


Abstract
Dynamic regulation is an effective strategy for fine-tuning metabolic pathways in order to maximize target product synthesis. However, achieving dynamic and autonomous up- and down-regulation of the metabolic modules of interest simultaneously, still remains a great challenge. In this work, we created an autonomous dual-control (ADC) system, by combining CRISPRi-based NOT gates with novel biosensors of a key metabolite in the pathway of interest. By sensing the levels of the intermediate glucosamine-6-phosphate (GlcN6P) and self-adjusting the expression levels of the target genes accordingly with the GlcN6P biosensor and ADC system enabled feedback circuits, the metabolic flux towards the production of the high value nutraceutical N-acetylglucosamine (GlcNAc) could be balanced and optimized in Bacillus subtilis. As a result, the GlcNAc titer in a 15-l fed-batch bioreactor increased from 59.9 g/l to 97.1 g/l with acetoin production and 81.7 g/l to 131.6 g/l without acetoin production, indicating the robustness and stability of the synthetic circuits in a large bioreactor system. Remarkably, this self-regulatory methodology does not require any external level of control such as the use of inducer molecules or switching fermentation/environmental conditions. Moreover, the proposed programmable genetic circuits may be expanded to engineer other microbial cells and metabolic pathways.
INTRODUCTION
The main objective of synthetic biology and metabolic engineering is to design and construct microbial cell factories, which could efficiently synthesize numerous valuable products including chemicals, biofuels, nutraceuticals or materials using renewable carbon sources from biomass such as glucose etc. (1–4). In order to enhance the metabolic flux into a target product, the synthesis modules are often strengthened at transcription or translation levels by engineering promoter or ribosomal binding site (RBS), while the competitive modules are generally knocked out or knocked down (5), in either static or dynamic manners. The static metabolic engineering, in which the expression of target genes is usually controlled by the constitutive genetic elements, may lead to metabolic imbalance, accumulation of intermediate products, and impairment of cell growth (6). Alternatively, the dynamic metabolic engineering, which aims at improving product synthesis by dynamic coordination of the metabolic networks with the help of native regulatory mechanisms from the cell, has been proposed and has found wide applications in the construction of microbial cell factories (7–10).
For implementing dynamic regulation, synthetic genetic circuits needs to be designed and constructed (11). This can be achieved by making a control switch that could regulate the related pathways by manually adding specific inducer or switching culture conditions such as temperature. For example, a metabolic toggle switch was built using two inducible promoters which were able to redirect the metabolic flux from cell growth to γ-aminobutyric acid synthesis at a desired time point (12). Other autonomous and dynamically regulated systems have been carried out by the aid of genetic modules such as metabolite-responsive biosensors or the pathway-independent quorum sensing (QS) systems. As an example, a genetic feedback loop was constructed by using FA/acyl-CoA biosensors derived from an allosteric transcription factor (aTF) FadR for the dynamic regulation of fatty acid-based products synthesis, and the regulation of fatty acid ethyl ester (FAEE) production by dynamic sensor-regulator system (DSRS) have been proved more efficient than static control mediated by a series of constitutive promoters (13). In addition, QS based switch circuits have been engineered as a broadly applicable tool to dynamically decouple cell growth and product synthesis because of its pathway-independent nature (14,15). However, autonomous and dynamic up-regulation and down-regulation on multiple targets simultaneously, which is widespread in natural regulatory networks, still needs further studies due to the current restrictions of tools and technologies (16).
Recently, the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) interference (CRISPRi) has been developed and used for transcription repression in virtue of dCas9, which was DNase inactivated and still could bind to specific site on DNA to block transcription by using the single guide RNA (sgRNA) (17). The regulation is easy to implement benefiting by its trans-acting mechanism, which avoided the genetic manipulation on the target genes. Instead, repression could be achieved after expressing dCas9 and sgRNA either integrated into the genome or in a plasmid. In addition, the regulation strength can be fine-tuned by adjusting the expression of dCas9 or sgRNA (18). The CRISPRi system has been widely used in the construction of genetic circuits due to its simplicity, specificity, and orthogonality (19). For example, combinatorial library of NOR gates were constructed in Saccharomyces cerevisiae using gRNAs based repression cascade (20). In another study, a set of 30 orthogonal NOT gates were built using the designed sgRNA-promoter pairs (21). In our previous work, a xylose induced CRISPRi system was constructed for dynamically knocking down competitive modules for the production of value-added nutraceutical N-acetylglucosamine (GlcNAc) in Bacillus subtilis (22). However, these systems were all manually switched by the addition of inducer molecules such as isopropyl β-D-1-thiogalactopyranoside (IPTG) and xylose. Recently, Moser et al. have developed three interesting Escherichia coli biosensors that respond to the consumption of feedstock (glucose), dissolved oxygen, and by-product accumulation (acetate), respectively, and achieved the signal conversion from activation to repression by using CRISPRi, sRNA, or proteases as an actuator (23). However, they only repressed poxB or pta in acetate synthetic pathway using gene circuits and did not perform dynamic activation simultaneously. Here, we attempted to combine CRISPRi with a metabolite-responsive biosensor to simultaneously achieve the autonomous dual-control (activation and repression) of targets genes. The regulation strengths of activation and repression could be adjusted to modulate the dynamic responsive range of the biosensor and such system would allow the engineering of multi-targets modules.
GlcNAc is a widely used nutraceutical for arthritic treatment and can be synthesized from glucose through multiple enzymatic steps including phosphorylation (from glucose to glucose-6-phosphate), isomerization (from glucose-6-phosphate to fructose-6-phosphate), amination (from fructose-6-phosphate to glucosamine-6-phospahte), acetylation (from glucosamine-6-phospahte to N-acetylglucosamine-6-phosphate) and dephosphorylation (24). In the synthesis pathway of GlcNAc, the intracellular concentration of intermediate metabolite glucosamine-6-phosphate (GlcN6P) is tightly regulated by the feedback inhibition of glmS ribozyme in B. subtilis (25), and the GlcNAc titer was greatly improved after removing the feedback inhibition by ribozyme deletion (24). GlcN6P is a key metabolic precursor of cell wall, and it also plays an important role in the synthesis of the other nutraceuticals such as hyaluronic acid or Lacto-N-Tetraose (24,26,27). There are three competing pathways, namely, glycolysis (EMP), pentose phosphate pathway (HMP), and peptidoglycan synthesis pathway (PSP) in the production of GlcNAc from glucose by B. subtilis. However, these pathways cannot be completely removed because they are all essential for normal cell growth (22). In order to construct a genetic feedback circuits to fine-tune the metabolic flux distribution between cell growth and GlcNAc synthesis, here we designed and built a series of GlcN6P-responsive biosensors, which could activate gene expression after sensing the intracellular GlcN6P. To construct these biosensors, we hacked the aTF GamR that is associated with amino sugar catabolism of B. subtilis (28). And on this basis, an autonomous dual-control (ADC) system, which could be used to dynamically activate and repress target genes expression at the same time, was constructed by coupling the GlcN6P-responsive biosensor with CRISPRi based NOT gates. A feedback loop was designed and constructed by repressing the competitive modules and activating the synthesis module of GlcNAc simultaneously using the GlcN6P-responsived ADC system (Figure 1A). The competitive modules of GlcNAc will be repressed with the control of the GlcN6P-responsive repression cascade and this further promoted the accumulation of GlcN6P from glucose. At the same time, the synthesis module will be activated by the biosensor to channel GlcN6P into GlcNAc. By fine-tuning the metabolic flux of the synthesis module and competitive modules with the ADC enabled feedback circuits and blocking the formation of the by-product acetoin, which is an overflow product from pyruvate (29), the GlcNAc titer in a 15-l bioreactor was significantly improved. In addition, the biosensors built here may be generally used for the dynamic regulation, directed evolution, and high-throughput screening in GlcN6P related metabolites producing cells. In addition, here the proposed programmable genetic biosensor-CRISPRi circuits may be used for the construction of the other microbial cell factories.
Figure 1.

GlcN6P biosensor based synthetic genetic circuits for the regulation of N-acetylglucosamine (GlcNAc) bioproduction in B. subtilis. (A) A feedback loop was constructed using the glucosamine-6-phosphate (GlcN6P) biosensors and CRISPRi based NOT gates. The GlcNAc synthesis was upregulated by the GlcN6P biosensor, and the competitive pathways were downregulated by the GlcN6P-responsive repression cascade. The by-product acetoin, which is an overflow product from pyruvate, was eliminated by knocking out alsSD. (B) Absorption and metabolism of GlcN and GlcNAc in B. subtilis. (C) Regulation of the GlcN and GlcNAc metabolic associated pathways in B. subtilis, which are controlled by the GlcN6P responsive allosteric transcription factor (aTF) NagR and GamR, respectively. (D) The GlcN6P biosensor was constructed based on the regulatory mechanism of GamR on promoter PgamA. GamR could bind to specific sequences (gamO1 and gamO2) when the intracellular GlcN6P concentration is low and block the binding of RNA polymerase to the promoter that is necessary for transcription initiation. When the intracellular GlcN6P concentration increases, the DNA-binding activity of GamR is antagonized by the allosteric regulation of GlcN6P, and RNA polymerase can bind to the promoter initiating transcription. Glc6P: glucose-6-phosphate, Fru6P: fructose-6-phosphate, ptsG: PTS system glucose-specific EIICB component, pgi: glucose-6-phosphate isomerase, glmS: glutamine-fructose-6-phosphate aminotransferase, GNA1: GlcN6P N-acetyltransferase, zwf: glucose-6-phosphate 1-dehydrogenase, pfkA: 6-phosphofructokinase, glmM: phosphoglucosamine mutase, alsS: acetolactate synthase, alsD: acetolactate decarboxylase, gamR: GlcN6P responsive transcriptional regulator.
