

Abstract
Background
Clostridioides difficile infection (CDI) might be complicated by the development of nosocomial bloodstream infection (n-BSI). Based on the hypothesis that alteration of the normal gut integrity is present during CDI, we evaluated markers of microbial translocation, inflammation, and intestinal damage in patients with CDI.
Methods
Patients with documented CDI were enrolled in the study. For each subject, plasma samples were collected at T0 and T1 (before and after CDI therapy, respectively), and the following markers were evaluated: lipopolysaccharide-binding protein (LPB), EndoCab IgM, interleukin-6, intestinal fatty acid binding protein (I-FABP). Samples from nonhospitalized healthy controls were also included. The study population was divided into BSI+/BSI- and fecal microbiota transplantation (FMT) +/FMT- groups, according to the development of n-BSI and the receipt of FMT, respectively.
Results
Overall, 45 subjects were included; 8 (17.7%) developed primary n-BSI. Markers of microbial translocation and intestinal damage significantly decreased between T0 and T1, however, without reaching values similar to controls (P < .0001). Compared with BSI-, a persistent high level of microbial translocation in the BSI+ group was observed. In the FMT+ group, markers of microbial translocation and inflammation at T1 tended to reach control values.
Conclusions
CDI is associated with high levels of microbial translocation, inflammation, and intestinal damage, which are still present at clinical resolution of CDI. The role of residual mucosal perturbation and persistence of intestinal cell damage in the development of n-BSI following CDI, as well as the possible effect of FMT in the restoration of mucosal integrity, should be further investigated.
Keywords: Clostridioides difficile infection, microbial translocation, nosocomial bloodstream infection, LPS, I-FABP, fecal microbiota transplantation
Clostridioides difficile infection (CDI) is associated with high level of microbial translocation, intestinal damage and inflammation, which persist despite clinical resolution of CDI. The role of residual mucosal perturbation in the pathogenesis of bloodstream infections following CDI should be investigated.
Clostridioides difficile infection (CDI) represents the leading cause of nosocomial diarrhea worldwide and is associated with growing rates of morbidity and mortality [1]. Apart from recurrences, CDI is complicated by the development of nosocomial bloodstream infections (n-BSIs) [2–4], with Candida spp. [2, 5, 6] and enteric bacteria as the leading pathogens [7–13]. Nevertheless, whether CDI predisposes subjects to subsequent BSI is still controversial, and neither the clinical link between CDI and n-BSI nor the exact pathogenesis has been demonstrated yet [11, 12]. Given that the observed n-BSIs have mostly been caused by fungi and bacteria belonging to enteric organisms, it was hypothesized that alteration of the normal gut integrity and microbial translocation from the gut to the systemic circulation may play a role as pathogenetic triggers for the development of n-BSI [2].
Microbial translocation, defined as the migration of bacteria or their products from the gut to the extraintestinal space and eventually to the systemic circulation, might be promoted by increased intestinal permeability induced by disruption of intestinal epithelial barrier function, intestinal bacterial overgrowth, and changes in the composition of bacterial microbes in the gut, all conditions present during CDI [13, 14]. Hence, the presence of circulating levels of lipopolysaccharide (LPS), a component of gram-negative bacterial cell membrane, induces production of host response molecules such as lipopolysaccharide-binding protein (LBP) and consumption of neutralizing antibodies against LPS endotoxin core antigen (antiendotoxin core antibody [EndoCab]).
Furthermore, C. difficile toxins are responsible for damage to the intestinal epithelial cells, which represents an additional key pathway implicated in the increased gut permeability and microbial translocation [15–17]. Fatty acid binding protein-2 (FABP-2), also known as intestinal (I)-FABP, is present in mature enterocytes, and it is released as soon as cell membrane integrity is compromised and subsequently appears in the circulation only after enterocyte injury [18, 19].
During the course of CDI, a significant increase in the levels of interleukin (IL)-6 has been reported [20], leading to the hypothesis that this cytokine plays a pivotal role in intestinal inflammation and systemic inflammatory response in CDI-infected subjects [20].
