

Abstract
Background
Deregulation of the nuclear factor of activated T cell (NFAT) pathway has been reported in several human cancers. Particularly, NFAT2 is involved in the malignant transformation of tumor cells and is identified as an oncogene. However, the role of NFAT2 in glioblastoma (GBM) is largely unknown.
Methods
The expression and prognostic value of NFAT2 were examined in the databases of the Repository of Molecular Brain Neoplasia Data and The Cancer Genome Atlas (TCGA) and clinical samples. The functional effects of silencing or overexpression of NFAT2 were evaluated in glioma stem cell (GSC) viability, invasion, and self-renewal in vitro and in tumorigenicity in vivo. The downstream target of NFAT2 was investigated.
Results
High NFAT2 expression was significantly associated with mesenchymal (MES) subtype and recurrent GBM and predicted poor survival. NFAT2 silencing inhibited the invasion and clonogenicity of MES GSC-enriched spheres in vitro and in vivo. NFAT2 overexpression promoted tumor growth and MES differentiation of GSCs. A TCGA database search showed that histone deacetylase 1 (HDAC1) expression was significantly correlated with that of NFAT2. NFAT2 regulates the transcriptional activity of HDAC1. Rescue of HDAC1 in NFAT2-knockdown GSCs partially restored tumor growth and MES phenotype. Loss of NFAT2 and HDAC1 expression resulted in hyperacetylation of nuclear factor-kappaB (NF-κB), which inhibits NF-κB–dependent transcriptional activity.
Conclusion
Our findings suggest that the NFAT2-HDAC1 pathway might play an important role in the maintenance of the malignant phenotype and promote MES transition in GSCs, which provide potential molecular targets for the treatment of GBMs.
Keywords: glioblastoma, HDAC1, mesenchymal, NF-κB, NFAT2
Key Points.
1. The NFAT2-HDAC1 pathway is important for the maintenance of the MES phenotype.
2. The NFAT2-HDAC1 pathway influences the acetylation and activity of NF-κB.
Importance of the Study.
MES transition, especially in GSCs, is usually associated with acquired resistance to radiochemotherapy and malignant progression. However, the intrinsic molecular mechanism of MES differentiation is still largely unclear. In this study, we demonstrate that NFAT2 is important for the maintenance of the MES phenotype. And HDAC1 was identified as the downstream target of NFAT2. By regulation of HDAC1 expression, NFAT2 influences the acetylation of NF-κB subunit p65 and affects the transcriptional activity of NF-κB, which further modulates the MES transition of GSCs. By modulating NF-κB activity, the NFAT2-HDAC1 pathway supports the malignant phenotype and promotes MES transition in GBMs. Therefore, inhibition of the NFAT2/HDAC1/NF-κB axis is an attractive therapeutic approach for GBMs, especially for the MES subtype.
Glioblastoma (GBM) is the most common and lethal primary brain tumor in adults.1 According to characteristic gene expression, GBM can be classified into 3 clinically relevant molecular subtypes: mesenchymal (MES), classical (CL), and proneural (PN).2,3 The MES subtype is the most aggressive and radioresistant, correlating with poor prognosis.4,5 Moreover, GBM often exhibits transcriptional plasticity and heterogeneity, resulting in the transition from one subtype to another.6 MES transition, especially in glioma stem cells (GSCs), is usually associated with acquired resistance to radiochemotherapy and malignant progression.6–8 However, the intrinsic molecular mechanism of MES differentiation is still largely unclear. Several inflammatory pathways, microenvironment factors, and epigenetic events might contribute to the MES phenotype in GBMs.4,9
Nuclear factor of activated T cells (NFAT) family members were first identified as transcription factors in T cells.10 By interaction with other pathways, including the histone deacetylase (HDAC) and nuclear factor-kappaB (NF-κB) pathways, NFATs regulate inflammatory reactions and immune responses.11,12 It has been revealed that NFATs play important roles in cancer initiation and progression.13 NFAT2, also known as NFATc1, is involved in cancer development. Dysregulation of NFAT2 activity inhibits cell differentiation and induces malignant transformation, suggesting that NFAT2 is an oncogene.14 Moreover, NFAT2 is related to acquiring stem cell–like properties and self-renewal capacity and promoting tumor cell epithelial-to-mesenchymal transition (EMT).14–16 Through EMT and gain of stemness, cancer cells become highly invasive and aggressive, leading to tumor metastasis, treatment resistance, and recurrence.17 Recently, abnormal activation of NFAT2 signaling has been found in the malignant progression of colon cancer,18 pancreatic cancer,19 and mammary cancer.20 However, the role of the NFAT2 pathway has not yet been elucidated in GBMs. NFAT2 may activate different signaling pathways and have distinct functions in different cancers.13 Therefore, in this study, we examined the effects of NFAT2 on the malignant phenotype of GSCs in vitro and in vivo, and we also investigated the downstream target of NFAT2.
Materials and Methods
Ethics
This study was approved by the institutional review board of The First Hospital of China Medical University (approval number 81472360). Written informed consent was obtained from each tumor tissue donor for the use of the tumor tissue and clinical data for future research. All animal experiments were performed under the supervision of the China Medical University Animal Ethics Committee.
GSC Isolation
Patient-derived GSCs (G03, G08, G10, G12, G13, and G23) were isolated, and neurosphere culture was performed as previously reported.21 Briefly, freshly resected clinical glioma tissues were dissociated into single cells and cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 with B27 (1:50), recombinant human (rh) basic fibroblast growth factor (20 ng/mL), and rh–epidermal growth factor (20 ng/mL; Gibco). Then, neurospheres were collected and grown in the neurosphere medium following the standard procedure. For GSC adherent cultures, a single-cell suspension was plated on laminin-coated culture plates along with standard GSC medium as reported by Pollard et al.22 For differentiation, GSCs were cultured in DMEM/F12 supplemented with 10% fetal bovine serum. The cancer stem cell nature of the GSCs was evaluated by functional assays of self-renewal and in vivo tumor formation. The expression of stem cell markers (cluster of differentiation [CD]133, CD44, and nestin) was detected by flow cytometry or immunofluorescence, and the multi-lineage differentiation capacity of GSCs was detected by immunofluorescence. CD44high and CD44low GSCs were isolated using magnetic cell sorting with CD44 microbeads (Miltenyi Biotec, #130-095-194). All of the GSCs analyzed were cultured fewer than 20 passages.
Plasmids
Short hairpin (sh)RNA–based silencing and overexpression of NFAT2 and HDAC1 were performed as previously described.21 NFAT2, HDAC1, and control shRNAs were purchased from GeneChem. Lentivirus-based vector was constructed for NFAT2 and HDAC1 overexpression (GeneChem). Null or inhibitor of kappaB alpha (IκBα) mutant (IκBαM, serine [S]32/S36) adenoviral particles were purchased from Vector Biolabs. The effectiveness of gene silencing and overexpression was detected by western blotting (WB).