MATERIALS AND METHODS
Chemicals and reagents
All chemicals were purchased from Sangon Biotech (Shanghai, China) unless otherwise specified. The GlcNAc and acetoin standards were purchased from Sigma-Aldrich (Shanghai, China). Restriction enzymes, DNA gel purification kit, and plasmid extraction kit were purchased from Thermo Scientific (Waltham, USA). PrimeSTAR HS DNA polymerase, and T4 DNA ligase were purchased from Takara Biomedical Technology (Beijing, China). Seamless Cloning Kit was purchased from Beyotime Biotechnology (Shanghai, China). Oligonucleotides were synthesized by GENEWIZ (Suzhou, China).
Strains, plasmids, and cultivation conditions
The strains and plasmids used in this study are listed in Table 1 and Supplementary Table S1, respectively. E. coli JM109, used for vector construction, was cultured in Luria-Bertani (LB) medium (10 g/l tryptone, 5 g/l yeast extract and 5 g/l NaCl). The following antibiotics were used for selections: ampicillin, 100 μg/ml; kanamycin, 50 μg/ml; chloromycetin, 5 μg/ml; zeocin, 30 μg/ml; and tetracycline, 20 μg/ml.
Table 1.
Strains used in this study
| Strain | Characteristics | Ref. |
|---|---|---|
| Escherichia coli JM109 | recA1, endA1, thi, gyrA96, supE44, hsdR17Δ (lac-proAB) /F’[traD36,proAB+, laclq, lacZΔ M15] | Lab stock |
| Bacillus subtilis 168 | trpC2 | Lab stock |
| BS01 | B. subtilis 168 derivate, ΔgamR::lox72 | This work |
| BS02 | B. subtilis 168 derivate, ΔnagBΔgamA::lox72 | This work |
| BS03 | BS01 derivate, ΔnagBΔgamA::lox72 | This work |
| BS13 | BS03 derivate, ΔamyE:: sgGFP, ΔlacA::gamR-Pvg6-dCas9, pP43-GFP | This work |
| BS23 | BS03 derivate, ΔamyE:: sgGFP, ΔlacA::gamR-PgamA-dCas9, pP43-GFP | This work |
| BS33 | BS03 derivate, ΔamyE:: sgGFP, ΔlacA::gamR-Psg2-dCas9, pP43-GFP | This work |
| BS34 | BS33 derivate, pHTcsg2m | This work |
| BNY | B. subtilis 168ΔnagPΔgamPΔgamAΔldhΔptaΔglcKΔpckAΔpyk::lox72, ΔnagAB::Pveg-yqaB, Pveg-glmS, pP43-GAN1 | (22) |
| BNX122 | BNY derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::xylR-PxylA-dCas9, | (22) |
| BNY0 | BNY derivate, eliminating plasmid pP43-GNA1, ΔgamR::lox72 | This work |
| BNX0 | BNX122 derivate, eliminating plasmid pP43-GNA1, ΔgamR::lox72 | This work |
| BNDR000 | BNY0 derivate, ΔlacA:: gamR-PgamA-dCas9, pP43-GNA1 | This work |
| BNDR010 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-Pvg6-dCas9, pP43-GNA1 | This work |
| BNDR020 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-PgamA-dCas9, pP43-GNA1 | This work |
| BNDR030 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-Psg2-dCas9, pP43-GNA1 | This work |
| BNDR001 | BNY0 derivate, ΔlacA:: gamR-PgamA-dCas9, pSTv-GNA1 | This work |
| BNDR011 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-Pvg6-dCas9, pSTv-GNA1 | This work |
| BNDR021 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-PgamA-dCas9, pSTv-GNA1 | This work |
| BNDR031 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-Psg2-dCas9, pSTv-GNA1 | This work |
| BNDR002 | BNY0 derivate, ΔlacA:: gamR-PgamA-dCas9, pSTg-GNA1 | This work |
| BNDR012 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-Pvg6-dCas9, pSTg-GNA1 | This work |
| BNDR022 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-PgamA-dCas9, pSTg-GNA1 | This work |
| BNDR032 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-Psg2-dCas9, pSTg-GNA1 | This work |
| BNDR003 | BNY0 derivate, ΔlacA:: gamR-PgamA-dCas9, pSTs-GNA1 | This work |
| BNDR013 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-Pvg6-dCas9, pSTs-GNA1 | This work |
| BNDR023 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-PgamA-dCas9, pSTs-GNA1 | This work |
| BNDR033 | BNY0 derivate, ΔamyE:: sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-Psg2-dCas9, pSTs-GNA1 | This work |
| BNDR040 | BNY0 derivate, P43-ptsG, P43-pgi, pP43-GNA1 | This work |
| BNDR041 | BNY0 derivate, P43-ptsG, P43-pgi, pSTv-GNA1 | This work |
| BNDR042 | BNY0 derivate, P43-ptsG, P43-pgi, pSTg-GNA1 | This work |
| BNDR043 | BNY0 derivate, P43-ptsG, P43-pgi, pSTs-GNA1 | This work |
| BNDR122 | BNDR022 derivate, ΔalsSD::lox72 | This work |
| BNDR222 | BNDR022 derivate, ΔthrC:: sgalsS1 | This work |
| BNDR322 | BNDR022 derivate, ΔthrC:: sgalsS2 | This work |
| BNDR422 | BNDR022 derivate, ΔthrC:: sgalsS3 | This work |
| BNDR522 | BNDR022 derivate, ΔthrC:: sgalsS4 | This work |
| BNDR622 | BNDR022 derivate, ΔthrC:: sgalsS5 | This work |
| BNDR100 | BNDR000 derivate, ΔalsSD::lox72 | This work |
DNA manipulation
The primers used for gene cloning are listed in Supplementary Table S2, and the sequences of genetic parts used in this study are listed in Supplementary Table S3. PCR and DNA digestion were performed according to the manufacturer's instruction. Ligation was constructed using the Seamless Cloning Kit, which facilitated the ligation of two or more DNA fragments through a 20 bp overlap. Escherichia coli JM109 was transformed with plasmids using heat shock, and B. subtilis was transformed by the modified method of Anagnostopoulos & Spizizen (Supplementary method) (30). A marker-free genome editing approach was used to perform gene knockout or overexpression in B. subtilis as previously reported (31).
The trap plasmid pHTa0 used for promoters testing was constructed by inserting egfp amplified from pP43-egfp into linearized pHT01 generated by PCR using prime pairs egfp-pHT01-F/egfp-pHT01-R and pHT01-li-F/pHT01-li-R. Promoters Pveg and PgamA were amplified from B. subtilis genome using primer pairs Pveg-pHTa0-R/Pveg-pHTa0-R and PgamA-pHTa0-F/PgamA-pHTa0-R, and then inserted between EcoRI and KpnI restriction sites of pHTa0 by Seamless Cloning resulting in pHTave and pHTaga, respectively. The hybrid promoters containing gamO1 in Pveg were produced by the one-step site-directed plasmid mutagenesis protocol (32) in the order shown in the Figure 2A. Briefly, mutagenesis was generated by inverse PCR of the plasmid using the corresponding primers, and the PCR product digested by DpnI was transformed into E. coli JM109. Colonies with related mutations were verified by PCR and confirmed by DNA sequencing. The hybrid promoters jointed by PgamA with different promoters was constructed by inserting the promoter fragment amplified from B. subtilis genome into linearized pHTaga generated by PCR using prime pairs PlepA-pHTaga-F/PlepA-pHTaga-R, PrelA-pHTaga-F/PrelA-pHTaga-R, PsrfA-pHTaga-F/PsrfA-pHTaga-R, Pveg-pHTaga-F/Pveg-pHTaga-R, and pHTa-pHTc-liF/pHTa-pHTc-liR in the order shown in the Figure 2A. Plasmids pHTcvg6,pHTcga, and pHTcsg2 were produced by inserting gamR with its promoter amplified from B. subtilis genome into linearized pHTavg6, pHTaga, and pHTasg2 generated by PCR using prime pairs gamR-pHTc-F/gamR-pHTc-R and pHTa-pHTc-liF/pHTa-pHTc-liR. The native promoter of gamR in plasmid pHTcsg2 was replaced by the promoter PxylA using primer pairs pHTc-pHTd-liF/pHTc-pHTd-liR and PxylA-pHTd-F/PxylA-pHTd-F and generated pHTdsg2. The egfp in plasmid pHTcsg2 was replaced by mcherry using primer pairs pHTcm-liF/pHTcm-liR and mcherry-pHTc-F/mcherry-pHTc-R and generated pHTcsg2m, and a N-terminal coding sequence of abrB was added into mCherry to improve its expression (33).
