

Abstract
Background
Despite increased understanding of the genetic events underlying pediatric high-grade gliomas (pHGGs), therapeutic progress is static, with poor understanding of nongenomic drivers. We therefore investigated the role of alterations in mitochondrial function and developed an effective combination therapy against pHGGs.
Methods
Mitochondrial DNA (mtDNA) copy number was measured in a cohort of 60 pHGGs. The implication of mtDNA alteration in pHGG tumorigenesis was studied and followed by an efficacy investigation using patient-derived cultures and orthotopic xenografts.
Results
Average mtDNA content was significantly lower in tumors versus normal brains. Decreasing mtDNA copy number in normal human astrocytes led to a markedly increased tumorigenicity in vivo. Depletion of mtDNA in pHGG cells promoted cell migration and invasion and therapeutic resistance. Shifting glucose metabolism from glycolysis to mitochondrial oxidation with the adenosine monophosphate–activated protein kinase activator AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) or the pyruvate dehydrogenase kinase inhibitor dichloroacetate (DCA) significantly inhibited pHGG viability. Using DCA to shift glucose metabolism to mitochondrial oxidation and then metformin to simultaneously target mitochondrial function disrupted energy homeostasis of tumor cells, increasing DNA damage and apoptosis. The triple combination with radiation therapy, DCA and metformin led to a more potent therapeutic effect in vitro and in vivo.
Conclusions
Our results suggest metabolic alterations as an onco-requisite factor of pHGG tumorigenesis. Targeting reduced mtDNA quantity represents a promising therapeutic strategy for pHGG.
Keywords: DIPG, gliomas, mitochondria, radiotherapy
Key Points.
1. MtDNA copy number is significantly reduced in pHGGs, leading to a glycolytic phenotype.
2. Targeting glucose metabolism significantly improves radiosensitivity of pHGG.
3. Targeting decreased mtDNA number may be a novel therapeutic strategy for pHGG.
Importance of the Study.
We report a novel finding that mtDNA copy number is significantly reduced in pHGGs, leading to a glycolytic phenotype that is strongly correlated with enhanced cell migration and invasion, therapeutic resistance, and in vivo tumorigenicity. To further exploit this finding, an orally bioavailable compound, DCA, was tested and exhibited potent antiglioma activity. The combination of DCA and the antidiabetic drug metformin triggered an energy crisis with a profound antitumor effect. The triple combination of DCA, metformin, and radiation was more effective against both patient-derived cell cultures and orthotopic xenografts. This strategy of triple combination has not been reported before, is demonstrated with clinically available compounds, has the potential for significant clinical impact, and is ripe for rapid translation to clinical trial.
Pediatric high-grade gliomas (pHGGs) are aggressive neoplasms accounting for ~20% of brain tumors in children. Among them, diffuse intrinsic pontine gliomas (DIPGs) and supratentorial HGGs are incurable malignancies for which no effective therapies exist. Radiation therapy (RT) has been considered the only standard treatment for DIPG, although its role is primarily palliative.1
Our understanding of the molecular underpinnings of pHGGs has increased dramatically over the last few years. The advent of next generation sequencing–based technologies has led to the discovery of highly recurrent aberrations in vital genetic drivers such as mutations in histone H3.3 or H3.1 (H3K27M) that occur in almost 80% of DIPG. However, H3K27M tumors typically harbor additional genetic aberrations, and modeling efforts using histone mutations alone have failed to promote transformation, suggesting that H3K27M by itself is insufficient to drive malignant transformation. Hitherto, mitochondrial dysfunction and its induced metabolic alterations, a major hallmark of cancer, have been understudied in these tumors. Mitochondria exert critical roles in energy metabolism, free radical production, and apoptosis. Mitochondria possess their own genome, mitochondrial (mt)DNA, which is present in hundreds to thousands of copies per mammalian cell.2 Diminished mtDNA copy number is closely associated with aggressive clinicopathological characteristics in several types of solid tumors.3 Mounting evidence has demonstrated that reduction in mtDNA number is sufficient to initiate neoplastic transformation and/or expedite cancer progression and metastatic potential of tumor cells.3 Collectively, these data reveal an intimate and inextricable link between mtDNA instability and tumorigenesis that may underlie the molecular basis for the Warburg effect.
HGGs, like most solid tumors, are highly glycolytic, producing large amounts of lactate as a metabolic by-product.4 Tumors with high rates of aerobic glycolysis are less responsive to chemo/RT and behave more aggressively.5 There are several approaches to targeting metabolically different cancer cells. A promising one is to target pyruvate dehydrogenase kinase (PDK) responsible for regulating mitochondrial activity.6 PDK is a gate-keeping mitochondrial enzyme that represses the conversion of pyruvate to acetyl-CoA and prevents the coupling of glycolysis to oxidative phosphorylation (OXPHOS) via inhibition of pyruvate dehydrogenase (PDH). Dichloroacetate (DCA) is a PDK inhibitor with the potential to reverse the Warburg effect by coupling glycolysis with OXPHOS, thereby inhibiting tumor cell growth.7 Two small phase I trials of DCA in adult patients with recurrent gliomas have highlighted the potential clinical activity of DCA.8,9 However, the dose of DCA achieved in adults is limited by a dose-dependent peripheral neuropathy. Importantly, this toxicity is age dependent and the DCA dose can be escalated further in children without any significant side effects.10 These findings suggest a potential role for DCA in pHGGs.
Here we show that mtDNA is depleted in pHGGs and may contribute to metabolic reprogramming. Targeting altered mtDNA content in pHGG cells with a tool compound, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), activated adenosine monophosphate–activated protein kinase (AMPK), reversed the Warburg effect and inhibited tumor growth. To develop a clinically tractable strategy, we tested 2 readily available metabolic modulators, DCA and metformin, in combination with the standard treatment RT, and for the first time we found a potent triple combination against a broad panel of in vitro and in vivo pHGG models.
Materials and Methods
Study Subjects and Samples
Thirty-five DIPGs (33 postmortem and 2 biopsy samples), 25 supratentorial HGGs, and 19 normal brain (the frontal lobes consisting primarily of gray matter) autopsy samples were obtained from the Hospital for Sick Children following approval by the institutional research ethics board. Informed consent was obtained for all patients or their guardians.
Quantification of mtDNA Copy Number
Total DNA from normal and tumor tissues, pHGG cultures were isolated by using a Qiagen DNeasy Blood & Tissue Kit and quantified on a NanoDrop spectrophotometer (Thermo Fisher Scientific). Quantitative (q)PCR was performed on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) to determine mtDNA copy number.
