


Abstract
IRF3, IRF5 and IRF9 are transcription factors, which play distinct roles in the regulation of antiviral and inflammatory responses. The determinants that mediate IRF-specific enhancer selection are not fully understood. To uncover regions occupied predominantly by IRF3, IRF5 or IRF9, we performed ChIP-seq experiments in activated murine dendritic cells. The identified regions were analysed with respect to the enrichment of DNA motifs, the interferon-stimulated response element (ISRE) and ISRE half-site variants, and chromatin accessibility. Using a machine learning method, we investigated the predictability of IRF-dominance. We found that IRF5-dominant regions differed fundamentally from the IRF3- and IRF9-dominant regions: ISREs were rare, while the NFKB motif and special ISRE half-sites, such as 5′-GAGA-3′ and 5′-GACA-3′, were enriched. IRF3- and IRF9-dominant regions were characterized by the enriched ISRE motif and lower frequency of accessible chromatin. Enrichment analysis and the machine learning method uncovered the features that favour IRF3 or IRF9 dominancy (e.g. a tripartite form of ISRE and motifs for NF-κB for IRF3, and the GAS motif and certain ISRE variants for IRF9). This study contributes to our understanding of how IRF members, which bind overlapping sets of DNA sequences, can initiate signal-dependent responses without activating superfluous or harmful programmes.
INTRODUCTION
The interferon regulatory factor (IRF) family is comprised of nine members (IRF1–IRF9) in mammals (1). IRFs play important roles, not only in interferon (IFN) induction, but also in cell development, cell-intrinsic antiviral responses, inflammation, and oncogenesis (1,2). Within the IRF family, IRF3, IRF5 and IRF9 have been identified as key regulators of various antiviral and inflammatory responses (1,2). Upon stimulation by specific pathways, IRF3 and IRF5 undergo posttranslational modifications (mainly phosphorylation), resulting in activation, nuclear translocation, dimerization or complex formation (1,3). IRF3 and IRF5 are phosphorylated by protein kinases, which are activated by signalling pathways of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) that signal via TRIF (TLR3 and TLR4) and MyD88 (e.g. TLR7 and TLR9), respectively (1,4). The binding of type I IFNs to their receptors results in the activation of a heterotrimeric transcriptional activator known as IFN-stimulated gene factor 3 (ISGF3), which consists of IRF9 and signal transducer and activator of transcription 1 (STAT1) and STAT2 (1,5). In addition to the canonical ISGF3, complexes containing IRF9 and either STAT1 or STAT2, but not both, also control gene expression (6,7). Notably, the IRF association domain (IAD) of IRF9 lacks the autoinhibitory element, explaining previous notions that activation by signal-induced phosphorylation may not be necessary for association of IRF9 with STAT2 (8,9). However, an early study suggested that IRF9 could be phosphorylated constitutively within the DNA-binding domain (DBD) in the absence of IFN stimuli (10).
IRF3, IRF5 and IRF9 regulate overlapping but distinct sets of target genes. IRF3 induces the production of many antiviral cytokines, including IFN-β, CCL5, CXCL9 and CXCL10 (1,11,12). IRF5 is involved in inflammatory responses, as demonstrated by impaired inflammatory cytokine production in Irf5-deficient mice (13,14). Besides these prominent roles, IRF3 and IRF5 contribute to other biological processes as well (15–18). In the ISGF3 complex, IRF9 is an essential mediator of IFN signalling and a transcriptional regulator of IFN-stimulated genes (ISG) involved in the establishment of the so-called antiviral state (19–21). Notably, a subset of ISGs, such as Ifit1 and Isg15, is also regulated directly by the TLR3 pathway via IRF3 (12,22). IRFs frequently co-bind with other TFs at the promoter regions or at distal enhancers of the regulated genes. The co-binding of IRFs and members of AP-1 and NF-κB families has been well documented (23,24). Human IFNB activation by Sendai virus infection demonstrates the central role of co-operative binding and synergy between NF-κB, IRF3/IRF7 and ATF-2/c-Jun in selective transcriptional activation (25,26). ChIP-seq analysis revealed an extensive collaboration of IRF3 and RelA in the antiviral responses (27). During viral infection, IRF3 and NF-κB drive both de novo polymerase recruitment and mediate the release of paused Pol II at their target sites (27). IRF5 binding also co-occurs frequently with RelA binding, at the promoter of genes that are strongly induced by LPS in macrophages (28).
Selectivity in gene activation is a well-documented phenomenon for IRFs, and gene-targeting studies performed on IRFs have revealed the markedly diverse roles played by these transcription factors (TFs) (29). Selective gene activation is critical for limiting potential superfluous or harmful transcriptional events. For example, after activation by inflammatory agents, IRF5 induces inflammatory cytokines, without activating antiviral ISGs. Similarly, type I IFN-activated ISGF3 establishes an antiviral state without activating the production of type I IFNs, because this would lead to an ‘IFN storm’ (30).
Dimers or trimers formed by IRF3, IRF5 and IRF9 regulate gene expression via indirect mechanisms or direct DNA binding (1). The indirect mechanisms and their relative contribution to gene regulation are not completely understood. In contrast, the mechanism of direct DNA binding and IRF-bound DNA sequences have been extensively investigated using protein binding microarrays (PBM), electrophoretic mobility shift assays (EMSA), and protein crystallization methods (26,31–33). DNA motifs, which are enriched in the binding regions, have been identified by ChIP-seq for many IRFs (11,27,28,34,35). The canonical binding sequence for IRF dimers is called the interferon-stimulated response element (ISRE, 5′-GAAANNGAAA-3′) (29,36–38). ISREs are occupied by IRF homo- or heterodimers, or by the ISGF3 complex, while a single molecule of IRF3, IRF5 and IRF9 binds to the ISRE half-site (5′-GAAA-3′).
Many DNA sequences have been identified, which are bound more efficiently by one IRF than another. Bases, which have been associated with IRF-specific binding, are localized in the 4-bp ISRE half-sites, in the 2-bp spacer between half-sites, or in the 5′ and 3′ flanking regions (31–33). The existence of shared and IRF-specific binding sequences provides a potential mechanism for the IRFs to regulate both common and dimer-specific genes (33). However, the contribution of these sequences to IRF-specific binding has not been evaluated by genome-wide ChIP-seq studies. In the current study, our starting assumption was that DNA sequences that play a role in enhancer selection by these IRFs and their dimerization and trimerization partners are enriched in the binding regions occupied dominantly by IRF3, IRF5 or IRF9. We also postulated that if chromatin accessibility and collaborating TFs contribute to IRF-specific binding, the binding regions would differ from each other in terms of chromatin status and the presence of co-binding TF DNA motifs. To test these hypotheses and to determine which features contribute to IRF-specific binding, we identified and characterized binding regions for IRF3, IRF5 and IRF9 in stimulated dendritic cells (DCs). Our analysis indicates that IRF-specificity in enhancer selection is mediated by DNA sequences occupied directly by IRFs, and additional features in a combinatorial fashion.
MATERIALS AND METHODS
Cells and ligands
A wild-type CD8+ dendritic cell line (MuTu1940) was established and cultured as described previously (22,39). For ChIP-seq and ChIP-qPCR experiments, CD8+ dendritic cells (DCs) were treated for 90 min with 5 μg/ml of high molecular weight polyinosinic–polycytidylic acid (pIC, InvivoGen), 1 mM class B CpG oligonucleotide 1826 (CpG, InvivoGen) or 100 U/ml interferon-β (IFN-β, Millipore) or a combination of these ligands. The binding sites of IRF3, IRF5 and IRF9 were determined in DCs stimulated by pIC, CpG and IFN-β, respectively. The binding of cRel and Junb was determined in CpG-stimulated and unstimulated DCs. For gene expression qPCR experiments, CD8+ DCs were treated for 1.5, 3, 6 or 12 h with 5 μg/ml pIC, 1 mM CpG or 100 U/ml IFN-β.
ChIP-seq and ChIP-qPCR
Chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) experiments were performed as previously described (40–42) with minor modifications. Briefly, 10–20 million DCs were crosslinked with 2 mM disuccinimidyl glutarate (DSG, ProteoChem) for 40 min and 1% methanol-free formaldehyde (Thermo Fisher Scientific) for 10 min. Cross-linking was stopped by the addition of glycine to a final concentration of 0.125 M for 10 min followed by addition of ChIP lysis buffer containing 150 mM NaCl, 1 mM EDTA pH 8, 20 mM Tris–HCl pH 8, 1% Triton X-100, 0.1% SDS with protease inhibitors (cOmplete Mini EDTA-free protease inhibitor cocktail, Roche). Chromatin was sheared with sonication (Diagenode Bioruptor Standard) and immunoprecipitated overnight using antibodies against IRF3 (D83B9, Cell Signaling), IRF5 (ab21689, Abcam), IRF9 (AF5629, R&D Systems), cRel (sc-71x, Santa Cruz Biotechnology) and Junb (sc-46x, Santa Cruz Biotechnology). Chromatin-antibody complexes were washed and eluted after being pulled down with magnetic beads (Protein A or G Dynabeads, Thermo Fisher Scientific). Eluted complexes were de-crosslinked overnight and purified using a NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel). ChIP-DNA was quantified using a Qubit fluorometer. For ChIP-seq experiments indexed cDNA libraries were prepared from 1 to 10 ng ChIP-DNA using a TruSeq ChIP Sample Preparation Kit (Illumina) according to manufacturer's instructions. Libraries were sequenced on Illumina NextSeq 500 or HiSeq 2500 platforms. For ChIP-qPCR experiments primers were designed for amplifying the promoter regions of the nine paradigm genes and enrichment was determined by qPCR relative to input using the ready-to-use hot start reaction mix, LightCycler 480 SYBR Green I Master (Roche) and the LightCycler 480 Instrument (Roche). Primer sequences are available in Supplementary Table S4.
