

Abstract
Lysine post-translational modifications (PTMs) play a crucial role in regulating diverse functions and biological processes of proteins. However, because of the large volumes of sequencing data generated from genome-sequencing projects, systematic identification of different types of lysine PTM substrates and PTM sites in the entire proteome remains a major challenge. In recent years, a number of computational methods for lysine PTM identification have been developed. These methods show high diversity in their core algorithms, features extracted and feature selection techniques and evaluation strategies. There is therefore an urgent need to revisit these methods and summarize their methodologies, to improve and further develop computational techniques to identify and characterize lysine PTMs from the large amounts of sequence data. With this goal in mind, we first provide a comprehensive survey on a large collection of 49 state-of-the-art approaches for lysine PTM prediction. We cover a variety of important aspects that are crucial for the development of successful predictors, including operating algorithms, sequence and structural features, feature selection, model performance evaluation and software utility. We further provide our thoughts on potential strategies to improve the model performance. Second, in order to examine the feasibility of using deep learning for lysine PTM prediction, we propose a novel computational framework, termed MUscADEL (Multiple Scalable Accurate Deep Learner for lysine PTMs), using deep, bidirectional, long short-term memory recurrent neural networks for accurate and systematic mapping of eight major types of lysine PTMs in the human and mouse proteomes. Extensive benchmarking tests show that MUscADEL outperforms current methods for lysine PTM characterization, demonstrating the potential and power of deep learning techniques in protein PTM prediction. The web server of MUscADEL, together with all the data sets assembled in this study, is freely available at http://muscadel.erc.monash.edu/. We anticipate this comprehensive review and the application of deep learning will provide practical guide and useful insights into PTM prediction and inspire future bioinformatics studies in the related fields.
Keywords: lysine post-translational modification, prediction model, sequence features, feature engineering, deep learning
Introduction
Post-translational modifications (PTMs) occurring at the lysine (K) residues of proteins, such as acetylation, glutarylation, glycation, malonylation, methylation, succinylation, sumoylation and ubiquitination, have been experimentally verified to play crucial roles in diverse biological processes [1–10]. To date, advances in experimental techniques have significantly assisted biologists, allowing them to identify various types of lysine PTMs. As a result, >20 lysine PTMs have been characterized, with annotations deposited in public databases, such as Phosphorylation Sites Plus (PhosphoSitePlus) [11] and Protein Lysine Modifications Database (PLMD) [1]. Given the prevalence and importance of lysine PTMs, aberrant modifications of lysine residues may result in a variety of human diseases [12]. Among different types of lysine PTMs, acetylation, glycation, ubiquitination and methylation have been extensively investigated. Lysine acetylation is characterized as an important regulator for diverse biological processes of proteins, such as protein stability [13], transcription [14, 15], protein–protein interactions [13] and cellular metabolism [16–18]. Glycation, another important type of lysine PTM, is formed via a chemical reaction between reducing sugar molecules and certain amino acids, including lysine, arginine and some certain N-terminal residues [19, 20]. It has been established that the accumulation of glycation products is associated with aging and pathogenesis of diabetes [21, 22]. Protein ubiquitination serves as an indicative signal for protein degradation [10, 23, 24]. In addition, the ubiquitin system is involved in regulating more fundamental cellular processes including gene transcription, DNA repair and replication, intracellular trafficking and virus particle budding [23, 25, 26], cellular transformation, immune response and inflammatory response [27]. Aside, small ubiquitin-like modifier (SUMO) proteins are structurally conserved with ubiquitins and play an essential role in the regulation of gene expression, DNA repair, chromosome assembly and cellular signaling [9, 28–30]. Likewise, methylation, a PTM involving the transfer of one, two or three methyl groups to the ε-amine of the lysine side chain, is also involved in a broad spectrum of biological and physiological processes, such as transcriptional and epigenetic regulation, cell metabolism and the development of human diseases [31–34].
Recently, three novel lysine PTMs were discovered and reported, including malonylation [6], succinylation [35, 36] and glutarylation [5], which share commonalities in terms of formed molecular structure but diversify into different regulated proteins and pathways [37]. More interestingly, a growing number of studies on the cross-talk of lysine PTMs have been published, benefiting from cumulative studies of lysine PTMs [3, 38]. The cross-talks within different types of lysine PTMs [39, 40], and across lysine PTMs and other PTMs [12], have demonstrated the significance of lysine PTMs and highlight the indispensable biological functions of lysine PTMs in diverse processes.
To date, many efforts have been dedicated to the investigation of cellular mechanisms of lysine PTMs, which is based on accurate identification of lysine PTM substrates and sites. Advances in lysine PTM research have driven continued computational studies for accurate prediction of lysine PTMs, so as to significantly reduce time and effort involved in experimental identifications. Compared with labor-intensive and time-consuming experimental characterization of lysine PTMs, computational prediction of lysine PTMs in proteins provides a useful and complementary approach to shortlist likely candidates for subsequent experimental validation. Thus far, a number of computational approaches have been developed and published for this purpose (Figure 1). Even though these methods can be generally categorized into two classes (i.e. machine learning-based and peptide similarity-based; Figure 2), they differ in a variety of aspects in terms of model construction, including training data set construction, features employed and software availability and utility [41]. Despite significant research efforts being devoted to the construction of computational methods for lysine PTM prediction, little work has been done to systematically summarize and evaluate state-of-the-art computational approaches, which could potentially shed a light on bottlenecks or missing features to improve algorithm design for lysine PTM prediction.
Figure 1.
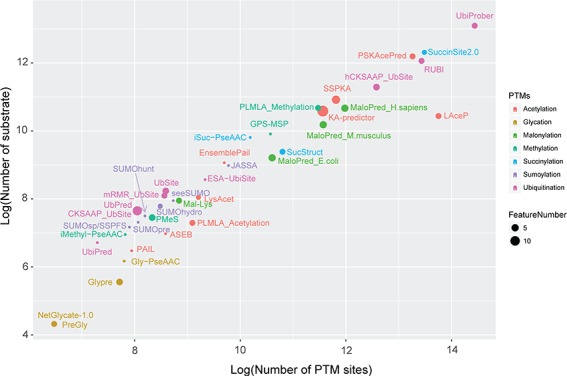
Bubble plots showing the sizes of data sets and the numbers of features for each method surveyed in this article. The x-axis shows the log2 value of the numbers of PTM sites, while the y-axis denotes the log2 of numbers of substrates. The dot size indicates the numbers of features calculated in individual method.
Figure 2.
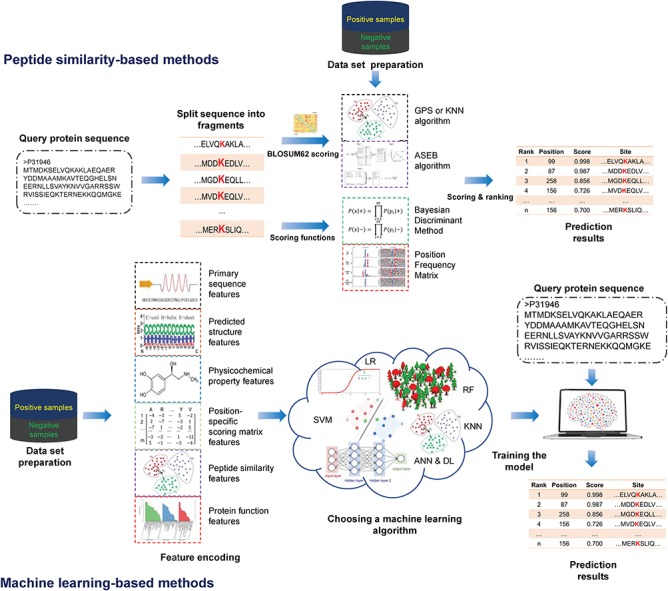
An overview of the current computational approaches for lysine PTM prediction. The methods can be roughly categorized into two classes: peptide similarity-based approaches (top half of the figure) and machine learning-based approaches (down half).
With this goal in mind, in this article, we first deliver a comprehensive survey regarding the state-of-the-art computational models. We discuss a wide range of aspects we investigated, including the core algorithms selected for individual methods, feature selection techniques employed, performance evaluation strategy and user experience. To the best of our knowledge, this is the most up-to-date work that systematically surveys and critically evaluates the state-of-the-art bioinformatics methods for lysine PTM prediction. Based on our survey and findings, we further propose a novel framework, MUscADEL (Multiple Scalable Accurate Deep Learner for lysine PTMs), based on the deep bidirectional long short-term memory recurrent neural networks (RNN) scheme [42, 43], to evaluate the power of deep learning in predicting eight lysine PTM types using protein sequence(s). Our empirical study shows that the proposed MUscADEL model has superior prediction performance, compared to current predictive models for lysine PTMs. In addition, we constructed a portal (http://muscadel.erc.monash.edu/) to facilitate online high-throughput prediction of lysine PTMs. We anticipate that our portal will serve as a useful computational tool for accurate identification of lysine PTMs, providing highly reliable candidates for subsequent experimental validation.
Materials and methods
State-of-the-art computational approaches for lysine PTM prediction
Recent decades have witnessed the development and proliferation of computational approaches, including for prediction of lysine PTM sites. Methods differ in a variety of aspects, including the training and test data sets, sequence/structural descriptors and physicochemical properties employed, feature selection techniques, targeted lysine PTM types, etc. In Table 1, we summarize 49 computational approaches for lysine PTM prediction according to algorithm selected, features employed, performance evaluation strategy and web server availability.
Table 1.