Cell Culture
Cell culture condition of normal human astrocytes (NHAs), pHGG, and DIPG cell lines as well as establishment of SF188 and SU-DIPG4 ρ 0 models are described in the Supplementary Methods.
Cell Proliferation Assay
Cell proliferation was assessed using alamarBlue assays and combination index was calculated by using the CalcuSyn.
Cell Migration and Invasion Assays
Cell migration and invasion were respectively assessed by wound healing and Boyden Chamber assays.
Phosphorylated PDH-E1α Detection and PDH Activity Assay
Western blotting was performed to examine the protein level of phosphorylated PDH subunit E1α (p-PDH-E1α). A PDH enzyme activity microplate assay kit (Abcam) was used to measure PDH activity.
L-Lactate Analysis
Intracellular lactate levels were measured using an L-Lactate Colorimetric Assay Kit (Abcam). An identical plate was set in parallel with the assay plate for protein quantification, and L-lactate levels were normalized to an equal amount of protein.
ATP Quantification
The ATP levels were determined by a CellTiter-Glo Luminescent Cell Viability Assay (Promega). An identical plate was set in parallel with the assay plate for protein extraction such that final ATP levels were normalized to an equal amount of protein.
Flow Cytometry
MitoSOX Red (Thermo Fisher Scientific) and dihydroethidium (Thermo Fisher Scientific) were used to measure mitochondrial and cytosol reactive oxygen species (ROS) production, respectively, according to the manufacturer’s instructions. JC-1 (Sigma) was used to measure mitochondrial membrane potential. Apoptosis was quantified by an Annexin-V–FLUOS Staining Kit (Roche).
Western Blotting
Cells were lysed with radioimmunoprecipitation assay supplemented with protease and phosphatase inhibitors. Proteins were separated via reducing 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and standard procedures as described previously11 were used.
Lentivirus Preparation and Infection
We obtained pGIPZ lentiviral vectors encompassing non-overlapping short hairpin (sh)RNA targeting the human mitochondrial transcription factor A (TFAM) gene or scrambled nonsilencing shRNA from the SPARC (SickKids Proteomics, Analytics, Robotics & Chemical Biology Centre) lentiviral core facility at the Hospital for Sick Children (originally from the Dharmacon GIPZ lentiviral shRNA libraries).
In Vivo Studies
The animal experiments were carried out following approval of the Animal Care and Ethics Committees of the Hospital for Sick Children (AUP#0267) and the University of New South Wales, Sydney (#16/11A).
Additional Methods
Additional methods can be found in the Supplementary Methods, including RNA extraction and quantitative reverse transcription PCR, cell cycle analysis, colony formation assays, extracellular flux assay, determination of sensitivity to temozolomide and irradiation, TFAM overexpression, measurement of ROS production and immunohistochemistry, primary antibodies, tumor engraftment, and in vivo efficacy experiments.
Statistical Analysis
Statistical analysis was performed with the GraphPad Prism 7.0b. Data are presented as the mean ± SD and all experiments were repeated at least twice in triplicate. An unpaired 2-tailed Student’s t-test and ANOVA were conducted for comparisons followed by a post-hoc Dunnett’s test or post-hoc Tukey’s test where appropriate. The clonogenic survival curve for each condition was fitted to a linear quadratic model (Y = exp-[A*X+B*X2]) according to a least squares (ordinary) fit. The surviving fractions of RT in combination with drugs have been normalized to drug treatments alone. The combination curves that are above, overlapping, or below the RT alone curve are defined as antagonistic, additive, or synergistic effect, respectively. Survival analysis was compared by the log-rank test. P-values <0.05 were considered statistically significant.
Results
MtDNA Copy Number Is Reduced in DIPG and pHGG
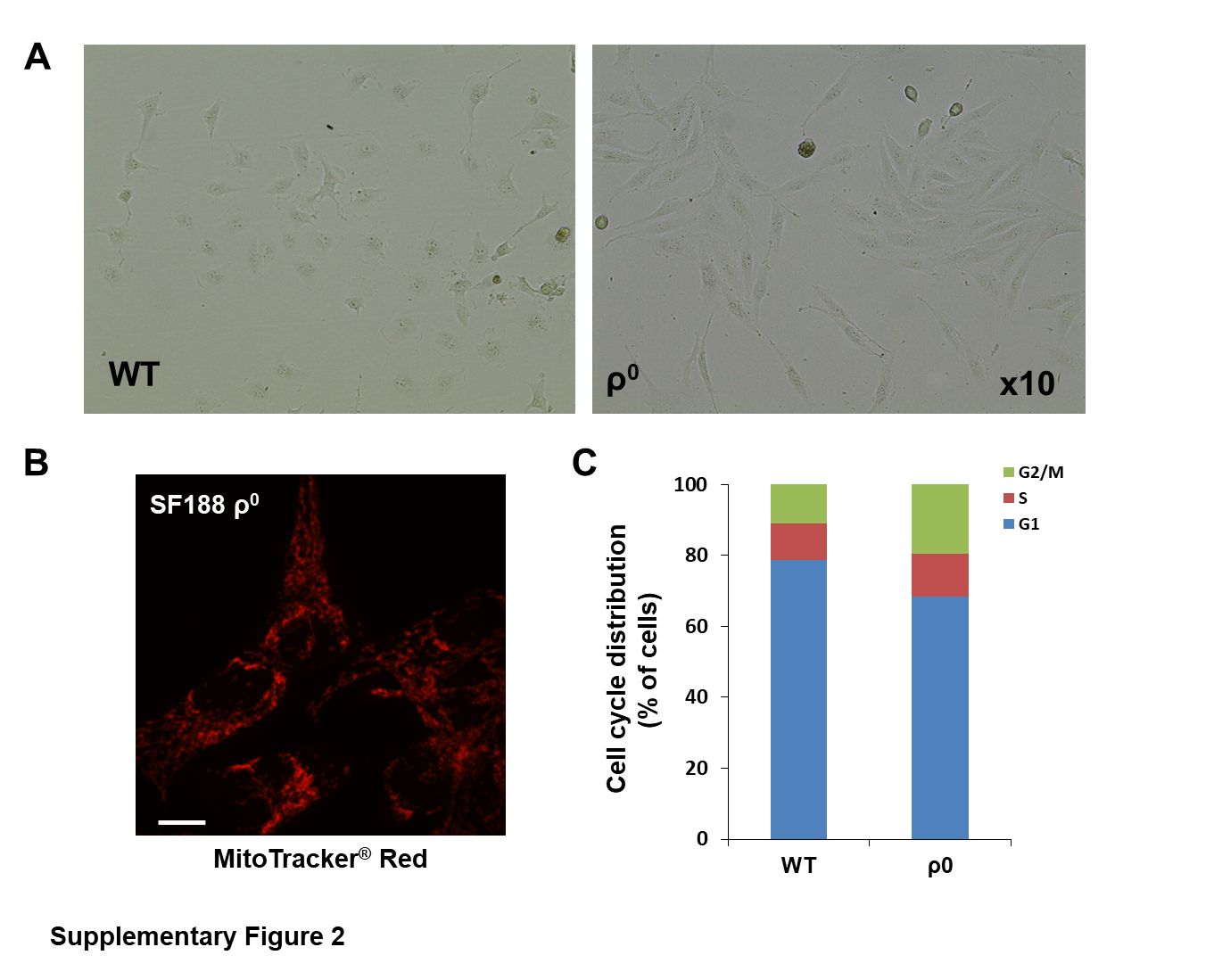
To investigate if alterations in mtDNA copy number occur and are implicated in pHGG gliomagenesis, we determined mtDNA levels in a total of 35 DIPGs and 25 supratentorial pHGGs using qPCR. Average mtDNA content was significantly reduced in tumor tissues compared with that in 19 normal brains (Fig. 1A). All DIPG and supratentorial cases carried lower mtDNA quantity than the median value of normal brains. In addition, mRNA expression of TFAM and PPARGC1A, 2 master regulators of mtDNA duplication and mitochondrial biogenesis, were downregulated in DIPG compared with paired normal controls (Supplementary Fig. 1).