Figure 2.

Design and construction of the GlcN6P-responsive hybrid promoters. (A) Construction flow diagram of hybrid promoters, which were obtained by inserting the GamR binding site gamO2 into a σA RNA polymerase recognized constitutive promoter Pveg or directly joining together the downstream part of −10 region of PgamA with the upstream part of −10 region of different constitutive promoters. (B) DNA sequence of hybrid promoters compared with the native promoters PgamAand Pveg. The bold and underlined sequences represent −10 and −35 regions. The GamR binding site gamO1 is shown with blue underline. The sequences in blue and italics represent another GamR binding site gamO2. Transcript start sites are colored red. The sequences with blue background represent the downstream part of −10 region of PgamA. The sequences with gray background represent the upstream part of −10 region of the constitutive promoter. (C) The strength of each promoter in strains with or without GamR, respectively. Numbers above each bar represent the dynamic ranges. Differences were determined by two-tailed Student's t-test. All data were the average of three independent studies with standard deviations.
To achieve the GlcN6P-responsive biosensors mediated transcription repression, primer pairs pLCx-dCas9-liF/pLCx-dCas9-liR and Biosen-dCas9-F/Biosen-dCas9-R were used to replace the xylose induced promoter xylR-PxylA in pLCx-dCas9 with gamR-Pvg6, gamR-PgamA and gamR-Psg2 resulting pLCv-dCas9, pLCg-dCas9, and pLCs-dCas9, respectively. To achieve the GlcN6P-responsive biosensors mediated GNA1 expression, promoter P43 used for GNA1 expression in pP43-GNA1 was replaced by Pvg6, PgamA and Psg2 respectively. However, these new constructed plasmids cannot be transformed into B. subtilis for some unknown reason. Hence, plasmid pSTOP1622 that possessed the same B. subtilis replicon with pP43-GNA1 was used as a vector for the expression of GNA1 under GlcN6P-responsive promoters. And primer pairs pSTOP1622-li-F/pSTOP1622-li-R, Pbiosen-pST-F/PgamA-pST-R, Pbiosen-pST-F/Psg2-pST-R, Pbiosen-pST-F/Pvg6-pST-R, and GNA1-pST-F/GNA1-pST-R were used to generate pSTv-GNA1, pSTg-GNA1, and pSTs-GNA1. The sgRNAs targeting to alsS were generated as previously reported (22), and were integrated into the genome of strain BNDR022 using the fusion segments of thrC homologous arms and corresponding sgRNA.
Fluorescence assays
The recombinant B. subtilis strains containing the fluorescence plasmids were pro-cultured in LB medium for 10 h and further inoculated into 200 μl LB medium with 1% proportion at 96-well black plates (corning 3603). The 96-well plates were subsequently cultured at 37°C with shaking at 900rpm. The following minimal media was also used for fluorescence assays: 54 g/l K2HPO4·3H2O, 30 g/l KH2PO4·3H2O, 5 g/l sodium citrate, 1 g/l MgSO4, 10 g/l K2SO4, 2 g/l glutamine, 5 μM MnCl2, 50 μM FerCl3, 1 mg/l vitamin B1, 0.1 g/l tryptophan and 4 g/l sucrose. GFP fluorescence (excitation, 490 nm; emission, 530 nm), mCherry fluorescence (excitation, 580 nm; emission, 610 nm), and optical density (absorbance at 600 nm) were measured at early stationary phase (∼22 h) using a microplate Multi-Mode Reader (BIOTEK, Cytation 3) directly (34). The background fluorescence of the strain without fluorescent protein expression (FPbg) and the background OD of the medium (ODbg) were corrected, and the relative fluorescence intensities was calculated equation (1) as follows (35).
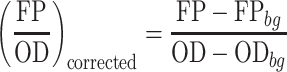 |
(1) |
Batch fermentation in shake flasks
During shake flask fermentations, the following fermentation medium was used: 6 g/l urea, 12 g/l yeast extract, 6 g/l tryptone, 12.5 g/l K2HPO4·3H2O, 2.5 g/l KH2PO4·3H2O, 3 g/l MgSO4, and 10 ml/l trace metal solution. The trace metal solution contained 4 g/l FeSO4·7H2O, 4 g/l CaCl2, 1 g/l MnSO4·5H2O, 0.4 g/l CoCl2·6H2O, 0.2 g/l NaMoO4·2H2O, 0.2 g/l ZnSO4·7H2O, 0.1 g/l AlCl3·6H2O, 0.1 g/l CuCl2·H2O and 0.05 g/l H3BO4. Glucose was sterilized separately, and added to the shake flask to a final concentration of 75 g/l. The engineered B. subtilis strains were streaked to LB plates, and single colonies were picked into 20 ml LB medium at 37°C with shacking at 220 rpm for 10 h in 250 ml shake flasks. The seed cultures were further inoculated into 50 ml fermentation medium with 5% proportion at 250 ml baffled flask and grown at 37°C with shaking at 220 rpm for 60 h, and three replicates were set for each strain. One milliliter of cell suspension was sampled every 12 h for OD600, glucose, GlcNAc, and by-product acetoin measurements. The following minimal media was also used for shake flask fermentations: 6 g/l urea, 54 g/l K2HPO4·3H2O, 30 g/l KH2PO4·3H2O, 5 g/l sodium citrate, 1 g/l MgSO4, 10 g/l K2SO4, 2 g/l glutamine, 5 μM MnCl2, 50 μM FerCl3, 1 mg/l vitamin B1, 0.1g/l tryptophan and 75 g/l glucose.
Fed-batch culture in 15-l bioreactor
The fermentation medium used for fed-batch culture consisted of 20 g/l yeast extract, 20 g/l tryptone, 10 g/l urea, 12.5 g/l K2HPO4·3H2O, 2.5 g/l KH2PO4 and 10 ml/l trace metal solution. The trace metal solution contained 4 g/l FeSO4·7H2O, 4 g/l CaCl2, 1 g/l MnSO4·5H2O, 0.4 g/l CoCl2·6H2O, 0.2 g/l NaMoO4·2H2O, 0.2 g/l ZnSO4·7H2O, 0.1 g/l AlCl3·6H2O, 0.1 g/l CuCl2·H2O and 0.05 g/l H3BO4. Seed culture was carried out in baffled 500-ml shake flasks containing 100 ml of LB medium at 37°C with shacking at 220 rpm for 12 h. The seed culture (300 ml) was inoculated into a 15-l fermenter (T&J-Dtype; T&J Bio-engineering Co., Ltd, Shanghai, China) with an initial 5.5 l of fermentation medium. The pH was kept at 7.0 via the addition of 29% NH3, and the temperature was maintained at 37°C. The aeration rate and agitation speed were 1.5 vvm and 900 rpm, respectively. The glucose concentration was maintained within 2–30 g/l by feeding with 750 g/l glucose, and the feeding rates were adjusted based on the change of glucose concentrations in the fermentation medium. A total of 1 l nitrogen source feeding solution contained 140 g yeast extract, 140 g tryptone, and 70 g urea was pumped into the fermenter at a rate of 30 ml/h when OD600 reached 30.
Analytical methods
Cell densities were monitored by measuring the optical density at 600 nm (OD600). The OD600 value was converted to dry cell weight (DCW) according to the equation 1OD600 = 0.35 g/l DCW . The concentrations of GlcNAc and the by-product acetoin in the fermentation broth were measured by high-performance liquid chromatography (HPLC) on an instrument (Agilent 1260) equipped with an HPX-87H column (Bio-Rad, Hercules, CA, USA) and a refractive index detector. Aqueous H2SO4 (5 mM) was used as the mobile phase at a flow rate of 0.6 ml/min at 40°C. Glucose was measured using a glucose-lactate analyzer (M100, Shenzhen Sieman Technology Co., Ltd, Shenzhen, China). The intracellular GlcN6P was measured using previously reported methods (36). Fluorescence characterization with cytometry was performed on a BD FACSAria III(BD, Franklin Lakes, NJ, USA). Cells diluted in PBS were run at a rate of 0.5 μl/s. The events were gated to reduce false events, and 50000 events were used for analysis after gating (Supplementary Figure S1). The relative transcription levels of the regulated genes were determined by RT-qPCR as previously described (22), and the rpsJ gene was used as the internal standard (37). All experiments were independently carried out at least three times.
Statistical analysis
All data were expressed as mean ± SD. Differences between two groups were determined by two-tailed Student's t-test, and one-way ANOVA followed by post-hoc Dunnett's test for multiple groups. Statistical significance is indicated as * for P < 0.05 and ** for P < 0.01, respectively.