Although biomarkers of microbial translocation, inflammation, and intestinal damage have been associated with multiple infective and noninfective diseases [13, 21], their role in CDI or in the pathogenesis of n-BSI after CDI is still unexplored.
Fecal microbiota transplantation (FMT) represents a highly effective strategy for the treatment of recurrent CDI [22, 23]. In addition, its possible role as an anti-infective therapeutic strategy has been recently hypothesized, likely through restoration of the intestinal microbiota barrier and modulation of systemic inflammation [24].
On the basis of these observations, this study was undertaken with the following objectives: (i) to evaluate the dynamic changes of circulating levels of LBP and EndoCab IgM (markers of microbial translocation), IL-6 (marker of inflammation), and I-FABP (marker of intestinal damage) in patients with CDI; (ii) to analyze whether these biomarkers are specifically modified in subjects who develop n-BSI within 60 days after the onset of CDI compared with those who do not develop n-BSI; and (iii) to investigate the effect of FMT on circulating levels of microbial translocation, inflammation, and intestinal damage biomarkers.
METHODS
Study Population
Over an 18-month period (January 2017–June 2018), patients with CDI hospitalized at the Azienda Ospedaliero-Universitaria Policlinico Umberto 1, Sapienza University (Rome), were enrolled in the study. Patients gave informed written consent, and the study protocol was approved by the local ethics committee. Exclusion criteria were age <18 years, CDI suspected but not confirmed, patients not willing to participate at the study. Subjects were further divided into those developing primary n-BSI (BSI+) and those not developing primary n-BSI (BSI-) during the 60-day follow-up period. For the comparative analysis, subjects with nonprimary BSI were included in the BSI- group. In addition, patients receiving FMT as a CDI-specific therapy (FMT+) were compared with those being treated with conventional therapies (FMT-). Demographic, clinical, and microbiology data from all patients were recorded in an electronic database. Nonhospitalized healthy subjects (defined as controls, n = 15) matched for age and sex were also included. Each subject of the control group was asked whether he/she suffered from diarrhea or other chronic intestinal issues or new-onset symptoms in the 30 days before the blood collection. In the presence of at least 1 of these conditions, subjects were excluded from the control group.
Definitions
CDI infection, severe CDI, recurrence, and failure were defined according to guidelines [23, 25]. Treatment of CDI was chosen by the Infectious Diseases specialists according to severity of disease, previous number of CDI episodes, and risk factors for recurrence and included oral vancomycin (125 mg every 6 hours for 10–14 days) ± intravenous metronidazole (500 mg every 6 hours for 10–14 days), fidaxomicin (200 mg every 12 hours for 10 days), FMT.
According to the institutional protocol, FMT was performed only in subjects with recurrent (>1 episode) CDI, who were willing to receive FMT as part of CDI treatment, without immunosuppression, and in the absence of technical contraindications to the procedure, as determined by the Gastroenterology consultant. Each subject who received FMT was treated with a high dosage of oral vancomycin (ie, 500 mg every 6 hours) for 4 days before the procedure, which was performed via retention enema. The reasons to exclude subjects from having FMT were an active BSI and being critically ill.
BSI was defined as a nosocomial infection (n-BSI) if it occurred >48 hours after admission to the hospital [26]. Primary BSI was defined as BSI occurring in patients without a recognized source of infection.
Markers of Microbial Translocation, Inflammation, and Intestinal Damage
For each subject, blood samples were collected at T0 (ie, before therapy or within 24 hours of the onset of CDI-specific therapy) and T1 (within 48 hours of the end of CDI-specific therapy), respectively.
In subjects who received FMT after a 4-day course of high-dose oral vancomycin, T0 and T1 were defined as immediately before and 10 days after the FMT procedure, respectively, in accordance with the institutional protocol on blood collection in patients undergoing FMT.