Fig. 1.

Elimination of mtDNA leads to increased migration, invasion, and therapeutic resistance in pHGG. (A) MtDNA quantity was significantly lower in DIPG and supratentorial pHGG specimens than normal brain. (B) Generation of a mutant pediatric glioblastoma SF188 cell line (ρ 0) with complete removal of mtDNA. (C) Quantitative PCR assays demonstrating SF188 ρ 0 cells had markedly reduced mtDNA content compared with WT counterparts. (D, E) MtDNA depletion significantly reduced proliferative capacity. (F) JC-1 staining showed that SF188 ρ 0 cells had a notably decreased mitochondrial membrane potential compared with WT controls. (G) A significant reduction in total cellular ATP level was detected in ρ 0 cells in comparison with parental WT cells. (H) MtDNA-depleted SF188 cells exhibited a ~3-fold increase in L-lactate levels compared with controls. (I, J) Wound healing and transwell cell invasion assays show that lower mtDNA amount strongly enhanced cell migration and invasion. Scale bar: 50 μm. (K, L) Decreasing mtDNA copy number rendered SF188 cells more resistant to temozolomide and radiation. *P < 0.05; **P < 0.01; ***P < 0.001.
MtDNA Depletion Leads to Increased Migration, Invasion, and Therapeutic Resistance in pHGG Cells
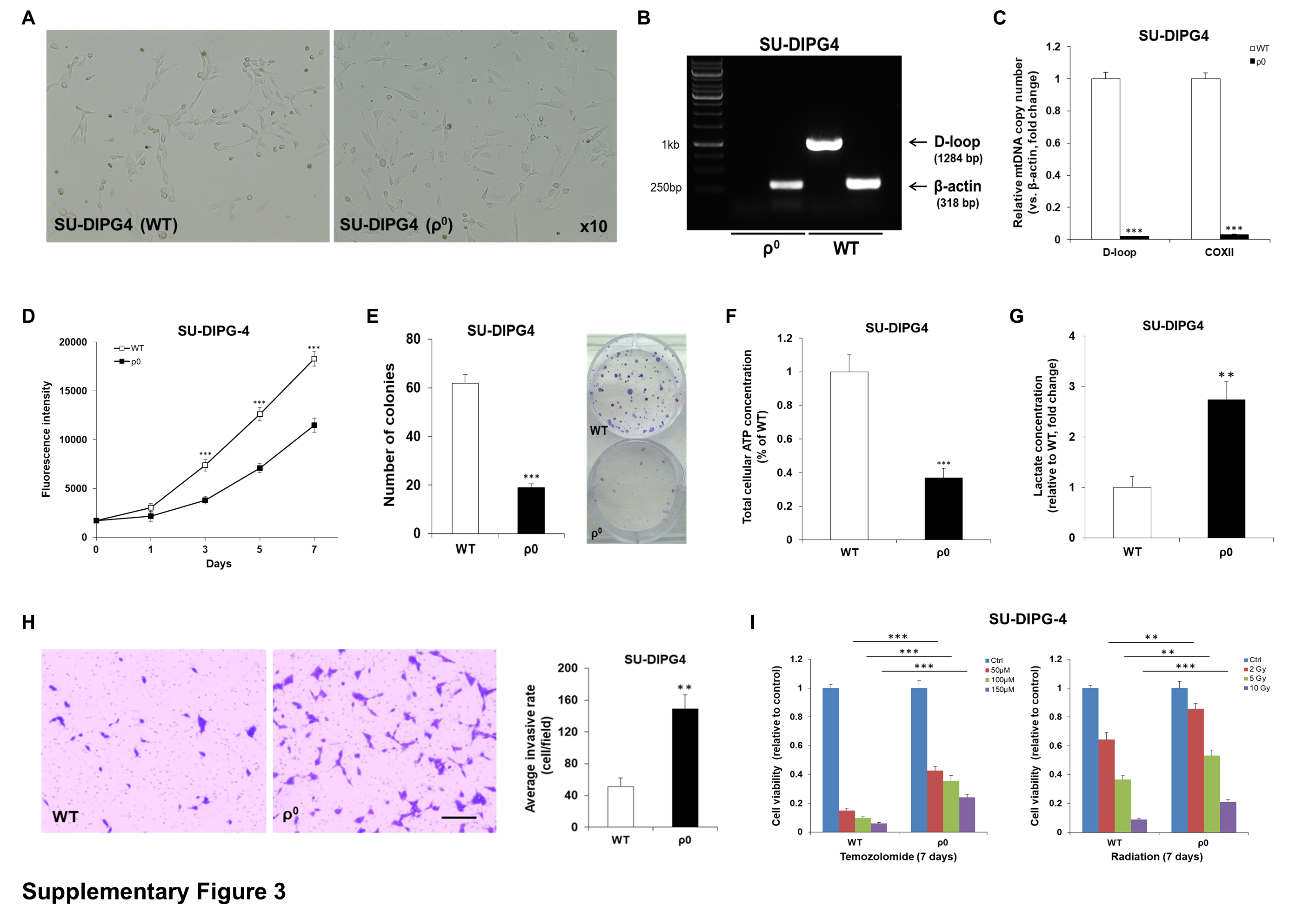
To elucidate the functional consequences of reducing mtDNA levels in established pHGG cell lines, a mutant pediatric glioblastoma SF188 ρ 0 cell line devoid of endogenous mtDNA was established through long-term exposure to low-dose EtBr (Fig. 1B, C). Relative to parental wild-type (WT) cells, ρ 0 cells exhibited morphological changes and had considerably reduced growth rate and cell cycle arrest at G2-M phase (Fig. 1D, E; Supplementary Fig. 2). Although mitochondria in ρ 0 cells remained present, there was a marked decline in mitochondrial membrane potential (ΔΨm) and intracellular ATP levels in mtDNA-depleted cells (Fig. 1F, G; Supplementary Fig. 2), which implies impaired basal mitochondrial function and suppressed energy production in mutant cells. SF188 ρ 0 cells secreted significantly higher levels of L-lactate (~3.2-fold increase), the final glycolytic product, than their WT counterparts (Fig. 1H), indicative of a glycolytic switch whereby knockdown cells favor energy metabolism by glycolysis over mitochondrial respiration. Complete knockout of mtDNA in SF188 cells not only led to enhanced cell migration and invasion (Fig. 1I, J), but significantly decreased their sensitivity to the alkylating agent temozolomide and radiation (Fig. 1K, L). These observations were further confirmed in a SU-DIPG4 ρ 0 model (Supplementary Fig. 3).