RESULTS AND DISCUSSION
Developing GlcN6P biosensors by hacking the aTF GamR in B. subtilis
The catabolic metabolism of GlcN and GlcNAc are both regulated by GlcN6P in B. subtilis (28,38). When either GlcN or GlcNAc enters the cell, GlcN6P and GlcANc6P are synthesized, respectively, via PTS (phosphotransferase system) (39). GlcNAc6P can be further deacetylated to GlcN6P, which can be used for the synthesis of peptidoglycan or enter glycolysis after deamination, generating frucose-6P (Fru6P) (Figure 1B). The expression of genes related to the catabolic metabolism of GlcN and GlcNAc is controlled by the GntR family regulators NagR and GamR, both of which are GlcN6P-responsive aTF (28) (Figure 1C). NagR can interact with GlcNAc6P except for GlcN6P (38), while GamR is unsensitive to GlcNAc6P and thus it was chosen to construct the GlcN6P biosensor. There are two binding sites of GamR in the promoter PgamA. One of them (gamO1: TAAATTCGTAATGACAA) overlapped the −10 region and +1 site of the promoter and another (gamO2: TAAATTGGTCATAACAA) exited 9 bp downstream of the +1 site (Figure 1D). When the intracellular GlcN6P concentration is low, GamR binds to these two sites in the promoter, which prevents RNA polymerase from binding to the promoter and represses the transcription of the downstream gene; When the intracellular GlcN6P concentration is high, the DNA-binding activity of GamR is abolished by the allosteric regulation of GlcN6P, which enables the binding of RNA polymerase to the promoter and turns on transcription (28,40). First, the activity of promoter PgamA with and without the binding of GamR, as indicated by the fluorescence intensity, was tested by putting it in front of GFP in the trap plasmid pHTa0 and transforming to strain B. subtilis 168 and BS01 (B. subtilis 168△gamR), respectively. The result shows that it possesses a dynamic range of 5.4 with or without the binding of GamR, and the full strength of the promoter PgamA without GamR binding is 2.27-fold that of the strength of a constitutive promoter Pveg that was recognizable by the native B. subtilis σA RNA polymerase (Figure 2B).
On the basis of the above work, 14 hybrid promoters were constructed for the purpose of getting more GlcN6P-reponsive biosensors with different dynamic responsive ranges. The general way to construct a hybrid promoter is inserting an aTF binding site into a constitutive promoter (13,41), hence the hybrid promoters were designed by inserting the GamR binding site gamO2 into the promoter Pveg (Figure 2B). These promoters were inserted in front of GFP in the trap plasmid pHTa0, which was then transformed to the GamR− and GamR+ strains, respectively. Promoter Pvg2 that was obtained by inserting gamO2 between −10 and −35 region had a very weak strength with almost no dynamic range; Promoters Pvg1 and Pvg3 that were obtained by adding gamO2 at the downstream of +1 site or at the upstream of −35 region were both stronger than Pveg, but did not have dynamic range; Adding another gamO2 into downstream of Pvg2 resulted in Pvg4, the strength of which was also very weak; Adding another gamO2 at downstream of +1 site of Pvg3 generated Pvg5 that processed similar strength with Pvg3 but had a weak dynamic range (0.3); Pvg6 that was acquired by inserting another gamO2 between −10 and −35 region of Pvg3 possessed a dynamic range of 3.6 and a similar full strength with Pveg; Although Pvg7 that was obtained by adding another gamO2 at the downstream of +1 site of Pvg6 possessed a higher dynamic range (4.6), its full strength was reduced by 28.4% compared with Pvg6. Taken together, the promoter Pvg6 was chosen for further study.
The two GamR binding sites gamO1 and gamO2 were both located in the downstream of the −10 region of promoter PgamA, therefore, hybrid promoters may also be constructed by directly joining together the downstream part of −10 region of PgamA with the upstream part of −10 region of different constitutive promoters (Figure 2B). Four constitutive promoters PrelA, PlepA, Pveg and a PsrfA mutant P10 (42,43) that possessed different strengths were joined with PgamA as mentioned above, resulting in Prg, Plg, Pvg and Psg, respectively (Figure 2B). The full strengths of these hybrid promoters were 21.2%, 74.1%, 91.6% and 3.67-fold that of the strength of Pveg, respectively (Figure 2C), which were coincident with the previously reported strengths of these constitutive promoters (42,43). In order to reduce the basal expression of promoter Psg, which possessed a 1.29-fold strength of that of Pveg in the GamR+ strain, extra gamO2 was added. Both Psg1 and Psg3 had a gamO2 addition between -10 and -35 region and possessed very weak strength, which was consistent with the previous result for the construction of Pvg2. Psg2 that was obtained by adding another gamO2 at upstream of −35 region had a similar full strength with Psg, but its dynamic range was improved to 13.4. The promoter PsrfA was previously classified as a quorum-sensing related promoter in B. subtilis (44), however, the expression profile over times of Psg2 was not significantly different from that of Pvg6 and PgamA (Supplementary Figure S2), perhaps as a result of the truncation and mutation performed here.
Construction of the GlcN6P-responsive autonomous dual-control system
To explore the dose-dependent characteristics of the GlcN6P-responsive promoters, the catabolic pathway of GlcN6P was blocked by knocking out simultaneously nagB and gamA, which could transform GlcN6P to Fru6P, in strains B. subtilis 168 and BS01(ΔgamR), resulting in strains BS02 and BS03, respectively. Thus, the intracellular GlcN6P in these strain can be controlled by adding GlcN into the media (Supplementary Figure S3A (45)). The plasmids pHTaga, pHTasg2 and pHTavg6 were transformed into the GamR+ strain BS02 (B. subtilis 168△nagB△gamA), and the biosensor plasmids containing both gamR and the GlcN6P-responsive promoters were transformed into the GamR− strain BS03 (B. subtilis 168△gamR△nagB△gamA). These strains were pre-cultured in LB medium for 10 h and further inoculated into LB medium with different concentrations of GlcN. The fluorescence intensity was determined at the early stationary phase, and the data were fitted to the equation (2) as follows:
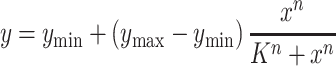 |
(2) |
where y is relative expression activity of the promoter (ymin and ymax are the minimum/maximum activities), x is the GlcN concentration, K is the threshold, and n is the cooperativity (46), and these function parameters are listed in Table 2. The initial extracellular concentration of the inducer was often used as input to fit the function because it was easy to control (46). Of course, the correlation between the intracellular and the extracellular concentrations of the inducer is also considered. Here although the intracellular GlcN6P changes with time, there are good correlations between intracellular GlcN6P and extracellular GlcN over the time frame that promoter characterization measurements are taken (Supplementary Figure S3A). Hence, we directly used the initial extracellular GlcN concentration as the input of the function, and that fit well to the equation. It is worth mentioning that the parameters presented in Table 2 are empirically-derived and cannot be interpreted as physical parameters, but it provides a good quantitative handle to compare relative strengths and dynamic ranges of the promoters tested. Whether the TF GamR was expressed either in the plasmid (Figure 3A) or in the genome (Supplementary Figure S4A), the strengths of all these promoters increased with the concentration of GlcN added into the media, and the highest strengths of these promoters were similar to their full strengths without GamR determined above. However, the response function parameters of these GlcN6P-responsive promoters were different with the change of GamR position (Table 2), suggesting that the dose-dependent characteristics correlated to the expression level of GamR, which is consistent with a previous study (16). The performance of the biosensor in minimal media was also tested, and the trends were the same as that in the complex media except for the decrease of the overall fluorescence intensities, which may result from the weaker protein expression in this barren environment (Supplementary Figure S4B).
Table 2.