A comprehensive summary of the reviewed predictors for lysine PTMs in this study
| Tool | PTM | Algorithm/category a | Feature selection | Data set size (PTM sites and proteins) | Evaluation strategy | Option of batch prediction | Web server | Adjustment of predictor thresholds | Stand-alone software and Platform | Window size | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PAIL | Acetylation | BDM | None | 246/89 | Jack-knife validation K(6, 8 and 10)-fold cross-validation | NA | No | NA | None | 13 | [75] |
| LysAcet | Acetylation | SVM | CfsSubsetEval | 591/264 | Jack-knife validation K(5 and 10)-fold cross-validation | NA | No | NA | None | 13 | [94] |
| PHOSIDA | Acetylation | SVM | None | 2392/NA | 10-fold cross-validation Independent test | Yes (maximum 20 protein sequences) | http://www.phosida.com/ | Continuous adjustment | None | 13 | [147] |
| EnsemblePail | Acetylation | SVM | None | 830/535 | 10-fold cross-validation Independent test | Yes | http://app.aporc.org/EnsemblePail/ | Low/medium/high | None | 19 | [115] |
| BRABSB | Acetylation | SVM | None | 2873/NA | 5-fold cross-validation Independent test | NA | No | NA | None | 15 | [148] |
| PLMLA | Acetylation | SVM | None | 546/157 | 10-fold cross-validation | Yes | http://app.aporc.org/EnsemblePail/ | Sp (0.5–0.9) | None | 13 | [113] |
| Methylation | 2842/1639 | Independent test | |||||||||
| PSKAcePred | Acetylation | SVM | IG | 9815/4682 | 10-fold cross-validation Independent test | Yes | http://bioinfo.ncu.edu.cn/inquiries_PSKAcePred.aspx | Sp (0.5–0.9) | None | 13 | [50] |
| ASEB | Acetylation | ASEB | None | 384/126 | Jack-knife validation Independent test | NA | No | NA | None | 17 | [47] |
| LAceP | Acetylation | LR | CfsSubsetEval | 13 810/6388 | 10-fold cross-validation Independent test | NA | No | NA | None | 21 | [72] |
| SSPKA | Acetylation | RF | mRMR | 3596/1936 | 5-fold cross-validation Independent test | NA | No | NA | None | 7 | [62] |
| iPTM-mLys | Acetylation | RF | None | 3991/NA | Jack-knife validation 5-fold cross-validation Independent test | Yes | http://www.jci-bioinfo.cn/iPTM-mLys | No | None | 27 | [114] |
| Crotonylation | 115/NA | ||||||||||
| Methylation | 127/NA | ||||||||||
| Succinylation | 1169/NA | ||||||||||
| KA-predictor | Acetylation | SVM | PCC | 3024/1537 | 5-fold cross-validation Independent test | Yes | No | No | Python | 19 | [52] |
| NetGlycate-1.0 | Glycation | ANN | None | 89/20 | k-fold cross-validation | Yes (maximum 2000 protein sequences) | http://www.cbs.dtu.dk/services/NetGlycate-1.0/ | No | None | 23 | [20] |
| PreGly | Glycation | SVM | mRMR | 89/20 | 10-fold cross-validation Jack-knife validation Independent test | NA | No | NA | None | 23 | [95] |
| BPB_GlySite | Glycation | SVM | None | 323/NA | 10-fold cross-validation Independent test | Yes | http://123.206.31.171/BPB_GlySite/ | No | None | 15 | [116] |
| Glypre | Glycation | SVM | mRMR | 210/47 | 10-fold cross-validation Independent test | None | No | No | Matlab source code | 31 | [149] |
| Gly-PseAAC | Glycation | SVM | None | 323/72 | K(6, 8 and 10)-fold cross-validation Jack-knife validation Independent test | Yes | http://app.aporc.org/Gly-PseAAC/ | No | None | 15 | [96] |
| Mal-Lys | Malonylation | SVM | None | 458/247 | 10-fold cross-validation Independent test | Yes (maximum 100 K file size) | http://app.aporc.org/Mal-Lys/ | No | None | 16 | [118] |
| MaloPred | Malonylation | SVM | IG | 8635/3384 | 10-fold cross-validation Independent test | Yes | http://bioinfo.ncu.edu.cn/MaloPred.aspx | Continuous adjustment | None | 25 | [51] |
| MeMo | Methylation | SVM | None | 156/NA | 7-fold cross-validation | NA | No | NA | None | 14 | [150] |
| BPB-PPMS | Methylation | SVM | None | 188/NA | 5-fold cross-validation Independent test | NA | No | NA | None | 11 | [151] |
| MASA | Methylation | SVM | None | 460/NA | k-fold cross-validation Independent test | NA | No | NA | None | 13 | [152] |
| PMeS | Methylation | SVM | None | 322/175 | 10-fold cross-validation Independent test | Yes | http://bioinfo.ncu.edu.cn/inquiries_PMeS.aspx | Sp (0.5–0.9) | Matlab source code | 15 | [119] |
| MethK | Methylation | SVM | None | 1259/NA | 5-fold cross-validation Independent test | Yes (maximum 2 M file size) | http://csb.cse.yzu.edu.tw/MethK/prediction.php | No | None | 15 | [120] |
| PSSMe | Methylation | SVM | None | 2110/NA | 10-fold cross-validation Independent test | Yes | http://bioinfo.ncu.edu.cn/PSSMe.aspx | Continuous adjustment | None | 19 | [121] |
| iMethyl-PseAAC | Methylation | SVM | None | 226/124 | Jack-knife validation Independent test | Yes | http://www.jci-bioinfo.cn/iMethyl-PseAAC | No | None | 11 | [153] |
| GPS-MSP | Methylation | GPS | None | 1521/962 | K(4, 6, 8 and 10)-fold cross-validation Jack-knife validation | Yes | http://msp.biocuckoo.org/online.php | All/low/medium/high | Windows/Linus/MacOS | 15 | [48] |
| iSuc-PseAAC | Succinylation | SVM | None | 1167/896 | Jack-knife validation Independent test | Yes | http://app.aporc.org/iSuc-PseAAC/ | No | None | 11 | [117] |
| iSuc-PseOpt | Succinylation | RF | None | 1167/896 | 10-fold cross-validation Independent test | Yes (maximum 100 protein sequences) | http://www.jci-bioinfo.cn/iSuc-PseOpt | No | None | 31 | [63] |
| pSuc-Lys | Succinylation | RF | None | 1167/896 | 5-fold cross-validation Independent test | NA | No | NA | None | 31 | [64] |
| SuccinSite2.0 | Succinylation | RF | None | 11 472/5080 | 5-fold cross-validation Independent test | Yes | http://biocomputer.bio.cuhk.edu.hk/SuccinSite2.0/ | No | None | 41 | [65] |
| SucStruct | Succinylation | Decision tree | None | 1782/670 | K(6, 8 and 10)-fold cross-validation Independent test | No | No | No | None | 31 | [108] |
| SUMOhydro | Sumoylation | SVM | None | 358/221 | Jack-knife validation Independent test | No | http://protein.cau.edu.cn/others/SUMOhydro/ | No | None | 25 | [112] |
| seeSUMO | Sumoylation | RF | None | 425/247 | 10-fold cross-validation Independent test | NA | No | NA | None | 20 | [154] |
| SUMOhunt | Sumoylation | RF | None | 293/181 | K(3–10)-fold cross-validation Self-consistency test Jack-knife validation | No | No | No | None | 21 | [111] |
| SUMOsp | Sumoylation | GPS | None | 239/144 | 5-fold cross validation Independent test | Yes (maximum 2 M file size) | http://sumosp.biocuckoo.org/online.php | All/low/medium/high | JAVA | 15 | [76] |
| GPS-SUMO | Sumoylation | GPS | None | 1059/594 | Jack-knife validation | Yes (maximum 2 M file size) | http://sumosp.biocuckoo.org/online.php | All/low/medium/high | JAVA | 15 | [49] |
| SUMOpre | Sumoylation | Statistical method | None | 268/159 | 5-fold cross-validation Independent test | NA | No | NA | None | NA | [155] |
| SSPFS | Sumoylation | KNN | mRMR | 239/144 | Jack-knife validation | No | No | No | None | 14 | [71] |
| SUMOsu | Sumoylation | SVM | RELIEFF | 381/NA | K(4, 6, 8 and 10)-fold cross-validation Jack-knife validation | No | No | No | None | 7 | [99] |
| JASSA | Sumoylation | PFM score | None | 877/505 | NA | Yes | http://www.jassa.fr/ | No | None | 21 | [122] |
| UbiPred | Ubiquitination | SVM | mRMR | 157/105 | Jack-knife validation Independent test | Yes (maximum 100 protein sequences) | http://www.ubpred.org/ | No | None | 21 | [97] |
| UbPred | Ubiquitination | RF | None | 265/201 | 100-fold cross-validation | No | http://www.ubpred.org/ | High/medium/low | Linus/Shell script | 25 | [123] |
| CKSAAP_UbSite | Ubiquitination | SVM | None | 263/203 | 100-fold cross-validation Independent test | No | http://protein.cau.edu.cn/cksaap_ubsite | High/low | Linux/Perl script | 27 | [60] |
| UbSite | Ubiquitination | SVM | None | 385/301 | 5-fold cross-validation Independent test | No | No | No | None | 41 | [156] |
| mRMR_UbSite | Ubiquitination | KNN | mRMR | 378/273 | Jack-knife validation Independent test | No | No | No | None | 21 | [70] |
| UbiProber | Ubiquitination | SVM | IG | 22 192/8750 | 5-fold cross-validation Independent test | Yes | http://bioinfo.ncu.edu.cn/ubiprober.aspx | Continuous adjustment | Windows/C# (.NET 4.0 framework) | 27 | [98] |
| RUBI | Ubiquitination | SVM/BRNN | None | 11 054/4273 | 10-fold cross-validation Independent test | Yes | http://protein.bio.unipd.it/rubi/ | No | None | 13 | [67] |
| hCKSAAP_UbSite | Ubiquitination | SVM | None | 6118/2500 | 5-fold cross-validation Independent test | No | http://protein.cau.edu.cn/cksaap_ubsite | High/low | Linux/C++ | 27 | [61] |
| ESA-UbiSite | Ubiquitination | SVM | IBCGA | 645/379 | 10-fold cross-validation Independent test | Yes | http://iclab.life.nctu.edu.tw/iclab_webtools/ESAUbiSite/ | No | None | 21 | [100] |
aFull names of the algorithms: NA, not available; ASEB, acetylation set enrichment based; SVM, support vector machine; BDM, Bayesian discriminant method; IG, information gain; LR, logistic regression; RF, random forest; mRMR, maximum relevance and minimum redundancy; PCC, Pearson correlation coefficient; ANN, artificial neural networks; GPS, group-based prediction system; KNN, k-nearest neighbor; PFM, position frequency matrix; BRNN, bidirectional recurrent neural networks; IBCGA, inheritable bi-objective combinatorial genetic algorithm.
To provide interested readers with some useful insights into the development of computational frameworks listed in Table 1, we coarsely clustered approaches into two categories based on adopted machine learning techniques (Figure 2). The first category of approaches is based on traditional machine learning algorithms using sequence-derived features. A majority of the computational methods listed in Table 1 adopted this strategy to build their predictive models, while diverse sequence-derived features were calculated and/or extracted from third-party software before training the models. Another crucial step prior to model construction is feature selection that aims to reduce the feature dimensionality and eliminate misleading features for better prediction performance. The second category of approaches is based on peptide similarity. Methods within this category usually calculate a similarity score between the testing peptide and peptides with experimentally annotated lysine PTMs, using a number of measures, such as the BLOcks SUbstitution Matrix (BLOSUM62) matrix [44] and position-specific scoring matrix (PSSM) [45]. In Table 1, acetylation set enrichment-based (ASEB) [46, 47], Methyl-group Specific Predictor based on Group-based Prediction System (GPS-MSP) [48] and Small Ubiquitin-like MOdifier predictor based on Group-based Prediction System (GPS-SUMO) [49] are representative models based on peptide similarity. It is worth noting that some methods use both strategies (i.e. machine learning algorithms and peptide similarity) to build their models. For example, Position-Specific lysine (K) Acetylation Predictor (PSKAcePred) [50], lysine Malonylation Predictor (MaloPred) [51], lysine (K) Acetylation predictor (KA-predictor) [52] and the k-nearest neighbor (KNN) score [51] all calculate the similarity of two peptides, which is subsequently used as input feature in machine learning algorithms.
Machine learning algorithms employed for predicting lysine PTMs
As mentioned in the section entitled state-of-the-art computational approaches for lysine PTM prediction, a majority of current computational approaches for lysine PTM prediction were built using well-established machine learning algorithms. These widely employed algorithms include support vector machine (SVM) [53], random forest (RF) [54], artificial neutral network (ANN) [55], KNN [56], logistic regression (LR) [57], etc. Based on our survey, SVM is the most commonly used machine learning algorithm and is often considered as the ‘method-of-choice’ for building computational models (Table 1).
Support vector machine
SVM aims to accurately classify samples by generating optimal hyperplanes based on the feature dimensionality of the training data [58, 59]. The resulting mapping formula generated by SVM is usually not interpretable but invariably yields to satisfactory classification/prediction performance. Therefore, SVM is usually the ‘first choice’ adopted in many bioinformatics studies [60, 61]. To date, a variety of kernels have been developed for SVM, for different classification scenarios, including Gaussian radial basis function (RBF), linear/polynomial/sigmoid kernel, etc. Among the SVM-based approaches reviewed in this study, RBF kernel was most commonly used, but we note users are suggested to choose kernel methods according to needs and questions of interest. Another point worth stressing is the choice of parameters. It is generally recommended to do an experimental optimization of SVM parameters prior to model construction, as different parameter configurations could change prediction performance dramatically.
Random forest
RF [54] is another well-established and widely employed algorithm, not only for lysine PTM prediction but also for many other bioinformatics applications [62–65]. RF is essentially an ensemble of a number of decision trees, T = 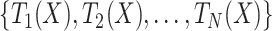 built on N random subsets of the training data, and the average prediction performance is usually reported in order to avoid over-fitting [54]. The obvious advantage of RF is its interpretability, as every decision tree consists of a number of ‘if…then…’ rules, which are fairly straightforward to explain. Such rules can potentially provide biologists with insights and knowledge discovery that would otherwise remain buried in the data. When applying RF, one should bear in mind that the number of decision trees is an important parameter and should be tested exhaustively based on the specific application or biological question, for optimal prediction performance.
built on N random subsets of the training data, and the average prediction performance is usually reported in order to avoid over-fitting [54]. The obvious advantage of RF is its interpretability, as every decision tree consists of a number of ‘if…then…’ rules, which are fairly straightforward to explain. Such rules can potentially provide biologists with insights and knowledge discovery that would otherwise remain buried in the data. When applying RF, one should bear in mind that the number of decision trees is an important parameter and should be tested exhaustively based on the specific application or biological question, for optimal prediction performance.