Limiting Dilution Neurosphere Formation Assay
The self-renewal capacity of GSCs was evaluated by the neurosphere formation assay as reported previously.21 In brief, neurospheres were dissociated and seeded into 96-well plates at 50, 100, 500, or 1000 cells per well. After 7 days, spheres with diameters larger than 50 μm were counted. Data were recorded and analyzed as previously described.21
Intracranial Xenografts
Female Bagg albino/c nude mice (6 wk old) were obtained from Beijing Vital River Laboratory Animal Technology and bred in laminar flow cabinets under specific pathogen-free conditions. Transfected GSCs (1 × 105 cells) were injected intracranially in anesthetized nude mice using a stereotaxic apparatus. We observed mice daily for neurological symptoms or death, and tumor growth was evaluated. When neurological symptoms were observed, mice were sacrificed by cervical spine dislocation. Mouse brains were collected for analysis as previously reported.21
Chromatin Immunoprecipitation Assays
Chromatin immunoprecipitation (ChIP) assays were performed using the EZ-ChIP Immunoprecipitation Kit (Millipore) as previously reported.21 Anti-NFAT2 (ab2796) and anti-HDAC1 (ab7028) antibodies were used for immunoprecipitation. PCR was performed using primer pairs for the NFAT2 binding site: forward 5′-ACAGTGCCTCTGGTGC-3′ and reverse 5′-CCAAACCCAACTCCC-3′. ChIP assays detecting the recruitment of HDAC1 to the chitinase 3-like protein 1 (YKL40) and CD44 promoters were performed as previously described by Bhat et al.23 The promoter regions of YKL40 and CD44 were determined using the National Center for Biotechnology Information gene database (https://www.ncbi.nlm.nih.gov/gene/?term) and Promoter 2.0 Prediction Server (http://www.cbs.dtu.dk/services/Promoter/). The promoters of YKL40 and CD44 were identified between −4000 bp upstream of the transcription start site and +100 bp downstream of the transcription start site. Ten PCR primers (Supplementary Table 2) were designed to examine the recruitment of HDAC1 to the YKL40 and CD44 promoters.
Additional details about the materials and methods are available in the Supplementary Materials.
Results
NFAT2 Is Upregulated in MES GBMs and Inversely Correlated with Survival
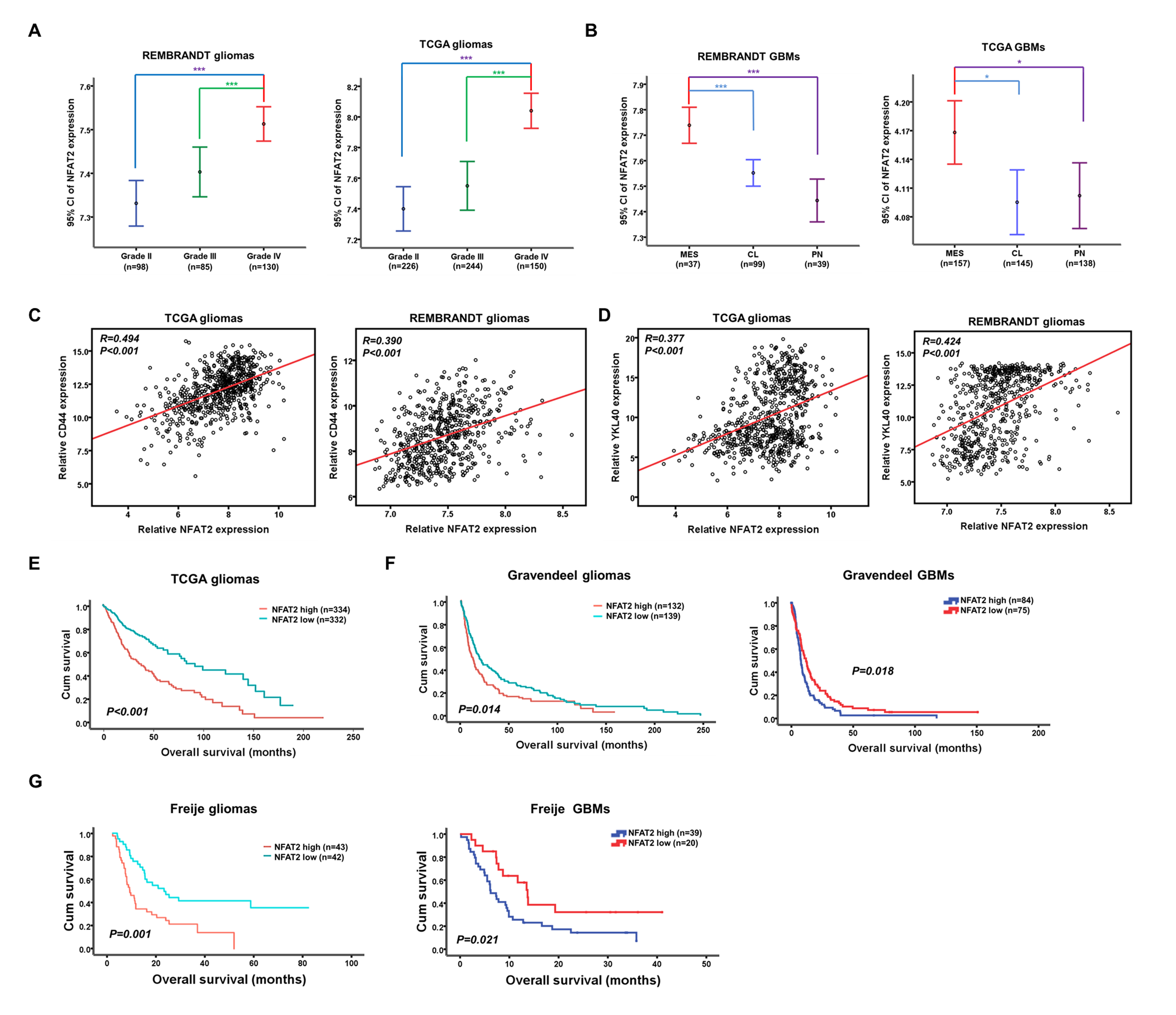
First, the expression of NFAT2 was examined in publicly available glioma datasets. We found that NFAT2 expression was associated with the histopathologic grade of glioma, and higher expression of NFAT2 was observed in grade IV gliomas in the databases of both the Repository of Molecular Brain Neoplasia Data (REMBRANDT) and The Cancer Genome Atlas (TCGA) (Fig. 1A and Supplementary Fig. 1A). Moreover, the level of NFAT2 expression was upregulated in the MES subtype of GBM compared with the CL and PN subtypes (Fig. 1B and Supplementary Fig. 1B). Interestingly, NFAT2 expression was higher in recurrent GBMs (Fig. 1C), which often exhibited an MES subtype.6,24 The expression of NFAT2 positively correlated with the expression of MES markers CD44 and YKL40 (Supplementary Fig. 1C, D). In addition, elevated expression of NFAT2 was associated with poorer survival in gliomas, GBMs, and MES GBMs according to several independent datasets (Fig. 1D–G and Supplementary Fig. 1E–G).
