tumor and matched normal samples. Then, GenoClone detects sSNV–SNP linkage by paired-end reads or long reads to infer the genotype of sSNVs. The observed VAFs are computed from short-read coverage. Finally, GenoClone integrates the genotype of sSNVs and their VAFs in the optimization model to infer subclones.
Methods
Somatic and germline mutation detection
Given the exome or whole-genome sequencing data from tumor and the matched normal samples, we can detect the mutations from both by the existing SNP calling methods. Next, we can determine the somatic and germline mutations by comparing the variant calling results from two samples. These two steps can be integrated (e.g. by VarScan2 [26]). Here, we used VarScan v2.4.2 and named the germline mutation as SNP and somatic mutation as sSNV below.
sSNV–SNP linkage
It is ideal to obtain the linkages among sSNVs for inferring the haplotypes of subclones. sSNVs are separated by long distance in the genome, so whole-genome sequencing by long reads is required to obtain such linkages among sSNVs. However, the whole-genome sequencing by long reads is of high cost and requires a large amount of DNA materials that the primary tumor samples may not provide. Instead of linkages among sSNVs, the linkage of sSNV and the adjacent SNP (termed sSNV–SNP linkage) can be detected by paired-end reads or long reads from either exome or whole-genome sequencing. Given an SNV, we use the adjacent SNPs to identify whether the SNV originates from one haplotype or both haplotypes. For example, one sSNV and one SNP is simultaneously detected on some paired-end reads, and the SNP on one haplotype is A and on the other haplotype is T. If the sSNV is only detected to be linked with A, then we could infer that the sSNV originates from one haplotype. If the sSNV is detected to be linked with A and T, then the sSNV originates from both haplotypes.
Optimization model to estimate the fractions of subclones
Suppose the tumors contain  subclones. Let
subclones. Let 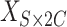 be the subclone matrix with the binary elements
be the subclone matrix with the binary elements  and
and 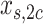 denoting whether
denoting whether 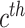 subclone contains the
subclone contains the 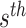 sSNV in its two haplotypes where
sSNV in its two haplotypes where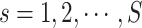 and
and 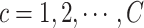 . Denote
. Denote 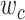 as the fraction of subclone
as the fraction of subclone 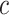 , then we have
, then we have
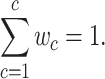 |
(1) |
Next, we can infer the true VAFs of sSNVs by the subclone matrix  and their fraction
and their fraction 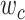
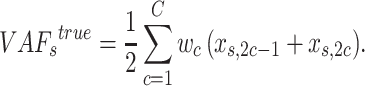 |
(2) |
However, the observed VAF is influenced by the sequencing depth: as the sequencing depth increases, the variance of observed VAF decreases. If the depth is large enough, then we could assume that the observed VAF is nearly the same as true VAF. According to this assumption, given the sequencing depth of the 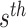 sSNV (termed
sSNV (termed 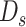 ), we minimize the difference between true and observed VAF
), we minimize the difference between true and observed VAF
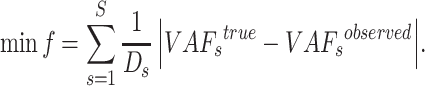 |
(3) |
Considering the sSNV–SNP linkages in the model, for the sSNV 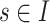 ,
, 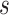 should occur in no more than one haplotype from each subclone, then
should occur in no more than one haplotype from each subclone, then
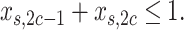 |
(4) |
For 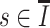 ,
, 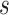 should occur on two haplotypes at least in one subclone, then we could get
should occur on two haplotypes at least in one subclone, then we could get
 |
(5) |
Therefore, we can construct an optimization model to obtain the feasible solution,
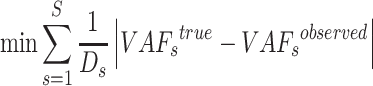 |
(6) |
subject to
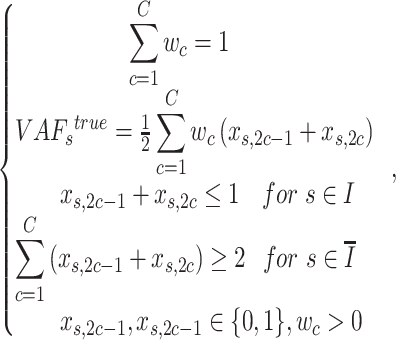 |
where set 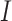 is for the sSNVs that originated from only one haplotype, and
is for the sSNVs that originated from only one haplotype, and 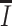 for the ones that originated from both. If all subclones can be inferred from the set
for the ones that originated from both. If all subclones can be inferred from the set 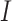 , then let
, then let  and we could obtain a simplified model
and we could obtain a simplified model
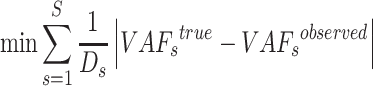 |
(7) |
subject to
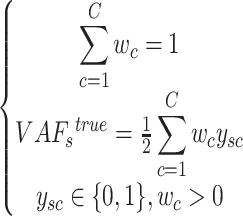 |
where 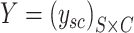 is a binary matrix.