Artificial neural network
An ANN usually contains multiple nodes as input and multiple layers to connect these input nodes, mimicking neurons and their functions/connectivity in human brains [66]. Among the reviewed predictors for lysine PTM prediction, NetGlycate-1.0 [20] and Rapid UBIquitination sites predictor (RUBI) [67] employed ANN as their core algorithm. A typical architecture of ANN is composed of three layers, including the input layer, the hidden layer and the output layer. However, ANNs can be more complex, with multiple hidden layers [68, 69]. Among the predictors using ANNs, NetGlycate-1.0 trained network algorithms using sequence input alone, with the relative position of lysine in the sequence as additional input. To determine the best result(s), all network combinations with 1, 2, 3, …, 20 hidden neurons and window sizes of 3–51 amino acid residues were investigated. This yielded a neural network with 5 hidden neurons and a window size of 23 [20].
k-nearest neighbors algorithm
KNN algorithm is another commonly employed unsupervised algorithm that clusters samples by calculating their similarities/distances [70]. Among the reviewed predictors for lysine PTM identification, Sumoylation Site Prediction base on Features Selection (SSPFS) [71] and Ubiquitination Site predictor based on mRMR feature selection (mRMR_UbSite) [70] were built using the KNN algorithm. Given the training data set 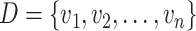 and a testing sample x, KNN [70] calculates the distances between x and all the instances in D. As a result, the query sample will be assigned to the same class as its nearest neighbor (shortest distance) in the training data set.
and a testing sample x, KNN [70] calculates the distances between x and all the instances in D. As a result, the query sample will be assigned to the same class as its nearest neighbor (shortest distance) in the training data set.
Logistic regression
Among the reviewed predictors, Lysine Acetylation Site Prediction (LAceP) [72] constructed an LR model for lysine PTM prediction. LR can be used to build a classification model for many prediction tasks [73, 74], which can be represented as [72]
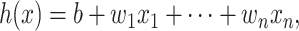 |
(1) |
where xi are the input features, wi are the weight parameters and b is the bias value. Given an unlabeled input x, the likelihood of x with the class label (a given PTM type) can be defined as
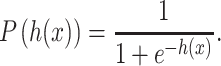 |
(2) |
Bayesian discriminant method
Among the reviewed predictors, the Bayesian discriminant method (BDM) was used to predict acetylation sites [75]. BDM assumes that all flanking residues around the PTM site are mutually independent. It then estimates the probability that an unlabeled input x belongs to the positive samples (P(x|+)) or the negative samples (P(x|-)).
Group-based prediction system
The basic premise of the group-based prediction system (GPS) scoring strategy is that similar peptides exhibit similar biochemical properties and functions [48]. To implement such hypothesis, a peptide with a PTM site is denoted as PEP(m, n), where m and n are the numbers of upstream and downstream residues around the PTM site, respectively. Then the BLOSUM62 [44] matrix is used to estimate the similarity between two peptides A and B:
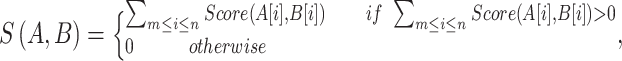 |
(3) |
where Score(A[i], B[i]) represents the substitution score of amino acids A[i] and B[i] in the BLOSUM62 matrix. Given a testing peptide PEP(m, n)u, the similarity score between PEP(m, n)u and each peptide with a PTM site (PEP(m, n)) is calculated, and the average score is reported. If the final score is higher than a threshold, the peptide PEP(m, n)u will be predicted to harbor a PTM site. Four distinct steps, including k-means clustering, motif length selection, weight training and matrix mutation [48], can be used to improve the performance of the GPS algorithm. Among the reviewed predictors in our study, GPS-MSP [48] and SUMOylation Site Prediction (SUMOsp) [49, 76] were developed based on the GPS scheme.
ASEB algorithm
The ASEB method, which was used to predict acetylation sites [47], is another peptide similarity-based scheme, developed to determine whether a given peptide could be acetylated or not by estimating the similarity and the significance of the peptide similarity with respect to known acetylation site sets [46, 47]. A typical ASEB algorithm is composed of three major steps: similarity calculation, enrichment score (ES) calculation and significance estimation of the ES. The details of the ASEB method are described as follows:
Input. An acetylated peptide set Sk containing N peptides and a pre-defined background peptide set Sb (i.e. including both acetylated and non-acetylated peptides) containing 10 000 randomly selected peptides.
Calculate similarity scores. The similarity score between an unknown peptide Pquery and each peptide in Sk ∪ Sb was calculated according to the BLOSUM62 matrix [44]. The similarity scores were normalized to [0, 1] and [−1, 0] for positive and negative scores, respectively. All the scores were ranked from highest to lowest.
Calculate ES. The ES reflects how enriched the Pquery in Sk was at the top of the ranked list. Suppose that ri is the similarity score between Pquery and peptide pi in Sk, R is the sum of |ri| for all pi ∈ Sk and RS is the running sum score. Then, walking down the list, the RS increased |ri|/R when encountering a peptide in Sk and decreased 1/10 000 when encountering a peptide in Sb. Finally, the maximum of the RS was taken as the ES.
Estimate significance of ES. A total of 9999 peptide sets with the same size as Sk were randomly generated from the background peptides and labeled Snull1 to Snull9999. The ES for each set was calculated and 10 000 ESs (ES(Sk), ES(Snull1), …, ES(Snull9999)) were ranked from high to low. Finally, the significance of ES(Sk) for Pquery was defined as L/10 000, and L is the rank of ES(Sk).
Features calculated and extracted for machine learning based predictors
To construct robust and accurate machine learning predictors for lysine PTM prediction, diverse features in terms of sequence, structure and function are extracted/calculated for individual proteins/peptides. Such features will then be encoded as numeric vectors for training a machine learning model. In this section, we summarize six major types of features based on our investigation of current computational approaches for lysine PTM prediction (Table 2). These major feature types include (1) protein primary sequence-derived features, (2) predicted protein structural features, (3) protein physicochemical properties, (4) protein PSSMs, (5) peptide similarity features and (6) protein functional annotations. Based on our survey, we collected the representative features for each type, together with their biological annotation and significance. We note that some features, such as predicted protein structural features and protein PSSMs, always require third-party software to generate outputs prior to feature calculation and encoding. This means calculating these types of features is usually time-consuming and the calculated features will significantly change depending on the parameter configurations of these independent tools. For example, the generated PSSMs largely depend on the searching database employed by Basic Local Alignment Search Tool (BLAST) programs [45, 77]. For protein blast, the proteome databases, such as SwissProt, Ref90, etc. [78–80], have been often selected as searching databases. Interested readers are therefore encouraged to consider parameters based on instructions of these computational tools. Extraction of protein functional annotations, which are mostly derived directly from experimental studies, also deserves careful consideration. Such annotations are often not complete and undergo frequent updates from time to time; moreover, these may not even be available for all proteins, even though protein functional annotations usually contribute significantly to the prediction performance [62, 81]. This presents a significant challenge when testing an unknown protein/peptide on the web server, as the functional annotations for this protein/peptide may developed computational tools in cases where such functional annotation information is not available, which do not require the protein functional annotations as the essential input features [82, 83]. Fortunately, many packages/web servers have been developed to calculate a variety of structural and physicochemical features, including Protein Features (PROFEAT) [84], Pseudo Amino Acid Composition (PseAAC) [85], protein in python (propy) [86], Pse-in-One [87], protr/ProtrWeb [88], Compound-Protein Interaction with R (Rcpi) [89], the Pseudo K-tuple Reduced Amino Acids Composition (PseKRAAC) [90], Position-Specific Scoring matrix-based featUre generator for Machine learning (POSSUM) [91] and iFeature [92]. After the feature-coding process, the initial feature set usually has a high dimensionality, which is not suitable for training a computational model. Therefore, feature selection is done as a next step, to reduce the dimensionality of the initial feature set, prior to constructing the computational models.
Table 2.
Different types of features employed by the reviewed approaches for lysine PTM site prediction
| Feature type | Feature | Biological interpretation | Reference |
|---|---|---|---|
| 1. Protein primary sequence-derived features | AAC | The AAC of the sequence fragments surrounding the PTM site. | [51, 95, 98, 99, 120, 121, 123] |
| Binary | The position-specific information of the amino acids surrounding ubiquitination sites. | [50, 51, 61, 67, 94, 112, 123, 147, 150, 152] | |
| PCP | The expression [XdZ] denotes the coupling pattern of residue types X and Z that are separated by d residues. | [94] | |
| PWAA | The order of residues around the PTM site. | [52, 113, 119] | |
| PSSC | The PSSC for a peptide of L residues is calculated by  , where Wij equals to 1 when amino acid i occurs in position j, 0 otherwise; while Sij denotes the information entropy of amino acid i in position j. , where Wij equals to 1 when amino acid i occurs in position j, 0 otherwise; while Sij denotes the information entropy of amino acid i in position j. |
[72] | |
| CKSAAP | The CKSAAP reflects the short-range interactions of residues within the sequence surrounding the PTM site. | [52, 60, 95, 96] | |
| LC | For the sites located in the N-terminal, C-terminal or the middle of a sequence, LC used 3-bit binary to encode this terminal information, i.e. N-terminal for (1, 0, 0), C-terminal for (0, 0, 1) and middle for (0, 1, 0). | [52] | |
| k-grams | The k-grams represent a pattern of k consecutive amino acids (or nucleic acids in context of DNA/RNA). | [118] | |
| 2. Predicted protein structural features | SS | This orthogonal binary coding scheme is used to transform the three types of SSs (helix, sheet and coil) into numeric vectors. In this work, helix, sheet and coil were encoded as (0, 0, 1), (1, 0, 0) and (0, 1, 0), respectively. | [52, 62, 96, 108, 113, 152] |
| ASA | The predicted protein ASA of the residues surrounding the PTM site. | [52, 62, 108, 119, 120, 152] | |
| Predicted HSE | HSE is used to measure the solvent exposure of a protein. | [52] | |
| Protein disordered region | The predicted protein disorder information of the residues surrounding the PTM site. | [52, 62, 67, 70, 99, 120, 123] | |
| LBA (local backbone angles) | The predicted torsion angles between neighboring amino acids surrounding the PTM site. | [123] | |
| 3. Protein physicochemical properties | AAindex | AAindex reflects the physicochemical properties of the amino acids surrounding the PTM site. | [50, 52, 61, 62, 70–72, 95–98, 100, 111, 118, 120, 121] |
| Charge-hyd | The charge and hydrophobicity ratio values of the amino acids surrounding the PTM site. | [123] | |
| EBGW | An encoding scheme for amino acid sequences based on the hydrophobicity and charged of each residue. | [52, 63, 64, 114, 153] | |
| ACC transformation | A combination of AC and CC measure, where CC measures the correlation of two different peptides between two residues separated by a distance of ‘lag’ along the sequence, and AC measures the correlation of the same property between two residues separated by a distance of lag along the sequence. | [52] | |
| Chou’s PseAAC | In additional to the traditional amino acid composition features, Chou’s PseAAC contains extra sequence-order information. | [52, 63, 64, 114, 153] | |
| PC-PseAAC | PC-PseAAC is an approach incorporating the contiguous local sequence-order information and the global sequence-order information into the feature vector of the protein sequence. | [52] | |
| VDWV | The normalized VDWV of the amino acid side chain of a peptide sequence. | [119] | |
| SPC | The SPC reflects the distribution of residues with the same unique characteristic of a peptide sequence. | [119] | |
| AGG | The AGG of a peptide sequence containing a PTM site. | [61] | |
| 4. Protein position-specific scoring matrices | PSSM (position-specific scoring matrix) | The PSSM reflects the evolutionary information and conservation of the amino acids surrounding the PTM site. | [51, 52, 62, 67, 70, 120, 123, 154, 156] |
| pbCKSAAP (profile-based composition of k-spaced amino acid pairs) | Comparing with traditional CKSAAP, pbCKSAAP is calculated based on the protein PSI-BLAST profile (i.e. the PSSM matrix). | [65] | |
| HH-profile (PSSM by HHblits) | HHblits is an open-source tool to build protein multiple-sequence alignments (MSAs) using profile hidden Markov models. | [52] | |
| Bi-profile | Two position-specific profiles, including positive position-specific profile and negative position-specific profile, can be generated through calculating the frequency of each amino acid at each position in the positive data set and negative data set, respectively. | [116, 151] | |
| PWM | Every row of matrix corresponds to one kind of amino acids and every column corresponds to the position in the sample fragments. | [115] | |
| BRABSB | BRABSB is used to extract the posterior probabilities for both positive and negative data sets (i.e. peptides with and without PTM sites in our scenario), respectively. | [148] | |
| TPM | TPM represents the transition rate of adjacent amino acids that is utilized as the transition probability of the Markov model. | [72] | |
| PSAAP | PSAAP is used to measure the conservation status for a specified location. | [117, 149] | |
| BLOSUM62 | The BLOSUM62 matrix is adopted to represent the protein primary sequence information reflecting the similarity of two sequence fragments. | [156] | |
| 5. Peptide similarity features | KNN score | The KNN encoding implies the clustering information (i.e. sequence similarity/distance) of peptides with PTM sites. | [50–52, 98, 121] |
| 6. Protein functional annotations | Functional features | The functional features are the protein biological annotations usually extracted from third-party biological databases. These features include but not limited to, BP, CC, MF, functional domain, signaling/metabolic pathway and protein–protein interaction, etc. | [62, 81] |
Abbreviations: AAC, amino acid composition; PCP, protein coupling patterns; PWAA, position weight amino acid composition; PSSC, position-specific symbol composition; CKSAAP, composition of k-spaced amino acids pairs; LC, location coding; SS, secondary structure; ASA, accessible solvent accessibility; HSE, half sphere exposure; LBA, local backbone angles; Charge-hyd, charge/hydrophobicity ratio; EBGW, encoding based on grouped weight; ACC, auto-cross covariance; AC, auto-covariance; CC, cross-covariance; PseAAC, pseudo-amino acid composition; PC-PseAAC, parallel correlation pseudo-amino acid composition; VDWV, van der Waals volume; SPC, sparse property coding; AGG, aggregation propensity; MSAs, multiple-sequence alignments; PWM, position weighted matrix; BRABSBI, bi-relative adapted binomial score Bayes; TPM, transition probability matrix; PSAAP, position-specific amino acid propensity; KNN, k-nearest neighbor; BP, biological process; CC, cellular component; MF, molecular function.