Markers of microbial translocation (lipopolysacharide-binding protein, Hycult Biotech; EndoCab IgM, Hycult Biotech), inflammation (IL-6, R&D Systems), and intestinal damage (intestinal fatty acid binding protein [I-FABP], R&D Systems) were evaluated using enzyme-linked immunosorbent assays in plasma. Briefly, after collection, blood was immediately centrifuged at 2000 rpm for 10 minutes and further frozen at –80°C until the assays were performed. LBP was expressed as ng/mL (sample diluted 1:2000), EndoCab IgM as MU/mL (sample diluted 1:100), IL-6 as pg/mL (sample undiluted), and I-FABP as pg/mL (sample diluted 1:5). Samples from healthy nonhospitalized subjects (controls, n = 15) matched for age and sex were also included and collected once.
Statistical Analysis
Results were expressed as mean (±SD) or median (interquartile range [IQR]) and as percentages for continuous and categorical variables, respectively. Categorical variables were compared by using the X2 or Fisher exact test, as appropriate, whereas continuous data were analyzed with the Student t test and the nonparametric Mann-Whitney test. Differences between T0 and T1 were evaluated by Wilcoxon test. Analysis of variance was used to compare the means of more than 2 groups. Statistical analyses were performed using GraphPad Prism, version 8, for Windows (Grafpad Software MacKiev).
RESULTS
Study Population
A total of 45 patients were included in the study. The mean age of the participants was 75 (±13.11) years, and one-third were females (n = 15) (Table 1). Two out of 45 (4.4%) were in the intensive care unit during admission, and 11/45 (24.4%) had central lines during hospitalization. The median time from the admission to CDI (IQR) was 12 (4–25) days. Specific therapy for CDI was oral vancomycin in 42/45 (93.3%), including those who received oral vancomycin before FMT (n = 6/45, 13.3%), intravenous metronidazole (always combined with oral vancomycin) in 8/45 (17.7%), and fidaxomicin in 6/45 (13.3%). In particular, fidaxomicin was given as a firstline therapy in 2 patients and after vancomycin failure in 4 subjects. As for the 60-day outcomes, clinical cure was observed in 38/45 (84.4%), failure in 4/45 (8.9%), and recurrence in 3/45 (6.7%). N-BSI was observed in 10/45 (22.2%) subjects: Candida albicans, n = 2; Gram-negatives, n = 4 (Klebsiella pneumoniae, n = 1; Escherichia coli, n = 1; Acinetobacter baumannii, n = 2); Enterococcus faecalis, n = 1; C. albicans, K. pneumoniae, E. faecalis, n = 1; methicillin-resistant Staphylococcus aureus (MRSA), n = 1; Staphylococcus hominis, n = 1. Among the n-BSI, 8 were considered primary BSIs, and 2 were attributable to secondary bacteremia and/or caused by microorganisms not normally present in the gut; thus, only 8/45 (17.7%) subjects were included in the BSI+ group, whereas subjects with nonprimary BSI were included in the non-BSI population (Supplementary Table 1). The median time from CDI and the development of primary n-BSI (IQR) was 20.5 (9.75–35.75) days (Supplementary Table 1). Among subjects with severe CDI (12/45, 26.6%), 2 (16.6%) developed primary n-BSI, whereas in nonsevere CDI (33/45, 73.4%) primary n-BSI was observed in 6 patients (18.2%).
Table 1.