Fig. 1.

Correlation between NFAT2 expression and the clinic-pathologic features of gliomas. (A) NFAT2 mRNA expression is shown according to the histopathologic grades of REMBRANDT and TCGA gliomas. (B) Messenger RNA expression of NFAT2 is shown according to the molecular subtypes of REMBRANDT and TCGA GBMs. (C) NFAT2 mRNA expression is compared between primary and recurrent TCGA GBMs. (D) Prognostic significance of NFAT2 in REMBRANDT gliomas. (E–G) The prognostic significance of NFAT2 in MES GBMs was tested using the REMBRANDT database (E), Lee Y dataset (F), and Freije dataset (G). *P < 0.05, ***P < 0.001.
Next, we examined the expression of NFAT2 in our clinical samples and patient-derived primary GSCs. Clinical information on the 6 patient-derived GSC cultures is presented in Supplementary Table 1. Hematoxylin and eosin (H&E) staining of the original patient tumors is shown in Supplementary Fig. 2A. More than 85% of G03, G12, and G13 GSCs were CD133 positive (PN GSC marker5), while over 80% of G08, G10, and G23 GSCs were CD44 positive (MES GSC marker4,5; Supplementary Fig. 2B). The enrichment of CD133+, CD44+, and nestin+ GSCs in the isolated neurospheres was confirmed by immunofluorescence (Supplementary Fig. 2C). The multi-lineage differentiation capacity of GSCs is shown in Supplementary Fig. 2D. In 46 clinical glioma samples (grade II: 10; grade III: 15; grade IV: 21), immunohistochemistry (IHC) (Fig. 2A) and WB (Fig. 2B) demonstrated that the expression of NFAT2 was positively correlated with tumor grade, and NFAT2 was upregulated and located mainly in the nuclei of high-grade gliomas. In patient-derived GSC-enriched cultures, the expression of NFAT2 was examined by WB. NFAT2 protein levels were positively correlated with MES markers YKL40 and CD44 and were negatively correlated with PN marker oligodendrocyte transcription factor 2 (Olig2) (Fig. 2C). The expression and intracellular localization of NFAT2 in different GSCs were also examined by immunofluorescence (Fig. 2D). Moreover, when we sorted GSCs into CD44high and CD44low subpopulations, higher NFAT2 expression was found in CD44high subfractions (Fig. 2E, F). Altogether, these data suggest that elevated NFAT2 expression is associated with the malignant phenotype of glioma and is clinically relevant in GBMs.
Fig. 2.

NFAT2 expression in primary GSCs. (A) Representative IHC images of NFAT2 expression in WHO grade IV, grade III, and grade II glioma clinical samples. Intense immunostaining was observed in the nuclei of grade IV glioma tissues. Scale bar = 50 μm. (B) Representative WB image of NFAT2 protein expression in WHO grade IV, grade III, and grade II glioma clinical samples. (C) Western blotting shows the expression of NFAT2, YKL40, CD44, and Olig2 in the indicated GSCs. (D) Immunofluorescence of NFAT2 in the primary GSCs. Scale bar = 25 μm. (E, F) The expression of NFAT2 in CD44high and CD44low GSCs was examined by real-time PCR (E) and WB (F). Results are presented as mean ± SD of triplicate samples from 3 independent experiments. *P< 0.05 and **P< 0.01.
NFAT2 Silencing Inhibits MES GSC-Enriched Tumor Sphere Growth In Vitro
To investigate the functional significance of elevated NFAT2 expression using 2 different shRNAs, NFAT2 was silenced in MES GSC-enriched G08 and G10 spheres. WB showed that NFAT2 silencing led to a decrease in CD44 and YKL40 expression, indicating the loss of the MES phenotype (Fig. 3A). NFAT2 knockdown remarkably inhibited cell proliferation over time (Fig. 3B) and promoted cell apoptosis (4.8–7.6-fold; Fig. 3C). Loss of NFAT2 expression also decreased GSC invasion as judged by the transwell invasion assays (Fig. 3D) and 3D spheroid-based invasion assays (Fig. 3E). Moreover, GSC spheres with NFAT2 knockdown grew much slower than control cells, showing marked reductions in sphere size (2.3–3.5-fold; Fig. 3F) and sphere formation (Fig. 3G). However, in shNFAT2-transfected PN GSC-enriched G03 and G12 spheres that had low basal NFAT2 levels, the growth and invasion were not significantly affected (Supplementary Fig. 3). These results suggest that NFAT2 is important for the MES phenotype and is preferentially required for the malignant growth of MES GSCs.
Fig. 3.

NFAT2 silencing inhibits MES GSC-enriched spheres clonogenicity in vitro. (A) Western blotting of NFAT2, CD44, and YKL40 in GSCs transfected with shRNA targeting NFAT2 (shNFAT2-1 or shNFAT2-2) or a control shRNA (shC). (B) Cell viability assay shows that NFAT2 knockdown markedly decreases the proliferation of G08 and G10 GSCs. (C) Targeting of NFAT2 via specific shRNAs significantly increases the apoptosis of G08 and G10 GSCs, as assessed with an assay by TUNEL (terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling). Scale bar = 50 μm. (D) Representative microphotographs showing the invasion of G08 and G10 GSCs in the presence of NFAT2-shRNA or control-shRNA using the Matrigel assay. Cells were allowed to invade the Matrigel-coated filters toward the lower compartment for 20 h. Scale bar = 50 μm. Histogram showing the quantification of invasive cells. (E) The 3D spheroid invasion assay demonstrates that NFAT2 knockdown significantly decreased the invasion of G08 and G10 neurospheres. Scale bar: 100 μm. (F) Representative images of G10 and G08 neurospheres transfected with shRNA targeting NFAT2. Histogram showing the quantification of neurosphere size. Scale bar = 50 μm. (G) Limiting dilution neurosphere formation assays of effect of NFAT2 silencing on GSC renewal. Results are presented as mean ± SD of triplicate samples from 3 independent experiments. *P < 0.05 and **P < 0.01.