Feature selection strategy
Highly dimensional feature sets usually contain noisy and misleading features that are detrimental to the prediction performance. For example, the Amino Acid index (AAindex) database [93] contains 544 physicochemical properties for each amino acid. For a peptide of 21 residues, the dimensionality of the AAindex features would be 11 424 (i.e. 21 × 544), and these 11424 features are not equally important to predict the PTM sites. Therefore, prior to model construction, feature selection is a nontrivial step that measures the importance of all the features and eliminates the less informative ones. Approximately, one-third of the machine learning approaches in Table 2 adopted the feature selection procedure. The most commonly applied feature selection techniques include Correlation-based Feature Subset selection (CfsSubsetEval) [72, 94], maximum relevance and minimum redundancy (mRMR) [62, 70, 71, 95–97], information gain (IG) [50, 51, 98], Pearson correlation coefficient (PCC) [52], RELIEFF [99] and inheritable bi-objective combinatorial genetic algorithm (IBCGA) [100].
Performance evaluation measures and strategies
Based on our investigation, five measures, including sensitivity (Sn), specificity (Sp), Matthew correlation coefficient (MCC), accuracy (Acc) and area under the curve (AUC), were widely used to estimate the prediction performance. Sn, Sp, MCC and Acc are defined as follows:
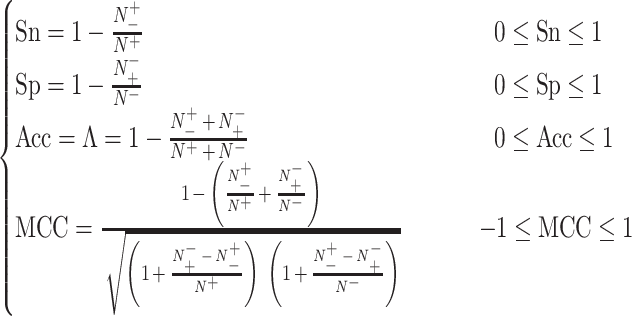 |
(4) |
The above intuitive metrics were derived in [101] based on the symbols introduced by Chou [102] in studying protein signal peptides, where 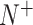 ,
, 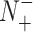 ,
, 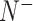 and
and 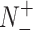 represent the numbers of positives, false positives, negatives and false negatives, respectively. The MCC values range from −1 to 1, where a coefficient of +1 means a perfect prediction and −1 indicates total disagreement between the prediction and observation [103–106]. The Acc value ranges from 0 to 1, with a higher Acc value indicating a better performance [103–106]. The AUC value is calculated based on the receiver-operating-characteristic (ROC) curve and takes values between 0 and 1, where the higher the AUC value, the better the prediction performance.
represent the numbers of positives, false positives, negatives and false negatives, respectively. The MCC values range from −1 to 1, where a coefficient of +1 means a perfect prediction and −1 indicates total disagreement between the prediction and observation [103–106]. The Acc value ranges from 0 to 1, with a higher Acc value indicating a better performance [103–106]. The AUC value is calculated based on the receiver-operating-characteristic (ROC) curve and takes values between 0 and 1, where the higher the AUC value, the better the prediction performance.
Evaluation strategy
Validation examination is important prior to applying a predictor [107], and three validation methods, including K-fold cross-validation test, jack-knife validation test and independent data set test, are often used to derive comparative metrics (values) among the reviewed predictors.
K-fold cross-validation test. In the K-fold cross-validation test, the data set is divided into K roughly equal parts, one part kept as validation data and the remaining K-1 parts used as training data. The training data are used to estimate the parameters of the model, while the validation data are used to compute all the performance metrics. The procedure is repeated K times, thus using each of the K parts as a test data [108].
Jack-knife validation test. Jack-knife validation test (also known as leave-one-out cross-validation test) is the most objective validation method [109] and provides unique results for a benchmark data set [110]. During the jack-knife process, one sample is selected to be the test data and the remaining are the training data. This procedure will be repeated N times in a data set with N samples.
Independent test. Among the reviewed predictors, the independent test is usually adopted to evaluate the performance of two or more predictors.
Among the reviewed predictors, most took one or two cross-validation tests combined with the independent test as their evaluation strategy. Moreover, the self-consistency test [111] is also a cross-validation scheme, which is not commonly used.
Software availability and usability
An important consideration for developing computational approaches is that they should be directly employed by biologists to facilitate target selection, experimental design and hypothesis generation and validation. Therefore, a user-friendly web server and/or a local executable of the proposed predictor should be ideally available along with the publication. Based on our survey of the predictors for lysine PTM prediction (Table 1), 43/49 (87.8%) predictors were made available as web servers and/or stand-alone software for high-throughput lysine PTM screening; however, about half of these web servers have been taken offline to date. Normally, user input, validation and prediction output are the main components of a web server/stand-alone tool and these should be designed carefully.
Specific to lysine PTM prediction, the design of a user-friendly web page should consider the following important aspects: (i) user input format, (ii) any parameter configurations and their explanations and (iii) validation of submitted data. Among the 28 predictors with available web servers, 24 servers permit multiple-sequence submission, and 4 predictors only allow users to submit one protein sequence a time. Among these 28 servers, 15 facilitate file uploading for sequence submission (generally, the file size is limited, which also limits the maximum number of protein sequences). ‘FASTA’ is the commonly used sequence format for online submission, with the exception of prediction of SUMOylation sites based on hydrophobic properties (SUMOhydro) [112], which requires users to submit sequences as plain text. The lysine PTM types can be selected on the web servers of Prediction of Lysine Methylation and Lysine Acetylation (PLMLA) [113] and identifying multiple Lysine PTM sites (iPTM-mLys) [114], which were designed to target multiple PTMs.
Data validation is another crucial step prior to server processing, in order to ensure the server is running smoothly. A fatal error usually occurs on a poorly designed server when the sequence format is illegal or the sequence contains uncommon residues. Besides, the escape characters contained in the FASTA headers in the submission should also be considered carefully, as they are usually interpreted in different ways by the backstage server and database system, depending on the programming languages employed.
A reasonable output design is crucial for the interpretability of prediction results. At least four aspects regarding the output, specific to PTM prediction, should be taken into consideration: (i) protein indicator (e.g. job ID, protein name, etc.), (ii) the position of predicted PTM sites in the protein sequence, (iii) the peptide containing the predicted PTM site and (iv) the prediction score/confidence. Among the predictors with available web servers, EnsemblePail [115], PSKAcePred [50], NetGlycate 1.0 [20], Glycation Sites prediction by using Bi-Profile Bayes (BPB_GlySite) [116], PLMLA [113], identify Succinylation sites by using Pseudo Amino Acid Composition (iSuc-PseAAC) [117], Mal_Lys [118], MaloPred [51], Prediction of Methylation Sites (PMeS) [119], Methylated lysine(K) sites predictor (MethK) [120], Prediction Species-Specific Methylation sites (PSSMe) [121], RUBI [67], GPS-MSP [48], SUMOhydro [112], SUMOsp [76], Succinylation Sites predictor (SuccinSite2.0) [65], Ubiquitination Sites predictor based on Composition of K-Spaced Amino Acid Pairs (CKSAAP_UbSite) [60], human Ubiquitination Sites predictor based on Composition of K-Spaced Amino Acid Pairs (hCKSAAP_UbSite) [61], Ubiquitination sites Prober (UbiProber) [98] and Ubiquitination Sites prediction based on Evolutionary Screening Algorithm (ESA_UbSite) [100] provide detailed output information including the PTM site and prediction score. BPB_GlySite [116], GPS_MSP [48], SUMOsp [76], SuccinSite2.0 [65], Joint Analyzer of Sumoylation Site and SIMs (JASSA) [122], CKSAAP_UbSite [60], hCKSAAP_UbSite [61] and RUBI [67] allow users to download the prediction results in ‘TEXT’ format for further analysis. Data visualization techniques can facilitate the systematic display of prediction results. An interactive web page can assist users to better understand the distribution of predicted PTM sites across the whole protein sequence. NetGlycate 1.0 [20], GPS_MSP [48], SUMOsp [76], SUMOhydro [112], JASSA [122], CKSAAP_UbSite [60] and hCKSAAP_UbSite [61] provide such a graphical data display. Another important functionality aspect is the possibility to revisit historical prediction results (based on the job ID). Among the reviewed predictors, SuccinSite2.0 [65], SUMOhydro [112], CKSAAP_UbSite [60], hCKSAAP_UbSite [61] and RUBI [67] have provided such functionality on their web servers.
Stand-alone tools are available for KA-predictor [52], Glycation sites Predictor (Glypre) [96], PMeS [119], GPS-MSP [48], SUMOsp [76], UbPred [123], CKSAAP_UbSite [60], UbiProber [98] and hCKSAAP_UbSite [61]—all of which are reviewed in this study. Providing detailed software installation instructions, with information about dependencies and runtime environment, is therefore strongly suggested, especially considering that it is generally challenging for biologists to use these stand-alone tools on their local machines. Among these stand-alone tools, GPS-MSP [48], SUMOsp [76] and UbiProber [98] provide a graphical user interface.
Development of the MUscADEL approach
Recent years have witnessed the rise of deep learning techniques and their use in various real-world applications such as image analysis [124], speech recognition [125] and natural language processing [126]. In addition, a number of bioinformatics studies have demonstrated the power of deep learning techniques in the prediction of protein secondary structures [127], protein disordered regions [128], ribonucleicacid (RNA) splicing [129], RNA-binding sites [130] and protein PTM sites (e.g. phosphorylation [131] and ubiquitylation [132]). The deep learning scheme, derived from ANN, has shown great capability to self-learn sparse representations, using multiple hidden layers; in contrast, conventional machine learning algorithms require experts to pre-define informative features [133] prior to model construction. Based on one typical deep learning model, the RNN [134], an extended version called ‘long short-term memory RNN (LSTM-RNN)’ has been proposed. The LSTM-RNN architecture consists of an input gate, a neuron with a self-recurrent connection, a forget gate and an output gate [42]. To evaluate the applicability of the deep learning scheme for lysine PTM prediction, we proposed the MUscADEL framework, based on a bidirectional LSTM-RNN algorithm. We constructed two predictive models, i.e. the full-sequence (Figure 3A) model and the sequence-fragment (Figure 3B) model. Both models contain five layers, including the input layer, the word embedding layer [126], the recurrent layer, the fully connected layer and the output layer. Please refer to the Supplementary Materials and Methods for the detailed description of these layers.
Figure 3.
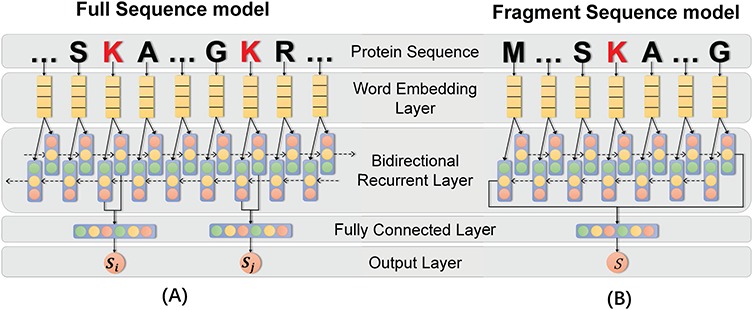
The flowchart of the proposed MUscADEL framework. In this study, two predictive models were constructed. The left (A) shows the full-sequence model, while the right (B) shows the sequence-fragment model. Both models contain five layers, including the input layer, the word embedding layer, the recurrent layer, the fully connected layer and the output layer.