ChIP-seq data analysis
The primary analysis of raw ChIP-seq reads was carried out using our ChIP-seq analysis pipeline (43). Briefly, Burrows-Wheeler Alignment Tool (bwa, (44)) was used to align the reads with the mm10 genome assembly, and Model-based Analysis of ChIP-Seq 2 (MACS2, (45)) was used for predicting peaks (binding regions) with the following specific parameters: qvalue cut-off (q) = 0.001 and subpeaks deconvolved within each peak (–call-summits). Artifacts were removed using the ENCODE blacklist (46). The identified peak summits were extended by ±100 bp to obtain binding regions. Consensus peak sets for each IRF contain the common peaks of the duplicates. By merging binding regions of IRF3, IRF5 and IRF9, a ‘unified IRF3/IRF5/IRF9 cistrome’ was generated. Coverage of predicted peaks (expressed as Reads Per Kilobase Million, RPKM) was calculated using bamtools, bedtools (coverageBed), and awk. For clustering, occupancy values were normalized by the median of values of each IRF. Integrative Genomics Viewer (IGV, Broad Institute) was used for data browsing and creating representative snapshots (47). Genome coverage files (BedGraphs) were converted into tdf files using igvtools with the ‘toTDF’ option. Normalized tag counts for read distribution (RD) histograms and heat maps were generated by HOMER (48) and then visualized by R or Java TreeView.
IDR analysis
Irreproducible Discovery Rate (IDR) analysis (49) was used to quantify the consistency between replicates. The software (https://github.com/nboley/idr) uses the narrowPeak files of MACS2 peak calling, calculates a q-value for each peak of the two datasets, and gives a cutoff value for separating low confidence regions from high confidence regions.
De novo motif discovery, PWM enrichment analysis, and the search for DNA sequences
200 bp long regions were used for motif enrichment analyses, performed by findMotifsGenome.pl (HOMER). P-values were calculated by comparing the target region enrichments with those of background sets generated by HOMER. Position weight matrices (PWMs) for ISRE, EICE, AICE1, NFKB, TRE, CRE and GAS motifs were obtained from the HOMER database (Supplementary Table S2). PWMs for IECS, AICE2 and TISRE were not available in the database; therefore, PWMs were generated by re-analyzing publicly available ChIP-seq datasets (Supplementary Table S2). The threshold values (cut-offs) for each motif were determined based on a set of randomly selected, size-matched genomic regions (random set). The 95th percentile value of each motif was used as a cut-off. Mapping of PWMs in the genome was performed by scanMotifGenomeWide.pl (HOMER). Calculation of motif scores in IRF binding regions was performed by annotatePeaks.pl (HOMER). For searching special DNA sequences in the clusters, lists of 6-mers and ISRE variants were generated. The DNA sequence of each binding region was obtained from the mouse genome (GRCm38/mm10) using bedtools (fastaFromBed). Using grep command, we searched for all 6-mers and ISRE variants and their reverse complements in all binding regions. The position of the GAANNGAAA sequence was determined genome-wide using the R package Biostrings.
ATAC-seq experiments and data analysis
Assay for Transposase Accessible Chromatin (ATAC) with high-throughput sequencing (ATAC-seq) was carried out in biological duplicates as described earlier with minor modifications (50). Cells were scraped and counted to achieve 10 000–15 000/ml in ice-cold PBS. Nuclei were isolated with ATAC lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1% IGEPAL). Nuclei were used for tagmentation from two biological replicates using a Nextera DNA Library Preparation Kit (Illumina). After tagmentation, DNA was purified with a MinElute PCR Purification Kit (QIAGEN). Tagmented DNA was amplified with Kapa Hifi Hot Start Kit (Kapa Biosystems) using nine PCR cycles. Amplified libraries were purified again with MinElute PCR Purification Kit. Fragment distribution of libraries was assessed with Agilent Bioanalyzer and libraries were sequenced on an Illumina HiSeq 2500 platform. Similarly to ChIP-seq analysis, primary analysis of the ATAC-seq raw reads was carried out using an analysis command line pipeline. Briefly, bwa was used to align the reads with the mm10 genome assembly using default parameters and MACS2 was used for predicting ATAC-seq peaks (q-value ≤ 0.001). Artifacts were removed using the ENCODE blacklist. Genome coverage files (BedGraphs) for visualization purposes were generated with makeUCSCfile.pl and then converted into tdf files using igvtools with the ‘toTDF’ option. IGV was used for data browsing and creating representative snapshots. For calculating the ratio of ATAC-positive regions, the overlap between the IRF binding regions and ATAC-seq peaks was determined using ‘bedtools intersect’. For the identification of ATAC-positive IRF-binding regions by ‘bedtools intersect’, we used the ATAC sample, which contained more identified ATAC-seq peaks.
Ternary diagrams
An R software package, ‘ggtern’, was used to create the ternary diagrams that show the ratios between the three variables (median normalized IRF3, IRF5 and IRF9 occupancy values). A given point on the ternary diagram represents one binding region, and the three input variables for a given point are the median-normalized occupancy values of the IRFs in the given region. Colour gradients for NFKB, TRE, CRE, GAS and ATAC are based on motif similarity scores or RPKM values.
PCA and machine learning
For Principal Component Analysis (PCA), the scikit-learn Python library was used, and two principal components were computed. The input for the PCA was a table containing the three IRFs’ occupancy values and enhancer features per peak. PCA was run on the three IRFs’ occupancy values for one plot, and on 11 enhancer features for the other. PCA was visualized with matplotlib, a Python library for 2D plotting. The Random Forest machine learning method was applied in Python using the RandomForestClassifier from the scikit-learn package. The motif scores for ten PWMs, ATAC-seq signals, and occurrence data concerning 37 variants of ISRE and 30 selected 6-mers were used as attributes of the binding regions. Various sets of these values were used as input variables for Random Forest, while cluster names were used as ‘class labels’. The Random Forest method not only provided prediction accuracy, but also reported the relative importance of the features (‘feature importance’ or ‘predictive power’). For selecting 6-mers with high predictive power, we performed nine Random Forest classification using the occurrence data of all 6-mers (n = 535) and running three analyses for each comparison (TLR3-dom. versus TLR5-dom., TLR3-dom. versus TLR9-dom. and TLR5-dom. versus TLR9-dom). The 6-mers were ranked based on their cumulative importance scores calculated from the nine scores. Based on additional test runs, we decided to use the ‘Top 30’ 6-mers, because accuracies were not increased significantly, when more than thirty 6-mers were used for the predictions.
ROC curves and AUC values
Receiver operating characteristic (ROC) curve analysis was used to evaluate the ability of different motifs to separate two datasets with the Random Forest classifier algorithm. The algorithm performed binary classification using the IRF dominant and IRF cistrome datasets pairwise, with the given motifs scores as its input features. The dataset was split into training and test sets. The analysis uses the scikit-learn Python library for both running the machine learning algorithm and calculating and visualizing the ROC curve and Area Under Curve (AUC) value. The classifier gives a probability estimate for both of the class labels, which shows what the probability of a given record from the test set is in that class. The ROC curve shows how the classifiers perform with different cutoff values of the prediction probabilities. The AUC measures the area under the ROC curve.
Gene sets
Genes belonging to various GO categories were downloaded from the Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org). The list of ‘antiviral cytokines’ contains the common genes of ‘defence response to virus’ (GO:0051607) and ‘cytokine activity’ (GO:0005125) GO categories. The list of ‘inflammatory program’ contains those genes of ‘inflammatory response’ (GO:0006954), which were also listed in the ‘cytokine activity’ (GO:0005125) or ‘DNA-binding transcription factor activity’ (GO:0003700) categories. The list of ‘antiviral ISGs’ was selected based on review articles on ISGs (20,21). Gene lists are supplied in the Supplementary Table S3.
Quantitative PCR (qPCR)
RNA was isolated using Trizolate reagent (UD-GenoMed). Reverse transcription was performed using a High Capacity cDNA Reverse Transcription Kit (Thermo Fischer Scientific) according to the manufacturer's instructions. Quantitative PCR (qPCR) was performed using the ready-to-use hot start reaction mix, LightCycler 480 SYBR Green I Master (Roche) and the LightCycler 480 Instrument (Roche). The comparative cycle threshold method was used to calculate expression relative to Rplp0. Primer sequences are supplied in Supplementary Table S4.
RESULTS
Clustering IRF3, IRF5 and IRF9 binding regions
We utilized a transformed murine DC line that exhibits most features of primary CD8+ DCs (22,39), but provides a significantly greater number of cells. IRF3, IRF5 and IRF9 are highly expressed at the protein and mRNA levels in this DC subset (Supplementary Figure S1A). To map IRF3, IRF5 and IRF9 binding regions, we performed ChIP-seq analyses after stimulating DCs for 90 minutes with polyinosinic-polycytidylic acid (pIC), synthetic oligodeoxynucleotides containing CpG motifs (CpG) or IFN-β, respectively. These ligands activate receptors, signalling pathways, and the corresponding IRFs very efficiently (e.g. pIC→TLR3→IRF3, CpG→TLR9→IRF5 and IFN-β→IFNAR1/2→IRF9). ChIP-seq experiments were performed with biological duplicates (Supplementary Figure S1B), resulting in the identification of 25 203, 17 435 and 25 040 peaks (binding regions) for IRF3, IRF5 and IRF9, respectively. We performed an IDR analysis (49) of the IRF ChIP-seq data sets (the results are shown in Supplementary Table S1). The comparison of our results with publicly available IRF3 and IRF5 ChIP-seq data sets (Supplementary Table S5, (11,28,34,51)) are shown in Supplementary Figure S1C.