NFAT2 Overexpression Promotes MES Transition and Growth of GSCs In Vitro
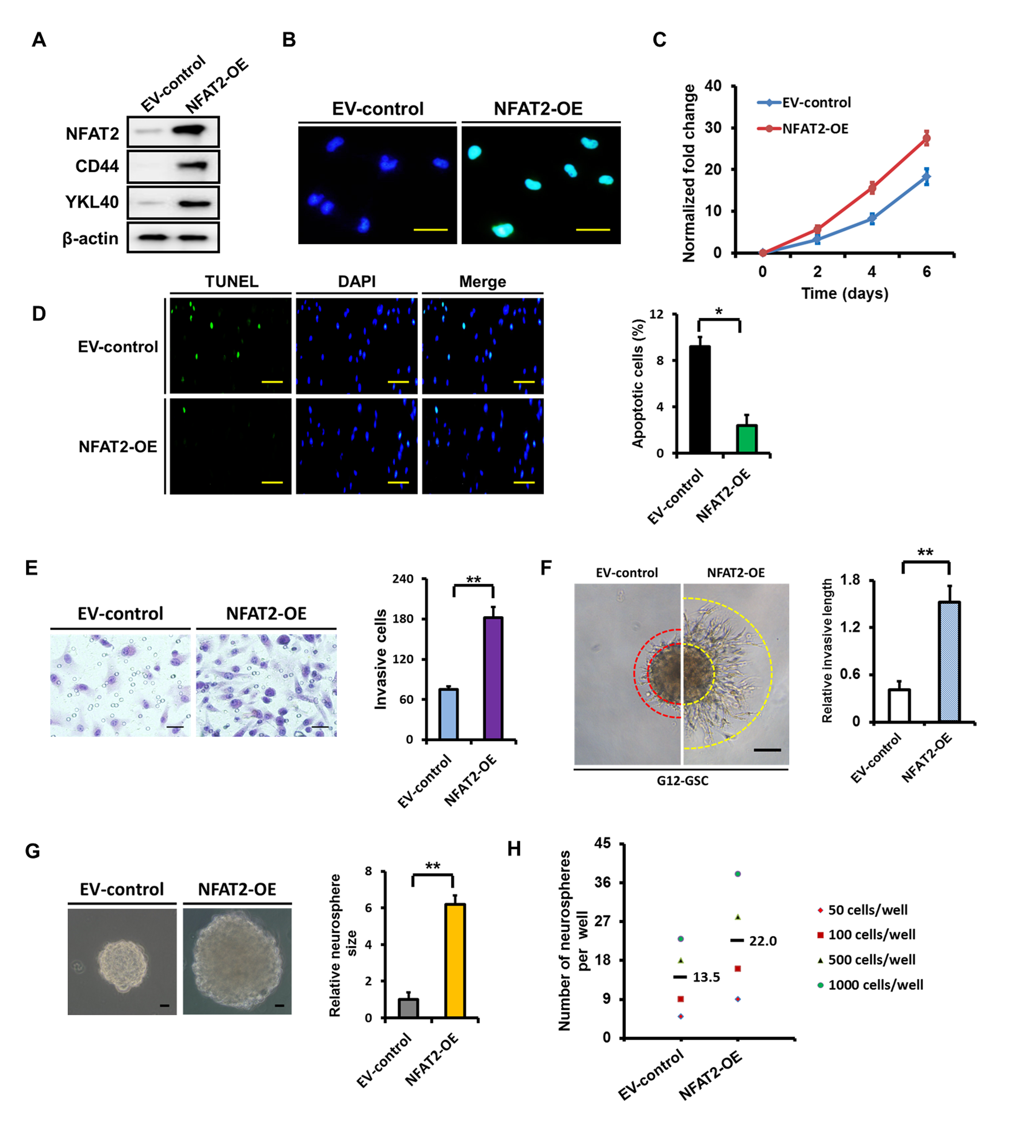
To further confirm its role, NFAT2 was overexpressed in G12 GSCs (Supplementary Fig. 4A, B). As shown in Supplementary Fig. 4A, NFAT2 overexpression increased the expression of CD44 and YKL40, showing the MES differentiation. NFAT2 upregulation resulted in an obvious increase in cell proliferation (Supplementary Fig. 4C) and decrease in cell apoptosis (2.8-fold; Supplementary Fig. 4D). In addition, NFAT2 overexpression significantly promoted cell invasion (2.4-fold; Supplementary Fig. 4E, F). Furthermore, upregulation of NFAT2 enhanced the growth of G12 spheres and resulted in a marked increase in sphere size (5.2-fold; Supplementary Fig. 4G) and sphere formation (Supplementary Fig. 4H). NFAT2-induced MES transition and malignant phenotype were also validated in G03 and G13 GSCs (Supplementary Fig. 5).