Construction of benchmarking and independent test data sets
In order to objectively evaluate the prediction performance among existing approaches and to build MUscADEL, we constructed eight lysine PTMs data sets for two mammalian species (i.e. Homo sapiens and Mus musculus) from the PhosphoSitePlus database and further mapped the protein sequences to human and mouse proteomes annotated by the UniProt database [78]. Even though ∼20 types of experimentally verified lysine PTMs are covered in the PhosphoSitePlus database [11], we only extracted associated sequences and constructed reliable prediction models for those eight PTM types that have >150 positive protein sequences in the data set. For each type of PTM, we applied the Cluster Database at High Identity with Tolerance (CD-HIT) program [135] to remove sequence redundancy using a threshold of 30%. All the data sets were then randomly divided into two subsets for model training via 5-fold cross-validation and independent test, respectively. Experimentally validated PTM sites, based on the annotation from the PhosphoSitePlus database, were taken as positive samples; the rest of lysine residues in the proteins were regarded as negative samples. The statistical summary of the selected types of lysine PTMs from the PhosphoSitePlus database and the positive and negative samples for each PTM type after the sequence redundancy reduction is provided in Table 3.
Table 3.
Statistical summary of benchmarking and independent test data sets constructed for eight lysine PTMs
| PTM type | 5-fold cross-validation | Independent test | ||
|---|---|---|---|---|
| Number of proteins | Number of PTM sites | Number of proteins | Number of PTM sites | |
| Acetylation | 3289 | 10 845 | 1645 | 5052 |
| Glutarylation | 124 | 432 | 31 | 123 |
| Glycation | 1621 | 3209 | 811 | 1530 |
| Malonylation | 1702 | 4300 | 425 | 1050 |
| Methylation | 902 | 1352 | 226 | 337 |
| Succinylation | 772 | 1728 | 193 | 382 |
| Sumoylation | 1286 | 3863 | 640 | 1890 |
| Ubiquitination | 4768 | 19 949 | 2385 | 9229 |
Results and discussion
Motif conservation analysis for eight types of lysine PTMs
To illustrate the distribution and preference of the flanking residues of lysine PTM sites, we examined the motif conservation for eight lysine PTM types using the Probability Logo Generator (pLogo) [136] algorithm. The logos of motif conservation of eight lysine PTM types are presented in Figure 4. The default ±4.08 were used as the thresholds of significantly overrepresented and underrepresented amino acids, respectively. Among these PTM types, sumoylation is the only type that demonstrated a clear motif pattern (Figure 4G), ψ-K-X-E (ψ is any hydrophobic amino acid, while X denotes any amino acid residue), which is consistent with previous reports [137, 138]. Lysine acetylation showed relatively distinguishable motif conservation patterns (Figure 4A), where the hydrophobic amino acids G and P predominately occurred on positions −1 and +1, and the charged residues K and R were most likely underrepresented on position −1. In contrast to sumoylation and acetylation, other PTM types did not show very obvious or indicative motif patterns. For lysine glutarylation, the only observation that we made is that the charged residues K and R were overrepresented at positions −10 and −8 (Figure 4B). For glycation, we can only conclude that the polar and hydrophobic residues (i.e. A, T, G, V and M) are likely overrepresented in the motifs; while charged residues, such as R, K, D and E, were usually underrepresented (Figure 4C). Figure 4D shows the motif conservation for lysine malonylation, where no polar residues were reported to be overrepresented in these motifs. Figure 4E illustrates that the charged residues, K and R, were overrepresented at positions −14, −8, +7, +12 and +15 for methylation. For succinylation, the charged residues K and R were overrepresented at positions −7, +5, +7 and +8. Another charged residue, D, was also found to be overrepresented at positions −2, +1 and +2 (Figure 4F). Last but not least, for ubiquitination, the charged residue R was found overrepresented at positions −13 ∼ −7, +6 ∼ +12 and +14; while another charged residue, K, was predominately found underrepresented at positions −6 ∼ −1 and +1 ∼ +5 (Figure 4H).
Figure 4.
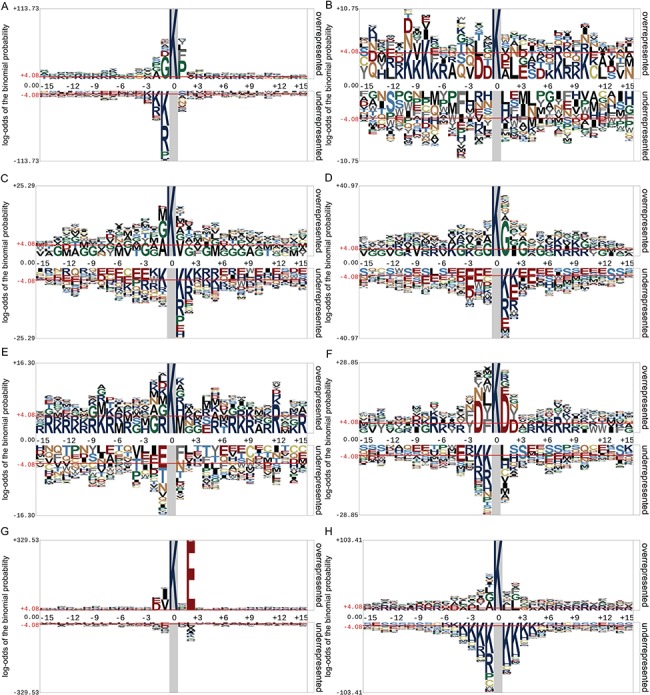
Motif conservation analysis of eight lysine PTM types including (A) acetylation, (B) glutarylation, (C) glycation, (D) malonylation, (E) methylation, (F) succinylation, (G) sumoylation and (H) ubiquitination. Plots were generated with pLogo and scaled better data visualization. The red horizontal lines on the sequence logos denote the P < 0.05 threshold.
Performance comparison between the full-sequence and fragment models of MUscADEL
We have built two models of MUscADEL based on the input, i.e. the full-sequence model and the fragment model. For the full-sequence model, the full-length protein sequences were used as input; for the fragment model, motifs with fixed window size with centered lysine residues were used. The full-sequence model considers both contributions of the full-length sequences and motifs surrounding the lysine PTM sites, while the fragment model only extracts the motif patterns with the lysine PTM sites. In our study, the window size was set to 27 residues to extract the motifs after testing the performance for a series of window sizes (i.e. 15, 19, 23, 27 and 31). We validated the prediction performances of these two models on training data sets for each type of PTMs. For each lysine PTM type, the model with the best performance was chosen by MUscADEL.
We first evaluated the prediction performance of the two MUscADEL models for each type of lysine PTM via 5-fold cross-validation, based on the training data set with the fixed Sp of 90%. Table 4 summarizes the performance comparison between the full-sequence and fragment models for individual types of lysine PTMs. It is clear from Table 4 that the full-sequence model outperformed the fragment model for predicting lysine acetylation, glycation, malonylation, methylation, sumoylation and ubiquitination. This means that, for these PTM types, both full-length sequence information and local motif information contributed to the prediction of PTM sites. For predicting glutarylation, the fragment model generally outperformed the full-sequence model, further indicating that the determinative sequence patterns of these PTM types can be capltured by studying the local motif environment surrounding the lysine PTM residues. In other words, the determination of these PTM sites may only be affected by the motif patterns surrounding the sites. Based on the performance demonstrated in Table 4, the final MUscADEL framework was composed by the fragment model for lysine glutarylation and full-sequence models for the remaining PTMs. In these eight types of PTMs, the data set sizes for glutarylation and methylation are quite small compared with that of other types of PTMs. In order to test whether MUscADEL can achieve better performance than traditional machine learning algorithms for glutarylation and methylation, we used the commonly used binary [60, 139, 140] and AAindex encodings [132] together with the RF and SVM algorithms. The number of decision trees was set to 1000, and the RBF kernel of the SVM was used. As for the AAindex encoding, we collected 544 physicochemical properties from the AAindex database (http://www.genome.jp/aaindex/) and retained 531 properties after removal of properties with ‘NA’ in the amino acid indices. We then calculated the performance for each property and selected the top 20 properties with the highest AUC values based on 5-fold cross-validation. Consequently, a peptide with 27 residues was converted into a vector of 540 = (27 × 20) dimensions using the AAindex encoding. The results show that MUscADEL performs better than traditional machine learning algorithms for glutarylation and methylation (Table S1).
Table 4.
The predictive performance of the full-sequence and fragment models based on the benchmark data sets via 5-fold cross-validation with a fixed Sp of 90%
| PTM type | Full-sequence model | Fragment model | ||||
|---|---|---|---|---|---|---|
| Sn (%) | MCC | AUC | Sn (%) | MCC | AUC | |
| Acetylation | 36.46 | 0.2057 | 0.754 | 31.62 | 0.1915 | 0.742 |
| Glutarylation | 18.29 | 0.0913 | 0.602 | 26.45 | 0.1733 | 0.739 |
| Glycation | 31.77 | 0.1282 | 0.705 | 16.67 | 0.0622 | 0.610 |
| Malonylation | 45.79 | 0.2544 | 0.822 | 40.36 | 0.2616 | 0.807 |
| Methylation | 28.80 | 0.0926 | 0.703 | 20.42 | 0.0960 | 0.626 |
| Succinylation | 33.98 | 0.1822 | 0.734 | 32.67 | 0.2001 | 0.748 |
| Sumoylation | 42.66 | 0.2326 | 0.764 | 36.25 | 0.2291 | 0.741 |
| Ubiquitination | 43.77 | 0.2968 | 0.799 | 37.11 | 0.2432 | 0.769 |
Performance comparison between MUscADEL and existing methods on the independent data set
We then compared the prediction performance of our proposed MUscADEL framework with that of existing computational tools for lysine PTM prediction, based on the independent data set. We selected the 19 methods with available tools/web servers to compare with our proposed MUscADEL framework. In addition, since MUscADEL is the only framework allowing for lysine glutarylation prediction, we only compared the prediction results for the other 7 PTMs with the 19 existing methods. To conduct stringent and objective comparison, we evaluated the prediction performance using our assembled independent testing data set.
To obtain the prediction results from the existing methods, we manually submitted the protein sequences in the independent data sets to their web servers or ran their corresponding executables locally. The performance comparison is illustrated in Figure 5. Overall, MUscADEL outperformed the existing prediction methods for all seven lysine PTM types. To name one example, among the methods for lysine ubiquitination prediction, MUscADEL achieved the best performance (AUC = 0.822), followed by RUBI (AUC = 0.746) and hCKSAAP_UbSite (AUC = 0.668). When focusing on the performance with low false positive rate (i.e. Sp = 90%), MUscADEL identified 49.2% ubiquitination sites, whereas RUBI accurately predicted only 34.4% of the ubiquitination sites with the same Sp threshold. MUscADEL also demonstrated superior prediction performance for other PTM types, in terms of overall performance and the performance specific to a low false positive rate. Taken together, the performance comparison using the independent testing data set clearly demonstrates that MUscADEL can accurately predict eight types of lysine PTMs, confirming the suspected power of deep learning techniques for protein PTM identification.
Figure 5.
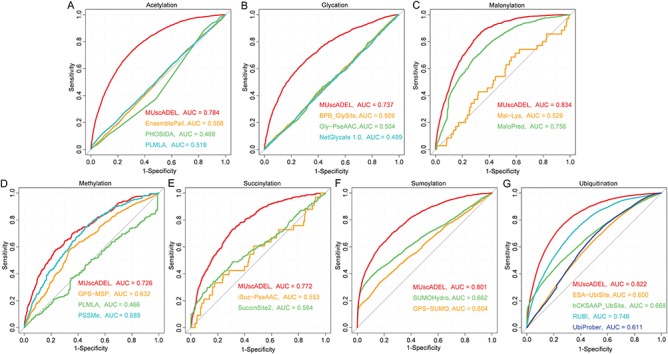
Performance comparison between MUscADEL and the existing methods for lysine PTM prediction. ROC curves and AUC values of MUscADEL and other predictors for (A) acetylation, (B) glycation, (C) malonylation, (D) methylation, (E) succinylation, (F) sumoylation and (G) ubiquitination.