After mapping binding regions for IRF3, IRF5 and IRF9, we determined the regions, which were differently bound by these IRFs. We generated a complete list of genomic regions (hereafter, referred to as the ‘unified IRF3/5/9 cistrome’) that were bound by at least one IRF (Figure 1A). We calculated the normalized occupancy values for IRF3, IRF5 and IRF9 and determined the ratio of occupancy values. A genomic region was considered IRF3-, IRF5- or IRF9-dominant if the normalized occupancy value of the corresponding IRF was at least twofold higher than the values for the other two IRFs (Figure 1B). If the occupancy values for IRF3, IRF5 and IRF9 on a given binding regions were similar (0.5 < ratio < 2), the region was considered to be a common binding region for IRF3, IRF5 and IRF9 (referred to as a ‘Common cluster’). We also determined three additional clusters (Shared clusters), in which the binding by one IRF was weaker than the other two IRFs (binding regions shared by ‘IRF3 and IRF5’, ‘IRF3 and IRF9’ and ‘IRF5 and IRF9’). Collectively, the unified IRF3/5/9 cistrome was classified into seven clusters based on IRF binding patterns (Figure 1A and B). IRF3-, IRF5- and IRF9-dominant binding regions are exemplified in Figure 1C by the cis-regulatory elements of Ifnb1, Il12b and Eif2ak2. These genes have been identified as IRF3, IRF5 and IRF9 target genes, respectively (18,26,52).
Figure 1.

Identification of binding regions occupied differently by IRF3, IRF5 and IRF9. (A) Read distribution plot of IRF3, IRF5 and IRF9 in a 2 kb window around the summit of the IRF peaks. For ChIP-seq experiments, murine CD8+ DCs were treated for 90 min with high molecular weight polyinosinic-polycytidylic acid (pIC), class B CpG oligonucleotide (CpG) or interferon-β (IFN-β). The binding sites of IRF3, IRF5 and IRF9 were determined in DCs stimulated for 90 minutes by pIC, CpG, and IFN-β, respectively. (B) The distribution of binding regions occupied by IRF3, IRF5 and IRF9 is shown on a ternary plot. The position of each dot in the interior of the triangle was calculated based on the normalized IRF3, IRF5 and IRF9 occupancy values. The seven IRF-binding clusters were classified based on the fold induction values. The normalized occupancy values in the dominant clusters exhibit at least two-fold differences relative to the two other IRFs. (C) The genome browser view of IRF3, IRF5 and IRF9 peaks in the 40 kb window around the TSS of three representative genes. Arrowheads indicate binding regions occupied in an IRF3-dominant (red), IRF5-dominant (blue) or IRF9-dominant (green) manner.
Identification of DNA motifs in the IRF3-, IRF5- and IRF9-dominant clusters
We analysed DNA motifs and sequences in the IRF3-, IRF5- and IRF9-dominant clusters using three different strategies: (i) de novo motif discovery, (ii) enrichment analysis of position weight matrices (PWMs) and (iii) a search for specific DNA sequences (Figure 2A). The three strategies have their own advantages and limitations and combining these approaches resulted in a more complex picture. For the sake of simplicity, we focused on the genomic regions, which were occupied dominantly by one of the three IRFs (e.g. IRF3-, IRF5- and IRF9-dominant clusters). However, we performed similar analyses for the three Shared and the Common clusters as well as for the entire IRF3, IRF5, IRF9 cistromes. The key results for these analyses are provided as supplementary data (Supplementary Figure S2).
Figure 2.

DNA motifs and sequences in the IRF-binding clusters. (A) A scheme showing the three different strategies for identification of DNA motifs and sequences in the clusters. Position weight matrices, PWMs. (B) Results of de novo motif discovery from the IRF3-, IRF5- and IRF9-dominant clusters. Motif frequencies (%) in the target (tg) and background set (bg) are shown. (C, D) PWM enrichment analysis in three IRF clusters. The enrichment was calculated relative to the random set. PWMs were obtained from the HOMER database or from re-analysed ChIP-seq datasets. (C) The enrichment of six PWMs for IRF-binding DNA sequences and (D) four PWMs for co-activated transcription factors are shown. (E) The distribution of NFKB, TRE, CRE and GAS motifs is shown on ternary plots. The position of each dot in the interior of the triangle was calculated based on the normalized IRF3, IRF5 and IRF9 occupancy values, while colours indicate motif scores. IRF binding regions with motif scores above the cut-offs are shown.
Using the first approach (de novo motif discovery), we found that the most enriched motifs in the IRF3- and IRF9-dominant clusters were DNA motifs that resembled ISRE, or a ‘mixture’ of ISRE and an Ets-IRF composite element (EICE) (53,54) (Figure 2B). Consistent with previous IRF5 ChIP-seq studies (28,34,55), de novo motif discovery failed to detect an ISRE motif in the IRF5-dominant cluster. However, an ISRE half-site motif and a weak IRF-Ets composite sequence (IECS) (56), were enriched in this cluster (Figure 2B). In addition to these IRF-binding motifs, several other motifs were identified, including the NF-κB-binding motif (κB or NFKB), the TPA response element (TRE), the cAMP response elements (CRE) and the gamma IFN activation site (GAS). These motifs are binding sites for TFs that are co-activated with IRFs in a pathway-specific manner. Upon TLR ligation, IRF3 or IRF5 are activated together with activating protein 1 (AP-1) members, which bind TRE or CRE (57), and members of the NF-κB family, which bind to κB sites (58). I-IFNs induce the formation of the ISGF3 complex together with STAT1 homodimers, which bind to GAS (59–61). The results of the de novo motif discovery for the other clusters, and for the entire IRF3, IRF5 and IRF9 cistromes are shown in Supplementary Figure S2A, B.
Due to the limitation of the de novo motif discovery method, similar motifs could be merged, eliminated, or underestimated. Moreover, a direct comparison of motif enrichment in different clusters is not feasible. Our second approach compared the enrichment of the same PWMs in the clusters side-by-side. We obtained PWMs from the HOMER database or from reanalysed ChIP-seq data (Supplementary Table S2). In addition to ISRE, EICE, IECS and the motifs for co-activated TFs, we investigated three longer motifs. Two composite elements for AP-1 and IRF (AICE1 and AICE2) were identified as binding sites for IRF4 and/or IRF8 and their co-binding TFs (62–66). A tripartite form of ISRE (called hereafter as ‘TISRE’), which was previously identified as binding sites for IRF1, IRF2 and IRF8 (35,38,67) was also included in the analysis. We determined the threshold (cut-off) values for each motif systematically, based on a set of randomly selected size-matched genomic regions (random set) (Supplementary Figure S2C). We then calculated the PWM enrichment relative to the random set (Figure 2C and D). We made the following four observations. First, as expected, ISRE was the most enriched motif in the IRF3- and IRF9-dominant cluster, while it was poorly enriched in the IRF5-dominant cluster (Figure 2C). Second, TISRE was especially frequent in the IRF3-dominant cluster. Third, the enrichment of the EICE motif was similar to the ISREs’ in the investigated clusters. These results do not imply necessarily that IRF3-dimers and ISGF3 (STAT1/STAT2/IRF9) bind EICE motif with a high frequency. More likely, IRF3-dimers and ISGF3 show a preference for accessible genomic regions that are primed by PU.1 and IRF8. These two TFs are lineage-determining TFs in this DC subtype (68), and together they bind EICE and IECS (56). Last, de novo motif discovery results and the enrichment analysis of PWMs for co-activated TFs agreed in most cases. Most co-activated TF motifs, which were detected with the low p-value by the de novo motif discovery, were enriched in the IRF dominant clusters; NFKB, TRE, and CRE motifs in the IRF3-dominant cluster, NFKB in the IRF5-dominant cluster, and GAS in the IRF9-dominant cluster (Figure 2D). The only discrepancy was observed in the IRF5-dominant cluster, where TRE and CRE motifs were enriched by de novo motif discovery, but not by PWM analysis. The differences in the enrichments are coming from the fact that de novo determined PWMs are similar but not identical to the PWMs obtained from the HOMER database (Figure 2B and D), and only de novo determined PWMs were enriched. We also investigated the distribution of binding regions, which contained sequences with high motif scores for co-activated TFs (Figure 2E). Since motif scores signify similarities between the identified DNA sequences and the used PWMs, these sites typically represent high affinity binding sites. We found that NFKB motifs with high scores were polarized in the IRF3- and IRF5-dominant clusters, while regions containing GAS motifs with high scores were specially enriched in the IRF9-dominant cluster. In the case of TRE and CRE motifs, a modest IRF3- and IRF5-biased polarization was detectable. PWM enrichment analysis for the other four clusters is shown in Supplementary Figure S2D. Previous studies demonstrated that many genes are co-regulated by I-IFN and NF-κB pathways (69,70). The low frequency of the NFKB motif in the IRF9-dominant cluster raised the question of what types of binding regions could be responsible for the cross-talk between I-IFN and NF-κB pathways. By analysing the frequency of NFKB motifs in the clusters (Supplementary Figure S2D) and the binding regions associated with genes co-regulated by I-IFN and NF-κB pathways, we concluded that binding regions occupied by IRF3, IRF5 and IRF9 (Common Cluster) play a prominent role in the cross-talk. (The list of co-regulated genes by I-IFN and NF-κB pathways was obtained from the study of Wienerroither et al. (69). Illustrative binding regions co-occupied by IRF9 and cRel associated with co-regulated genes are shown in Supplementary Figure S2E.)