NFAT2 Affects the Tumorigenicity and MES Differentiation of GSCs In Vivo
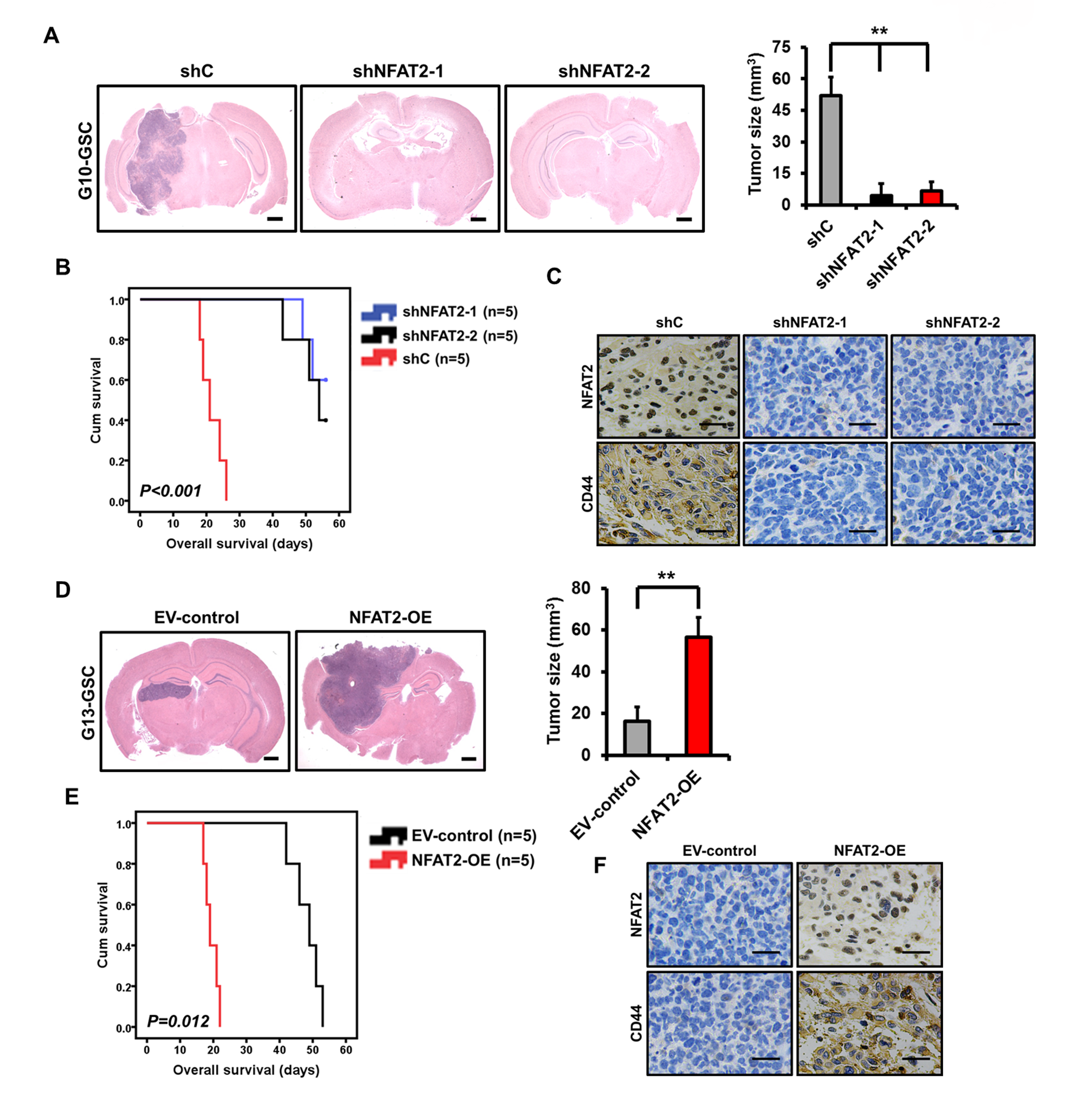
To determine the role of NFAT2 in GSC tumorigenesis and MES transition in vivo, the transfected G08, G10, G12, or G13 GSCs were injected into the brains of mice. Compared with control shRNA, silencing of NFAT2 significantly inhibited the intracranial tumor growth of G08 GSCs (4.7–7.5-fold; Fig. 4A) and prolonged survival time (mean survival: 34.4 ± 4.5 days vs 109.6 ± 10.6 and 102.2 ± 9.9 days, respectively; Fig. 4B). IHC from in vivo tumors showed that NFAT2-knockdown GSCs expressed lower levels of CD44 than did control GSC-induced tumors (Fig. 4C). Similar in vivo effects were observed in NFAT2-knockdown G10 GSCs (Supplementary Fig. 6A–C). Meanwhile, NFAT2 overexpression significantly enhanced the intracranial tumor growth of G12 GSCs (2.3-fold; Fig. 4D) and diminished survival periods (mean survival: 53.2 ± 5.8 days vs 24.6 ± 4.0 days; Fig. 4E). Moreover, intracranial tumors formed by NFAT2-overexpression G12 expressed higher levels of CD44 than did control G12-induced tumors (Fig. 4F), indicating an NFAT2-driven MES transition in vivo. The in vivo effects of NFAT2 overexpression were also confirmed in G13 GSCs (Supplementary Fig. 6D, F).
Fig. 4.

NFAT2 regulates GSC growth in vivo. (A) H&E-stained brain sections of mice after intracranial transplantation of G08 GSCs transfected with shC, shNFAT2-1, or shNFAT2-2. Brains were harvested at 15 days after transplantation. NFAT2 silencing significantly inhibited tumor growth in vivo. Scale bars = 1 mm. (B) Kaplan–Meier survival curves of mice injected with G08 GSCs transfected with shC, shNFAT2-1, or shNFAT2-2. (C) IHC staining of NFAT2 and CD44 in intracranial tumors derived from G08 GSCs transfected with shC, shNFAT2-1, or shNFAT2-2. Scale bar = 25 μm. (D) H&E-stained brain sections of mice after intracranial transplantation of G12 GSCs transfected with the NFAT2 overexpression plasmids or empty vector. Brains were harvested at 15 days after transplantation. NFAT2 overexpression markedly enhanced tumor growth in vivo. Scale bars = 1 mm. (E) Kaplan–Meier survival curves of mice injected with G12 GSCs transfected with the NFAT2 overexpression plasmids or empty vector. (F) IHC staining of NFAT2 and CD44 in intracranial tumors derived from G12 GSCs transfected with the NFAT2 overexpression plasmids or empty vector. Scale bar = 25 μm. *P < 0.05, **P < 0.01.
NFAT2 Regulates the Expression of HDAC1 in GSCs
Next, we sought to identify potential downstream targets of NFAT2. According to the database of TCGA, the expression of NFAT2 was significantly correlated with the expression of HDAC1 (R = 0.692, P < 0.001; Fig. 5A). The correlation was validated in the REMBRANDT database (Supplementary Fig. 7A). Consistent with NFAT2 expression, the expression of HDAC1 was also significantly upregulated in the MES subtype (Fig. 5B and Supplementary Fig. 7B) and recurrent GBMs (Supplementary Fig. 7C). The expression of HDAC1 positively correlated with the expression of the MES markers CD44 and YKL40 (Supplementary Fig. 7D, E). In addition, higher levels of HDAC1 expression were associated with poorer survival in gliomas and GBMs according to the databases of TCGA and REMBRANDT (Fig. 5C and Supplementary Fig. 7F–H). And similar to NFAT2, the expression of HDAC1 was higher in CD44high GSCs than in CD44low GSCs (Fig. 5D, E).
Fig. 5.