Response function parameters
| Figure | Promoter | y min | y max | K | n | R 2 | Equation |
|---|---|---|---|---|---|---|---|
| S5A | Pvg6 | 410.6±7.48 | 2205.8±52.54 | 1.9±0.11 | 3.3±0.33 | 0.994 | (2) |
| S5A | PgamA | 747.3±25.97 | 5317.4±37.45 | 1.5±0.05 | 3.8±0.25 | 0.999 | (2) |
| S5A | Psg2 | 547.9±17.47 | 8234.6±31.09 | 1.2±0.04 | 3.0±0.18 | 0.999 | (2) |
| S5B | Pvg6 | 45.8±0.68 | 308.8±3.79 | 1.4±0.07 | 5.5±0.61 | 0.998 | (2) |
| S5B | PgamA | 48.3±3.61 | 544.1±4.77 | 0.92±0.01 | 2.9±0.11 | 0.999 | (2) |
| S5B | Psg2 | 29.2±4.05 | 1171.4±17.47 | 1.4±0.04 | 3.8±0.19 | 0.998 | (2) |
| 3A | Pvg6 | 108.6±6.56 | 2309.2±113.79 | 1.9±0.13 | 2.1±0.15 | 0.992 | (2) |
| 3A | PgamA | 179.2±92.75 | 5444.4±114.62 | 1.6±0.09 | 2.5±0.25 | 0.996 | (2) |
| 3A | Psg2 | 78.6±11.05 | 8471.1±202.69 | 1.5±0.08 | 2.7±0.15 | 0.997 | (2) |
| 3B | Pvg6 | 6851.3±40.26 | 15091.8±131.70 | 1.0±0.01 | 2.9±0.25 | 0.999 | (3) |
| 3B | PgamA | 1643.6±38.58 | 15210.5±136.52 | 0.64±0.009 | 2.7±0.07 | 0.999 | (3) |
| 3B | Psg2 | 139.6±11.03 | 14821.8±138.88 | 0.64±0.02 | 3.2±0.13 | 0.999 | (3) |
| 3C | Psg2 | 49.6±2.97 | 15319.7±94.13 | 0.89±0.010 | 4.2±0.07 | 0.999 | (2) |
| 3C | Psg2 | 87.1±8.43 | 14969.5±55.50 | 0.66±0.008 | 3.9±0.10 | 0.999 | (3) |
Response data of activation or repression were fitted to equation (2) or equation (3), respectively. Explanation about these parameters are provided in the main text. The parameters presented in the table are empirically-derived and cannot be interpreted as physical parameters, but that provide a good quantitative handle to compare relative strengths and dynamic ranges of the promoters tested.
Figure 3.

GlcN6P biosensors mediated transcription activation and repression. (A) The dose-dependent characteristics of the GlcN6P biosensors in strain BS03 (B. subtilis 168ΔgamRΔnagBΔgamA). GlcN was added into the media for improving the intracellular GlcN6P concentration. (B) The response of GlcN6P-responsive repression cascade constructed by the CRISPRi based NOR gate. (C) Verification of the function of the GlcN6P-responsive autonomous dual-control (ADC) system. Promoter Psg2 was used both for the expression of mCherry and dCas9. Flow cytometry scatter plots shows GFP and mCherry fluorescence regulated by the ADC system, and the induction of the GlcN6P biosensor promotes the switch from the GFP+ to the mCherry+ state. The means of GFP and mCherry were calculated and marked in the figure. All data were the average of three independent studies with standard deviations.
In order to further explore the effects of the TF gamR expression on the biosensor response, the native promoter of gamR in the biosensor plasmid pHTcsg2 was replaced by a xylose induced promoter PxylA, and xylose was added to regulate gamR expression. As shown in Supplementary Figure S4C, the response range of the GlcN6P biosensor could be up-regulated by increasing gamR expression, which is consistent with the above results. When xylose was not added into the media, the leakage expression of promoter PxylA is enough to give promoter Psg2 a certain dynamic range; When 10 g/l xylose was added to strongly induce gamR expression, the response threshold value was close to the GlcN concentration that can achieve fully induced in the above study (5 mM). This seems to indicate that an enhanced expression of gamR may increase the response threshold value and reduce the sensitivity of the biosensors. However, this characteristic could increase the flexibility when these GlcN6P biosensors are used for directed evolution, high-throughput screening, or dynamic regulation.
For the construction of sensitive cell factories, responsive repression is needed in addition to responsive activation (47). For instance, the two regulators mediated layered dynamic regulation with activation and repression have been employed for glucaric acid synthetic in E. coli (16). In addition, single regulator based dual regulation was also reported by coupling some negative regulation elements such as RNAi and asRNAs (48,49). In order to achieving the autonomous dual-control, we attempted to construct a GlcN6P-responsive repression cascade by coupling the above created GlcN6P-responsive promoters with CRISPRi based NOT gate (22). In order to achieve this aim, the xylose induced promoter PxylA used for dCas9 expression in plasmid pLCx-dCas9 was replaced by Pvg6, PgamA and Psg2, resulting in pLCv-dCas9, pLCg-dCas9, and pLCs-dCas9, respectively. These plasmids were linearized and integrated into the genome of strain BS03 for the verification of the repression effect by using the constitutive expression GFP as an indicator, and the data were fitted to the equation (3) as follows:
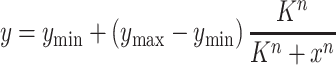 |
(3) |
where y is relative expression activity of the promoter, x is the GlcN concentration, ymin is the basal expression, ymax is the peak values, K is the threshold, and n is the cooperativity (21), and these function parameters are listed in Table 2. As mentioned above, here the extracellular GlcN concentration was used as the input of the function as well. It was reported that high-level dCas9 expression is toxic to E. coli and will lead to abnormal cell morphology (50), but that was not observed by using dCsa9 as a repression cascade in our study (Supplementary Figure S5), which may be due to the low expression level with the integrated expression in the genome.
As shows in Figure 3B, the repression strength was increased with the increase of GlcN concentration, and the repression effect showed a positive correlation with the strength of these promoters (Pvg6 < PgamA < Psg2). Then the function of GlcN6P-responsive ADC system was tested by putting the GlcN6P activation and repression cascades into the same cell (Figure 3C). The fluorescent proteins mCherry and GFP were used as indicators for activation and repression, respectively, and the data were fitted to equation (2) and equation (3), respectively. In addition, flow cytometry analysis was performed to confirm the gradually change from GFP+ to mCherry+ state with the increase of regulation intensity. The means of GFP and mCherry measured by flow cytometry correlated well with the results of microplate reader (Supplementary Figure S6). These results suggest that the GlcN6P-responsive ADC system was successfully constructed by the combination of the GlcN6P biosensors with the CRISPRi based NOT gate. As shown in Figure 3C, bi-modality was observed in flow cytometry analysis, which reflected the metabolite heterogeneity of intracellular GlcN6P at low extracellular GlcN concentrations (51). This also means that the GlcN6P biosensor could distinguish these heterogenous populations and has the ability to perturb the metabolism within individual cells. In addition, flow cytometry analysis of the biosensor in the strains with enhanced GlcN6P synthesis showed a single shifted single peak in fluorescence, which further confirmed that the bi-modality came from metabolite heterogeneity of GlcN6P rather than the characteristics of the biosensor (Supplementary Figure S7).
Reprograming the GamR regulon for the dynamic regulation of GlcNAc synthesis
In our previous study, a xylose induced CRISPRi system was used to relieve carbon catabolite repression of B. subtilis and promote GlcNAc synthesis from glucose and xylose (22). However, using the mixture of glucose and xylose will increase the cost of industrial fermentation because the price of pure xylose is higher than glucose. Fermentation with cellulosic hydrolysate that are mainly composed of xylose and glucose could reduce the cost, however further work is needed to improve the tolerance of the strains to the toxic compounds present in the hydrolysates (52). Moreover, it has been found that both the amount of added xylose and the addition time had a great influence on cell growth and GlcNAc synthesis when dCas9 was controlled by the xylose induced promoter PxylA (22). In order to solve these problems, in this work, the manually controlled xylose induced system was updated into an autonomous controlled dynamic regulation system by reprograming the GamR regulon from GlcN catabolism with the above constructed ADC system.
The strain BNY that was previously used for the application of xylose induced CRISPRi system (22) was chosen as the starting strain for the implementation of GlcN6P-responsive dynamic regulation. To facilitate subsequent operations, the GNA1 (glucosamine-6-phosphate N-acetyltransferase) expression plasmid was removed and the native gamR was knocked out, resulting in the strain BNY0. In order to implement dynamic regulation of GlcNAc synthesis, feedback circuits were constructed using the GlcN6P-responsive ADC system. First, the repression cascade composed of dCas9 controlled by different GlcN6P-responsive promoters with gamR and sgRNAs array, targeting to zwf (glucose-6-phosphate dehydrogenase), pfkA (phosphofructokinase), and glmM (phosphoglucosamine mutase) that control the major competitive modules of GlcNAc synthesis and constructed in the previous study (22), were integrated into the genome of BNY0 to achieve the dynamic repression of competitive modules of GlcNAc synthesis and control the accumulation of GlcN6P. Then GNA1, which is the key enzyme for GlcNAc overproduction (53), was selected as another regulation target by the GlcN6P-responsive promoters to realize the dynamic activation of GlcNAc synthesis module and adjust the consumption of GlcN6P. A total of 16 engineered strains were obtained by combining the competitive modules and the synthesis modules of GlcNAc to coordinate the accumulation and consumption of GlcN6P and promote the efficient synthesis of GlcNAc from glucose (Figure 4A). The relative transcription levels of the target genes on each strain were measured by qPCR (Supplementary Figure S8).
Figure 4.

Dynamic regulation of GlcNAc synthesis by the GlcN6P-responsive ADC system. (A) Schematic of the GlcN6P biosensors mediated dynamic regulation on the pathways associated with GlcNAc synthesis. Effects of introduction of the feedback loop on cell growth (B, C), GlcNAc production (D) and by-product acetoin production (E). CK: control strain that only possessed dCas9 without sgRNAs. ST: statically upregulating ptsG and pgi (The promoters of ptsG and pgi in strain BNY0 were both replaced with P43, and GNA1 was expressed by P43, Pvg6, PgamA and Psg2, respectively). (F) Shake flask fermentations using minimal media.