Variants of ISRE and ISRE half-sites in the IRF3-, IRF5- and IRF9-dominant clusters
Using our third approach (search for specific DNA sequences), we were able to discriminate similar DNA sequences and determine their enrichments separately. We searched for 6-mers and ISRE variants that were distributed differently among the clusters. We investigated 6-mers instead of 4-mers because IRF binding is affected by 5′ and 3′ flanking bases of the 4bp long ISRE half-sites (31). We generated a list of all 6-mers, which contained the canonical 5′-GAAA-3′ and two extra bases (5′-NNGAAA-3′, 5′-NGAAAN-3′ or 5′-GAAANN-3′). In the 5′-GAAA-3′ sequence, one mismatch was allowed (Supplementary Table S6). We determined the frequency of these 6-mers (n = 535) in the clusters (Figure 3A). We identified 6, 68 and 10 sequences that occurred at least 1.5 times more frequently in the IRF3-, IRF5 and IRF9-dominant clusters, respectively, compared to the other two clusters (Supplementary Table S6 and Figure S3A). Notably, the enrichment of many 6-mers was consistent with the results of the previous in vitro studies. PBM data indicated that sequences that contain 5′-GGAAAC-3′ are bound with higher affinity by IRF3 compared to IRF5, IRF6 or IRF9 (31,33). Consistent with this observation, we found that GGAAAC is more enriched in the IRF3-dominant cluster (first plot on Figure 3B and Supplementary Figure S3A). PBM experiments revealed that IRF5 is more tolerant of the ISRE half-site at position 3 and 4 (5′-GAAA-3′) than IRF7 (33) or certain other IRFs (31). We detected enrichment of several 6-mers that differed at these positions from the canonical 5′-NGAAAN-3′ core sequence (for example, CGACAC, CGAGAC, CGAACC and CGAAGC) in the IRF5-dominant cluster (second and third ternary plots on Figure 3B, and Supplementary Figure S3A). Finally, in PBM experiments, TGAAAC was bound with higher affinity by IRF9 compared to IRF3, IRF5 or IRF6 (31). This and some other 6-mers, such as GAAACT, were especially enriched in the IRF9-dominant clusters (fourth plot on Figure 3B, and Supplementary Figure S3A).
Figure 3.

Frequency and distribution of extended ISRE half-sites (6-mers) and ISRE variants in the IRF-binding clusters. (A) The frequency of 6-mers in IRF-binding clusters is shown on a heat map. (B) The distribution of binding regions containing the indicated 6-mers is shown on ternary plots. (C) The frequency of ISRE variants in IRF-binding clusters is shown on a heat map. (D) The distribution of binding regions containing the indicated ISRE variants is shown on ternary plots. On panel B and D the position of each dot in the interior of the triangle was calculated based on the normalized IRF3, IRF5, and IRF9 occupancy values. (E) A heat map showing the frequency of selected 6-mers (n = 13), which were more frequent in the IRF5-dominant cluster, than in the IRF3-, or IRF9-dominant clusters. (F) ROC curves and AUC values as determined in the indicated pair-wise comparisons. Occurrence data of 13 selected 6-mers, which were most frequent in the IRF5-dominant cluster, were used as input features for the analyses. TPR, true positive rate; FPR, false positive rate.
We also searched for specific ISRE variants (n = 37) that were selected based on previous studies (31–33,38) or our 6-mer analysis (Figure 3C, D, Supplementary Figure S3B and Table S6). We identified 20 and 4 ISRE variants that occurred at least 1.5 times more frequently in the IRF3- and IRF9-dominant clusters, respectively, compared to the other two clusters. We could not detect any ISRE variants, which occurred more frequently in the IRF5-dominant cluster than in the other two clusters.
Consistent with the 6-mer results, we found that GGAAANNGAAA occurred more frequently in the IRF3-dominant cluster relative to the other clusters (first plot on Figure 3D). This sequence has been identified as the EIRE motif (3,71). The ISREs in which half-sites were separated by a 3 bp spacer, and ISREs with two extra AA at the 5′ position, also occurred more frequently in this cluster (Supplementary Figure S3B and second plot on Figure 3D).
Notably, ISRE variants (e.g. 5′-GAAANNGAGA-3′ and 5′-GAGANNGAGA-3′), which contained ISRE half-sites preferentially bound by IRF5 (e.g. GAGA) did not occur more frequently in the IRF5-dominant cluster (third ternary plot on Figure 3D and Supplementary Figure S3B).
EMSA and protein crystallization experiments suggest that, due to additional contacts between DNA and STATs, the ISGF3 complex has sequence requirements that extend beyond the consensus ISRE motif (8,32) and the detailed consensus for ISGF3 is 5′-WBVGGAAANNGAAACT-3′ (W = A or T; B = C or G or T; V = A or C or G). Of note, the special sequence requirement for ISGF3 does not necessarily imply that these sequences are bound more efficiently by IRF9 than other IRFs. Notably, we found that 5′-GAAANNGAAACT-3′ (fourth on Figure 3D), but not GGAAANNGAAA (first plot on Figure 3D) or WBVNGAAANNGAAA (Supplementary Table S6), were enriched in the IRF9-dominant cluster. The binding regions with the entire detailed consensus for ISGF3 (5′-WBVGGAAANNGAAACT-3′) were also enriched, but less than 1% (55 out of 7373) of IRF9-dominant binding regions (Supplementary Figure S3B) contained these ISRE variants.
ROC (Receiver Operating Characteristics) analysis is frequently used for assessing a classification model's performance (72). We determined ROC curves, and calculated AUC (Area Under the Curve) values in pair-wise comparisons for the three IRF-dominant subsets and three IRF cistromes. The following three types of input features were used: (i) motif strength of the ISRE motif (similarity score for the ISRE PWM from HOMER database), (ii) occurrence data of selected 6-mers (n = 13), which were most frequent in the IRF5-dominant cluster and (iii) occurrence data of selected ISRE variants (n = 7), which were most frequent in the IRF3-dominant cluster (Figure 3E, F and Supplementary Figure S3C, D). Because AUC represents the degree or measure of separability, the higher the AUC, the better the machine learning model is at predicting the class label in pair-wise comparisons. We found that ISRE motif scores could not separate IRF3-dominant vs. IRF9-dominant clusters or IRF3 versus IRF9 cistromes (low AUC values). In contrast, ISRE motif scores could separate IRF5-dominant and IRF5 cistrome from the other clusters/cistromes (higher AUC values, Supplementary Figure S3C). These data are consistent with the enrichment analyses: the frequency of ISRE is higher in the IRF3- and IRF9- dominant clusters, and IRF3 and IRF9 cistromes compared to the IRF5-dominant cluster and the IRF5 cistrome. As expected, the occurrence data of 6-mers most frequent in the IRF5-dominant cluster could not separate IRF3-dominant vs. IRF9-dominant clusters and IRF3 versus IRF9 cistromes (low AUC values). In contrast, these 6-mers could separate IRF5-dominant clusters from the other clusters (higher AUC values, Figure 3F). When occurrence data of selected ISRE variants (n = 7) were used for AUC analysis for the comparison of IRF3-dominant vs. IRF5- or IRF9-dominant clusters AUC values were higher (0.69 and 0.63). The AUC value was lower (0.59) in the IRF5-dominant vs. IRF9-dominant comparison. These results mirror the differences in the frequencies of these ISRE variants (e.g. IRF3-dominant > IRF9-dominant > IRF5-dominant cluster.
Collectively, certain 6-mers and ISRE variants were distributed differently in IRF-binding clusters; therefore, these DNA sequences most likely contribute to IRF-specific binding. None of the ISRE variants occurred exclusively in IRF-specific clusters. Thus, these sequences alone could not determine specificity.
Prior to stimulation, many IRF3- and IRF9-dominant binding regions have low accessibility
Previous studies revealed that a majority of the binding of stimulus-regulated TFs occurs at pre-existing enhancer-like accessible regions (73,74). A smaller percentage of TFs bind to ‘latent’ or ‘de novo’ enhancers defined as regions that acquire histone marks characteristic of enhancers only after stimulation (73,74). The recruitment of co-activators by TFs occupying pre-existing or de novo enhancers typically increases the acetylation of H3 and H4 histone tails and/or chromatin accessibility. IRF3 can promote nucleosome remodelling and/or the opening of previously inaccessible genomic regions in LPS-stimulated macrophages (75). Chromatin opening by ISGF3 has also recently been studied (76,77). Au-Yeung and Horvath found that stimulation with I-IFN decreased promoter-associated histones (e.g. H2A.Z), consistent with the observed nucleosome reorganization identified at ISG promoters. To our knowledge, the ability of IRF5 to reshape the chromatin landscape has not been investigated in genome-wide studies. Because different signaling pathways activate both overlapping and unique sets of ‘latent’ or ‘de novo’ enhancers (74), chromatin accessibility could be an important determinant for IRF-specific enhancer selection. Therefore, we investigated the accessible chromatin regions by performing ATAC-seq (Assay for Transposase Accessible Chromatin with high- throughput sequencing (50)) experiments in un-stimulated (UT) DCs. Based on these results, we calculated the percentages of accessible regions in the IRF cistromes and clusters (Figure 4A), and we determined the position of accessible regions relative to IRF peaks and motifs (Supplementary Figure S4B). We also investigated the distribution of IRF-binding regions with high ATAC-seq signals (Supplementary Figure S4C)
Figure 4.