is a binary matrix. 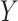 is similar to
is similar to  and thus can also be referred to a subclone matrix. Then, the diploid inference is converted to haplotype inference, which is the novelty of the model. The sSNV–SNP linkage information could obtain the sets
and thus can also be referred to a subclone matrix. Then, the diploid inference is converted to haplotype inference, which is the novelty of the model. The sSNV–SNP linkage information could obtain the sets 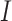 and
and  and we only use the sSNVs from set
and we only use the sSNVs from set 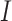 to simplify the calculation in the next section.
to simplify the calculation in the next section.
Solve the model by Monte Carlo optimization
For the simplified model, since 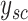 is an integer and
is an integer and 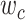 is a positive real number, the optimization problem is a mixed-integer problem. Mixed-integer linear programming (MILP) problems are NP-hard to solve [27, 28]. In our model, the constraints are quadratic, which makes the problem more difficult. Here we apply the Monte Carlo optimization method to solve the model by fixing the number of subclones:
is a positive real number, the optimization problem is a mixed-integer problem. Mixed-integer linear programming (MILP) problems are NP-hard to solve [27, 28]. In our model, the constraints are quadratic, which makes the problem more difficult. Here we apply the Monte Carlo optimization method to solve the model by fixing the number of subclones:
Step 1: Initialization. Given the maximal number of subclones 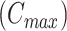 and let
and let 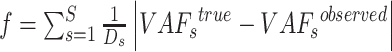 ,
, 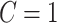 , then
, then 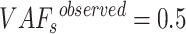 .
.
Step 2: Update 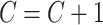 ;. Randomly generate
;. Randomly generate 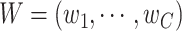 for 10 000 times from uniform distribution
for 10 000 times from uniform distribution  and normalize them by
and normalize them by 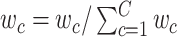 .
.
Step 3: For each 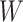 , calculate all the sums of elements from
, calculate all the sums of elements from  denoting the all subsets of W, and then assign
denoting the all subsets of W, and then assign 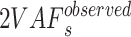 to the nearest sums to obtain
to the nearest sums to obtain 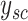 and further
and further 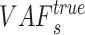 . The objective function
. The objective function  for
for 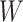 is also calculated.
is also calculated.
Step 4: Obtaining 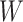 by minimizing all
by minimizing all  , if
, if 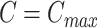 , stop; otherwise, go to Step 2.
, stop; otherwise, go to Step 2.
Usually, we set the 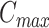 as 10 in the calculation. If needed, the user can modify
as 10 in the calculation. If needed, the user can modify 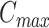 when many subclones are expected.
when many subclones are expected.
Subclones inference by balancing model goodness and the numbers of subclones
As the number of subclones is an important parameter in the model, we need to evaluate the goodness of the model to find the optimal number of subclones. Suppose the number of fragments is large enough in the sequencing library construction and then 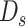 fragments are randomly selected for
fragments are randomly selected for 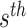 sSNV where
sSNV where 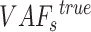 of these fragments contains alternative allele. Assume that this random process follows a binomial distribution, we have
of these fragments contains alternative allele. Assume that this random process follows a binomial distribution, we have
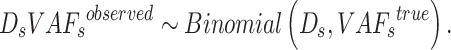 |
Furthermore, we estimate the standard variance of 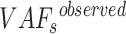 as
as
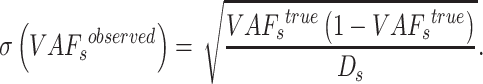 |
(8) |
The true VAF is not larger than 0.5, so we use the maximal  and the median
and the median 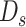 to obtain an estimation of
to obtain an estimation of 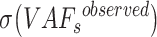 .
.