Characteristics of the General Population
| Population | Global (n = 45) | BSI+ a (n = 8) | BSI- (n = 37) |
|---|---|---|---|
| Age, mean ± SD, y | 75 ± 13.11 | 70 ± 14.26 | 77 ± 9.75 |
| Sex (M/F) | 30/15 | 6/2 | 24/5 |
| Risk factors, No. (%) | |||
| Previous hospitalization | 40 (88.8) | 7 (87.5) | 33 (89.1) |
| Previous antibiotics | 37 (82.2) | 7 (87.5) | 30 (81) |
| PPI | 30 (66.6) | 7 (87.5) | 23 (62.1) |
| Beta-blockers | 14 (31.1) | 4 (50) | 10 (27) |
| Major surgery | 2 (4.4) | 1 (12.5) | 1 (2.7) |
| Immunodepression | 5 (11.1) | 2 (25) | 3 (8.1) |
| ICU during admission, No. (%) | 2 (4.4) | 1 (12.5) | 1 (2.7) |
| Central lines during hospitalization, No. (%) | 11 (24.4) | 2 (25) | 9 (24.3) |
| Time from the admission to CDI, median (IQR), d | 12 (4–25) | 16.5 (4.5–28) | 12 (1.5–23) |
| Clinical presentation, No. (%) | |||
| Diarrhea | 45 (100) | 8 (100) | 37 (100) |
| Fever | 13 (28.8) | 1 (12.5) | 12 (32.4) |
| Leukocytosis | 15 (33.3) | 2 (25) | 13 (35.1) |
| Severity of disease, No. (%) | |||
| Mild–moderate | 33 (73.3) | 6 (75) | 27 (72.9) |
| Severe | 12 (26.6) | 2 (25) | 10 (27.1) |
| Ribotype, No. (%) | |||
| 027 | 8 (17.7) | 1 (12.5) | 7 (18.9) |
| 078 | 3 (6.6) | 0 (0) | 3 (8.1) |
| Therapy, No. (%) | |||
| Vancomycin | 42 (93.3) | 7 (87.5) | 36 (97.2) |
| Metronidazole | 8 (17.7) | 0 (0) | 8 (21.6) |
| Fidaxomicin | 6 (13.3) | 1 (12.5) | 5 (13.5) |
| FMT | 6 (13.3) | 0 (0) | 6 (16.2) |
| Outcome, 60 d, No. (%) | |||
| Clinical cure | 38 (84.4) | 7 (87.5) | 31 (83.8) |
| Failure | 4 (8.9) | 0 (0) | 4 (10.8) |
| Relapse | 3 (6.7) | 1 (12.5) | 2 (5.4) |
Abbreviations: BSI, bloodstream infection; CDI, Clostridioides difficile infection; FMT, fecal microbiota transplantation; ICU, intensive care unit; IQR, interquartile range; PPI, proton pump inhibitor.
aOnly primary BSIs, defined as BSIs occurring in patients without a recognized source of infection, were considered for the comparison of microbial translocation, inflammation, and intestinal damage markers between subjects developing n-BSI and subjects not developing n-BSI.
In subjects developing primary n-BSI (n = 8), the median time between the beginning and end of CDI-specific therapy (IQR) was 14 (12.5–14) days; 1 subject (12.5%) received fidaxomicin after vancomycin treatment failure, and 1 (12.5%) subject received fidaxomicin as firstline therapy (Supplementary Table 1). In the BSI- group (n = 37), the median time between the beginning and end of therapy (IQR) was 13 (9–14) days; 3 subjects (6.6%) received fidaxomicin after vancomycin treatment failure, and 1 subject (2.7%) received fidaxomicin as firstline therapy. No patients receiving FMT developed a subsequent n-BSI.
A total of 15 controls were also included in the study. The mean age of the controls was 71 (±15.56) years, and 4/15 (26.6%) were females. None of the controls suffered from diarrhea or other chronic intestinal issues or new-onset symptoms in the 30 days before blood collection.
Markers of Microbial Translocation, Inflammation, and Intestinal Damage During the Course of CDI
Overall, a high degree of microbial translocation, inflammation, and intestinal damage was observed during the course of CDI, which lowered from the beginning to the end of therapy but did not reach values similar to controls.
In particular, the median LPB plasma levels at T0 were significantly higher than those of controls (29 960 vs 14 357 ng/mL; P ≤ .0001). At the end of therapy (T1), there was a significant reduction in LPB levels (21 160 ng/mL; P < .0002), but values were still not comparable to controls (Figure 1A).
Figure 1.

Markers of microbial translocation (A and B), inflammation (C), and intestinal damage (D) in patients (n = 45) during the course of Clostridioides difficile infection (CDI). Horizontal lines and whiskers represent median and the 25th and 75th percentile values, respectively. *P < .01; **P < .001. Abbreviations: EndoCab IgM, antiendotoxin core antibody IgM; I-FABP, intestinal fatty acid binding protein; LBP, lipopolysaccharide-binding protein; T0, at the beginning of CDI-specific therapy; T1, at the end of CDI-specific therapy.