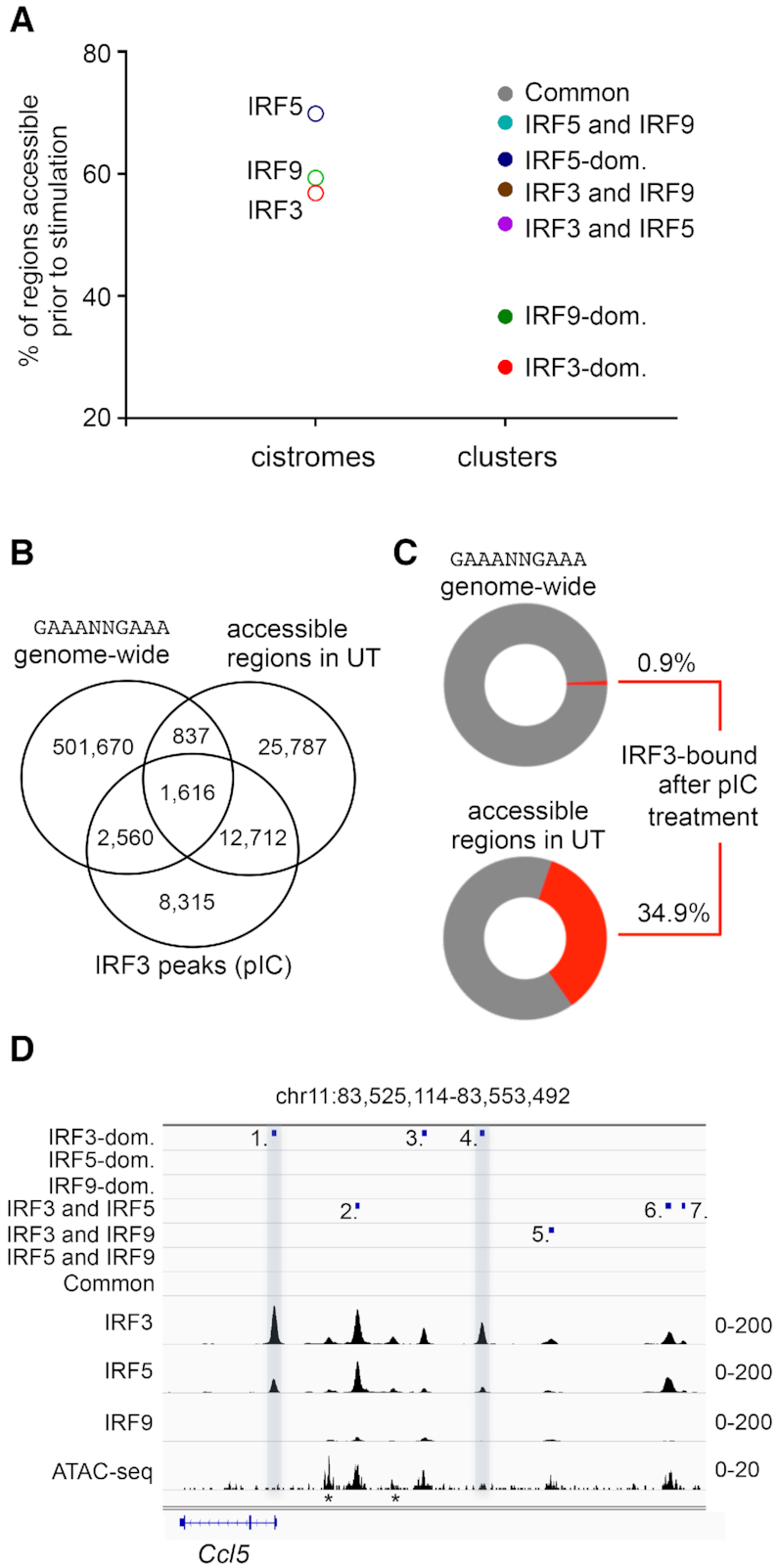
Chromatin accessibility in the IRF cistromes and IRF-binding clusters. (A) The frequency of ATAC-seq positive regions in unstimulated (UT) DCs is shown for the IRF cistromes and IRF-binding clusters. (B) The overlap between the canonical ISRE (GAAANNGAAA) sequence, the accessible genomic regions in unstimulated (UT) DCs, and IRF3-binding regions in pIC-stimulated DCs is shown on a Venn diagram. (C) Percentages of the canonical ISRE (GAAANNGAAA) sequence and accessible genomic regions in unstimulated (UT) DCs, which were bound by IRF3 after pIC-treatment. (D) The genome browser view of the binding of IRF3, IRF5 and IRF9, and the ATAC-seq signal. The positions of mapped binding regions are also shown. Two regions (nos. 1 and 4), which had low or no accessibility prior-to-stimulation and were occupied dominantly by IRF3 upon stimulation, are highlighted. Regions indicated with an asterisk (*) were not identified as IRF binding regions, most likely due to the strict peak calling strategy.
Regarding the cistromes, we found that the difference was moderate (∼13%) between the lowest and highest frequencies (IRF3: 56.8% versus IRF5: 69.8%). The overlap between IRF binding regions, the accessible genomic regions and the canonical ISRE (GAAANNGAAA) sequence was also determined to evaluate the impact of accessibility together with a high-affinity IRF-binding motif (Figure 4B and Supplementary Figure S4A). We found that only a minority of the canonical ISRE sequences (<1%) were occupied by IRF3 upon activation in vivo. In contrast, ∼35% of the regions, which were accessible before stimulation, were occupied by IRF3 after stimulation (Figure 4C). Of note, the exact ratio of accessible regions with or without any ISRE motifs could not be determined, because the presence of other ISRE variants was not investigated.
Regarding the IRF-binding clusters, we detected much larger differences (∼50%) between the lowest and highest frequencies (IRF3-dominant: 23.8% vs. Common: 73.8%). Notably, the lowest percentages of accessible regions were detected in the IRF3- and IRF9-dominant clusters (28.3% and 36.6% respectively; Figure 4A). We found that the IRF3-dominant regions, which were not accessible prior to stimulation, were often accompanied by fully accessible enhancers. The diversity of binding regions is exemplified by the cis-regulatory elements localized upstream of Ccl5 in Figure 4D. In contrast to IRF3 and IRF9, we found that the percentage of accessible regions was relatively high in the IRF5-dominant cluster (62.4%).
Collectively, these observations suggest that chromatin accessibility is a critical determinant for all the investigated IRFs. A majority of the IRF3, IRF5 and IRF9 binding events occurs at pre-existing accessible regulatory regions. Our data suggest that IRF3 and IRF9 (in the ISGF3 complex) more frequently occupy ‘latent’ or ‘de novo’ enhancers than IRF5. Notably, the finding that ATAC-negative regions were especially enriched in the IRF3- and IRF9-dominant regions suggests that the ‘chromatin barrier’ contributed to IRF3- and IRF9-specific, but not to IRF5-specific, enhancer selection, by inhibiting the binding of other IRFs. Our observations are consistent with previous findings related to other TFs indicating that chromatin features play active roles in shaping the selective transcriptional responses (78).
Machine learning predictions of IRF dominance
To investigate the relationship between enhancer features and IRF dominance, we used two approaches: a principal component analysis (PCA) and a machine learning approach called Random Forest (79,80). As expected, the three clusters were completely separated by PCA when IRF3, IRF5 and IRF9 occupancy values were used as input variables (Figure 5A, left panel). Using 10 motif scores and ATAC signals as inputs for PCA, the separation was not complete. Most binding regions in the IRF5-dominant cluster were separated from the binding regions in the IRF3- and IRF9-dominant clusters. In contrast, IRF3 and IRF9-dominant clusters appeared as partially overlapping populations (Figure 5A, right panel).
Figure 5.

Analysis of IRF-dominant binding regions by principal component analysis (PCA) and machine learning. (A) PCA of IRF3-, IRF5- and IRF9-dominant clusters based on IRF occupancy values (on the left) and 11 enhancer features (10 motif scores and ATAC-seq signal, on the right). Data points representing individual peaks are colour-coded based on their original cluster labels. (B) A scheme showing the pair-wise comparisons for the Random Forest method. (C) The accuracy of prediction by the Random Forest method using different sets of input variables. The scores for IRF-binding motifs, motifs for co-activated TFs, ATAC-signals, and occurrence data concerning variants of ISRE and 6-mers, or all features were used as input variables for the method. The number of features used for various Random Forest analyses is indicated. The accuracy indicates the percentage of correct predictions (where the original and predicted cluster labels matched) relative to all predictions.
After PCA, we used Random Forest, which combines the predictions of simple decision trees using a voting system. One of the most common applications of machine learning methods is to predict the ‘class label’ of any given entity after a learning phase based on several attributes. Using Random Forest, we investigated how accurately the IRF-dominance could be predicted using different sets of features. We performed pairwise comparisons (Figure 5B), using the cluster label (e.g. IRF3-, IRF5-, or IRF9-dominant) of each binding region as ‘class label’. The motif scores for ten PWMs, ATAC-signals, and occurrence data concerning 37 variants of ISRE and 30 selected 6-mers were used as attributes of the binding regions (Supplementary Table S7). The accuracy of the prediction was calculated based on the ratio of correct vs. all predictions. A prediction was correct, when original and predicted class labels agreed. We found that prediction accuracy was higher when more features were included for the prediction (Figure 5C). The class labels were predicted with high accuracy (>80%) when the IRF5-dominant cluster was compared to the other two dominant clusters. The class label prediction was less accurate (∼70%) for the comparison between IRF3-dominant and IRF9-dominant clusters. The results of the two methods confirmed each other; they indicated that the IRF5-dominant cluster differed markedly from the other two clusters. Based on the enhancer features, IRF3- and IRF9-dominant clusters were less separated from each other.