Then we define the goodness for a given number of subclones 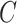 as
as
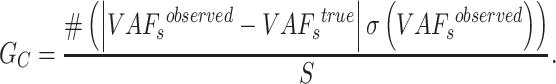 |
(9) |
The term  can be calculated for each
can be calculated for each  for
for 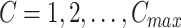 in the Monte Carlo optimization and the optimal number of subclones is selected by balancing the goodness and the number of subclones. By default, we select the number of subclones
in the Monte Carlo optimization and the optimal number of subclones is selected by balancing the goodness and the number of subclones. By default, we select the number of subclones 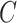 if
if 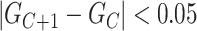 .
.
Implementation of GenoClone
As illustrated in Figure 1, the alignments (‘bam’ files) of the sequencing data of tumor and the matched normal samples are entered. Firstly, the somatic mutations (sSNVs) and germline mutations (SNPs) are called by VarScan2 [26]. Secondly, GenoClone uses the paired-end short reads or long reads sequencing to detect the sSNV–SNP linkages for the genotype of sSNVs. Simultaneously, GenoClone calculates VAFs of all sSNVs via short reads. Thirdly, GenoClone integrates the VAFs and their genotypes into the above optimization model that is solved by the Monte Carlo optimization.
Data set
The diversity of intra-tumor subclones in breast cancer is associated with cancer progression and therapeutic resistance (6). We analyzed 389 breast invasive carcinoma samples from The Cancer Genome Atlas (TCGA) by GenoClone (Supplementary Table S1). For each sample, the exome sequencing data from tumor and the matched normal were used. In addition, the exome sequencing data of different aliquots from two TCGA-BRCA samples (TCGA-A7-A13E and TCGA-A7-A13D) were also used to study the heterogeneity among different aliquots.
Results
Subclone inference by GenoClone
We applied GenoClone to a simulation data to evaluate the performance by the following three metrics: (i) correct identification of the number of subclones, (ii) the fractions of subclones and (iii) accuracy (i.e. the proportion of sSNVs correctly identified in each subclones). The simulation data contained four subclones with different fractions (0.05, 0.15, 0.35 and 0.45) and all sSNV-SNP linkages were known (see Supplementary Materials, Supplementary Figure S1). We examined the difference between true and observed VAFs and the goodness (see definition of ‘goodness’ in Method) with respect to the number of subclones (Figure 2). As the number of subclones increased from one to four, the difference between true and observed VAFs decreased dramatically (Figure 2A) and the goodness increased significantly (Figure 2B). Until the number of subclones was increased from four to five, the improvement of the goodness was not smaller than 0.05. Therefore, GenoClone identified four subclones that matched exactly to the simulation. The estimated fractions of subclones were 0.0748, 0.1418, 0.3427 and 0.4406, which were very close to the true values (Figure 3A). The accuracy of each subclone identified by GenoClone were 68.18%, 69.70%, 78.79% and 84.85%, respectively (Figure 3B). The subclones with low fractions (e.g. 0.05 and 0.15) showed relatively poorer accuracy because the low fractions might be sensitive to the process of solving the optimization problem in GenoClone (Supplementary Figure S2 and see Methods).
Figure 2.

Subclone inference by examining the difference of true and observed VAFs and goodness in the simulation. As the number of subclones increases, (A) the difference of true and observed VAFs decreased and (B) the goodness increases. The changes are significant until the number of subclones increases to four.
Figure 3.

Comparison of GenoClone, PyClone and SciClone in the simulation. (A) The black bar represents the true fractions of the four subclones. The fractions of the subclones inferred by GenoClone (red bars) are closer to the true ones than PyClone (green bars) and SciClone (blue bars). (B) The accuracy of sSNV composition identification of each subclone by GenoClone, SciClone and Pyclone. GenoClone showed the highest accuracy except the Subclone 1.
Influence of sSNV–SNP linkage ratio for subclone inference
In the simulation above, we assumed all sSNV–SNP linkage were known, while we often have only a subset of sSNVs linked with SNPs in real data, as the other sSNVs are far from SNPs. To investigate the influence of sSNV–SNP linkages for subclone inference, we define linkage ratio as the fraction of sSNVs linked with SNPs. Given a linkage ratio, we repeated 1000 simulations. We tested different linkage ratios from 20% to 90% (see Supplementary Materials). For the linkage ratio of 20%, only 1.2% tests failed to find the correct numbers of subclones. Moreover, the estimated fractions of subclones were close to the true values (Figure 4). As the linkage ratio increased, the standard deviation of the estimated fractions decreased. Interestingly, the most significant improvement occurred as the linkage ratio increased from 20% to 40% and then the improvement became marginal. Therefore, more linkage information were helpful to estimate subclone fractions with less variance, while the linkage ratio of 40% rather than 100% may be high enough to generate the optimal output.