When we assessed the degree of consumption of neutralizing antibodies against LPS endotoxin core antigen (EndoCab IgM) during the course of CDI, the median values at T0 were significantly lower than those of controls (30.31 vs 173.4 MU/mL; P < .0001). Although there was a significant increase at T1 (31.72 MU/mL; P = .0103), EndoCab IgM values remained lower than those observed in controls (Figure 1 B).
The median circulating levels of IL-6 at T0 were significantly higher than in controls (37.09 vs 1.82 pg/mL; P < .0001). Although a reduction of IL-6 levels was observed at the end of therapy (26.6 pg/mL; P = .16), values remained considerably higher than those of controls (Figure 1 C).
As for the degree of intestinal damage occurring during the course of CDI, the median values were similarly elevated at T0 and T1 (1171 and 1002 pg/mL), respectively, and were significantly higher than those of controls (P < .0001) (Figure 1 D).
Microbial Translocation, Inflammation, and Intestinal Damage in Patients Developing BSI During the Follow-up Period
Markers of Microbial Translocation (LBP and EndoCab IgM)
The extent of microbial translocation, expressed by high levels of LBP and low levels of EndoCab IgM, did not differ between the BSI+ and BSI- groups at the beginning and at the end of therapy. Interestingly, although microbial translocation remained constantly high between the beginning and end of therapy in subjects developing primary n-BSI (from 27 955 to 24 175 ng/mL and from 42.78 to 31.43 MU/mL for LBP and EndoCab IgM, respectively), it showed a statistically significant reduction in subjects not developing BSI (from 30 960 to 20 950 ng/mL; P = .0002; and from 28.86 to 34.46 MU/mL; P = .0006; for LBP and EndoCab IgM, respectively). Both groups at the end of therapy showed a significantly higher level of microbial translocation than controls (Figure 2A, B).
Figure 2.

Markers of microbial translocation (A and B), inflammation (C), and intestinal damage (D) in patients who developed BSI (n = 8) after Clostridioides difficile infection (CDI), in comparison with subjects not who did not develop BSI (n = 37). Horizontal lines and whiskers represent median and the 25th and 75th percentile values, respectively. *P < .01; **P < .001. Abbreviations: EndoCab IgM, antiendotoxin core antibody IgM; I-FABP, intestinal fatty acid binding protein; LBP, lipopolysaccharide-binding protein; T0, at the beginning of CDI-specific therapy; T1, at the end of CDI-specific therapy.
Marker of Inflammation (IL-6)
No differences were observed between the 2 groups as for median IL-6 circulating levels at the beginning and end of the therapy. Although both groups showed a decrease in IL-6 values from the beginning to the end of therapy (from 35.16 to 24.68 pg/mL in BSI+ and from 39.01 to 29.49 pg/mL in BSI-), this difference was not significant. However, at the end of therapy, both groups of subjects exhibited a higher grade of inflammation than controls (Figure 2 C).
Intestinal Damage (I-FABP)
The extent of intestinal damage during the course of CDI was constantly high and did not differ between the 2 groups. Interestingly, the intestinal damage tended to increase between the beginning and the end of therapy in subjects developing primary n-BSI (from 754.5 to 1002 pg/mL; P = .48), whereas in subjects not developing n-BSI a decrease in circulating I-FABP levels was observed (from 1171 to 1002 pg/mL; P = .53). At the end of therapy, the levels of I-FABP were significantly higher than those of controls in both groups (Figure 2 D).
Markers of Microbial Translocation, Inflammation, and Intestinal Damage in Patients Receiving FMT as a CDI-Specific Therapy
Microbial Translocation (LBP and EndoCab IgM)
Subjects receiving fecal microbiota transplantation had a lower level of microbial translocation than those treated with other therapies, both at the beginning and at the end of therapy. Notably, at the end of therapy subjects in the FMT+ group tended to have LBP circulating values similar to those observed in controls (P = .1), whereas subjects treated with other therapies maintained a significantly higher degree of LBP than controls (P < .0001). As for the levels of circulating EndoCab IgM, both groups had values significantly lower than controls (Figure 3A, B).