Reduced mtDNA Copy Number Enhances Tumorigenicity In Vivo
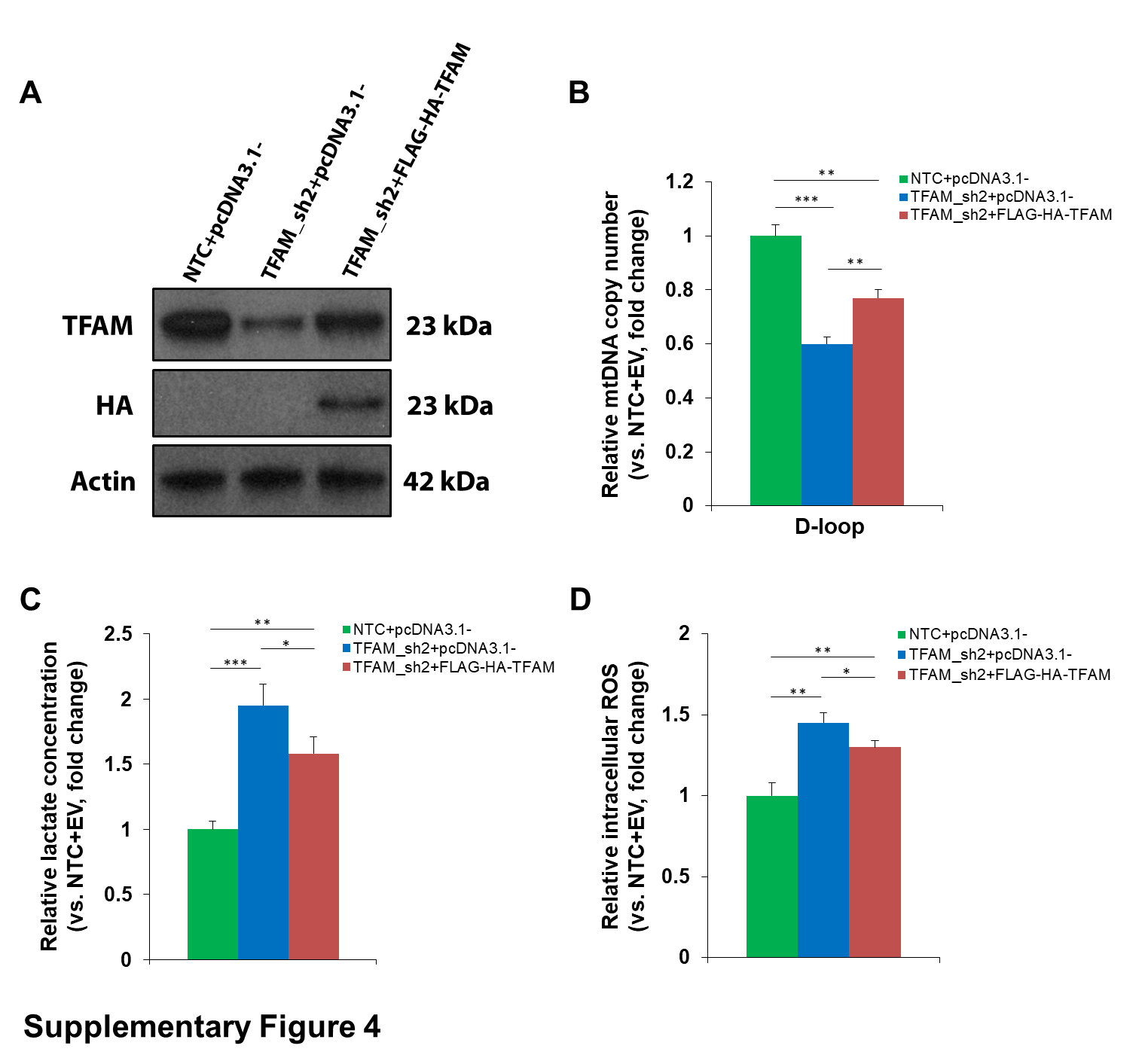
While the ρ 0 model provides important proof-of-principle information, the almost complete loss of mtDNA in this model is not physiologic. Moreover, since EtBr is a strong mutagen, there is a possibility that the phenotypes observed in ρ 0 models may not be solely attributed to mtDNA depletion. To test the transformative potential of reduced mtDNA number, and to more accurately mimic the situation seen in primary human tumors, we downregulated mtDNA content to pHGG-like levels in immortalized (i)NHA cells through stable knockdown of TFAM using an shRNA approach. TFAM-deficient iNHAs possessed reduced amounts of TFAM as anticipated, with a partial loss of mitochondrial respiratory chain components cytochrome c oxidase subunit II (COXII) and NADH dehydrogenase ubiquinone 1 beta subcomplex subunit 2 (NDUFB2) (Fig. 2A) and an increased level of L-lactate (Fig. 2B). TFAM-silenced cells had mildly depleted mtDNA quantity by ~30–40% (shRNA1: 69.8%, shRNA2: 62.6%, vs controls) and grew at a slightly lower rate compared with controls (Fig. 2C, D). Expression of exogenous TFAM was able to partially rescue the phenotypes of TFAM knockdown iNHAs including L-lactate and ROS production (Supplementary Fig. 4). MtDNA-depleted iNHAs and controls were implanted orthotopically into the frontal cortex of NSG mice. Compared with the control arm, mice bearing TFAM knockdown cells had a significantly shortened median survival (MS) (shRNA1: 126 days, shRNA2: 105 days; P < 0.01; Fig. 2E), supporting the oncogenic potential of altering mtDNA number. Hematoxylin and eosin (H&E) staining of iNHA xenografts with TFAM inhibition revealed pathological features of pHGGs, including hypercellularity and an invasive edge, compared with the control group (Fig. 2F). Quantitative PCR assays confirmed that mtDNA copy number in TFAM knockdown xenografts was significantly lower than that in iNHA control cells (Fig. 2G).