Figure 4.

Subclone fraction estimation with different sSNV–SNP linkage ratios in the simulation. The true fractions of the four subclones are 0.05, 0.15, 0.35 and 0.45 (dashed lines). The fractions estimated by GenoClone with different sSNV-SNP linkage ratios are close to the true values. As the linkage ratio increases, the variance of fraction estimation decreases.
Comparisons with PyClone and SciClone
Furthermore, we compared the performance of GenoClone with the existing tools PyClone and SciClone on the same simulation data (see Supplementary Materials) [19, 21]. PyClone inferred the cellular abundance for each sSNVs, based on which sSNVs were clustered. Pyclone yielded six clusters of sSNVs with their centroids of abundance at 0.0666, 0.2041, 0.3714, 0.2558, 0.5498 and 0.4721 (Supplementary Figure S3). Therefore, PyClone was not able to predict the number of subclones correctly and thus was also not able to estimate the fractions accurately. We tried to convert six clusters of sSNVs to four subclones manually by considering the size of cluster and their abundance and the fractions of four subclones could be obtained as 0.0666, 0.2041, 0.3579 and 0.3714 (see Supplementary Materials, Figure 3A). Except Subclone 1, the fraction estimations of the other subclones by PyClone were of much higher errors than GenoClone. The similar result was also observed in the comparison of the accuracy between GenoClone and PyClone (Figure 3B). Overall, GenoClone showed better performance of predicting the number of subclones and estimating of the fractions of subclones, as well as more accurate identification of sSNV composition in each subclone.
SciClone yielded four clusters of sSNVs and their fractions were estimated as 0.0382, 0.2148, 0.3666 and 0.3804 (Figure 3A). The average estimation error of subclone fractions by GenoClone was much smaller than SciClone (0.0124 versus 0.0407). In addition, the averaged accuracy of sSNV composition in each subclone predicted by SciClone (51.5%) was much lower than GenoClone (75.2%) (Figure 3B). Although SciClone predicted the same number of subclones, GenoClone showed advantages in estimating the fractions of subclones and identifying sSNV composition of each subclone.
Subclone inference of breast cancer
GenoClone detected two subclones in nearly all (388) the samples (Figure 5A), while the only outlier contained one subclone. Therefore, intra-tumor heterogeneity existed extensively in breast cancer. However, the goodness of 11.31% samples (44/389) was smaller than 0.7, which inferred the possible underestimation of the numbers of subclones. It may be caused by the lack of enough sSNV–SNP linkages (P-value = 7.75e-9, Student’s t test, Supplementary Figure S4). Thus, we defined that these samples contained two or more subclones (termed Group 1). In addition, 42.93% samples (167/389) with goodness higher than 0.85 were defined to contain exactly two subclones (termed Group 2). According to American Joint Committee on Cancer (AJCC), higher proportion of the samples in Group 1 was at Stage I than Group 2, while Group 1 was slightly lower than Group 2 at Stage III (Supplementary Figure S5). Therefore, the samples containing more subclones (i.e. Group 1) may increase the probability of obtaining metastasis at the early stage, while at the late stage the samples with less subclones (i.e. Group 2) may dominate to expose. We used 167 samples of Group 2 in the analysis below as they had higher goodness.
Figure 5.

Subclone inference of 389 TCGA-BRCA samples. (A) The histogram of the goodness for 389 TCGA-BRCA samples. The goodness of about half of the samples [167 (42.93%) of 389] is greater than 0.85. (B) The mean and variance of true and observed VAFs for 167 samples. The red box highlights the samples with mean < 0.045 and variance < 0.0015.