As shown in Figure 4D, strain DR022, in which both dCas9 and GNA1 genes were expressed by the moderate GlcN6P-responsive promoter PgamA, produced the highest GlcNAc titer of 28.0 ± 0.62 g/l with a yield of 0.37 ± 0.008 g/g glucose in shake flask, increased by 53.0% compared with the control strain BNDR000 (18.3 ± 0.81 g/l and 0.24 ± 0.011 g/g), and the by-product acetoin decreased by 30.3% (Figure 4E). This result suggests that appropriate regulatory intensity is needed both for weakening competitive modules (glycolysis module, pentose phosphate module, and peptidoglycan synthesis module) and strengthening GlcNAc synthesis module. Introducing the repression cascade controlled by the GlcN6P biosensors delayed the exponential growth phase (Supplementary Figure S9), because these weakened competitive modules are necessary for the normal cell growth; while expressing dCas9 by the strong GlcN6P-responsive promoter Psg2 had the greatest inhibition on cell growth (DCW decreased by 52.1% at 24 h and 59.5% at 36 h, respectively) (Figures 4B and C), accompanied by a sharp decrease in GlcNAc titer (65.6%) (Figure 4D). However, for the strains containing the weak GlcN6P-responsive promoter Pvg6 or the moderate GlcN6P-responsive promoter PgamA controlled repression cascade, the DCW at 24 h decreased by 28.8% and 29.6% (Figure 4B), with 12.6% and 20.6% increases in GlcNAc titer, respectively (Figure 4D).
For strains BNDR002 and BNDR003, in which only the GlcNAc synthesis module was dynamically activated by the moderate promoter PgamAor the strong promoter Psg2, the exponential growth phase was delayed (Supplementary Figure S9) and DCW at 24 h decreased by 69.3% and 78.9%, respectively (Figure 4A), suggesting that pulling the flux too strongly into GlcNAc synthesis module caused the undersupply of intracellular GlcN6P. However, further introduction of the repression cascade controlled by the weak or the moderate GlcN6P-responsive promoters recovered cell growth (strains BNDR012, BNDR022, BNDR013 and BNDR023), proving that weakening these competition modules promoted the accumulation of GlcN6P, and the variation tendency of intracellular GlcN6P was consistent with these observations (Supplementary Figure S3B). To assess the individual contributions of the loop, four control strains (BNDR040, BNDR041, BNDR042 and BNDR043) have been constructed by replacing the promoters of ptsG and pgi with P43 in strain BNY0, in which glmS had been statically upregulated using promoter Pveg, and expressing GNA1 by P43, Pvg6, PgamA and Psg2, respectively (Figure 4 and Supplementary Figure S9). When all of the genes in GlcNAc synthesis (ptsG, pgi, glmS, and GNA1) were upregulated in a static way (strain BNDR040), both cell growth and glucose consumption became very slow, with a similar GlcNAc titer (18.7 ± 1.01 g/l) and a longer fermentation period (60 h) compared with strain BNDR000 in which only glmS and GNA1 were upregulated in a static way. When GNA1 was controlled by the strong GlcN6P-responsive promoter Psg2, upregulating pgi, ptsG, and glmS in a static way (strain BNDR043) cannot solve the growth impairment as seen in strain BNDR003. However, when GNA1 was controlled by the moderate GlcN6P-responsive promoters PgamA, upregulating pgi, ptsG and glmS in a static way (strain BNDR042) recovered the cell growth, while the GlcNAc titer was lower than the strain with the GlcN6P-responsived repression cascades (BNDR022) (25.1 ± 0.22 g/l to 28.0 ± 0.62 g/l). These results approve that both dynamic activation and repression in the loop are necessary for the proper function of the synthetic genetic circuits.
Shake flask fermentations were also performed in minimal media, in which 6 g/l urea and 75 g/l glucose was added to meet the need of nitrogen and carbon for GlcNAc synthesis (Figure 4F). The growth of the starting strain BNDR000 was slow and weak in this media, and the DCW did not reach the top (5.0 ± 0.32 g/l) until 106 h with 15.0 ± 0.98 g/l GlcNAc. Upregulating ptsG and pgi by P43 in a static way (strain BNDR040) can improve the maximum DCW without improving the growth rate, while the fermentation time was still over 96 h. In addition, the strain BNDR042 in which GNA1 was regulated by PgamA had a better growth and higher GlcNAc titer than strain BNDR040 in which GNA1 was expressed by P43. Furthermore, the strain BNDR022 installed with the synthetic genetic circuits can use up glucose in 48 h although the growth was slightly slower than that in the complex medium, and the GlcNAc titer can reach 22.8 ± 0.19 g/l at 48 h. These results suggested that implementing dynamic regulation using the synthetic genetic circuits could better coordinate the expression of exogenous and endogenous pathways, which make the engineered strain perform well even in the nutrient-poor minimal media. In addition, the plasmid stability over time span has been checked, and the strain BNDR022 that installed the ADC system enable genetic circuits possess better plasmid stability than the starting stain BNSR000, which may be a result of the reduced metabolic burden by applying dynamic regulation (Supplementary Figure S10).
In short, our results indicate that it is effective to dynamically regulate the synthesis of GlcNAc by reprograming the GamR regulon into a synthetic feedback circuits using the GlcN6P responsive biosensors-CRISPRi coupled ADC system. However, some work still needs to be done in the future to further improve the programmable biological functionalities of this ADC system. First, the structural modification and expression regulation on the aTF GamR may be used to further optimize responsive strengths of biosensors, based on the previously described fundamental design principles and relevant research results (46,54,55). In addition, more complex genetic circuits can be designed and built to improve the accuracy and sensibility of the biosensors (11,56,57). Moreover, the transport mechanism of GlcNAc needs to be explored, and this process can be also strengthened dynamically for the improvement of efflux efficiency, which have been proven useful in a previous study regarding lysine production in Corynebacterium glutamicum (58).
Scale-up production of GlcNAc in a 15-l fermenter
To test the stability and robustness of the synthetic genetic circuits in a larger scale bioreactor, the strain BNDR022, which can produce 28.0 ± 0.62 g/l GlcNAc with a yield of 0.37 ± 0.008 g/g glucose in shake flask, was used for scaling up the production of GlcNAc in a 15-l fermenter. Because the by-product acetoin, which will affect the subsequent separation and purification process, still accumulated up to 10.1 g/l in shake flask culture, alsSD that controlled the synthesis of acetoin was first either knocked out or knocked down. Five sgRNAs that targeted to different positions on alsS, which is the first gene of the alsSD operon, were integrated into the genome of BNDR022, resulting in the strains BNDR222, BNDR322, BNDR422, BNDR522 and BNDR622, respectively. The strain BNDR122 was obtained by knocking out alsSD directly. Acetoin was not detected after knocking out or knocking down alsSD, but the GlcNAc titers reduced significantly with the reduced growth in these strains, which was caused by the decrease of media pH in shake flask (Figure 5). For scale-up production of GlcNAc in a 15-l fermenter, the pH is kept at 7.0 by the addition of NH4OH, so no negative effect in cell growth would be expected. As no obvious differences were found on cell growth and GlcNAc synthesis among the alsSD knocked-out and knocked-down strains in shake flasks, the strains BNDR122 was chosen for the fed-batch fermentation, as well as the parental strains BNDR000 and BNDR022. In addition, strain BNDR100 that was obtained by knocking out alsSD of strain BNDR000 was also performed fermentation to rule out the effects of alsSD deletion. Figure 6A–D shows the time courses of the GlcNAc production in fed-batch culture by the strains BNDR000, BNDR100, BNDR022, and BNDR122, respectively. Glucose was maintained between 2 and 30 g/l by feeding with concentrated glucose (750 g/l). The starting strain BNDR000 (BNY0ΔlacA::gamR-PgamA-dCas9, pP43-GNA1) can produce 59.9 g/l GlcNAc with a yield of 0.21 g/g glucose and by-product acetoin accumulated to 36.0 g/l. The strain BNDR100 (BNDR000ΔalsSD) could produce 81.7 g/l GlcNAc with a yield of 0.26 g/g glucose. For strain BNDR022 (BNY0ΔamyE::sgzwf1-sgpfkA2-sgglmM2, ΔlacA::gamR-PgamA-dCas9, pSTg-GNA1) installed with the biosensor-CRISPRi genetic circuits, the GlcNAc titer and yield increased by 62.1% and 57.1% to 97.1 g/l and 0.33 g/g glucose, respectively, with by-product acetoin decreased by 35.8% to 23.1 g/l. After further completely blocking acetoin formation, the strain BNDR122 (BNDR022ΔalsSD) could produce 131.6 g/l GlcNAc, which is 2.19-fold that of the starting strain BNDR000 and represents the highest GlcNAc titer ever reported, and the yield of GlcNAc increased to 0.38 g/g glucose.
Figure 5.