Genes encoding antiviral cytokines, inflammatory genes, and antiviral ISGs in DCs are often associated with IRF3-, IRF5- and IRF9-dominant regions
To investigate the biological relevance of the IRF3-, IRF5-, and IRF9-dominant clusters, we examined the genomic landscapes of three gene-sets: (i) antiviral cytokines, (ii) cytokines and TFs of the inflammatory program and (iii) antiviral ISGs. Many genes in these sets have been identified as targets of IRF3, IRF5 and IRF9, respectively (1,12–14, 21). We compiled three gene sets based on Gene Ontology (GO) annotations and review articles (Supplementary Table S3). The gene lists for antiviral cytokines, inflammatory program, and antiviral ISGs contained 25, 119 and 63 genes, respectively (Figure 6A, B and Supplementary Table S3). Binding regions were determined and counted separately for each gene in the 20-kb regions around the transcription start sites (TSS ± 10 kb). We found that in the 20-kb window of antiviral cytokines, the IRF3-dominant cluster was especially enriched (Figure 6B). In the 20-kb window of genes involved in the inflammatory program, the IRF5-dominant and ‘IRF3 and IRF5’ clusters were over-represented, while in the TSS ± 10 kb window of antiviral ISGs, the IRF9-dominant and the ‘IRF3 and IRF9’ clusters occurred most frequently (Figure 6B).
Figure 6.

Relationship between IRF binding regions and transcriptional programs. (A) A scheme showing the steps for the enrichment analysis. (B) The enrichment of IRF-binding clusters in 20-kb regions around the transcription start site (TSS) of the genes belonging to three transcriptional programs. (C) Radar graphs showing the median of values for seven features in IRF3- (red), IRF5- (blue), and IRF9-dominant (green) binding regions. The values were calculated from the entire clusters (left) or from the peaks in 20-kb regions around the TSS of the genes belonging to the three transcriptional programs (right). (D) The most characteristic patterns of the IRF3-, IRF5- and IRF9-dominant binding regions associated with the three gene sets. The symbols indicate the ratios of the score of the given motif or ATAC-seq signal relative to the median of the unified IRF3/IRF5/IRF9 cistrome: ratio less than 0.8 (–), between 0.8 and 1.25 (≈), between 1.25 and 2 (+), and higher than 2 (++). (E) The genome browser view of the binding of IRF3, IRF5 and IRF9. Three genes (TSS ±10kb) are shown for antiviral cytokines (top), inflammatory program (middle), and antiviral ISGs (bottom). (F) Induction kinetics of the same genes were determined by qPCR in the DC line stimulated with pIC, CpG and IFN-β. Expression was normalized relative to Rplp0. Mean and SD of three independent experiments are shown.
We also analysed the pattern of enhancer features for 28 IRF3-, 62 IRF5- and 45 IRF9-dominant binding regions, which were localized in the TSS ± 10 kb window of antiviral cytokines, inflammatory genes, and antiviral ISGs, respectively (Figure 6C). The analysis revealed that IRF-specific binding regions associated with specific gene-sets (Figure 6C, right panel) had patterns similar to the entire cluster in most cases (Figure 6C, left panel) with respect to the following seven key features: ISRE, TISRE, NFKB, TRE, CRE, GAS and ATAC-signal (Figure 6D).
Finally, based on their biological significance, we selected genes, which were associated with IRF3, IRF5- or IRF9-dominant peaks in their promoter regions (TSS ± 1 kb; Figure 6E and Supplementary Figure S5A). Using qPCR, we investigated whether they were induced most efficiently by ligands and pathways that favoured the corresponding IRFs. We found that selected antiviral cytokines, inflammatory genes, and antiviral ISGs were regulated most efficiently by pIC, CpG and IFN-β, respectively (Figure 6F). Because genes could be regulated by several enhancers and signalling pathways could activate several TFs simultaneously, the dominance of the given pathway is not necessarily mediated dominantly or exclusively by these regions and/or these IRFs. However, in many cases their contribution was certain. For example, in the case of antiviral ISGs, their TSS ± 10 kb window contained very few other enhancers (Figure 6E). Collectively, our results suggest that the identified and characterized regions, which were occupied by predominantly IRF3, IRF5 or IRF9, are important sites for signal specific transcriptional responses.
To investigate the interactions (competitions and co-operations) between IRFs or signaling pathways in our system, we performed a series of ChIP-qPCR experiments (Supplementary Figure S5B). In these experiments, the DCs were stimulated with either single agonists or a combination of ligands. We found that IRF3 binding to the IRF3-dominant regions of antiviral cytokines was inhibited by all combined treatments. The IRF5 binding to the IRF5-dominant regions of inflammatory genes was increased when the pIC + CpG combination was used. However, in other combinations, the IRF5 binding was inhibited. Because IRF5 was activated by both TLR3 and TLR9 agonists (Supplementary Figure S5B), the increased IRF5 binding may be caused by cooperation between the two pathways for IRF5 activation, and not by interaction between IRF3 and IRF5. Finally, the binding of IRF9 to IRF9-dominant regions of antiviral ISGs was similar when IFN-β was used alone or in combination with CPG, suggesting that the TLR9 pathway cannot inhibit IRF9 activation or binding. In all other ligand combinations, IRF9 binding was inhibited. Collectively, these results suggest that competition/cooperation between IRFs (and/or other TFs) and cross-talk between signalling pathways markedly influence the binding of IRF3, IRF5 and IRF9.
DISCUSSION
IRF3, IRF5 and IRF9 play distinct but partially overlapping roles in the regulation of antiviral cytokines, inflammatory responses, and cell-intrinsic antiviral immunity. The first premise for this ‘division of labour’ is that IRFs differ from each other in their DNA-binding domain (DBD) and IRF-association domains (IADs), which are responsible for interactions with other TFs (3). The second premise is that cis-regulatory elements of IRF-regulated genes contain features that differentially affect IRF binding. Recent high-throughput studies have highlighted the features beyond primary DNA sequence motifs that modulate binding specificity (81). The same TF and external stimulus regulate overlapping but distinct sets of genes in different cell types. For example, some NF-κB target genes are induced to a similar extent in both macrophages and B cells, while many others are preferentially activated in one cell type or the other (24). Similarly, responses induced by I-IFN are cell type-dependent (77). These cell type-specific responses are due to several phenomena, e.g. differentially expressed receptors, differences in the signalling pathways and in the repertoires of inducible enhancers (24,77). Our study suggests that DNA motifs occupied directly by IRFs and additional features (e.g. binding of co-activated TFs and chromatin accessibility) are involved in mediating IRF-specificity. Of note, the regions within IRF-dominant clusters were diverse, and not all regions shared the ‘prototypic pattern’ of features (Figure 6D). The diversity of the regions and the fact that none of the features occurred exclusively in the clusters indicate that a series of features in a combinatorial fashion, rather than exclusively highly polarized DNA sequences, mediate IRF-specific binding.
In this study, we identified and compared the IRF3-, IRF5-, and IRF9-dominant regions. From the three clusters, the IRF5-dominant cluster fundamentally differed from the IRF3- and IRF9-dominant clusters. IRF5 could occupy genomic loci via three potential mechanisms: (i) binding as a homo- or heterodimer to ISREs, (ii) binding as a monomer and (iii) indirect binding. The fact that ISRE motif was enriched in the ‘IRF3 and IRF5’ and Common clusters (Supplementary Figure S2D) suggests that at least a subset of IRF5 proteins binds as dimers occupying canonical ISREs. The observations that an ISRE half-site (but not ISRE) was identified by our de novo motif discovery in the IRF5 cistrome (Supplementary Figure S2B) and the IRF5 logo from PBM study resembles a half-site logo with a single core 5′-GAAA-3′ element (33) do not necessarily mean that IRF5 binds dominantly or exclusively as monomers. As suggested by Andrilenas et al. (33) the shorter logo represents a true dimer site but results from an IRF5 preference to engage more strongly to one ISRE half-site. In other words, in PBMs, IRF5 binds as a dimer with an asymmetric half-site preference (33). The last mechanism suggests that IRF5 does not bind DNA directly, and its binding site is determined by other TF(s). This has been shown for IRF3, which can function as a signal-specific cofactor to activate the transcription of a set of NF-κB-dependent genes, without binding to an ISRE (1,82). The contribution of indirect binding for IRF5-mediated gene regulation awaits further experimental confirmation.
We found that IRF5-dominant cluster had three characteristic features. First, most of the IRF5-dominant regions do not contain ISRE motifs. A recent study demonstrated that bases other than C at the 5′ and 3′ positions relative to the half-site (5′-CGAAAC-3′) could prevent IRF5 binding to the virus response elements of IFN promoters (33). Our analysis revealed that not only ISRE variants, which contained bases other than C at these positions (e.g. GAAANGGAAA), but all ISRE variants were underrepresented in the IRF5-dominant cluster (Figure 3C). Second, special ISRE half-sites, such as 5′-GAGA-3′ and 5′-GACA-3′, were enriched in the cluster. Further studies are needed to reveal the premise and molecular mechanism by which IRF5 binds to special 6-mers and whether IRF5 proteins could bind these ISRE half-sites as monomers. Last, the high frequency of the NFKB motif in the IRF5-dominant cluster was consistent with the observation that IRF5 binding appears to rely in large part on its interactions with NF-κB (28).