Figure 3.

Markers of microbial translocation (A and B), inflammation (C), and intestinal damage (D) in patients who received FMT (n = 6) as a Clostridioides difficile infection (CDI)–specific therapy, in comparison with subjects who did not receive FMT (n = 39). Horizontal lines and whiskers represent median and the 25th and 75th percentile values, respectively. *P < .01; **P < .001. Abbreviations: EndoCab IgM, antiendotoxin core antibody IgM; FMT, fecal microbiota transplantation; I-FABP, intestinal fatty acid binding protein; LBP, lipopolysaccharide-binding protein; T0, at the beginning of CDI-specific therapy; T1, at the end of CDI-specific therapy.
Inflammation (IL-6)
Median circulating IL-6 levels were significantly lower in subjects receiving fecal microbiota transplantation than those treated with other therapies at the beginning and at the end of therapy (8.32 vs 29.01 pg/mL and 5.21 vs 30.66 pg/mL, respectively), with a statistically significant difference especially at the end of treatment (P = .0005). Interestingly, after fecal microbiota transplantation, the degree of inflammation was similar to that observed in controls (5.21 vs 1.82 pg/mL) (Figure 3 C).
Intestinal Damage (I-FABP)
The extent of intestinal damage during the course of CDI did not differ between the 2 groups. At the end of therapy, the levels I-FABP were significantly higher than those of controls in both groups (Figure 3 D).
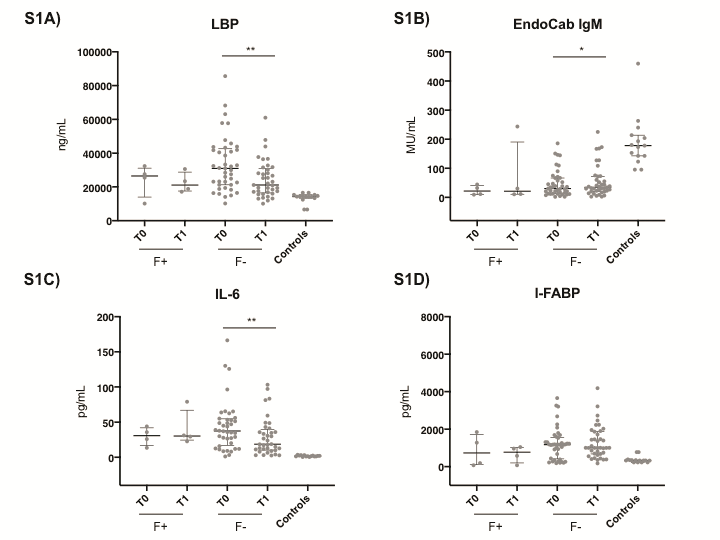
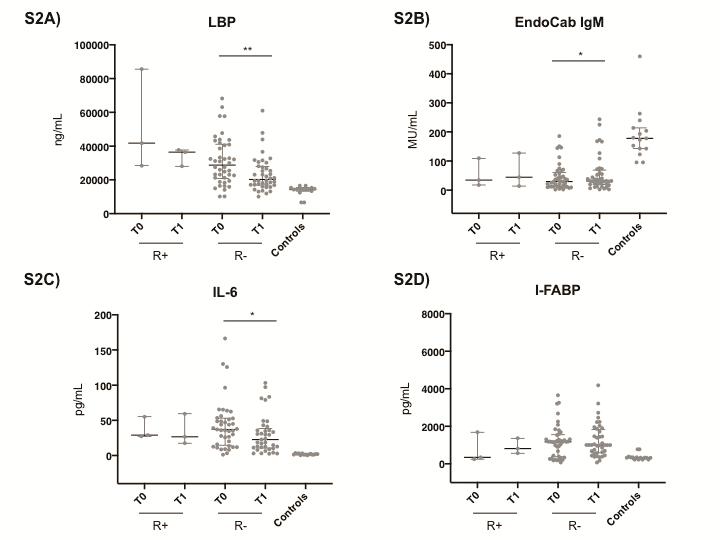
The distribution of markers of microbial translocation, inflammation, and intestinal damage in patients experiencing failure (n = 4) and CDI recurrence (n = 3) at the 60-day follow-up is shown in Supplementary Figures 1 and 2, respectively.