Subclones inference with non-SNP-linked sSNVs
While the SNP-linked sSNVs were informative for inferring subclones, the non-SNP-linked sSNVs may be important to study tumorigenesis and tumor evolution. Therefore, the non-SNP-linked sSNVs should be also considered in the analysis. We assigned the non-SNP-linked sSNVs to each subclone according to the difference between the fractions of subclones and the corresponding observed VAFs (see Supplementary Materials). After including both types of sSNVs in the analysis by GenoClone, the goodness of each sample was still higher than 0.85, except that two samples went down to 0.84 and 0.83 (Supplementary Figure S6A). Furthermore, in 155 samples, the average of the difference between true and observed VAFs was smaller than 0.045 and the variance was smaller than 0.0015 (Figure 5B). For example, the distribution of  of the tumor TCGA-EW-A1P7 centered at zero (Supplementary Figure S6B). Thus, GenoClone showed consistent and unbiased results when including non-SNP-linked sSNVs. Both SNP-linked and non-SNP-linked sSNVs were included in the analysis below to investigate the role of sSNVs in tumor evolution.
of the tumor TCGA-EW-A1P7 centered at zero (Supplementary Figure S6B). Thus, GenoClone showed consistent and unbiased results when including non-SNP-linked sSNVs. Both SNP-linked and non-SNP-linked sSNVs were included in the analysis below to investigate the role of sSNVs in tumor evolution.
sSNVs in tumor evolution of breast cancer
A total of 39 604 sSNVs were detected from 167 breast cancer samples (Supplementary Table S2). For each sample, the sSNVs that occurred in all subclones were defined as early mutants, and the sSNVs that occurred in only one subclone were defined as late mutants. Next, we studied the correlation between sSNVs and the tumor evolution of breast cancer. For example, the sSNV chr3_179234297_AG located at PIK3CA that had been shown as an oncogene in ovarian cancer [29] and high mutation frequency at PIK3CA was also found in breast cancer [30, 31]. This sSNV occurred in 17 samples and occurred in only one subclone in most samples (16 of 17). Therefore, it may likely emerge in the late stage of tumor evolution. In addition, another sSNV of PIK3CA, chr3_179218303_GA, was similar as it also occurred in one subclone of 11 of 12 samples containing it. These evidences implied that mutations of PIK3CA occurred at the late stage of tumor progression, which was consistent with the recent studies in human cancers [32, 33]. Another sSNV, chr10_26224072_GT, occurred in both subclones of three samples while a total of seven samples contained it. Thus, this sSNV might be the early mutant so it is associated with breast cancer occurrence. More interestingly, 24 sSNVs were found at TP53, which is a well-known tumor suppressor gene and driver gene in many types of human tumors [34, 35], including four sSNVs that occurred at both subclones. It showed a possibility that the majority of TP53 mutations occurred at the late stage of tumor evolution, while a few may occur at the early stage (Supplementary Table S3).
Similarities of sSNVs composition among subclones
To investigate the biological significance of subclones, we studied their similarities of sSNVs composition by Fisher’s exact test (see Supplementary Materials). In the majority [123 (73.6%) of 167] of the samples, significant similarity (P-value < 0.01) was found between two subclones. However, two subclones were different (P-value > 0.1) in 15.5% (23/167) of the samples (Supplementary Figure S7), which was likely due to significant difference of cancer progression in these samples. The difference existed in two subclones of the same sample demonstrated the importance of studying the tumor heterogeneity within tumors.
Next, we studied the similarities among subclones from all the samples by clustering analysis (Figure 6A). Although 97.6% pairs of subclones were different (P-value > 0.1), a cluster of 24 subclones with significant similarities were found (black solid-line box in Figure 6A and B). Within this cluster, the most common sSNV chr2_55679588_CA occurred in 10 subclones, followed by chr7_92040995_GT and chr6_83237608_CA occurring in eight subclones, chr19_16483788_TG, chr3_138269901_CA, chr2_169891311_CT, chr10_26224072_GT and chr10_32932369_CA in seven subclones, etc. Moreover, the panel of sSNVs chr2_55679588_CA, chr6_83237608_CA, chr19_16483788_TG and chr3_138269901_CA occurred in 83.33% (20/24) of subclones. Furthermore, all the subclones were not metastasis and most were Her2 negative (83.3%) and ER positive (70.8%) [31]. These results from the 24-subclone cluster provided useful information for the research of cancer progression and precision medicine.
Figure 6.

Similarities among the subclones from 167 TCGA-BRCA samples. (A) The heatmap of similarity of mutual subclone pairs identified from 167 TCGA-BRCA samples. The P-value of each pair of subclones was calculated by the Fisher’s exact test. The biggest cluster is highlighted by the black solid-line box and the other two clusters are highlighted by the white dashed-line box. (B) The sSNVs in the biggest clusters. The sSNVs occurred at least four times are shown. Chr2_55679588_CA occurs in 10 samples. Chr2_55679588_CA represents the sSNV at chromosome 2 and its position is 55679588 with reference C to A.