NFAT2 regulates the expression of HDAC1 in GSCs. (A) The expression of HDAC1 was significantly correlated with that of NFAT2 in TCGA gliomas. (B) The mRNA expression of HDAC1 is shown according to molecular subtypes of TCGA GBMs. (C) The prognostic significance of HDAC1 is examined in TCGA gliomas. (D, E) The expression of HDAC1 in CD44high and CD44low GSCs was examined by real-time PCR (D) and WB (E). (F) The effect of NFAT2 knockdown on the expression of HDAC1 was examined by western blotting in G08 and G10 GSCs. (G) The effect of NFAT2 overexpression on the expression of HDAC1 was examined by western blotting in G12 GSCs. (H, I) Double-labeled immunofluorescence staining demonstrates that NFAT2-knockdown is accompanied by reduced HDAC1 expression in G08 (H), while NFAT2 overexpression is accompanied by increased HDAC1 expression in G12 (I). Scale bar = 25 μm. (J, K) Effect of NFAT2 on HDAC1 promoter activities. Knockdown of NFAT2 in G08 significantly suppresses the luciferase activity driven by the wildtype HDAC1 promoters compared with control-shRNA. Mutating NFAT2 binding sites markedly decreases promoter activity compared with the wildtype promoter (J). NFAT2 overexpression in G12 enhances luciferase promoter activities (K). (L) Binding of NFAT2 to HDAC1 promoters. Binding of NFAT2 is suppressed when NFAT2 is knocked down in G08, while binding is enhanced when NFAT2 is overexpressed in G12. Results are presented as mean ± SD of triplicate samples from 3 independent experiments. *P < 0.05, ***P < 0.001.
As shown in Figure 5F–I, WB and double-labeled immunofluorescence demonstrated that NFAT2 silencing remarkably suppressed the expression of HDAC1 in MES GSC-enriched G08 and G10 spheres. In PN GSC-enriched G03 and G12 spheres with low basal NFAT2 and HDAC1 levels, shNFAT2 transfection did not significantly influence HDAC1 levels (Supplementary Fig. 7I). However, NFAT2 overexpression markedly increased the expression of HDAC1 in PN GSC-enriched G12, G03, and G13 spheres (Fig. 5G–I and Supplementary Fig. 7I). To determine whether NFAT2 regulated HDAC1 at the transcriptional level, luciferase reporter assays were designed so that the HDAC1 promoters were cloned upstream of the luciferase. NFAT2 silencing markedly decreased luciferase activity in G08 GSCs with wildtype promoters (Fig. 5J), and overexpression of NFAT2 significantly enhanced HDAC1 promoter activity in G12, G03, and G13 GSCs (Fig. 5K and Supplementary Fig. 7J). Promoter mutation in the NFAT2 binding site also markedly reduced luciferase activity (Fig. 5J–K and Supplementary Fig. 7J). ChIP assays showed that NFAT2 bound to the promoter of HDAC1 in G08 GSCs, and following NFAT2 knockdown, binding was significantly suppressed. Meanwhile, NFAT2 overexpression increased the binding of NFAT2 to the promoter of HDAC1 in G12, G03, and G13 GSCs (Fig. 5L and Supplementary Fig. 7K). Thus, these results indicate that NFAT2 enhances the promoter activities of HDAC1 in GSCs.
In addition, loss of HDAC1 markedly decreased CD44 and YKL40 expression, inhibited proliferation, and induced apoptosis (Supplementary Fig. 8A–C) in G08 and G10 GSCs. HDAC1-silenced G08 and G10 GSCs showed a substantial reduction of invasion (Supplementary Fig. 8D, E) and diminished self-renewal ability (Supplementary Fig. 8F, G), suggesting that HDAC1 contributes to the maintenance of the MES phenotype and tumor growth.
Rescue of HDAC1 in NFAT2-Silenced GSCs Partially Restores Tumor Growth
To investigate whether NFAT2 regulates the GSC malignant phenotype in part by modulating HDAC1 expression, we stably reexpressed HDAC1 in NFAT2-silenced GSCs. Rescuing HDAC1 in NFAT2-knockdown G08 GSCs resulted in restored expression of CD44 and YKL40 (Fig. 6A). When we reexpressed HDAC1, the proliferation (Fig. 6B and Supplementary Fig. 9A), invasion (Supplementary Fig. 9B), and self-renewal capacities (Fig. 6C, D) of NFAT2-silenced GSCs were partially restored. Moreover, reexpression of HDAC1 rescued tumor growth in vivo and resulted in shorter survival periods of tumor-bearing mice compared with NFAT2-silenced G08 transfected with an empty vector (Fig. 6E, F). Rescuing HDAC1 in NFAT2-knockdown G08 also restored the expression of CD44 in vivo (Fig. 6G). Similar results were obtained when HDAC1 was rescued in NFAT2-silenced G10 and G23 GSCs (Supplementary Figures 9C–H and 10A–C). These findings suggest that NFAT2-regulated MES differentiation is largely HDAC1 dependent.
Fig. 6.

Rescue of HDAC1 in NFAT2-silenced GSCs partially restores clonogenicity in vitro and in vivo. (A) Rescuing HDAC1 in NFAT2-silenced G08 restores the expression of CD44 and YKL40 in vitro. (B) Rescue of HDAC1 partially restores proliferation compared with an empty vector control in NFAT2-silenced G08. (C) Reexpression of HDAC1 rescues neurosphere growth compared with an empty vector control in NFAT2-silenced G08. Scale bar = 25 μm. (D) Neurosphere formation after HDAC1 is rescued in NFAT2-silenced G08. Neurosphere formation capacity is significantly increased after HDAC1 rescue. (E) NFAT2 silencing in G08 inhibits the tumor growth in vivo. Rescuing HDAC1 in NFAT2-silenced G08 reverts tumor growth in vivo. (F) NFAT2 silencing in G08 prolongs the survival of tumor-bearing mice. Reexpression of HDAC1 in NFAT2-silenced G08 reverts the survival time. (G) Rescuing HDAC1 in NFAT2-silenced G08 reverts the expression of CD44 in vivo. Scale bar = 25 μm. (H) Gene set enrichment analysis plots of NF-κB pathway signatures in high NFAT2/HDAC1 expression versus low NFAT2/HDAC1 expression TCGA gliomas. Normalized enrichment score (NES) and false discovery rate (FDR) are shown in the plot. (I–K) HDAC1 reexpression in NFAT2-silences G08 inhibits hyperacetylation of p65 (I), restores the ability of p65 to bind κB-DNA (J), and recovers NF-κB–dependent transcriptional activity (K). (L) A working model of mesenchymal transition mediated by NFAT2/HDAC1/NF-κB pathway in gliomas. Results are presented as mean ± SD of triplicate samples from 3 independent experiments. *P < 0.05, **P < 0.01.