Correlations Among Markers of Microbial Translocation, Inflammation, and Intestinal Damage During the Course of CDI
The extent of microbial translocation (expressed as high LBP and low EndoCab IgM levels, respectively) correlated with the degree of systemic inflammation, especially at T0, whereas a higher level of IL-6 at the beginning of therapy negatively influenced the levels of neutralizing antibodies against LPS endotoxin core antigen at the end of therapy. Furthermore, the high levels of microbial translocation, inflammation, and intestinal damage at the onset of CDI positively correlated with the degree of these biomarkers at the end of CDI-specific therapy (Table 2).
Table 2.
Correlation Analyses of MT, IN, and ID Biomarkers in 45 Patients During the Course of CDI
| LBP T0 | EndoCab IgM T0 | IL-6 T0 | I-FABP T0 | LBP T1 | EndoCab IgM T1 | IL-6 T1 | I-FABP T1 | CRP | CRE | |
|---|---|---|---|---|---|---|---|---|---|---|
| LBP T0 | – | –0.23 0.11 | 0.52 0.0002 | 0.0128 0.9392 | 0.42 0.004 | –0.18 0.22 | 0.12 0.43 | 0.10 0.54 | 0.18 0.25 | –0.30 0.05 |
| EndoCab IgM T0 | – | – | –0.38 0.01 | 0.06 0.71 | –0.15 0.32 | 0.82 <0.0001 | –0.13 0.39 | 0.02 0.86 | –0.04 0.77 | 0.17 0.28 |
| IL-6 T0 | – | – | – | –0.05 0.75 | 0.13 0.37 | –0.40 0.006 | 0.28 0.06 | 0.04 0.8 | –0.02 0.9 | |
| I-FABP T0 | – | – | – | – | 0.12 0.45 | –0.17 0.29 | –0.04 0.77 | 0.71 <0.0001 | 0.04 0.8 | –0.30 0.09 |
| LBP T1 | – | – | – | – | – | 0.07 0.63 | 0.60 <0.0001 | –0.06 0.70 | 0.02 0.89 | 0.03 0.85 |
| EndoCab IgM T1 | – | – | – | – | – | – | –0.19 0.20 | –0.21 0.21 | –0.08 0.6 | 0.15 0.35 |
| IL-6 T1 | – | – | – | – | – | – | – | –0.08 0.61 | 0.03 0.84 | –0.10 0.54 |
| I-FABP T1 | – | – | – | – | – | – | – | – | 0.10 0.58 | –0.31 0.09 |
The first lines are Spearman correlation coefficient values; the second lines are P values. Values in bold represent statistically significant correlations (P < .05).
Abbreviations: CDI, Clostridioides difficile infection; CRE, creatinine; CRP, C-reactive protein; EndoCab IgM, antiendotoxin core antibody IgM; I-FABP, intestinal fatty acid binding protein; ID, intestinal damage; IN, inflammation; LBP, lipopolysaccharide-binding protein; MT, microbial translocation; T0, at the beginning of CDI-specific therapy; T1, at the end of CDI-specific therapy.