Fig. 2.

Partial loss of mtDNA in iNHAs through shRNA-mediated silencing of TFAM promoted tumorigenesis. (A) Western blotting of TFAM and 2 key subunits of OXPHOS complexes, COXII and NDUFB2, in TFAM knockdown iNHAs and shRNA controls, with actin probed as a loading control. (B) Partial depletion of mtDNA greatly enhanced the production of L-lactate (~2-fold) in iNHAs relative to corresponding controls. (C) Stable knockdown of TFAM resulted in a ~30–40% decrease in relative mtDNA copy number in iNHAs compared with scrambled controls. (D) TFAM knockdown cells proliferated at a lower rate compared with controls. (E) NSG mice carrying TFAM-deficient iNHA orthotopic xenografts displayed enhanced tumorigenicity with a significantly worse survival time compared with the control group (n = 5/group). (F) Representative H&E staining of TFAM-deficient xenograft tumors showed some pathological characteristics of DIPG/pediatric high-grade astrocytoma compared with the control. Scale bar: 0.1 mm. (G) Quantitative PCR assays demonstrating a significantly lower mtDNA number in TFAM knockdown xenografts than iNHA non-targeting control (NTC) cells. *P < 0.05; **P < 0.01; ***P < 0.001.
Restoring mtDNA Number via AMPK Activation Is Cytotoxic to pHGG Cells
Recent studies showed that mitochondrial dysfunction can be therapeutically modulated by AMPK activation through pharmacological stimulation of mitochondrial biogenesis.12 To explore this possibility in pHGG, we treated patient-derived SU-DIPG4 and SF188 cells with serial concentrations (50 µM to 1 mM) of AICAR, an AMP-mimetic AMPK agonist. AICAR produced specific cytotoxic effects on pHGG cells in a time-dependent manner but with no apparent inhibitory effect on NHAs (Fig. 3A). The greatest growth inhibition was observed at concentration 250 μM, which also induced apoptosis in SU-DIPG4 (48 h) and SF188 cells (72 h) as measured by cleaved poly(ADP-ribose) polymerase (c-PARP) and reduction of myeloid cell leukemia 1 (MCL1), an anti-apoptotic B-cell lymphoma 2 (Bcl-2) family member (Fig. 3B). Mechanistically, treatment with 250 μM AICAR significantly downregulated L-lactate production (Fig. 3C), and this led to elevated mtDNA copies in SU-DIPG4 and SF188 cells (Fig. 3D). Western blotting analysis showed AICAR treatment activated AMPK signaling and increased expression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and TFAM in these cells (Fig. 3E). To validate these results, we assessed OXPHOS content following AICAR administration by examining 5 pivotal subunits of electron transport chain complexes I–V. In both lines, we observed a gradual time-dependent increase in their expression levels after AICAR exposure (Fig. 3F).
Fig. 3.

AICAR leads to cytotoxic effects on pHGG cells through AMPK activation, elevating mtDNA content and increasing levels of OXPHOS complex content via the PGC-1α/TFAM axis. (A) AICAR significantly reduced the viability of SU-DIPG4 and SF188 cells, while leaving nontransformed NHAs largely unaffected. (B) Exposure to AICAR triggered apoptosis in SU-DIPG4 and SF188 cells. (C) L-lactate concentration was significantly reduced by AICAR treatment in SU-DIPG4 and SF188 cells. (D) SU-DIPG4 and SF188 cells treated with 250 µM AICAR at various time points contained a significantly increased mtDNA content compared with controls. (E, F) AICAR treatment (250 µM) time-dependently increased expression of mitochondrial biogenesis modulators, PGC-1α and TFAM, induced activation of the AMPK signaling pathway, and markedly upregulated respiratory complex levels. *P < 0.05; **P < 0.01; ***P < 0.001.
Restoring Mitochondrial Metabolism by Targeting PDK1 Leads to Reduced Tumor Growth and Enhances Radiosensitivity
While AICAR has activity against pHGG cells, it is only an experimental tool compound and the blood–brain barrier (BBB) limits entry of AICAR into the CNS and leads to inadequate exposure within tumor tissues.13 We therefore examined the efficacy of DCA, a clinically available PDK inhibitor that also reverses the Warburg effect, stimulates OXPHOS, is rapidly absorbed through oral administration and widely distributed in vivo, and readily crosses the BBB. As a repurposed drug, DCA has been shown to achieve millimolar concentrations in serum in clinical trials and is routinely tested in in vitro culture systems at similar concentrations.14,15
To confirm the inhibitory effect of DCA on PDK in pHGG, the levels of p-PDH-E1α and PDH activity were measured. Treatment of DIPG cells for 8 h with DCA led to a reduction in phosphorylation of PDH-E1α, indicating that DCA treatment led to activation of PDH via PDK inhibition (Fig. 4A). This was supported by a PDH activity assay showing that DCA treatment under the same conditions led to a potent increase in PDH activity (Fig. 4B).
Fig. 4.