NFAT2-HDAC1 Signaling Regulates NF-κB Pathway Activity
Gene set enrichment analysis using the datasets of TCGA (Fig. 6H) or the Chinese Glioma Genome Atlas (www.cgga.org.cn;Supplementary Fig. 10D) showed a positive association with NF-κB pathway signatures in NFAT2 and HDAC1 high expression gliomas. In addition, in MES GSC-enriched spheres, immunoprecipitation indicated that NFAT2 silencing increased the level of acetylated lysine residues in p65 (Fig. 6I and Supplementary Fig. 10E). Meanwhile, NFAT2 knockdown suppressed the ability of p65 to bind κB-DNA, as shown by electrophoretic mobility shift assay (Fig. 6J and Supplementary Fig. 10F). In transcriptional assays using a luciferase reporter controlled by NF-κB binding sites, NFAT2 silencing inhibited NF-κB activity (Fig. 6K and Supplementary Fig. 10G). However, rescuing HDAC1 in NFAT2-knockdown GSCs inhibited hyperacetylation of p65, restored the ability of p65 to bind κB-DNA, and recovered NF-κB–dependent transcriptional activity (Fig. 6I–K and Supplementary Fig. 10E–G). Therefore, NFAT2-HDAC1 signaling affects NF-κB pathway activity by regulation of p65 acetylation in MES GSCs. However, in PN GSC-enriched G03 and G12 spheres, transfection of shNFAT2 did not significantly affect NF-κB activity (Supplementary Fig. 10H), indicating that the NFAT2/HDAC1/ NF-κB pathway specifically functions in the maintenance of MES GSCs. Next, we blocked NF-κB activation by introducing a dominant-negative IκBαM in NFAT2/HDAC1-upregulated G03 and G12 GSCs. Inhibition of NF-κB activity largely reduced NFAT2/HDAC1-induced YKL40 and CD44 expression (Supplementary Fig. 10I). Thus, our data suggest that NFAT2/HDAC1-mediated MES differentiation depends on NF-κB activity.
A previous study showed the repression of YKL40 by NF-κB via the recruitment of HDAC1 to the YKL40 promoter in the glioma cell line SNB-75.23 However, this effect occurred in a strict cell-specific manner, and in other cell types NF-κB promoted YKL40 expression, and HDAC1 was not recruited to the YKL40 promoter.23 Using ChIP assays we did not detect the recruitment of HDAC1 to the YKL40 or CD44 promoters in MES GSC-enriched G08 and G10 spheres with high basal HDAC1 or in NFAT2-HDAC1 upregulated G12 GSCs (Supplementary Fig. 10J, K). Therefore, given our results and the established molecular link between NF-κB activation and MES differentiation in GSCs,4,6 NF-κB and HDAC1 may play completely different roles in GSCs and SNB-75 cells.
Discussion
Despite the current development of multimodal therapy, the prognosis of GBM is still poor due to gradual malignant progression and accompanied treatment resistance.1 An important characteristic that equips GBMs with the capability to adapt to antitumor therapy is MES differentiation.4,6 During MES transition, GBMs become more aggressive and exhibit a more malignant phenotype.5 Therefore, understanding the molecular mechanisms that control MES differentiation and promote malignant transformation in GBMs holds the promise of improving the efficacy of therapeutic strategies.
Previous studies have evaluated the role of some NFAT family members in glioma biology,25–27 and NFAT1 has been shown to mediate the adhesion and migration of GBM cells.26 Nevertheless, the function of NFAT2 in glioma cells, especially in GSCs, is still largely unknown. Since different NFATs may play different roles in tumor development and progression,13,14 investigating the role of NFAT2 in GSCs and exploring the underlying molecular mechanism is a worthy endeavor. In this study, we demonstrated that NFAT2 is important for the malignant growth of GSCs. Consistently, one previous study showed that NFAT2 activation enhances the invasion of U251 glioma cells.28 In addition, sequential transcriptional activation of NFAT2 and c-Myc mediates cell growth promoted by transforming growth factor beta in several types of cancer cells.18 Constitutive NFAT2 activation induces a transformed phenotype in 3T3-L1 fibroblasts, showing the oncogenic potential of NFAT2.29 Therefore, NFAT2 may play a crucial role in regulating cancer initiation and progression. Furthermore, we found that NFAT2 signaling maintains the MES phenotype of GSCs. Previous studies associated inflammatory pathways with the MES phenotype4,9 and found that NFAT2 is at the intersection of cancer and inflammation.13 Activation of NFAT2 signaling upregulates a number of cytokines and cytokine receptors and establishes an inflammatory microenvironment for tumor development.15 Consistent with our study, NFAT2 has been identified as a central regulator of pancreatic cancer cell plasticity, and NFAT2 drives EMT reprogramming and maintains pancreatic cancer cells in a stem cell–like state.30
As regulators of inflammation and immunity, NFAT, HDAC, and NF-κB family members often collaborate with each other in physiological processes and immunologic disorders.11,12 For example, NFAT2 has been shown to interact with HDAC1 in the regulation of interleukin (IL)-2 and IL-10 expression in lymphocytes.31,32 NFAT2 and NF-κB synergistically activate CD154 gene transcription in aggressive B-cell lymphomas.33 We found that in GSCs, HDAC1 is the downstream target of NFAT2. By regulation of HDAC1 expression, NFAT2 may influence the acetylation of NF-κB subunit p65 and affect the transcriptional activity of NF-κB, which further modulates the MES transition (Fig. 7K). A previous study showed that downregulation of HDAC1 inhibited proliferation, prevented invasion, and induced apoptosis in glioma cells.34 Interestingly, knockdown of HDAC1 also suppressed the expression of EMT transcription factors TWIST1 and SNAIL, decreased the mesenchymal marker matrix metalloproteinase 9, and increased the epithelial marker E-cadherin,34 suggesting that HDAC1 may contribute to glioma cell EMT. It has been demonstrated that NF-κB signaling promotes the malignant phenotype and drives mesenchymal transition in GBMs.4,35 However, mutation or amplification of individual NF-κB subunits is rare in GBMs, indicating that the elevated NF-κB activity is likely the result of deregulation of the pathway.35 Moreover, HDAC1 may potentially modulate the acetylation status of NF-κB subunits and impact NF-κB activity in malignancies such as GBM and embryonic carcinoma.36,37 HDAC1 inhibitors have been shown to repress NF-κB activity and induce cancer cell apoptosis.36,37 Our results suggest a tight connection between NFAT2/HDAC1 signaling and NF-κB activity (Fig. 6L), emphasizing the role of inflammatory factors in the MES transition and malignant progression of GBMs.
Conclusion
Our findings suggest that by modulating NF-κB activity, the NFAT2-HDAC1 pathway might play an important role in the maintenance of the malignant phenotype in GBMs. Inhibition of the NFAT2/HDAC1/NF-κB axis is an attractive therapeutic approach for GBMs, especially for the MES subtype.
Funding
This work was supported by grants from the National Natural Science Foundation of China (nos. 81772653, 81402045, and 81602209).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Professor Baoman Li from the Laboratory of Metabolic Brain Diseases, Institute of Metabolic Disease Research and Drug Development, China Medical University, for technological support.