Thoughts for current predictors and future perspectives
Because of the limited availability of lysine PTM data sets, some predictors could only use small data sets to train their models, resulting in unsatisfactory prediction performance when tested with the independent test data set. For example, the CKSAAP_UbSite [60], one of our previous works for protein ubiquitination prediction published in 2011, was built based on the Saccharomyces cerevisiae ubiquitination data set, with only 263 ubiquitination sites. Because of the limited training data, CKSAAP_UbSite did not achieve the prediction performance as reported in the original study [92] according to a later independent test [141]. Fortunately, with the advances of mass spectrometry and high-throughput sequencing, large volumes of PTMs are being identified and the corresponding databases are updated frequently. On the other hand, some previously discarded PTM sites are now experimentally verified PTM sites. This means the predictors built on the old versions of the database are not reliable, as the old database contains a number of false negative samples. To keep the published predictor up-to-date, it is therefore suggested that models are retrained once up-to-date data sets become available.
Data redundancy is an important issue to consider prior to model construction. Redundant data result in overfit models with respect to the current training data set, leading to poor scalability and lack of robustness on independent test data sets. Based on our survey, 38 out of 49 methods for lysine PTM prediction have conducted sequence redundancy removal prior to model construction. CD-HIT [135] and clusters protein or DNA sequences based on pairwise matches found using the BLAST algorithm (blustclust) [45] are the most widely used approaches to remove sequence redundancy. The threshold of sequence identity used in these two methods is normally 30%. Another issue to consider is the ratio of positive and negative samples in the training data set. Normally, negative samples (i.e. non-PTM sites) significantly outnumber the positive samples (i.e. PTM sites). Therefore, a balanced training data set should be generated, constructing reliable and nonbiased predictive models. To do so, a random selection is usually conducted, to choose a number of negative samples equal to the number of positive samples. Then the random selection can be repeated and the average performance reported.
A number of machine learning algorithms have been successfully applied to lysine PTM prediction (Table 1). To further improve the prediction performance, we would like to make two suggestions. First, to import multi-omics data to build a systems biology model; for example, the ASEB [46] approach for lysine acetylation prediction was built based on the results of gene set enrichment analysis [142]. The analysis started with DNA microarray data that were subsequently used to detect coordinated changes of expression in a group of functionally related genes and to find the putative functions of the long noncoding RNAs. Second, to consider ensemble methods, for example, our proposed ZincExplorer, for protein zinc binding sites, integrates the outputs from three predictors (i.e. an SVM predictor, a cluster-based predictor and a template-based predictor). Those experiments demonstrated that prediction performance was significantly improved by integrating the outputs of multiple predictors [143]. Another good example is the prediction for secreted proteins of type III and IV secretion systems by integrating the prediction outcomes from SVMs, RF and LR models [144].
An important goal of this review and survey paper is to provide practical and useful guidance for researchers to facilitate the identification and validation of PTM sites by experimental methods. The predicted proteins by computational methods such as those reviewed in this paper are just potential or putative modified substrates and would need to be verified by experimental methods. Consequently, users need to be cautious when interpreting the prediction results generated by the bioinformatics predictors.
Moreover, the prediction of PTM substrates is much more complicated than the prediction of PTM sites, as the short sequence motifs surrounding the PTM sites are often insufficient to provide full Sp for determining modification events, especially in vivo [145]. In fact, various ‘contextual factors’ such as the physical association, co-occurrence in the genome and co-expression of the enzymes (e.g. kinases in the case of phosphorylation events) and substrates are currently not considered but will likely contribute to determining if in vivo modifications occur. A purely sequence-based predictor such as MUscADEL will only be able to predict modifications that may occur under ideal conditions (e.g. in a test tube) but that may not happen in vivo. Thus, inclusion of ‘contextual factors’ will potentially improve the accuracy of in vivo PTM predictions [145]. However, a major challenge is that some of these ‘contextual factors’ are extremely difficult to obtain and experimentally validate. Accordingly, their usage in predictive models is not possible at present. Therefore, in MUscADEL we only considered sequence information to construct the prediction model, since such information is readily accessible. Importantly, we find that by considering the sequence information, one can achieve reasonably good prediction performance, stressing both high practicality and usefulness. We will consider including ‘contextual factors’ into machine learning models during the development of an upgraded version of MUscADEL in our future work, if and when such quality and quantity of data become available.
Additional complexity in predicting PTM substrates and sites originates from PTM subtypes. Such subtypes may be too similar for a purely sequence-based machine learning model to be able to identify any differences between them and their associated modification sites. Moreover, often models are trained on PTM data that do not differentiate between PTM subtypes. Consequently, such trained models will also not be able to predict if a substrate or site is modified by one or the other subtype. To provide one example, SUMO1 and SUMO2/3 are two sumoylation subtypes. SUMO2 and SUMO3 are highly similar, while both have moderate similarity with SUMO1. SUMO2 and SUMO3 can form heterodimeric chains (i.e. SUMO-chains), while SUMO1 is singly attached to protein substrates. Thus, from a structural perspective, SUMO2/3 will have similar but still different Sp for substrate recognition in contrast to SUMO1 because of the larger size of the SUMO2/3 complex compared to SUMO1. Interestingly, using mass spectrometry, a recent study identified 14 869 potential SUMO2/3 sites in 3870 human proteins [146]. Therefore, in principle, such data can now be used to train a SUMO2/3-specific predictor, though a similar data set would be needed to train a SUMO1-specific predictor. In sum, with increasing availability of subtype data, the challenging task of subtype-specific PTM prediction will likely be overcome in the future. Lastly, as shotgun proteomics is often error-prone, the 14 869 sites can also only be considered potential candidates, and experimental validation by small-scale and conventional experiments remains necessary.
Conclusion
Characterization of the lysine PTM sites is an important topic, which can increase our understanding of lysine PTM molecular mechanisms and related biological processes. A number of computational methods have been developed to accurately predict different types of lysine PTMs, hoping to accelerate and complement experimental discoveries. To assist interested readers to better understand the methodologies and development of these approaches, we systematically surveyed 49 studies revolving around the prediction of eight types of lysine PTMs, including acetylation, glutarylation, glycation, malonylation, methylation, succinylation, sumoylation and ubiquitination. We coarsely categorized the reviewed predictors into two types: machine learning-based methods and peptide similarity-based methods. We then carefully reviewed these computational approaches in terms of their core algorithm, sequence and structure features, feature selection technique, evaluation strategy and software utility. Based on our investigation, we described limitations of current computational approaches for lysine PTM prediction and provided insights into data set construction, model updates and performance improvements. Following our survey, we evaluated the feasibility of applying deep learning techniques to lysine PTM prediction. We built a novel computation model, MUscADEL, based on the ‘long short-term memory recurrent neural network’ learning scheme. For each type of lysine PTM, we constructed two models, i.e. the full-sequence and fragment models. Five-fold cross-validation and independent testing demonstrated that MUscADEL outperformed current machine learning-based approaches, demonstrating the potential power of the deep learning scheme in protein PTM prediction. We anticipate that our survey will provide useful insights and guidance into the development of novel computational approaches for lysine PTM prediction and that MUscADEL will inspire future works on accurate predictions of protein PTMs using a deep learning framework.
Key Points:
Lysine post-translational modifications (PTMs) play important roles in a myriad of diverse biological processes. This study serves as a comprehensive survey of current methods for lysine PTM prediction, particularly in terms of model construction and evaluation.
We propose a new deep learning model, termed Multiple Scalable Accurate Deep Learner for lysine PTMs (MUscADEL), to improve the prediction of eight different types of lysine PTMs. Experimental results demonstrate the superior performance of MUscADEL compared to existing methods.
A web portal (http://muscadel.erc.monash.edu/) has been made available to facilitate online high-throughput prediction of lysine PTMs.
We demonstrate the predictive power of deep learning-based models in lysine PTM prediction.
Supplementary Material
Zhen Chen is an assistant professor at the School of Basic Medical Science, Qingdao University, China. His research interests include protein bioinformatics, machine learning and analysis of next-generation sequencing.
Xuhan Liu was a bioinformatician at Departments of Information Technology, Beijing Oriental Yamei Gene Technology Institute Co. Ltd., Beijing, China before August 2017. Currently he is a PhD student at Drug Discovery and Safety, Leiden Academic Centre for Drug Research, Leiden University, The Netherlands. Since his graduation from China Agricultural University, he has been involved in several bioinformatics projects related to protein function, drug discovery, machine learning and data mining. His research interests are deep learning applications for drug discovery.
Fuyi Li received his bachelor and master degrees in software engineering from the Northwest A&F University, China. He is currently a PhD student at the Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia. His research interests are bioinformatics, computational biology, machine learning and data mining.
Chen Li received his PhD degree in bioinformatics from Monash University, Australia. He is currently an NHMRC CJ Martin Early Career Research fellow at the Institute of Molecular Systems Biology, ETH Zürich, Switzerland and the Monash Biomedicine Discovery Institute, Monash University, Australia. His research interests include systems immunology, computational proteomics, immunopeptidomics, systems biology and data mining.
Tatiana Marquez-Lago is an associate professor at the Department of Genetics and the Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham (UAB) School of Medicine, USA. Her research interests include multi-scale modeling and simulations, artificial intelligence, bioengineering and systems biomedicine. Her interdisciplinary laboratory studies stochastic gene expression, chromatin organization, antibiotic resistance reversal in bacteria and host–microbiota interactions in complex diseases.
André Leier is currently an assistant professor at the Department of Genetics, UAB School of Medicine, USA. He is also an associate scientist in the UAB Comprehensive Cancer Center. He received his PhD in Computer Science (Dr rer. nat.), University of Dortmund, Germany. He conducted postdoctoral research at the Memorial University of Newfoundland, Canada, The University of Queensland, Australia, and ETH Zürich, Switzerland. His research interests are in biomedical informatics and computational and systems biomedicine.
Tatsuya Akutsu received his DEng degree in information engineering in 1989 from University of Tokyo, Japan. He has been appointed as a professor since 2001 in the Bioinformatics Center, Institute for Chemical Research, Kyoto University, Japan. His research interests include bioinformatics and discrete algorithms.
Geoffrey I. Webb received his PhD degree in computer science in 1987 from La Trobe University. He is the director of the Monash Centre for Data Science and a professor in the Faculty of Information Technology at Monash University. His research interests include machine learning, data mining, computational biology and user modeling.
Dakang Xu began his career as a medical clinician and completed his MD and PhD in Japan. He undertook his postdoc training at the Monash Institute of Medical Research, Monash University, Australia. He is a professor in the Shanghai Jiaotong University School of Medicine, China. His team has used a combination of genetically modified mouse models of disease, cell signaling, bioinformatics analyses and clinical applications, to understand the mechanisms of cellular regulation in cancer, immunity and inflammation.
Alexander Ian Smith completed his PhD at Prince Henry’s Institute Melbourne and Monash University, Australia. He is the vice-provost (research and research infrastructure) of Monash University. He is also a professorial fellow in the Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology at Monash University where he runs his research group. His research applies proteomics technologies to study the proteases involved in the generation and metabolism of peptide regulators involved in both brain and cardiovascular function.
Lei Li received his master degree from Nanjing University of Aeronautics and Astronautics, China, and PhD from Nanyang Technological University, Singapore. He is a professor at the School of Basic Medical Science, Qingdao University, China. His research interests include bioinformatics, systems biology and proteomics.
Kuo-Chen Chou received his DSc degree in 1984 from Kyoto University, Japan. He is the founder and chief scientist of Gordon Life Science Institute. He is also a distinguished high-impact professor and advisory professor of several universities. Professor Chou’s research interests are in computational biology and biomedicine, protein structure prediction, low-frequency internal motion of protein/DNA molecules and its biological functions, diffusion-controlled reactions of enzymes as well as graphic rules in enzyme kinetics and other biological systems.
Jiangning Song is a senior research fellow and group leader at the Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia. He is a member of the Monash Centre for Data Science, Faculty of Information Technology and an associate investigator of the ARC Centre of Excellence in Advanced Molecular Imaging, Monash University. His research interests primarily focus on data mining, data analytics, machine learning, pattern recognition and their applications to bioinformatics, computational biomedicine, enzyme engineering, functional genomics, medical informatics and image processing.
Funding
This work was financially supported by grants from the Australian Research Council (LP110200333 and DP120104460), the Young Scientists Fund of the National Natural Science Foundation of China (31701142), the National Natural Science Foundation of China (31770821), the National Health and Medical Research Council of Australia (NHMRC) (4909809), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965), a major inter-disciplinary research project awarded by Monash University and the collaborative research program of the Institute for Chemical Research, Kyoto University (2018-28). C.L. is currently supported by an NHMRC CJ Martin Early Career Research Fellowship (1143366). T.T.M.L. and A.L.’s work was supported in part by the Informatics Institute of the School of Medicine at the University of Alabama at Birmingham.