In addition to the cluster above, we found two smaller clusters (white dashed-line box in Figure 6A). An sSNV at PIK3CA, ch3_17923497_AG, occurred in all subclones of the first cluster of 14 subclones (Supplementary Figure S8A). Similarly, the other sSNV at PIK3CA, ch3_179218303_GA, occurred in all subclones of the second cluster of 14 subclones (Supplementary Figure S8B). Thus, the two sSNVs could be target driver mutations for these subclones.
Subclone inference in aliquots of tumors
In order to further analyze the intra-tumor heterogeneity of breast cancer sample, we applied GenoClone to three aliquots of the tumor TCGA-A7-A13E. At least three subclones were found in each aliquot (Figure 7). All three subclones in the second vial (01B-06D-A272-09) did not contain the sSNV chr17_7675088_CT, which is located at TP53 (Figure 7A) and was shared by two aliquots of the first vial (01A-11D-A272-09 and 01A-11D-A12Q-09). This implied the heterogeneity between different vials of this sample. More interestingly, significant difference of the subclones was also detected even within the same vial of the sample. In the first vial, four subclones were found in the aliquot 01A-11D-A12Q-09 (Figure 7C), while three subclones in the other aliquot 01A-11D-A272-09 (Figure 7B). Furthermore, we found that the VAF of chr17_7675088_CT was 0.2051 in the 01A-11D-A272-09 and was increased to 0.3113 in 01A-11D-A12Q-09. Therefore, the aliquot 01A-11D-A12Q-09 might occur in a later phase of tumor evolution than 01A-11D-A272-09.
Figure 7.

Subclone inference from three aliquots of the tumor TCGA-A7-A13E by examining the difference of true and observed VAFs and goodness. (A) The second vial ‘B’ and both (B) and (C) are from the first vial ‘A’. Three subclones are predicted in both (A) and (B) and four subclones in (C). When the number of subclones increase from three to four, the improvement of goodness of (B) and (C) are 0.039 and 0.069, individually.
In addition to TCGA-A7-A13E, we applied GenoClone to another tumor TCGA-A7-A13D containing two aliquots (01A-13D-A272-09 and 01A-13D-A12Q-09) from the same vial. Similarly, different numbers of subclones were found in the two aliquots (Supplementary Figure S9). The sSNV chr17_7674870_CA at TP53 was detected in both aliquots. Moreover, the aliquot 01A-13D-A272-09 that contained less subclones (Supplementary Figure S9A) had smaller VAF of this sSNV (0.5882) than the other aliquot 01A-13D-A12Q-09 (VAF = 0.7307, Supplementary Figure S9B). If the sSNV chr17_7674870_CA at TP53 correlates with tumor evolution, then the aliquot 01A-13D-A12Q-09 may emerge in a later evolution stage than the other aliquot.
Discussion
Deciphering tumor heterogeneity is very helpful for understanding the causes and consequences of tumor evolution [9] and finally assists therapeutic treatment and personal medicine [36, 37]. Recent studies identified subclones by VAFs without utilizing genotype information [19, 21]. GenoClone integrates the genotype of sSNVs and VAFs in an optimization model innovatively, so as to achieve better characterization of tumor heterogeneity. Compared to the existing methods, GenoClone showed superior performance of inferring the number of subclones, estimating subclone fractions and identifying sSNVs composition of each subclone. If the fraction of subclones is equal, GenoClone could infer the number of subclones and their fractions but nearly impossible to assign sSNVs to each subclone (Supplementary Table S4). PyClone and SciClone also obtained similar result. This situation may be solved by single-cell sequencing. Therefore, the re-analysis of the publicly available data by GenoClone may provide more accurate understanding of heterogeneity of the samples, as paired-end reads can detect the genotype of sSNVs. By GenoClone, we showed the extensive intra-tumor heterogeneity of 389 breast cancer samples in TCGA. Further analysis also revealed the possible roles of a few sSNVs in tumor evolutions, especially the ones at the oncogene PIK3CA and the tumor suppressor gene TP53. Interestingly, similarity analysis of sSNVs composition among all subclones found from 167 TCGA-BRCA samples identified three subclone clusters, which contained a panel of sSNVs. These results may further benefit the treatment and drug design for large population. In addition, the heterogeneity in different aliquots of the same samples showed that the subclones did not uniformly distribute within the tumor. Therefore, in order to obtain a better understanding of tumor evolution, we needed to study the heterogeneity in the whole tumor rather than a fraction of sample.