DCA induced cytotoxic and radiosensitizing effect on DIPG cells by shifting glucose metabolism from glycolysis to OXPHOS. (A) DCA treatment at 10 mM decreased the level of phosphorylated PDH-E1α in DIPG cells. (B) DCA treatment increased the PDH activity in HSJD-DIPG007. (C) DCA treatment induced an increase in oxygen consumption rate (OCR) and a decrease in extracellular acidification rate (ECAR) dose-dependently in HSJD-DIPG007. (D) DCA induced a dose-dependent decrease in mitochondrial reserve respiratory capacity in HSJD-DIPG007. (E) PDK1 is overexpressed in DIPG cultures compared with NHAs. (F) DCA selectively inhibited the proliferation of DIPG cells. (G) DCA treatment augmented RT-induced DNA DSBs. (H) DCA in combination with RT synergistically inhibited clonogenicity of DIPG cells. *P < 0.05; **P < 0.01; ***P < 0.001.
In response to DCA treatment, there was a dose-dependent increase in OXPHOS and a dose-dependent decrease in glycolysis (Fig. 4C, Supplementary Fig. 5), implying that DCA shifted glucose metabolism by coupling glycolysis with OXPHOS. By sequentially injecting oligomycin, FCCP (carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone), the mixture of antimycin and rotenone, a complete mitochondrial profile was further revealed (Supplementary Fig. 5). Because DCA treatment did not affect the maximal respiratory capacity of tumor cells, the mitochondrial reserve respiratory capacity of DCA-treated cells significantly decreased in a dose-dependent manner attributed to an increase in the oxygen consumption rate (Fig. 4D).
To examine the clinical tractability of targeting glycolysis with DCA in DIPG, we compared the effect of DCA on DIPG versus normal cells. First, we assessed PDK1 expression in a panel of primary DIPG cultures and NHAs, and found that PDK1 levels were higher in most of the DIPG cultures than in NHAs (Fig. 4E). We next treated a panel of DIPG lines with a dose range of DCA. DCA was effective in inhibiting the growth of DIPG neurospheres dose-dependently, with significant inhibition of cell proliferation at doses that had no significant effect on noncancerous cells (Fig. 4F). RT is the only standard treatment for pHGG, and we hypothesized that activation of OXPHOS and subsequent ROS production could potentiate DNA damaging effects of RT.16 DCA treatment markedly elevated the level of RT-induced double strand breaks (DSBs) (Fig. 4G), evidenced from examining the level of phosphorylation of histone γ-H2AX, a key step in nucleation of the DNA repair complex at DSBs.17 A clonogenic assay showed that the effect of RT was significantly augmented by the application of DCA, with the combined treatment leading to a synergistic inhibition of clonogenicity in DIPG cells (Fig. 4H).
The Combination of DCA and Metformin Synergistically Inhibits DIPG Growth via Modulation of the AMPK/Mammalian Target of Rapamycin Pathway
DCA treatment led to a shift of glucose metabolism from glycolysis to OXPHOS, which suggests that targeting the increase in OXPHOS in combination may lead to an enhanced antitumor effect. Metformin is one of the most widely used biguanides for treatment of type II diabetes. As a well-documented activator of AMPK and inhibitor of mitochondrial complex I, metformin has been shown to have anticancer activity.18 The DCA/metformin combination synergistically reduced proliferation across a panel of DIPG cultures, with a combination index less than 0.8 across a broad dose range for all cultures tested (Fig. 5A, Supplementary Fig. 6A), but spared noncancerous MRC5 cells (Supplementary Fig. 6B). Cell cycle analysis revealed a small but significant increase at the G2-M phase in the combination treated arm compared with vehicle treated controls (Fig. 5B). To understand the synergistic effect of the combination therapy, we examined ROS production, and an increase in mitochondrial ROS was detected as early as 1 h after each treatment (Fig. 5C), followed by a significant increase in cytosolic ROS after 3 h (Fig. 5C), with the combination arm leading to a higher level of ROS compared with monotherapy. This led to a significantly higher level of ROS-induced DSBs relative to single agent treatments (Fig. 5D, Supplementary Fig. 7A). When treatment extended to 24 and 48 h, mitochondrial membrane depolarization (Fig. 5E) and apoptosis (Fig. 5F, Supplementary Fig. 7B) occurred, with the combination having a greater capacity to induce mitochondrial dysfunction and caspase-mediated cell death than either agent alone.
Fig. 5.

Dual targeting of glucose metabolism with DCA and metformin leads to proliferative arrest and apoptosis in DIPG cells. (A) DCA/metformin combination synergistically inhibited cell proliferation. The combination index (CI) was calculated using CalcuSyn and CI < 1, = 1 and >1 was defined as synergistic, additive, and antagonistic effects, respectively. (B) DCA/metformin combination induced a decrease in the proportion of cells in G1 phase and an increase in G2-M phase. DCA/metformin combination induced higher levels of ROS production (C), H2AX (D), mitochondrial depolarization (E), and apoptosis (F), compared with each treatment alone. (G) DCA attenuated metformin-induced L-lactate production. (H) DCA/metformin combination further decreased cellular ATP level compared with monotherapy. (I) DCA/metformin combination activated the AMPK pathway. *P < 0.05; **P < 0.01; ***P < 0.001.
By inhibiting mitochondrial complex I, metformin treatment stimulated glycolytic rates, as reflected by a pronounced increase in L-lactate production that may favor tumor cell growth through the Warburg effect. However, the addition of DCA sufficiently offset metformin-induced lactate fermentation (Fig. 5G). To assess the effect of combination therapy on energy production, intracellular ATP levels in DIPG cells were quantified. As depicted in Fig. 5H, DCA did not alter ATP levels significantly, while metformin significantly decreased ATP levels. When treated with the combination, intracellular ATP levels of DIPG cells further decreased significantly, indicating that an energy crisis was triggered. The reduction of the intracellular ATP level corresponded with activation of the AMPK pathway, including upregulation of p-AMPK; inhibition of phosphorylated mammalian target of rapamycin (mTOR) and p-P70S6K, two AMPK downstream targets that are frequently activated in cancers, including gliomas; and induction of LC3AB, a marker of autophagy (Fig. 5I, Supplementary Fig. 7C).