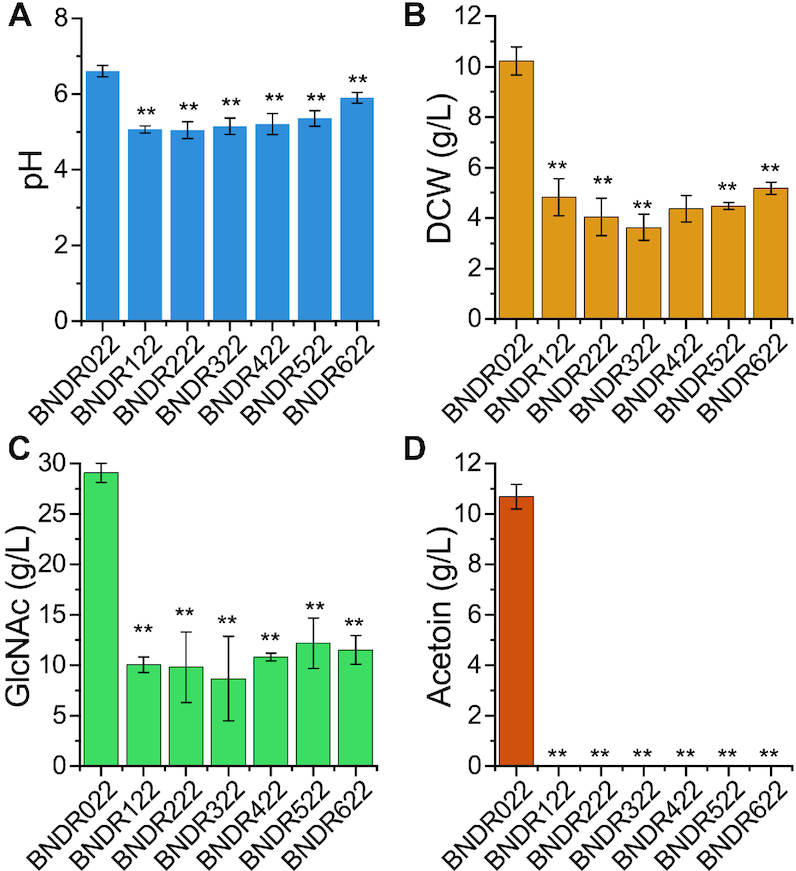
Blocking by-product acetoin formation by knocking out or knocking down alsSD. Effects of knocking out or knocking down alsSD on media pH (A), cell growth (B), GlcNAc synthesis (C), and acetoin formation (D). All data were the average of three independent studies with standard deviations. The * and ** indicate P < 0.05 and P < 0.01 relative to control strain BNDR022, respectively.
Figure 6.

Production of GlcNAc in a 15-l fermenter. Fed-batch fermentation of strain BNDR000 (A), BNDR100 (B), BNDR022 (C), and BNDR122 (D) in a 15-l fermenter. The glucose concentration was maintained within 2–30 g/l by feeding with 750 g/l glucose, and a total of 1 l nitrogen source feeding solution contained 140 g yeast extract, 140 g tryptone and 70 g urea was pumped into the fermenter at a rate of 30 ml/h when OD600 reached 30.
CONCLUSIONS
In this work, a GlcN6P-responsive biosensor-CRISPRi coupled ADC system was designed and developed in B. subtilis. Then, a genetic feedback circuits was constructed to dynamically up-regulate the GlcNAc synthesis module and down-regulate the competitive modules simultaneously, leading to a significantly increased GlcNAc titer. Compared with the previously reported control systems, the proposed ADC system can perform the up-regulation and down-regulation of synthetic modules at the same time without the need of inducer addition or switches on the fermentation conditions. In addition, we proved that the ADC system functions in a robust and stable manner in a large bioreactor, producing the higher titer ever described of the important nutraceutical GlcNAc. Together, our results suggest that the microbial cell factory installed with a suitable ADC system can achieve a high-level of production performance without complex processes control, which will be useful for the fine-tuning of synthetic modules in the other microbial species and metabolic pathways.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2018YFA0900300]; National Natural Science Foundation of China [31622001, 31671845, 31600068, 31930085]; Fundamental Research Funds for the Central Universities [JUSRP51713B]; Natural Science Foundation of Jiangsu Province [BK20160176]; 111 Project [111-2-06]; Postgraduate Research & Practice Innovation Program of Jiangsu Province [KYCX18_1802]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cordova L.T., Alper H.S.. Central metabolic nodes for diverse biochemical production. Curr. Opin. Chem. Biol. 2016; 35:37–42. [DOI] [PubMed] [Google Scholar]
- 2. Liu L., Guan N., Li J., Shin H., Du G., Chen J.. Development of GRAS strains for nutraceutical production using systems and synthetic biology approaches: advances and prospects. Crit. Rev. Biotechnol. 2017; 37:139–150. [DOI] [PubMed] [Google Scholar]
- 3. Zhou Y.J., Kerkhoven E.J., Nielsen J.. Barriers and opportunities in bio-based production of hydrocarbons. Nat. Energy. 2018; 3:925–935. [Google Scholar]
- 4. Luo X., Reiter M.A., D’Espaux L., Wong J., Denby C.M., Lechner A., Zhang Y., Grzybowski A.T., Harth S., Lin W. et al.. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature. 2019; 567:123–126. [DOI] [PubMed] [Google Scholar]
- 5. Holtz W.J., Keasling J.D.. Engineering static and dynamic control of synthetic pathways. Cell. 2010; 140:19–23. [DOI] [PubMed] [Google Scholar]
- 6. Cress B.F., Trantas E.A., Ververidis F., Linhardt R.J., Koffas M.A.. Sensitive cells: Enabling tools for static and dynamic control of microbial metabolic pathways. Curr. Opin. Biotechnol. 2015; 36:205–214. [DOI] [PubMed] [Google Scholar]
- 7. Lalwani M.A., Zhao E.M., Avalos J.L.. Current and future modalities of dynamic control in metabolic engineering. Curr. Opin. Biotechnol. 2018; 52:56–65. [DOI] [PubMed] [Google Scholar]
- 8. Shen X., Wang J., Li C., Yuan Q., Yan Y.. Dynamic gene expression engineering as a tool in pathway engineering. Curr. Opin. Biotechnol. 2019; 59:122–129. [DOI] [PubMed] [Google Scholar]
- 9. Xu P. Production of chemicals using dynamic control of metabolic fluxes. Curr. Opin. Biotechnol. 2018; 53:12–19. [DOI] [PubMed] [Google Scholar]
- 10. Rugbjerg P., Sommer M.O.A.. Overcoming genetic heterogeneity in industrial fermentations. Nat. Biotechnol. 2019; 37:869–876. [DOI] [PubMed] [Google Scholar]
- 11. Xia P.-F., Ling H., Foo J.L., Chang M.W.. Synthetic genetic circuits for programmable biological functionalities. Biotechnol. Adv. 2019; 37:107393. [DOI] [PubMed] [Google Scholar]
- 12. Soma Y., Fujiwara Y., Nakagawa T., Tsuruno K., Hanai T.. Reconstruction of a metabolic regulatory network in Escherichia coli for purposeful switching from cell growth mode to production mode in direct GABA fermentation from glucose. Metab. Eng. 2017; 43:54–63. [DOI] [PubMed] [Google Scholar]
- 13. Zhang F., Carothers J.M., Keasling J.D.. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat. Biotechnol. 2012; 30:354–359. [DOI] [PubMed] [Google Scholar]
- 14. Gupta A., Reizman I.M.B., Reisch C.R., Prather K.L.J.. Dynamic regulation of metabolic flux in engineered bacteria using a pathway-independent quorum-sensing circuit. Nat. Biotechnol. 2017; 35:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim E., Woo H.M., Tian T., Yilmaz S., Javidpour P., Keasling J.D., Lee T.S.. Autonomous control of metabolic state by a quorum sensing (QS)-mediated regulator for bisabolene production in engineered E. coli. Metab. Eng. 2017; 44:325–336. [DOI] [PubMed] [Google Scholar]
- 16. Doong S.J., Gupta A., Prather K.L.J.. Layered dynamic regulation for improving metabolic pathway productivity in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:2964–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A. et al.. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013; 154:442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fontana J., Dong C., Ham J.Y., Zalatan J.G., Carothers J.M.. Regulated expression of sgRNAs tunes CRISPRi in E. coli. Biotechnol. J. 2018; 13:1800069. [DOI] [PubMed] [Google Scholar]
- 19. Didovyk A., Borek B., Tsimring L., Hasty J.. Transcriptional regulation with CRISPR-Cas9: Principles, advances, and applications. Curr. Opin. Biotechnol. 2016; 40:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gander M.W., Vrana J.D., Voje W.E., Carothers J.M., Klavins E.. Digital logic circuits in yeast with CRISPR-dCas9 NOR gates. Nat. Commun. 2017; 8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang S., Voigt C.A.. Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res. 2018; 46:11115–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu Y., Chen T., Liu Y., Lv X., Li J., Du G., Ledesma-Amaro R., Liu L.. CRISPRi allows optimal temporal control of N-acetylglucosamine bioproduction by a dynamic coordination of glucose and xylose metabolism in Bacillus subtilis. Metab. Eng. 2018; 49:232–241. [DOI] [PubMed] [Google Scholar]
- 23. Moser F., Espah Borujeni A., Ghodasara A.N., Cameron E., Park Y., Voigt C.A.. Dynamic control of endogenous metabolism with combinatorial logic circuits. Mol. Syst. Biol. 2018; 14:e8605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niu T., Liu Y., Li J., Koffas M., Du G., Alper H.S., Liu L.. Engineering a glucosamine-6-phosphate responsive glmS ribozyme switch enables dynamic control of metabolic flux in Bacillus subtilis for overproduction of N-acetylglucosamine. ACS Synth. Biol. 2018; 7:2423–2435. [DOI] [PubMed] [Google Scholar]