References
- 1. Xu H, Zhou J, Lin S, et al. PLMD: an updated data resource of protein lysine modifications. J Genet Genomics 2017;44:243–50. [DOI] [PubMed] [Google Scholar]
- 2. Du Y, Zhai Z, Li Y, et al. Prediction of protein lysine acylation by integrating primary sequence information with multiple functional features. J Proteome Res 2016;15:4234–44. [DOI] [PubMed] [Google Scholar]
- 3. Xu HD, Wang LN, Wen PP, et al. Site-specific systematic analysis of lysine modification crosstalk. Proteomics 2018;18:e1700292. [DOI] [PubMed] [Google Scholar]
- 4. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol 2015;16:258–64. [DOI] [PubMed] [Google Scholar]
- 5. Tan M, Peng C, Anderson KA, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab 2014;19:605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peng C, Lu Z, Xie Z, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics 2011;10:M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lanouette S, Mongeon V, Figeys D, et al. The functional diversity of protein lysine methylation. Mol Syst Biol 2014;10:724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishida Y, Rardin MJ, Carrico C, et al. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol Cell 2015;59:321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 2007;8:947–56. [DOI] [PubMed] [Google Scholar]
- 10. Welchman RL, Gordon C, Mayer RJ. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol 2005;6:599–609. [DOI] [PubMed] [Google Scholar]
- 11. Hornbeck PV, Zhang B, Murray B, et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 2015;43:D512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nie Q, Gong XD, Liu M, et al. Effects of crosstalks between sumoylation and phosphorylation in normal cellular physiology and human diseases. Curr Mol Med 2017;16:906–13. [DOI] [PubMed] [Google Scholar]
- 13. Polevoda B, Sherman F. The diversity of acetylated proteins. Genome Biol 2002;3: reviews0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Das C, Kundu TK. Transcriptional regulation by the acetylation of nonhistone proteins in humans—a new target for therapeutics. IUBMB Life 2005;57:137–49. [DOI] [PubMed] [Google Scholar]
- 15. Glozak MA, Sengupta N, Zhang X, et al. Acetylation and deacetylation of non-histone proteins. Gene 2005;363:15–23. [DOI] [PubMed] [Google Scholar]
- 16. Kim SC, Sprung R, Chen Y, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 2006;23:607–18. [DOI] [PubMed] [Google Scholar]
- 17. Spange S, Wagner T, Heinzel T, et al. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009;41:185–98. [DOI] [PubMed] [Google Scholar]
- 18. Zhao S, Xu W, Jiang W, et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010;327:1000–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bidasee KR, Zhang Y, Shao CH, et al. Diabetes increases formation of advanced glycation end products on sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004;53:463–73. [DOI] [PubMed] [Google Scholar]
- 20. Johansen MB, Kiemer L, Brunak S. Analysis and prediction of mammalian protein glycation. Glycobiology 2006;16:844–53. [DOI] [PubMed] [Google Scholar]
- 21. Nicolls MR. The clinical and biological relationship between type II diabetes mellitus and Alzheimer’s disease. Curr Alzheimer Res 2004;1:47–54. [DOI] [PubMed] [Google Scholar]
- 22. Munch G, Gerlach M, Sian J, et al. Advanced glycation end products in neurodegeneration: more than early markers of oxidative stress? Ann Neurol 1998;44:S85–8. [DOI] [PubMed] [Google Scholar]
- 23. Pickart CM. Ubiquitin enters the new millennium. Mol Cell 2001;8:499–504. [DOI] [PubMed] [Google Scholar]
- 24. Chau V, Tobias JW, Bachmair A, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 1989;243:1576–83. [DOI] [PubMed] [Google Scholar]
- 25. Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2001;2:195–201. [DOI] [PubMed] [Google Scholar]
- 26. Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 2009;33:275–86. [DOI] [PubMed] [Google Scholar]
- 27. Schwartz AL, Ciechanover A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med 1999;50:57–74. [DOI] [PubMed] [Google Scholar]
- 28. Hay RT. SUMO: a history of modification. Mol Cell 2005;18:1–12. [DOI] [PubMed] [Google Scholar]
- 29. Muller S, Hoege C, Pyrowolakis G, et al. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol 2001;2:202–10. [DOI] [PubMed] [Google Scholar]
- 30. Seeler JS, Dejean A. Nuclear and unclear functions of SUMO. Nat Rev Mol Cell Biol 2003;4:690–9. [DOI] [PubMed] [Google Scholar]
- 31. Wang Z, Tang WH, Cho L, et al. Targeted metabolomic evaluation of arginine methylation and cardiovascular risks: potential mechanisms beyond nitric oxide synthase inhibition. Arterioscler Thromb Vasc Biol 2009;29:1383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mastronardi FG, Wood DD, Mei J, et al. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci 2006;26:11387–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suzuki A, Yamada R, Yamamoto K. Citrullination by peptidylarginine deiminase in rheumatoid arthritis. Ann N Y Acad Sci 2007;1108:323–39. [DOI] [PubMed] [Google Scholar]
- 34. Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell 2006;126:257–68. [DOI] [PubMed] [Google Scholar]
- 35. Xie Z, Dai J, Dai L, et al. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics 2012;11:100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Z, Tan M, Xie Z, et al. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol 2011;7:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirschey MD, Zhao Y. Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol Cell Proteomics 2015;14:2308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Noort V, Seebacher J, Bader S, et al. Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol Syst Biol 2012;8:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caron C, Boyault C, Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays 2005;27:408–15. [DOI] [PubMed] [Google Scholar]
- 40. Hunter T, Sun H. Crosstalk between the SUMO and ubiquitin pathways. Ernst Schering Found Symp Proc 2008;1:1–16. [DOI] [PubMed] [Google Scholar]
- 41. Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol 2011;273:236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput 1997;9:1735–80. [DOI] [PubMed] [Google Scholar]
- 43. Schuster M, Paliwal KK. Bidirectional recurrent neural networks. IEEE Trans Signal Process 1997;45:2673–81. [Google Scholar]
- 44. Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 1992;89:10915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li T, Du Y, Wang L, et al. Characterization and prediction of lysine (K)-acetyl-transferase specific acetylation sites. Mol Cell Proteomics 2012;11:M111.011080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang L, Du Y, Lu M, et al. ASEB: a web server for KAT-specific acetylation site prediction. Nucleic Acids Res 2012;40:W376–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deng W, Wang Y, Ma L, et al. Computational prediction of methylation types of covalently modified lysine and arginine residues in proteins. Brief Bioinform 2017;18:647–58. [DOI] [PubMed] [Google Scholar]
- 49. Zhao Q, Xie Y, Zheng Y, et al. GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res 2014;42:W325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suo SB, Qiu JD, Shi SP, et al. Position-specific analysis and prediction for protein lysine acetylation based on multiple features. PLoS One 2012;7:e49108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang LN, Shi SP, Xu HD, et al. Computational prediction of species-specific malonylation sites via enhanced characteristic strategy. Bioinformatics 2017;33:1457–63. [DOI] [PubMed] [Google Scholar]
- 52. Wuyun Q, Zheng W, Zhang Y, et al. Improved species-specific lysine acetylation site prediction based on a large variety of features set. PLoS One 2016;11:e0155370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cortes C, Vapnik V. Support-vector networks. Mach Learn 1995;20:273–97. [Google Scholar]
- 54. Breiman L. Random Forests. Mach Learn 2001;45:5–32. [Google Scholar]
- 55. McCulloch WS, Pitts W. A logical calculus of the ideas immanent in nervous activity. Bull Math Biophys 1943;5:115–33. [PubMed] [Google Scholar]
- 56. Altman NS. An introduction to kernel and nearest-neighbor nonparametric regression. Am Stat 1992;46:175–85. [Google Scholar]
- 57. Freedman AD. Statistical Models: Theory and Practice, Technometrics, Vol. 48. New York: Cambridge University Press; 2005, 315–15. [Google Scholar]
- 58. Vapnik VN. The Nature of Statistical Learning Theory. New York: Springer., 1995. [Google Scholar]
- 59. Vapnik VN. An overview of statistical learning theory. IEEE Trans Neural Netw 1999;10:988–99. [DOI] [PubMed] [Google Scholar]
- 60. Chen Z, Chen YZ, Wang XF, et al. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PLoS One 2011;6:e22930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen Z, Zhou Y, Song J, et al. hCKSAAP_UbSite: improved prediction of human ubiquitination sites by exploiting amino acid pattern and properties. Biochim Biophys Acta 2013;1834:1461–7. [DOI] [PubMed] [Google Scholar]
- 62. Li Y, Wang M, Wang H, et al. Accurate in silico identification of species-specific acetylation sites by integrating protein sequence-derived and functional features. Sci Rep 2014;4:5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jia J, Liu Z, Xiao X, et al. iSuc-PseOpt: identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training data set. Anal Biochem 2016;497:48–56. [DOI] [PubMed] [Google Scholar]
- 64. Jia J, Liu Z, Xiao X, et al. pSuc-Lys: predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J Theor Biol 2016;394:223–30. [DOI] [PubMed] [Google Scholar]
- 65. Hasan MM, Khatun MS, Mollah MNH, et al. A systematic identification of species-specific protein succinylation sites using joint element features information. Int J Nanomedicine 2017;12:6303–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang S-C. Artificial neural network. In: Interdisciplinary Computing in Java Programming. Boston, MA: Springer US, 2003, 81–100. [Google Scholar]
- 67. Walsh I, Di Domenico T, Tosatto SC. RUBI: rapid proteomic-scale prediction of lysine ubiquitination and factors influencing predictor performance. Amino Acids 2014;46:853–62. [DOI] [PubMed] [Google Scholar]
- 68. Kavuncuoglu H, Kavuncuoglu E, Karatas SM, et al. Prediction of the antimicrobial activity of walnut (Juglans regia L.) kernel aqueous extracts using artificial neural network and multiple linear regression. J Microbiol Methods 2018;148:78–86. [DOI] [PubMed] [Google Scholar]
- 69. Wu K, Wei GW. Quantitative toxicity prediction using topology based multitask deep neural networks. J Chem Inf Model 2018;58:520–31. [DOI] [PubMed] [Google Scholar]
- 70. Cai Y, Huang T, Hu L, et al. Prediction of lysine ubiquitination with mRMR feature selection and analysis. Amino Acids 2012;42:1387–95. [DOI] [PubMed] [Google Scholar]
- 71. Lu L, Shi XH, Li SJ, et al. Protein sumoylation sites prediction based on two-stage feature selection. Mol Divers 2010;14:81–6. [DOI] [PubMed] [Google Scholar]
- 72. Hou T, Zheng G, Zhang P, et al. LAceP: lysine acetylation site prediction using logistic regression classifiers. PLoS One 2014;9:e89575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Song J, Li F, Leier A, et al. PROSPERous: high-throughput prediction of substrate cleavage sites for 90 proteases with improved accuracy. Bioinformatics 2018;34:684–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li F, Li C, Marquez-Lago TT, et al. Quokka: a comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics 2018, (doi: https:10.1093/bioinformatics/bty522). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li A, Xue Y, Jin C, et al. Prediction of Nepsilon-acetylation on internal lysines implemented in Bayesian discriminant method. Biochem Biophys Res Commun 2006;350:818–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xue Y, Zhou F, Fu C, et al. SUMOsp: a web server for sumoylation site prediction. Nucleic Acids Res 2006;34:W254–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pearson WR. BLAST and FASTA similarity searching for multiple sequence alignment. Methods Mol Biol 2014;1079:75–101. [DOI] [PubMed] [Google Scholar]
- 78. The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res 2018;46:2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pundir S, Martin MJ, O’Donovan C. UniProt Protein Knowledgebase. Methods Mol Biol 2017;1558:41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res 2017;45:D158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cai Y, He J, Lu L. Predicting sumoylation site by feature selection method. J Biomol Struct Dyn 2011;28:797–804. [DOI] [PubMed] [Google Scholar]
- 82. Song J, Wang H, Wang J, et al. PhosphoPredict: a bioinformatics tool for prediction of human kinase-specific phosphorylation substrates and sites by integrating heterogeneous feature selection. Sci Rep 2017;7:6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li F, Li C, Wang M, et al. GlycoMine: a machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics 2015;31:1411–9. [DOI] [PubMed] [Google Scholar]