The Efficacy of Triple Combination Against DIPG
Given that DCA/metformin combination ultimately induced DSBs, a principle effect of RT, we hypothesized that DCA/metformin may have a more potent radiosensitizing effect. After treatment with combination, DIPG cells became more sensitive to RT compared with monotherapy, with the triple combination showing the greatest reduction in surviving fraction (Fig. 6A) and the highest level of γ-H2AX (Fig. 6B). The triple combination was further assessed using a xenograft orthotopic mouse model bearing patient-derived HSJD-DIPG007 cells. Compared with vehicle treated control (58 days), metformin did not significantly improve the survival (56 days), while DCA alone (64 days) prolonged MS by 6 days (P < 0.05). RT, as a single treatment, extended the MS to 66 days (P < 0.001). When combined with RT, the antitumor efficacy was significantly augmented by either DCA (69 days, P < 0.01 vs RT alone) or metformin (73 days, P < 0.001 vs RT alone). Strikingly, the longest survival benefit was observed in the triple combination arm (83 days) compared with all other treatment arms (Fig. 6C). The histological analysis at day 42 post tumor inoculation confirmed tumor formation and indicated significant reduction in the proliferation of tumor cells in the treatment groups, as reflected in the number of positive cells for the proliferation marker Ki-67 (Fig. 6D). Immunohistochemical staining of γ-H2AX also showed that the triple combination produced the greatest DNA damage in xenografted tumors compared with other groups (Fig. 6E).
Fig. 6.

DCA in combination with metformin enhances radiosensitivity of DIPG both in vitro and in vivo. (A) The clonogenicity of DIPG cells was maximally impaired by the triple combination. (B) The triple combination induced the highest level of DNA DSBs. (C) The triple combination treated orthotopic model of DIPG bearing HSJD-DIPG007 showed the longest median survival compared with other groups (n = 10/group). (D, E) The triple combination induced the lowest proliferation index (Ki-67) and the highest level of DNA DSBs (H2AX). Scale bar: 50 μm. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
In contrast to normal cells, glucose metabolism in cancer cells is often shifted from mitochondrial oxidation toward glycolysis during tumor progression. Consistent with this effect, we identified significantly reduced mtDNA content in the vast majority of pHGGs compared with normal brain. Since aberrant mtDNA quantity is a major contributor to compromised mitochondrial activity, lower mtDNA amount indicates that these tumors undergo a metabolic transition toward aerobic glycolysis in order to adapt to the dynamic metabolic demands. Our models with mtDNA removal demonstrate that mtDNA loss not only triggered a marked increase in L-lactate secretion, but significantly enhanced cell migration and invasion, resistance to chemo/RT, and in vivo tumorigenicity. Our work suggests for the first time that change in mtDNA content is not solely a bystander event but a crucial player in the complex network mediating the Warburg effect in pHGG. MtDNA copy number alteration has been studied in adult gliomas; however, results are inconsistent. Liang et al19,20 reported that both low-grade gliomas and HGGs had markedly increased mtDNA number compared with nonneoplastic brain tissues. In contrast, several recent studies showed that mtDNA quantity was reduced in adult gliomas, lower mtDNA content was associated with poorer prognosis, and the lowest mtDNA was seen in glioblastomas.21,22 It is possible that decreased mtDNA number may be a general phenomenon of both pediatric and adult gliomas.
Several mechanisms may be involved in mtDNA depletion in pHGG. First, TFAM and PPARGC1A mRNA transcripts were found to be downregulated in DIPG. Further investigation in a larger cohort is warranted to confirm its association with reduced mtDNA number. Second, our SNP6.0 data showed that there was a minor copy number deletion in 2 key regulators of mtDNA replication and mitochondrial biogenesis, TWNK (5 of 45 DIPGs) and GABPB1 (6 of 45 DIPGs).23 Third, decreased mtDNA number may stem from somatic mutations in the D-loop regulatory region, which could perturb binding affinities of nuclear factors involved in mtDNA duplication and transcription, leading to lower mtDNA copies.3 Fourth, mtDNA number was reported to range across various brain regions due to a different energy requirement.24 Since our normal brain tissues originated from a neuronal rich anatomical location, different mtDNA amount and mitochondrial function between neural and glial tumor cells might be a confounding factor for our observation in this study. Lastly, recent evidence showed that hypoxia may be a characteristic feature of pHGG including DIPG,25,26 which might contribute to higher rates of mitophagy and subsequently deplete mtDNA content. It was reported that hypoxia-inducible factor 1 (HIF1)–dependent expression of Bcl-2 nineteen-kilodalton interacting protein 3 is essential for mitophagy, and hypoxia also activates autophagy through HIF1-independent pathways.27
Cellular metabolism is controlled by an intricate web of energy sensors involving AMPK. The AMPK activator AICAR exhibited potent antitumor efficacy in pHGG cells by inhibiting glycolysis and leading to an increase in mtDNA copy number and respiratory complex levels. Interestingly, TFAM was identified as a selective target of translation mediated by eukaryotic initiation factor 4E binding proteins,28 indicating that it may also be a target of mTOR. As we have shown that AMPK activation significantly inhibited mTOR and its downstream signaling, the restoration of TFAM induced by AICAR may partially be due to mTOR inhibition following AMPK activation. The effect of AICAR on TFAM, PGC-1α, and AMPK signaling was not in a dose-dependent manner at least in the 2 pHGG lines we tested. This finding is in accordance with previous studies showing that AMPK-independent mechanisms (eg, inhibition of lipogenesis, acting on Akt phosphorylation and HIF1α content, degradation of the critical G2M phosphatase cdc25c) may also participate in the growth suppressive action of AICAR, depending on specific tumor models.29,30 Due to the low BBB penetration of AICAR, we investigated another approach to reversing the Warburg effect using the PDK inhibitor DCA. Importantly, the majority of primary patient DIPGs express high levels of PDK1, supporting this as a potential therapeutic target in these tumors. DCA is an orphan drug that has been long used as an investigational treatment for various mitochondrial disorders. When administered orally, DCA is rapidly absorbed with almost 100% bioavailability and concentrates in mitochondria.31 Most relevant to pHGGs, DCA is a clinically available drug that is well tolerated in children.32
DCA treatment led to higher levels of DSBs likely through ROS-dependent machinery, suggesting that combining it with RT may be an effective strategy. We observed improved radiosensitivity of DIPG cells when combined with DCA treatment. This effect was even greater when a third agent, metformin, was added. The metabolic stress induced by this combination was further confirmed by the activation of the AMPK pathway. More importantly, the DSBs induced by DCA/metformin also synergized with RT, with the triple combination leading to a more potent antitumor effect both in vitro and in vivo. Although DCA and metformin have been demonstrated as radiosensitizers individually via different mechanisms,33,34 this is the first time that dual targeting of glucose metabolism has been shown to be a more potent radiosensitizing strategy for pHGGs. As metformin and DCA can both readily cross the BBB, this treatment is readily translatable to the clinic and warrants testing in a disease setting with no known curative treatments.