GenoClone used the variant calling results from the existing tool VarScan2, which outputs the sSNVs with VAFs larger than a default threshold. Thus, GenoClone only identifies the large subclones with VAFs above the threshold. Small subclones can be detected if the threshold of VAF is relaxed. When we analyzed 389 TCGA-BRCA samples by GenoClone, two subclones with reasonably high VAFs were found in most samples because of this restriction. This result still demonstrated that tumor heterogeneity is widespread.
Because GenoClone utilizes sSNV-SNP linkage, it reduces the uncertainty of the genotype of sSNVs. As the sSNV–SNP linkage ratio increases, the estimation variance of subclone fractions decreases. However, GenoClone could also handle the low-coverage data by minimizing the difference of true and observed VAFs to reduce the influence of the variance of VAFs. While low sequencing coverage and the long distance between some sSNVs and the adjacent SNPs may result in low ratios of sSNV–SNP linkages, GenoClone showed robust performance in different linkage ratios. As a user-friendly tool, GenoClone defined the model goodness to evaluate the reliability of subclone inference so that the results can be used and interpreted appropriately in the downstream analysis. In addition, like PyClone, GenoClone assumes that sSNVs occur and remain in the descendant cells, while GenoClone does not require that sSNVs occur only once. As copy number variation (CNV) is also important for subclone formation and evolution, the integration of CNV data to GenoClone may obtain more accurate subclone inference in the future study. We downloaded the CNVs of the matched samples from TCGA and found no significant difference between GenoClone output with and without removing the sSNVs from CNV regions (Supplementary Figure S10). The heterogeneity may exist in different aliquots of the same sample, so the CNVs data may not be helpful in the subclone inference from exome sequencing data. Moreover, GenoClone may use the linkage information to distinguish the homozygous and heterozygous CNVs. GenoClone can be applied to both exome and whole-genome sequencing data. Both paired-end reads (e.g. Illumina) and Third Generation Sequencing long reads (e.g. Pacbio and Oxford Nanopore Technologies) can be used.
Key Points
We develop the bioinformatics tool GenoClone that first integrates the genotype of sSNVs and VAF to infer subclones and tumor heterogeneity.
We showed superior performance of GenoClone over the existing tools in three ways: (i) infer the number of subclones correctly, (ii) estimate subclone fractions accurately and (iii) identify sSNVs composition of each subclone accurately.
By GenoClone, we analyzed 389 TCGA breast cancer samples and showed the widespread heterogeneity of the samples. The further analysis revealed the possible roles of a few sSNVs in tumor evolution, including the ones at the oncogene PIK3CA and tumor suppressor gene TP53.
We discovered three significant clusters of subclones that were identified from different TCGA breast cancer samples, which would benefit the research of common variants in breast cancer and the following studies of precision treatment.
We also showed the heterogeneity among different aliquots of the same samples.
Supplementary Material
Acknowledgements
We thank Donghai Dai, MD, PhD, at the University of Iowa Carver College of Medicine, for discussion.
Meng Zou is currently an instructor at the School of Mathematics and Statistics, Huazhong University of Science and Technology. He was a postdoctoral scholar at the Department of Internal Medicine, University of Iowa.
Rui Jin is a PhD student at the Department of Statistics, University of Iowa.
Kin Fai Au is currently an associate professor at the Department of Biomedical Informatics, Ohio State University. His research focuses on the method development of Third Generation Sequencing. He was an associate professor at the Department of Internal Medicine, University of Iowa.
Funding
National Human Genome Research Institute [R01HG008759 to K.F.A and M.Z.]; Department of Internal Medicine, University of Iowa institutional fund to K.F.A. and M.Z.; Department of Biomedical informatics, Ohio State University institutional fund to K.F.A.