- 84. Rao HB, Zhu F, Yang GB, et al. Update of PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res 2011;39:W385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shen HB, Chou KC. PseAAC: a flexible web server for generating various kinds of protein pseudo amino acid composition. Anal Biochem 2008;373:386–8. [DOI] [PubMed] [Google Scholar]
- 86. Cao DS, Xu QS, Liang YZ. propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013;29:960–2. [DOI] [PubMed] [Google Scholar]
- 87. Liu B, Liu F, Wang X, et al. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res 2015;43:W65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Xiao N, Cao DS, Zhu MF, et al. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 2015;31:1857–9. [DOI] [PubMed] [Google Scholar]
- 89. Cao DS, Xiao N, Xu QS, et al. Rcpi: R/Bioconductor package to generate various descriptors of proteins, compounds and their interactions. Bioinformatics 2015;31:279–81. [DOI] [PubMed] [Google Scholar]
- 90. Zuo Y, Li Y, Chen Y, et al. PseKRAAC: a flexible web server for generating pseudo K-tuple reduced amino acids composition. Bioinformatics 2017;33:122–4. [DOI] [PubMed] [Google Scholar]
- 91. Wang J, Yang B, Revote J, et al. POSSUM: a bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics 2017;33:2756–8. [DOI] [PubMed] [Google Scholar]
- 92. Chen Z, Zhao P, Li F, et al. iFeature: a Python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018;34:2499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kawashima S, Pokarowski P, Pokarowska M, et al. AAindex: amino acid index database progress report 2008. Nucleic Acids Res 2008;36:D202–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Li S, Li H, Li M, et al. Improved prediction of lysine acetylation by support vector machines. Protein Pept Lett 2009;16:977–83. [DOI] [PubMed] [Google Scholar]
- 95. Liu Y, Gu W, Zhang W, et al. Predict and analyze protein glycation sites with the mRMR and IFS methods. Biomed Res Int 2015;2015:561547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhao X, Zhao X, Bao L, et al. Glypre: in silico prediction of protein glycation sites by fusing multiple features and support vector machine. Molecules 2017;22:1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tung CW, Ho SY. Computational identification of ubiquitylation sites from protein sequences. BMC Bioinformatics 2008;9:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chen X, Qiu JD, Shi SP, et al. Incorporating key position and amino acid residue features to identify general and species-specific ubiquitin conjugation sites. Bioinformatics 2013;29:1614–22. [DOI] [PubMed] [Google Scholar]
- 99. Yavuz AS, Sezerman OU. Predicting sumoylation sites using support vector machines based on various sequence features, conformational flexibility and disorder. BMC Genomics 2014;15(suppl 9):S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang JR, Huang WL, Tsai MJ, et al. ESA-UbiSite: accurate prediction of human ubiquitination sites by identifying a set of effective negatives. Bioinformatics 2017;33:661–8. [DOI] [PubMed] [Google Scholar]
- 101. Chen W, Feng PM, Lin H, et al. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res 2013;41:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chou KC. Prediction of signal peptides using scaled window. Peptides 2001;22:1973–9. [DOI] [PubMed] [Google Scholar]
- 103. Feng PM, Chen W, Lin H, et al. iHSP-PseRAAAC: identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal Biochem 2013;442:118–25. [DOI] [PubMed] [Google Scholar]
- 104. Su ZD, Huang Y, Zhang ZY, et al. iLoc-lncRNA: predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 2018, (doi: https:10.1093/bioinformatics/bty508) [DOI] [PubMed] [Google Scholar]
- 105. Chen W, Feng P, Yang H, et al. iRNA-3typeA: identifying three types of modification at RNA’s adenosine sites. Mol Ther Nucleic Acids 2018;11:468–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhang J, Feng P, Lin H, et al. Identifying RNA N(6)-methyladenosine sites in Escherichia coli genome. Front Microbiol 2018;9:955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chou KC, Shen HB. Recent progress in protein subcellular location prediction. Anal Biochem 2007;370:1–16. [DOI] [PubMed] [Google Scholar]
- 108. Lopez Y, Dehzangi A, Lal SP, et al. SucStruct: prediction of succinylated lysine residues by using structural properties of amino acids. Anal Biochem 2017;527:24–32. [DOI] [PubMed] [Google Scholar]
- 109. Chou KC, Zhang CT. Prediction of protein structural classes. Crit Rev Biochem Mol Biol 1995;30:275–349. [DOI] [PubMed] [Google Scholar]
- 110. Hajisharifi Z, Piryaiee M, Mohammad Beigi M, et al. Predicting anticancer peptides with Chou’s pseudo amino acid composition and investigating their mutagenicity via Ames test. J Theor Biol 2014;341:34–40. [DOI] [PubMed] [Google Scholar]
- 111. Ijaz A. SUMOhunt: combining spatial staging between lysine and SUMO with random forests to predict SUMOylation. ISRN Bioinform 2013;2013:671269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chen YZ, Chen Z, Gong YA, et al. SUMOhydro: a novel method for the prediction of sumoylation sites based on hydrophobic properties. PLoS One 2012;7:e39195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shi SP, Qiu JD, Sun XY, et al. PLMLA: prediction of lysine methylation and lysine acetylation by combining multiple features. Mol Biosyst 2012;8:1520–7. [DOI] [PubMed] [Google Scholar]
- 114. Qiu WR, Sun BQ, Xiao X, et al. iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics 2016;32:3116–23. [DOI] [PubMed] [Google Scholar]
- 115. Xu Y, Wang XB, Ding J, et al. Lysine acetylation sites prediction using an ensemble of support vector machine classifiers. J Theor Biol 2010;264:130–5. [DOI] [PubMed] [Google Scholar]
- 116. Ju Z, Sun J, Li Y, et al. Predicting lysine glycation sites using bi-profile bayes feature extraction. Comput Biol Chem 2017;71:98–103. [DOI] [PubMed] [Google Scholar]
- 117. Xu Y, Ding YX, Ding J, et al. iSuc-PseAAC: predicting lysine succinylation in proteins by incorporating peptide position-specific propensity. Sci Rep 2015;5:10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Xu Y, Ding YX, Ding J, et al. Mal-Lys: prediction of lysine malonylation sites in proteins integrated sequence-based features with mRMR feature selection. Sci Rep 2016;6:38318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Shi SP, Qiu JD, Sun XY, et al. PMeS: prediction of methylation sites based on enhanced feature encoding scheme. PLoS One 2012;7:e38772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lee TY, Chang CW, Lu CT, et al. Identification and characterization of lysine-methylated sites on histones and non-histone proteins. Comput Biol Chem 2014;50:11–8. [DOI] [PubMed] [Google Scholar]
- 121. Wen PP, Shi SP, Xu HD, et al. Accurate in silico prediction of species-specific methylation sites based on information gain feature optimization. Bioinformatics 2016;32:3107–15. [DOI] [PubMed] [Google Scholar]
- 122. Beauclair G, Bridier-Nahmias A, Zagury JF, et al. JASSA: a comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics 2015;31:3483–91. [DOI] [PubMed] [Google Scholar]
- 123. Radivojac P, Vacic V, Haynes C, et al. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 2010;78:365–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Krizhevsky A, Sutskever I, Hinton GE. ImageNet classification with deep convolutional neural networks. In: Proceedings of the 25th International Conference on Neural Information Processing Systems. Lake Tahoe, NV, USA: Curran Associates Inc.2012, 1097–105. [Google Scholar]
- 125. Graves A, Mohamed A-r, Hinton G. Speech recognition with deep recurrent neural networks. ArXiv: 1303.5778. 2013. [Google Scholar]
- 126. Maas AL, Daly RE, Pham PT, et al. Learning word vectors for sentiment analysis. In: Proceedings of the 49th Annual Meeting of the Association for Computational Linguistics: Human Language Technologies—Volume 1. Portland, Oregon: Association for Computational Linguistics; 2011, 142–50. [Google Scholar]
- 127. Heffernan R, Yang Y, Paliwal K, et al. Capturing nonlocal interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers and solvent accessibility. Bioinformatics 2017;33:2842–9. [DOI] [PubMed] [Google Scholar]
- 128. Hanson J, Yang Y, Paliwal K, et al. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics 2017;33:685–92. [DOI] [PubMed] [Google Scholar]
- 129. Leung MK, Xiong HY, Lee LJ, et al. Deep learning of the tissue-regulated splicing code. Bioinformatics 2014;30:i121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zhang S, Zhou J, Hu H, et al. A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res 2016;44:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wang D, Zeng S, Xu C, et al. MusiteDeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017;33:3909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. He F, Bao L, Wang R, et al. A multimodal deep architecture for large-scale protein ubiquitylation site prediction. In: 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2017, p. 108–13.
- 133. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521:436–44. [DOI] [PubMed] [Google Scholar]
- 134. Gal Y, Ghahramani Z. A theoretically grounded application of dropout in recurrent neural networks. In: Proceedings of the 30th International Conference on Neural Information Processing Systems. Barcelona Spain: Curran Associates Inc.2016, 1027–35. [Google Scholar]
- 135. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006;22:1658–9. [DOI] [PubMed] [Google Scholar]
- 136. O’Shea JP, Chou MF, Quader SA, et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nat Methods 2013;10:1211–2. [DOI] [PubMed] [Google Scholar]
- 137. Rodriguez MS, Dargemont C, Hay RT. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J Biol Chem 2001;276:12654–9. [DOI] [PubMed] [Google Scholar]
- 138. Sampson DA, Wang M, Matunis MJ. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J Biol Chem 2001;276:21664–9. [DOI] [PubMed] [Google Scholar]
- 139. Song J, Burrage K, Yuan Z, et al. Prediction of cis/trans isomerization in proteins using PSI-BLAST profiles and secondary structure information. BMC Bioinformatics 2006;7:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Song J, Tan H, Shen H, et al. Cascleave: towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics 2010;26:752–60. [DOI] [PubMed] [Google Scholar]
- 141. Chen Z, Zhou Y, Zhang Z, et al. Towards more accurate prediction of ubiquitination sites: a comprehensive review of current methods, tools and features. Brief Bioinform 2015;16:640–57. [DOI] [PubMed] [Google Scholar]
- 142. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chen Z, Wang Y, Zhai YF, et al. ZincExplorer: an accurate hybrid method to improve the prediction of zinc-binding sites from protein sequences. Mol Biosyst 2013;9:2213–22. [DOI] [PubMed] [Google Scholar]
- 144. An Y, Wang J, Li C, et al. Comprehensive assessment and performance improvement of effector protein predictors for bacterial secretion systems III, IV and VI. Brief Bioinform 2018;19:148–61. [DOI] [PubMed] [Google Scholar]
- 145. Linding R, Jensen LJ, Ostheimer GJ, et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007;129:1415–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hendriks IA, Lyon D, Su D, et al. Site-specific characterization of endogenous SUMOylation across species and organs. Nat Commun 2018;9:2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Gnad F, Ren S, Choudhary C, et al. Predicting post-translational lysine acetylation using support vector machines. Bioinformatics 2010;26:1666–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Shao J, Xu D, Hu L, et al. Systematic analysis of human lysine acetylation proteins and accurate prediction of human lysine acetylation through bi-relative adapted binomial score Bayes feature representation. Mol Biosyst 2012;8:2964–73. [DOI] [PubMed] [Google Scholar]
- 149. Xu Y, Li L, Ding J, et al. Gly-PseAAC: identifying protein lysine glycation through sequences. Gene 2017;602:1–7. [DOI] [PubMed] [Google Scholar]
- 150. Chen H, Xue Y, Huang N, et al. MeMo: a web tool for prediction of protein methylation modifications. Nucleic Acids Res 2006;34:W249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Shao J, Xu D, Tsai SN, et al. Computational identification of protein methylation sites through bi-profile Bayes feature extraction. PLoS One 2009;4:e4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Shien DM, Lee TY, Chang WC, et al. Incorporating structural characteristics for identification of protein methylation sites. J Comput Chem 2009;30:1532–43. [DOI] [PubMed] [Google Scholar]
- 153. Qiu WR, Xiao X, Lin WZ, et al. iMethyl-PseAAC: identification of protein methylation sites via a pseudo amino acid composition approach. Biomed Res Int 2014;2014:947416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Teng S, Luo H, Wang L. Predicting protein sumoylation sites from sequence features. Amino Acids 2012;43:447–55. [DOI] [PubMed] [Google Scholar]
- 155. Xu J, He Y, Qiang B, et al. A novel method for high accuracy sumoylation site prediction from protein sequences. BMC Bioinformatics 2008;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Lee TY, Chen SA, Hung HY, et al. Incorporating distant sequence features and radial basis function networks to identify ubiquitin conjugation sites. PLoS One 2011;6:e17331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.