In summary, our study suggests that there is a disruption of energy metabolism in pHGG favoring glycolysis over OXPHOS, and reveals a potential tumorigenic role played by metabolic reprogramming via reduced mtDNA content and enhanced glycolysis. Reversing this metabolic phenotype potently improves the radiosensitivity of pHGGs through induction of oxidative stress. This anti-metabolic strategy may open a window of opportunity for designing more efficacious treatment for these devastating diseases.
Supplementary Material
Supplementary data are available at Neuro-Oncology online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was funded by grants from The DIPG Collaborative, Kids Cancer Project, Canadian Cancer Society Research Institute, the Australian National Health and Medical Research Council, and the Benny Wills Brain Tumour Research Program. H.S. was supported by the Sydney West Radiation Oncology Network and M.Y. by the Garron Family Cancer Centre.
Conflict of interest statement. The authors have declared that no conflicts of interest exist.
Authorship statement. The entire study was conceived by HS, MY, CH, and DZ. The design and performance of the experiments were supervised by CH and DZ. The majority of the experiments were performed by HS and MY, and the manuscript and figures were prepared and revised by HS, MY, CH, and DZ. Experiments were performed by MT, CC, SJ, JL, SR, YC, and RS.
References
- 1. Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol. 2012;2:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yu M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011;89(3-4):65–71. [DOI] [PubMed] [Google Scholar]
- 4. Zhou Y, Zhou Y, Shingu T, et al. . Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem. 2011;286(37):32843–32853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sattler UG, Meyer SS, Quennet V, et al. . Glycolytic metabolism and tumour response to fractionated irradiation. Radiother Oncol. 2010;94(1):102–109. [DOI] [PubMed] [Google Scholar]
- 6. Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990;4(14):3224–3233. [DOI] [PubMed] [Google Scholar]
- 7. Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. 2013;3:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michelakis ED, Sutendra G, Dromparis P, et al. . Metabolic modulation of glioblastoma with dichloroacetate. Sci Trans Med. 2010;2(31):31ra34. [DOI] [PubMed] [Google Scholar]
- 9. Dunbar EM, Coats BS, Shroads AL, et al. . Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs. 2014;32(3):452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abdelmalak M, Lew A, Ramezani R, et al. . Long-term safety of dichloroacetate in congenital lactic acidosis. Mol Genet Metab. 2013;109(2):139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen H, Decollogne S, Dilda PJ, et al. . Dual-targeting of aberrant glucose metabolism in glioblastoma. J Exp Clin Cancer Res. 2015;34:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov. 2013;12(6):465–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marangos PJ, Loftus T, Wiesner J, et al. . Adenosinergic modulation of homocysteine-induced seizures in mice. Epilepsia. 1990;31(3):239–246. [DOI] [PubMed] [Google Scholar]
- 14. Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug? Biochim Biophys Acta. 2014;1846(2):617–629. [DOI] [PubMed] [Google Scholar]
- 15. Chu QS, Sangha R, Spratlin J, et al. . A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs. 2015;33(3):603–610. [DOI] [PubMed] [Google Scholar]
- 16. Vignard J, Mirey G, Salles B. Ionizing-radiation induced DNA double-strand breaks: a direct and indirect lighting up. Radiother Oncol. 2013;108(3):362–369. [DOI] [PubMed] [Google Scholar]
- 17. Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24(4):679–686. [DOI] [PubMed] [Google Scholar]
- 18. Pernicova I, Korbonits M. Metformin—mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143–156. [DOI] [PubMed] [Google Scholar]
- 19. Liang BC. Evidence for association of mitochondrial DNA sequence amplification and nuclear localization in human low-grade gliomas. Mutat Res. 1996;354(1):27–33. [DOI] [PubMed] [Google Scholar]
- 20. Liang BC, Hays L. Mitochondrial DNA copy number changes in human gliomas. Cancer Lett. 1996;105(2):167–173. [DOI] [PubMed] [Google Scholar]
- 21. Correia RL, Oba-Shinjo SM, Uno M, Huang N, Marie SK. Mitochondrial DNA depletion and its correlation with TFAM, TFB1M, TFB2M and POLG in human diffusely infiltrating astrocytomas. Mitochondrion. 2011;11(1):48–53. [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y, Qu Y, Gao K, et al. . High copy number of mitochondrial DNA (mtDNA) predicts good prognosis in glioma patients. Am J Cancer Res. 2015;5(3):1207–1216. [PMC free article] [PubMed] [Google Scholar]
- 23. Buczkowicz P, Hoeman C, Rakopoulos P, et al. . Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46(5):451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuke S, Kubota-Sakashita M, Kasahara T, Shigeyoshi Y, Kato T. Regional variation in mitochondrial DNA copy number in mouse brain. Biochim Biophys Acta. 2011;1807(3):270–274. [DOI] [PubMed] [Google Scholar]
- 25. Blandin A-F, Durand A, Litzler M, et al. . HGG-12. Hypoxia seems to be frequently upregulated in the pediatric high grade glioma and DIPG. Neuro Oncol. 2017;19(Suppl 4):iv25. [Google Scholar]
- 26. Yeom KW, Lober RM, Nelson MD Jr, Panigrahy A, Blüml S. Citrate concentrations increase with hypoperfusion in pediatric diffuse intrinsic pontine glioma. J Neurooncol. 2015;122(2):383–389. [DOI] [PubMed] [Google Scholar]
- 27. Zhang H, Bosch-Marce M, Shimoda LA, et al. . Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28. Morita M, Gravel SP, Chénard V, et al. . mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18(5):698–711. [DOI] [PubMed] [Google Scholar]
- 29. Liu X, Chhipa RR, Pooya S, et al. . Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A. 2014;111(4):E435–E444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo D, Hildebrandt IJ, Prins RM, et al. . The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci U S A. 2009;106(31):12932–12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stacpoole PW. The pharmacology of dichloroacetate. Metabolism. 1989;38(11):1124–1144. [DOI] [PubMed] [Google Scholar]
- 32. Stacpoole PW, Gilbert LR, Neiberger RE, et al. . Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 2008;121(5):e1223–e1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zannella VE, Dal Pra A, Muaddi H, et al. . Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res. 2013;19(24):6741–6750. [DOI] [PubMed] [Google Scholar]
- 34. Shen H, Hau E, Joshi S, Dilda PJ, McDonald KL. Sensitization of glioblastoma cells to irradiation by modulating the glucose metabolism. Mol Cancer Ther. 2015;14(8):1794–1804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.