Conflict of interest statement. MW and SH designed the study. YS, DT, YJ, ZW, RW, and MW performed the experimental work. YS, DT, YJ, ZW, RW, and SH performed data analyses. YS, DT, YJ, ZW, RW, MW, and SH produced the main draft of the text and the figures. All authors have seen, corrected, and approved the final manuscript.
References
- 1. Weller M, van den Bent M, Tonn JC, et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017;18(6):e315–e329. [DOI] [PubMed] [Google Scholar]
- 2. Wang Q, Hu B, Hu X, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32(1):42–56.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Behnan J, Finocchiaro G, Hanna G. The landscape of the mesenchymal signature in brain tumours. Brain. 2019;142(4):847–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bhat KPL, Balasubramaniyan V, Vaillant B, et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24(3):331–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mao P, Joshi K, Li J, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A. 2013;110(21):8644–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moreno M, Pedrosa L, Paré L, et al. GPR56/ADGRG1 inhibits mesenchymal differentiation and radioresistance in glioblastoma. Cell Rep. 2017;21(8):2183–2197. [DOI] [PubMed] [Google Scholar]
- 7. Cheng P, Wang J, Waghmare I, et al. FOXD1-ALDH1A3 signaling is a determinant for the self-renewal and tumorigenicity of mesenchymal glioma stem cells. Cancer Res. 2016;76(24):7219–7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. [DOI] [PubMed] [Google Scholar]
- 9. Verhaak RG, Hoadley KA, Purdom E, et al. ; Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science. 1988;241(4862):202–205. [DOI] [PubMed] [Google Scholar]
- 11. Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32(7):335–343. [DOI] [PubMed] [Google Scholar]
- 12. Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357(6380):695–697. [DOI] [PubMed] [Google Scholar]
- 13. Shou J, Jing J, Xie J, et al. Nuclear factor of activated T cells in cancer development and treatment. Cancer Lett. 2015;361(2):174–184. [DOI] [PubMed] [Google Scholar]
- 14. Robbs BK, Cruz AL, Werneck MB, Mognol GP, Viola JP. Dual roles for NFAT transcription factor genes as oncogenes and tumor suppressors. Mol Cell Biol. 2008;28(23):7168–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tripathi P, Wang Y, Coussens M, et al. Activation of NFAT signaling establishes a tumorigenic microenvironment through cell autonomous and non-cell autonomous mechanisms. Oncogene. 2014;33(14):1840–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horsley V, Aliprantis AO, Polak L, Glimcher LH, Fuchs E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell. 2008;132(2):299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Singh G, Singh SK, König A, et al. Sequential activation of NFAT and c-Myc transcription factors mediates the TGF-beta switch from a suppressor to a promoter of cancer cell proliferation. J Biol Chem. 2010;285(35):27241–27250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buchholz M, Schatz A, Wagner M, et al. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006;25(15):3714–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Quang CT, Leboucher S, Passaro D, et al. The calcineurin/NFAT pathway is activated in diagnostic breast cancer cases and is essential to survival and metastasis of mammary cancer cells. Cell Death Dis. 2015;6:e1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang Y, Song Y, Wang R, et al. NFAT1-mediated regulation of NDEL1 promotes growth and invasion of glioma stem-like cells. Cancer Res. 2019;79(10):2593–2603. [DOI] [PubMed] [Google Scholar]
- 22. Pollard SM, Yoshikawa K, Clarke ID, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4(6):568–580. [DOI] [PubMed] [Google Scholar]
- 23. Bhat KP, Pelloski CE, Zhang Y, et al. Selective repression of YKL-40 by NF-kappaB in glioma cell lines involves recruitment of histone deacetylase-1 and -2. FEBS Lett. 2008;582(21-22):3193–3200. [DOI] [PubMed] [Google Scholar]
- 24. Wang J, Cazzato E, Ladewig E, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48(7):768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chigurupati S, Venkataraman R, Barrera D, et al. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010;70(1):418–427. [DOI] [PubMed] [Google Scholar]
- 26. Lee JV, Berry CT, Kim K, et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca2+-NFAT signaling. Genes Dev. 2018;32(7-8):497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu H, Zheng J, Liu X, et al. Transcription factor NFAT5 promotes glioblastoma cell-driven angiogenesis via SBF2-AS1/miR-338-3p-mediated EGFL7 expression change. Front Mol Neurosci. 2017;10:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang L, Wang Z, Li J, Zhang W, Ren F, Yue W. NFATc1 activation promotes the invasion of U251 human glioblastoma multiforme cells through COX-2. Int J Mol Med. 2015;35(5):1333–1340. [DOI] [PubMed] [Google Scholar]
- 29. Neal JW, Clipstone NA. A constitutively active NFATc1 mutant induces a transformed phenotype in 3T3-L1 fibroblasts. J Biol Chem. 2003;278(19):17246–17254. [DOI] [PubMed] [Google Scholar]
- 30. Singh SK, Chen NM, Hessmann E, et al. Antithetical NFATc1-Sox2 and p53-miR200 signaling networks govern pancreatic cancer cell plasticity. EMBO J. 2015;34(4):517–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miyajima C, Itoh Y, Inoue Y, Hayashi H. Positive regulation of interleukin-2 expression by a pseudokinase, tribbles 1, in activated T cells. Biol Pharm Bull. 2015;38(8):1126–1133. [DOI] [PubMed] [Google Scholar]
- 32. Alrefai H, Muhammad K, Rudolf R, et al. NFATc1 supports imiquimod-induced skin inflammation by suppressing IL-10 synthesis in B cells. Nat Commun. 2016;7:11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pham LV, Tamayo AT, Yoshimura LC, Lin-Lee YC, Ford RJ. Constitutive NF-kappaB and NFAT activation in aggressive B-cell lymphomas synergistically activates the CD154 gene and maintains lymphoma cell survival. Blood. 2005;106(12):3940–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang XQ, Bai HM, Li ST, et al. Knockdown of HDAC1 expression suppresses invasion and induces apoptosis in glioma cells. Oncotarget. 2017;8(29):48027–48040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cahill KE, Morshed RA, Yamini B. Nuclear factor-κB in glioblastoma: insights into regulators and targeted therapy. Neuro Oncol. 2016;18(3):329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shu G, Tang Y, Zhou Y, Wang C, Song JG. Zac1 is a histone acetylation-regulated NF-κB suppressor that mediates histone deacetylase inhibitor-induced apoptosis. Cell Death Differ. 2011;18(12):1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li ZY, Li QZ, Chen L, et al. Histone deacetylase inhibitor RGFP109 overcomes temozolomide resistance by blocking NF-κB-dependent transcription in glioblastoma cell lines. Neurochem Res. 2016;41(12):3192–3205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.