References
- 1. Nowell PC. The clonal evolution of tumor cell populations. Science 1976;194(4260):23–8. [DOI] [PubMed] [Google Scholar]
- 2. Greaves M, Maley CC. Clonal evolution in cancer. Nature 2012;481(7381):306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 2012;12(5):323–34. [DOI] [PubMed] [Google Scholar]
- 4. Bedard PL, Hansen AR, Ratain MJ, et al. Tumour heterogeneity in the clinic. Nature 2013;501(7467):355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Szöllösi J, Balázs M, Feuerstein BG, et al. ERBB-2 (HER2/neu) gene copy number, p185HER-2 overexpression, and intratumor heterogeneity in human breast cancer. Cancer Res 1995;55(22):5400–7. [PubMed] [Google Scholar]
- 6. Polyak K. Heterogeneity in breast cancer. J Clin Invest 2011;121(10):3786–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koren S, Bentires-Alj M. Breast tumor heterogeneity: source of fitness, hurdle for therapy. Mol Cell 2015;60(4):537–46. [DOI] [PubMed] [Google Scholar]
- 8. Zardavas D, Baselga J, Piccart M. Emerging targeted agents in metastatic breast cancer. Nat Rev Clin Oncol 2013;10(4):191–210. [DOI] [PubMed] [Google Scholar]
- 9. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochimica et Biophysica Acta (BBA)-Reviews on. Cancer 2010;1805(1):105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet 2010;11(1):31–46. [DOI] [PubMed] [Google Scholar]
- 11. Koboldt Daniel C, Steinberg Karyn M, Larson David E, et al. The next-generation sequencing revolution and its impact on genomics. Cell 2013;155(1):27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol 2008;26(10):1135–45. [DOI] [PubMed] [Google Scholar]
- 13. Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature 2011;472(7341):90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19(5):455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ning L, Liu G, Li G, et al. Current challenges in the bioinformatics of single cell genomics. Front Oncol 2014;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hou Y, Song L, Zhu P, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 2012;148(5):873–85. [DOI] [PubMed] [Google Scholar]
- 17. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012;481(7382):506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nik-Zainal S, Van Loo P, Wedge DC, et al. The life history of 21 breast cancers. Cell 2012;149(5):994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roth A, Khattra J, Yap D, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods 2014;11(4):396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hajirasouliha I, Mahmoody A, Raphael BJ. A combinatorial approach for analyzing intra-tumor heterogeneity from high-throughput sequencing data. Bioinformatics 2014;30(12):i78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miller CA, White BS, Dees ND, et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol 2014;10(8):e1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee J, Müller P, Sengupta S, et al. Bayesian feature allocation models for tumor heterogeneity. In: Statistical Analysis for High-Dimensional Data. Cham: Springer, 2016, 211–32. [Google Scholar]
- 23. Lo YD, Chan KA, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2(61):61ra91. [DOI] [PubMed] [Google Scholar]
- 24. El-Maarri O, Seoud M, Coullin P, et al. Maternal alleles acquiring paternal methylation patterns in biparental complete hydatidiform moles. Hum Mol Genet 2003;12(12):1405–13. [DOI] [PubMed] [Google Scholar]
- 25. Marshall T, Slate J, Kruuk L, et al. Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol 1998;7(5):639–55. [DOI] [PubMed] [Google Scholar]
- 26. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22(3):568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chakrabarty K. Test scheduling for core-based systems using mixed-integer linear programming. IEEE Trans Comput Des Integr Circuits Syst 2000;19(10):1163–74. [Google Scholar]
- 28. Li D, Sun X. Nonlinear Integer Programming, Vol. 84 New York: Springer Science & Business Media, 2006. [Google Scholar]
- 29. Shayesteh L, Lu Y, Kuo W-L, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet 1999;21(1):99–102. [DOI] [PubMed] [Google Scholar]
- 30. Bachman KE, Argani P, Samuels Y, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther 2004;3(8):772–5. [DOI] [PubMed] [Google Scholar]
- 31. Network CGA. Comprehensive molecular portraits of human breast tumours. Nature 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304(5670):554–4. [DOI] [PubMed] [Google Scholar]
- 33. Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004;3(10):1221–4. [DOI] [PubMed] [Google Scholar]
- 34. Lowe SW, Schmitt EM, Smith SW, et al. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993;362(6423):847–9. [DOI] [PubMed] [Google Scholar]
- 35. Nigro JM, Baker SJ, Preisinger AC, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989;342(6250):705–8. [DOI] [PubMed] [Google Scholar]
- 36. Heppner GH, Miller BE. Tumor heterogeneity: biological implications and therapeutic consequences. Cancer and Metastasis Rev 1983;2(1):5–23. [DOI] [PubMed] [Google Scholar]
- 37. Longo DL. Tumor heterogeneity and personalized medicine. N Engl J Med 2012;366(10):956–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.