IRF3- and IRF9-dominant regions were more similar to each other, and the IRF3 versus IRF9 dominance was less predictable (Figure 5C). There are several potential reasons for the lower accuracy in prediction by machine learning. Clustering itself may be inaccurate due to differences in ChIP efficiency or other reasons. Certain non-characterized features may also be required for accurate predictions. Such ‘missing features’ may include the number of motifs in a given region, the distance between the different types of motifs, the occupancy values of TFs, the presence of additional motifs, or other variants of ISRE and 6-mers. A more complex analysis in the future may reveal other features that are needed for more accurate predictions. However, we uncovered patterns, which favour either IRF3-dominant or IRF9-dominant binding (Figures 2 and 6C).
In the IRF3-dominant cluster, we found that many of the binding regions had low accessibility prior to stimulation. The presence of higher frequency NFKB, AICE (which may serve as the binding site for IRF and AP-1), TRE and CRE motifs in the cluster suggests that IRF3 requires the collaborative binding of other co-activated TFs to ‘open’ the chromatin. Alternatively, tandem binding of three or four IRF3 or other IRF(s) to TISRE, which are also enriched in the cluster, may be sufficient for nucleosome removal. The IRF3-dominant binding regions are not bound by IRF9 with high affinity, most likely because NF-κB and AP-1 are not induced in the I-IFN pathway.
We found that many IRF9-dominant regions contain both ISRE and GAS motifs, without other motifs (Supplementary Figure S4B). Although the consensus sequence of 5′-WBVGGAAANNGAAACT-3′ (32) and its variants were enriched in the IRF9-dominant cluster, their frequencies were low (Supplementary Figure S3B). However, the enrichment of the GAS motif (Figure 2D, E) suggests that the STAT1 homodimers activated by I-IFN signalling are important determinants for IRF9-dominant binding.
Collectively, this research highlights the need for integrated analysis of in vitro and in vivo binding data, as well as computational analysis to understand specific enhancer selection by members of IRFs or other TF families. This study contributes to our understanding of how IRF members can initiate tailored responses to viral particles, inflammatory agents, and type I interferons without activating superfluous or harmful transcriptional programs in cells.
DATA AVAILABILITY
ChIP-seq and ATAC-seq datasets have been deposited in GEO under accession number GSE125340.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge Karen Uray, Ivan Uray, László Huzsvai, Szilvia Benkő, Szilárd Póliska and István Szatmári for discussions, technical supports and comments on the manuscript. Library preparation and sequencing were performed at the Genomic Medicine and Bioinformatic Core Facility, Department of Biochemistry and Molecular Biology, Faculty of Medicine, University of Debrecen (Hungary) and Analytical Genomics Core Facility at the Sanford-Burnham Prebys, Medical Discovery Institute, Orlando (U.S.A.).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Hungarian Scientific Research Fund [NN125613 to L.Sz.]; Bolyai Research Scholarship of the Hungarian Academy of Sciences (to L.Sz.); Economic Development and Innovation Operational Programme [GINOP, 3Z5SBKC0FUNO119] to the Genomic Medicine and Bioinformatics Core Facility, Department of Biochemistry and Molecular Biology, Faculty of Medicine, University of Debrecen; G.N. is supported by the Hungarian Scientific Research Fund (OTKA) [PD 124843]; Bolyai Research Scholarship of the Hungarian Academy of Sciences; ÚNKP-19-4-DE-173 New National Excellence Program of the Ministry of Human Capacities. Funding for open access charge: Hungarian Scientific Research Fund, NN125613 and Education and Research Support Department, University of Debrecen.
Conflict of interest statement. None declared.
REFERENCES
- 1. Honda K., Taniguchi T.. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006; 6:644–658. [DOI] [PubMed] [Google Scholar]
- 2. Savitsky D., Tamura T., Yanai H., Taniguchi T.. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol. Immunother. 2010; 59:489–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mancino A., Natoli G.. Specificity and function of IRF family transcription factors: insights from genomics. J. Interf. Cytokine Res. 2016; 36:462–469. [DOI] [PubMed] [Google Scholar]
- 4. Zhao G.N., Jiang D.S., Li H.. Interferon regulatory factors: at the crossroads of immunity, metabolism, and disease. Biochim. Biophys. Acta - Mol. Basis Dis. 2015; 1852:365–378. [DOI] [PubMed] [Google Scholar]
- 5. Fu X.Y., Kessler D.S., Veals S.A., Levy D.E., Darnell J.E.. ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:8555–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Majoros A., Platanitis E., Kernbauer-Hölzl E., Rosebrock F., Müller M., Decker T.. Canonical and non-canonical aspects of JAK-STAT signaling: lessons from interferons for cytokine responses. Front. Immunol. 2017; 8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Platanitis E., Demiroz D., Schneller A., Fischer K., Capelle C., Hartl M., Gossenreiter T., Müller M., Novatchkova M., Decker T.. A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nat. Commun. 2019; 10:2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rengachari S., Groiss S., Devos J.M., Caron E., Grandvaux N., Panne D.. Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E601–E609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paul A., Tang T.H., Ng S.K.. Interferon regulatory factor 9 structure and regulation. Front. Immunol. 2018; 9:1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Veals S.A., Santa Maria T., Levy D.E.. Two domains of ISGF3 gamma that mediate protein-DNA and protein-protein interactions during transcription factor assembly contribute to DNA-binding specificity. Mol. Cell Biol. 1993; 13:196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tong A.-J., Liu X., Thomas B.J., Lissner M.M., Baker M.R., Senagolage M.D., Allred A.L., Barish G.D., Smale S.T.. A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell. 2016; 165:165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doyle S.E., Vaidya S.A., O’Connell R., Dadgostar H., Dempsey P.W., Wu T.T., Rao G., Sun R., Haberland M.E., Modlin R.L. et al.. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002; 17:251–263. [DOI] [PubMed] [Google Scholar]
- 13. Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T., Kano S.I., Honda K., Ohba Y., Mak T.W. et al.. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005; 434:243–249. [DOI] [PubMed] [Google Scholar]
- 14. Krausgruber T., Blazek K., Smallie T., Alzabin S., Lockstone H., Sahgal N., Hussell T., Feldmann M., Udalova I.A.. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011; 12:231–238. [DOI] [PubMed] [Google Scholar]
- 15. Yanai H., Chen H. -m., Inuzuka T., Kondo S., Mak T.W., Takaoka A., Honda K., Taniguchi T.. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:3402–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Savitsky D.A., Yanai H., Tamura T., Taniguchi T., Honda K.. Contribution of IRF5 in B cells to the development of murine SLE-like disease through its transcriptional control of the IgG2a locus. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:10154–10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steinhagen F., Mcfarland A.P., Rodriguez L.G., Tewary P., Jarret A., Savan R., Klinman D.M.. IRF-5 and NF-κB p50 co-regulate IFN-β and IL-6 expression in TLR9-stimulated human plasmacytoid dendritic cells. Eur. J. Immunol. 2013; 43:1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Negishi H., Yanai H., Nakajima A., Koshiba R., Atarashi K., Matsuda A., Matsuki K., Miki S., Doi T., Aderem A. et al.. Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat. Immunol. 2012; 13:659. [DOI] [PubMed] [Google Scholar]
- 19. Suprunenko T., Hofer M.J.. The emerging role of interferon regulatory factor 9 in the antiviral host response and beyond. Cytokine Growth Factor Rev. 2016; 29:35–43. [DOI] [PubMed] [Google Scholar]
- 20. Schoggins J.W., Rice C.M.. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011; 1:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. MacMicking J.D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 2012; 12:367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Széles L., Meissner F., Dunand-Sauthier I., Thelemann C., Hersch M., Singovski S., Haller S., Gobet F., Fuertes Marraco S.A., Mann M. et al.. TLR3-Mediated CD8 + dendritic cell activation is coupled with establishment of a cell-intrinsic antiviral state. J. Immunol. 2015; 195:1025–1033. [DOI] [PubMed] [Google Scholar]
- 23. Platanitis E., Decker T.. Regulatory networks involving STATs, IRFs, and NFκB in inflammation. Front. Immunol. 2018; 9:2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Glass C.K., Natoli G.. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016; 17:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smale S.T. Selective transcription in response to an inflammatory stimulus. Cell. 2010; 140:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Panne D., Maniatis T., Harrison S.C.. An atomic model of the interferon-β enhanceosome. Cell. 2007; 129:1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Freaney J.E., Kim R., Mandhana R., Horvath C.M.. Extensive cooperation of immune master regulators IRF3 and NFκB in RNA Pol II recruitment and pause release in human innate antiviral transcription. Cell Rep. 2013; 4:959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saliba D.G., Heger A., Eames H.L., Oikonomopoulos S., Teixeira A., Blazek K., Androulidaki A., Wong D., Goh F.G., Weiss M. et al.. IRF5:RelA interaction targets inflammatory genes in macrophages. Cell Rep. 2014; 8:1308–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tamura T., Yanai H., Savitsky D., Taniguchi T.. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008; 26:535–584. [DOI] [PubMed] [Google Scholar]