- 25. Klein D.J. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science. 2006; 313:1752–1756. [DOI] [PubMed] [Google Scholar]
- 26. Baumgärtner F., Conrad J., Sprenger G.A., Albermann C.. Synthesis of the human milk oligosaccharide lacto-N-Tetraose in metabolically engineered, plasmid-free E. coli. ChemBioChem. 2014; 15:1896–1900. [DOI] [PubMed] [Google Scholar]
- 27. Westbrook A.W., Ren X., Oh J., Moo-Young M., Chou C.P.. Metabolic engineering to enhance heterologous production of hyaluronic acid in Bacillus subtilis. Metab. Eng. 2018; 27:558–563. [DOI] [PubMed] [Google Scholar]
- 28. Gaugué I., Oberto J., Plumbridge J.. Regulation of amino sugar utilization in Bacillus subtilis by the GntR family regulators, NagR and GamR. Mol. Microbiol. 2014; 92:100–115. [DOI] [PubMed] [Google Scholar]
- 29. Ma W., Liu Y., Shin H., Li J., Chen J., Du G., Liu L.. Metabolic engineering of carbon overflow metabolism of Bacillus subtilis for improved N-acetyl-glucosamine production. Bioresour. Technol. 2018; 250:642–649. [DOI] [PubMed] [Google Scholar]
- 30. Anagnostopoulos C., Spizizen J.. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 1961; 81:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yan X., Yu H.J., Hong Q., Li S.P.. Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl. Environ. Microbiol. 2008; 74:5556–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu H., Naismith J.H.. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008; 8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tian R., Liu Y., Chen J., Li J., Liu L., Du G., Chen J.. Synthetic N-terminal coding sequences for fine-tuning gene expression and metabolic engineering in Bacillus subtilis. Metab. Eng. 2019; 55:131–141. [DOI] [PubMed] [Google Scholar]
- 34. Yang S., Du G., Chen J., Kang Z.. Characterization and application of endogenous phase-dependent promoters in Bacillus subtilis. Appl. Microbiol. Biotechnol. 2017; 101:4151–4161. [DOI] [PubMed] [Google Scholar]
- 35. Peters G., De Paepe B., De Wannemaeker L., Duchi D., Maertens J., Lammertyn J., De Mey M.. Development of N-acetylneuraminic acid responsive biosensors based on the transcriptional regulator NanR. Biotechnol. Bioeng. 2018; 115:1855–1865. [DOI] [PubMed] [Google Scholar]
- 36. Liu Y., Link H., Liu L., Du G., Chen J., Sauer U.. A dynamic pathway analysis approach reveals a limiting futile cycle in N-acetylglucosamine overproducing Bacillus subtilis. Nat. Commun. 2016; 7:11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu Z., Yang S., Yuan X., Shi Y., Ouyang L., Jiang S., Yi L., Zhang G.. CRISPR-assisted multi-dimensional regulation for fine-tuning gene expression in Bacillus subtilis. Nucleic Acids Res. 2019; 47:e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fillenberg S.B., Grau F.C., Seidel G., Muller Y.A.. Structural insight into operator dre-sites recognition and effector binding in the GntR/HutC transcription regulator NagR. Nucleic Acids Res. 2015; 43:1283–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deutscher J., Francke C., Postma P.W.. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 2006; 70:939–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jain D. Allosteric control of transcription in GntR family of transcription regulators: a structural overview. IUBMB Life. 2015; 67:556–563. [DOI] [PubMed] [Google Scholar]
- 41. Koch M., Pandi A., Borkowski O., Cardoso Batista A., Faulon J.-l.. Custom-made transcriptional biosensors for metabolic engineering. Curr. Opin. Biotechnol. 2019; 59:78–84. [DOI] [PubMed] [Google Scholar]
- 42. Cheng J., Guan C., Cui W., Zhou L., Liu Z., Li W., Zhou Z.. Enhancement of a high efficient autoinducible expression system in Bacillus subtilis by promoter engineering. Protein Expr. Purif. 2016; 127:81–87. [DOI] [PubMed] [Google Scholar]
- 43. Guiziou S., Sauveplane V., Chang H.-J., Clerté C., Declerck N., Jules M., Bonnet J.. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res. 2016; 44:gkw624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang S., Hou Y., Chen X., Liu L.. Kick-starting evolution efficiency with an autonomous evolution mutation system. Metab. Eng. 2019; 54:127–136. [DOI] [PubMed] [Google Scholar]
- 45. Gaugué I., Oberto J., Putzer H., Plumbridge J.. The use of amino sugars by Bacillus subtilis: presence of a unique operon for the catabolism of glucosamine. PLoS One. 2013; 8:e63025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Meyer A.J., Segall-Shapiro T.H., Glassey E., Zhang J., Voigt C.A.. Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol. 2019; 15:196–204. [DOI] [PubMed] [Google Scholar]
- 47. Xu P., Li L., Zhang F., Stephanopoulos G., Koffas M.. Improving fatty acids production by engineering dynamic pathway regulation and metabolic control. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:11299–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Williams T.C., Averesch N.J., Plan M., Winter G., Vickers C.E., Nielsen L.K., Krömer J.O., Lekieffre N., Winter G., Vickers C.E. et al.. Quorum-sensing linked RNAi for dynamic pathway control in Saccharomyces cerevisiae. Metab. Eng. 2015; 29:124–134. [DOI] [PubMed] [Google Scholar]
- 49. Yang Y., Lin Y., Wang J., Wu Y., Zhang R., Cheng M., Shen X., Wang J., Chen Z., Li C. et al.. Sensor-regulator and RNAi based bifunctional dynamic control network for engineered microbial synthesis. Nat. Commun. 2018; 9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cho S., Choe D., Lee E., Kim S.C., Palsson B., Cho B.K.. High-level dCas9 expression induces abnormal cell morphology in Escherichia coli. ACS Synth. Biol. 2018; 7:1085–1094. [DOI] [PubMed] [Google Scholar]
- 51. Schmitz A.C., Hartline C.J., Zhang F.. Engineering microbial metabolite dynamics and heterogeneity. Biotechnol. J. 2017; 12:1700422. [DOI] [PubMed] [Google Scholar]
- 52. Glebes T.Y., Sandoval N.R., Gillis J.H., Gill R.T.. Comparison of genome-wide selection strategies to identify furfural tolerance genes in Escherichia coli. Biotechnol. Bioeng. 2015; 112:129–140. [DOI] [PubMed] [Google Scholar]
- 53. Ma W., Liu Y., Lv X., Li J., Du G., Liu L.. Combinatorial pathway enzyme engineering and host engineering overcomes pyruvate overflow and enhances overproduction of N-acetylglucosamine in Bacillus subtilis. Microb. Cell Fact. 2019; 18:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. De Paepe B., Maertens J., Vanholme B., De Mey M.. Chimeric LysR-type transcriptional biosensors for customizing ligand specificity profiles toward flavonoids. ACS Synth. Biol. 2019; 8:318–331. [DOI] [PubMed] [Google Scholar]
- 55. Mannan A.A., Liu D., Zhang F., Oyarzún D.A.. Fundamental design principles for transcription-factor-based metabolite biosensors. ACS Synth. Biol. 2017; 6:1851–1859. [DOI] [PubMed] [Google Scholar]
- 56. Hoynes-O’Connor A., Moon T.S.. Programmable genetic circuits for pathway engineering. Curr. Opin. Biotechnol. 2015; 36:115–121. [DOI] [PubMed] [Google Scholar]
- 57. Kim S.K., Kim S.H., Subhadra B., Woo S.-G., Rha E., Kim S.-W., Kim H., Lee D.-H., Lee S.-G.. A genetically encoded biosensor for monitoring isoprene production in engineered Escherichia coli. ACS Synth. Biol. 2018; 7:2379–2390. [DOI] [PubMed] [Google Scholar]
- 58. Zhou L.B., Zeng A.P.. Engineering a lysine-ON riboswitch for metabolic control of lysine production in Corynebacterium glutamicum. ACS Synth. Biol. 2015; 4:1335–1340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.