- 30. Ourthiague D.R., Birnbaum H., Ortenlof N., Vargas J.D., Wollman R., Hoffmann A.. Limited specificity of IRF3 and ISGF3 in the transcriptional innate-immune response to double-stranded RNA. J. Leukoc. Biol. 2015; 98:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Badis G., Berger M.F., Philippakis A.A., Talukder S., Gehrke A.R., Jaeger S.A., Chan E.T., Metzler G., Vedenko A., Chen X. et al.. Diversity and complexity in DNA recognition by transcription factors. Science. 2009; 324:1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmid S., Mordstein M., Kochs G., García-Sastre A., TenOever B.R.. Transcription factor redundancy ensures induction of the antiviral state. J. Biol. Chem. 2010; 285:42013–42022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Andrilenas K.K., Ramlall V., Kurland J., Leung B., Harbaugh A.G., Siggers T.. DNA-binding landscape of IRF3, IRF5 and IRF7 dimers: implications for dimer-specific gene regulation. Nucleic Acids Res. 2018; 46:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chow K.T., Driscoll C., Loo Y.-M., Knoll M., Gale M. Jr.. IRF5 regulates unique subset of genes in dendritic cells during West Nile virus infection. J. Leukoc. Biol. 2018; 105:411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mancino A., Termanini A., Barozzi I., Ghisletti S., Ostuni R., Prosperini E., Ozato K., Natoli G.. A dual cis-regulatory code links IRF8 to constitutive and inducible gene expression in macrophages. Genes Dev. 2015; 29:394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levy D.E., Kessler D.S., Pine R., Reich N., Darnell J.E.. Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev. 1988; 2:383–393. [DOI] [PubMed] [Google Scholar]
- 37. Tanaka N., Kawakami T., Taniguchi T.. Recognition DNA sequences of interferon regulatory factor 1 (IRF-1) and IRF-2, regulators of cell growth and the interferon system. Mol. Cell Biol. 1993; 13:4531–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fujii Y., Shimizu T., Kusumoto M., Kyogoku Y., Taniguchi T., Hakoshima T.. Crystal structure of an IRF-DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J. 1999; 18:5028–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fuertes Marraco S.A., Grosjean F., Duval A., Rosa M., Lavanchy C., Ashok D., Haller S., Otten L.A., Steiner Q.G., Descombes P. et al.. Novel murine dendritic cell lines: A powerful auxiliary tool for dendritic cell research. Front. Immunol. 2012; 3:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barish G.D., Yu R.T., Karunasiri M., Ocampo C.B., Dixon J., Benner C., Dent A.L., Tangirala R.K., Evans R.M.. Bcl-6 and NF- B cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010; 24:2760–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Daniel B., Nagy G., Hah N., Horvath A., Czimmerer Z., Poliska S., Gyuris T., Keirsse J., Gysemans C., Van Ginderachter J.A. et al.. The active enhancer network operated by liganded RXR supports angiogenic activity in macrophages. Genes Dev. 2014; 28:1562–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Siersbaek M.S., Loft A., Aagaard M.M., Nielsen R., Schmidt S.F., Petrovic N., Nedergaard J., Mandrup S.. Genome-wide profiling of peroxisome proliferator-activated receptor in primary epididymal, inguinal, and brown adipocytes reveals depot-selective binding correlated with gene expression. Mol. Cell Biol. 2012; 32:3452–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barta E. Command line analysis of ChIP-seq results. EMBnet. J. 2011; 17:13–17. [Google Scholar]
- 44. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nussbaum C., Myers R.M., Brown M., Li W. et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Consortium T.E.P., Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J. et al.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thorvaldsdóttir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. et al.. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012; 22:1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J.. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015; 109:doi:10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Uhlenhaut N.H., Barish G.D., Yu R.T., Downes M., Karunasiri M., Liddle C., Schwalie P., Hübner N., Evans R.M.. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol. Cell. 2013; 49:158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nakaya T., Sato M., Hata N., Asagiri M., Suemori H., Noguchi S., Tanaka N., Taniguchi T.. Gene induction pathways mediated by distinct IRFs during viral infection. Biochem Biophys Res Commun. 2001; 283:1150–1156. [DOI] [PubMed] [Google Scholar]
- 53. Brass A.L., Zhu A.Q., Singh H.. Assembly requirements of PU.1-Pip (IRF-4) activator complexes: Inhibiting function in vivo using fused dimers. EMBO J. 1999; 18:977–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eisenbeis C.F., Singh H., Storb U.. Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev. 1995; 9:1377–1387. [DOI] [PubMed] [Google Scholar]
- 55. De S., Zhang B., Shih T., Singh S., Winkler A., Donnelly R., Barnes B.J.. B cell-intrinsic role for IRF5 in TLR9/BCR-induced human B cell activation, proliferation, and plasmablast differentiation. Front. Immunol. 2018; 8:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tamura T., Thotakura P., Tanaka T.S., Ko M.S.H., Ozato K.. Identification of target genes and a unique cis element regulated by IRF-8 in developing macrophages. Blood. 2005; 106:1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eferl R., Wagner E.F.. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer. 2003; 3:859–868. [DOI] [PubMed] [Google Scholar]
- 58. Siggers T., Chang A.B., Teixeira A., Wong D., Williams K.J., Ahmed B., Ragoussis J., Udalova I.A., Smale S.T., Bulyk M.L.. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol. 2012; 13:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. DECKER T., KOVARIK P., MEINKE A.. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J. Interf. Cytokine Res. 1997; 17:121–134. [DOI] [PubMed] [Google Scholar]
- 60. Horvath C.M. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 2000; 25:496–502. [DOI] [PubMed] [Google Scholar]
- 61. Ivashkiv L.B., Donlin L.T.. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014; 14:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Glasmacher E., Agrawal S., Chang A.B., Murphy T.L., Zeng W., Vander Lugt B., Khan A.A., Ciofani M., Spooner C.J., Rutz S. et al.. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science. 2012; 338:975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tussiwand R., Lee W.L., Murphy T.L., Mashayekhi M., Kc W., Albring J.C., Satpathy A.T., Rotondo J.A., Edelson B.T., Kretzer N.M. et al.. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature. 2012; 490:502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li P., Spolski R., Liao W., Wang L., Murphy T.L., Murphy K.M., Leonard W.J.. BATF–JUN is critical for IRF4-mediated transcription in T cells. Nature. 2012; 490:543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ciofani M., Madar A., Galan C., Sellars M., MacE K., Pauli F., Agarwal A., Huang W., Parkurst C.N., Muratet M. et al.. A validated regulatory network for Th17 cell specification. Cell. 2012; 151:289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Iwata A., Durai V., Tussiwand R., Briseño C.G., Wu X., Grajales-Reyes G.E., Egawa T., Murphy T.L., Murphy K.M.. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat. Immunol. 2017; 18:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mould A.W., Morgan M.A.J., Nelson A.C., Bikoff E.K., Robertson E.J.. Blimp1/Prdm1 functions in opposition to Irf1 to maintain neonatal tolerance during postnatal intestinal maturation. PLoS Genet. 2015; 11:e1005375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Belz G.T., Nutt S.L.. Transcriptional programming of the dendritic cell network. Nat. Rev. Immunol. 2012; 12:101–113. [DOI] [PubMed] [Google Scholar]
- 69. Wienerroither S., Shukla P., Farlik M., Majoros A., Stych B., Vogl C., Cheon H.J., Stark G.R., Strobl B., Müller M. et al.. Cooperative transcriptional activation of antimicrobial genes by STAT and NF-κB pathways by concerted recruitment of the mediator complex. Cell Rep. 2015; 12:300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Park S.H., Kang K., Giannopoulou E., Qiao Y., Kang K., Kim G., Park-Min K.H., Ivashkiv L.B.. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat. Immunol. 2017; 18:1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Meraro D., Gleit-Kielmanowicz M., Hauser H., Levi B.-Z.. IFN-stimulated gene 15 is synergistically activated through interactions between the myelocyte/lymphocyte-specific transcription factors, PU.1, IFN regulatory factor-8/IFN consensus sequence binding protein, and IFN regulatory factor-4: characterization of a new subtype of IFN-stimulated response element. J. Immunol. 2002; 168:6224–6231. [DOI] [PubMed] [Google Scholar]
- 72. Fawcett T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006; 27:861–874. [Google Scholar]
- 73. Kaikkonen M.U., Spann N.J., Heinz S., Romanoski C.E., Allison K.A., Stender J.D., Chun H.B., Tough D.F., Prinjha R.K., Benner C. et al.. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell. 2013; 51:310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ostuni R., Piccolo V., Barozzi I., Polletti S., Termanini A., Bonifacio S., Curina A., Prosperini E., Ghisletti S., Natoli G.. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013; 152:157–171. [DOI] [PubMed] [Google Scholar]
- 75. Ramirez-Carrozzi V.R., Braas D., Bhatt D.M., Cheng C.S., Hong C., Doty K.R., Black J.C., Hoffmann A., Carey M., Smale S.T.. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 2009; 138:114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Au-Yeung N., Horvath C.M.. Histone H2A.Z suppression of interferon-stimulated transcription and antiviral immunity is modulated by GCN5 and BRD2. iScience. 2018; 6:68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mostafavi S., Yoshida H., Moodley D., Leboité H., Rothamel K., Raj T., Ye C.J., Chevrier N., Zhang S.Y., Feng T. et al.. Parsing the interferon transcriptional network and its disease associations. Cell. 2016; 164:564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Smale S.T., Tarakhovsky A., Natoli G.. Chromatin contributions to the regulation of innate immunity. Annu. Rev. Immunol. 2014; 32:489–511. [DOI] [PubMed] [Google Scholar]
- 79. Angermueller C., Pärnamaa T., Parts L., Stegle O.. Deep learning for computational biology. Mol. Syst. Biol. 2016; 12:878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li J., Ching T., Huang S., Garmire L.X.. Using epigenomics data to predict gene expression in lung cancer. BMC Bioinformatics. 2015; 16:S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Inukai S., Kock K.H., Bulyk M.L.. Transcription factor–DNA binding: beyond binding site motifs. Curr. Opin. Genet. Dev. 2017; 43:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Leung T.H., Hoffmann A., Baltimore D.. One nucleotide in a κB site can determine cofactor specificity for NF-κB dimers. Cell. 2004; 118:453–464. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq and ATAC-seq datasets have been deposited in GEO under accession number GSE125340.