Discussion
CDI is the most common cause of nosocomial infectious diarrhea in hospitalized patients, with up to 35% of patients having recurrent infections and a non-negligible percentage of subjects developing n-BSI [1]. However, even though there is increasing evidence that CDI might be complicated by n-BSI, with Candida spp. and enteric bacteria as the leading pathogens [2, 3, 5], the possible link between these infections is still a matter of debate [11, 12]. Indeed, from a clinical point of view, being aware of the possibility of this coinfection, physicians might be more vigilant for early recognition and treatment of n-BSI, especially during the first 4 weeks after CDI diagnosis, as this complication is associated with high mortality rates [2]. On the other hand, from a pathogenetic standpoint, it appears essential to demonstrate whether there is a condition predisposing to the passage of bacteria or fungi from the gut into the systemic circulation, with the hypothesized mechanism residing mostly on the possible alteration of mucosal integrity occurring during CDI.
Microbial translocation, which is defined as the passage of indigenous bacteria from the gastrointestinal tract through the lamina propria to the mesenteric lymph nodes and other organs, occurs as a consequence of the perturbation of the normal mucosal integrity, and the extent of microbial translocation rate differs between microorganisms, with gram-negative enteric bacilli translocating more than gram-positive and obligately anaerobic bacteria, respectively [14, 27–29].
Based on these considerations, the present study was aimed at examining whether microbial translocation, inflammation, and intestinal damage biomarkers are altered in subjects with CDI and, accordingly, revealing whether there is an increased susceptibility of patients to subsequent n-BSI. Indeed, here we have demonstrated for the first time that CDI is characterized by high levels of microbial translocation, inflammation, and intestinal damage and that the residual high mucosal perturbation and intestinal cell damage are still present at clinical resolution of the infection.
In fact, at the beginning of CDI (ie, before or within 24 hours of the onset of CDI-specific therapy), high levels of microbial translocation, inflammation, and intestinal damage were observed, which lowered at the end of treatment. However, these biomarkers, although significantly reduced from the beginning to the end of therapy, did not reach values comparable to those of controls, thus demonstrating that mucosal perturbation and intestinal damage persist despite the clinical resolution of CDI.
Apart from subjects with CDI, in critically ill patients the development of a systemic inflammatory response related to surgery, ischemia-riperfusion syndrome after major (ie, cardiothoracic) surgeries, and the use of extracorporeal circulation, all conditions related to a lower rate of intestinal perfusion, have been associated with a certain extent of microbial translocation, suggesting that microbial translocation does not occur infrequently and might be involved in the pathogenesis of infective and noninfective conditions [30–32].
When considering the microbial translocation, inflammation, and intestinal damage levels in subjects developing n-BSI in the 60 days after CDI, we noted that patients in the BSI+ group maintained a high degree of mucosal alteration and microbial translocation, which appeared to persist to a greater extent than those in the BSI- group.
Our findings are in line with recent murine models suggesting that the loss of barrier function during CDI predisposes to bacterial translocation and systemic dissemination [33–36], whereas noninfective gut disorders such as inflammatory bowel disease and antibiotic-induced diarrhea have been associated with anaerobic and Klebsiella oxytoca bacteremia [37, 38], respectively, indicating microbial translocation and intestinal damage as the common causal mechanisms [39].
With regard to the intestinal damage, an increase of I-FABP from the beginning to the end of therapy in the BSI+ group has been demonstrated, although it was not statistically significant [16, 40]; if confirmed in future better-powered cohorts, this might indicate that the CD-induced cell damage still present even at the end of therapy might favor the passage of microorganisms through an impaired intestinal barrier. In contrast, the observed high level of intestinal damage even in the BSI- group is possibly explained by the fact that oral vancomycin, which possesses an intrinsic toxicity toward enterocytes, has been given in almost all patients with CDI [41–43].
CDI toxins activate immune cells to produce pro-inflammatory cytokines such as TNF-alpha and IL-6, leading to a robust inflammatory response, which represents the hallmark of CDI pathogenesis [15]. In our cohort, both the BSI+ and BSI- groups showed higher levels of IL-6 than controls at the end of therapy, suggesting that the degree of inflammation might not be involved in the development of n-BSI. In contrast, the high level of inflammation at the beginning of CDI-specific therapy might influence the recovery rate of neutralizing antibodies against LPS endotoxin core antigen at the clinical resolution of CDI, as highlighted by the correlation analyses (Table 2).
It should be acknowledged that our study cohort is too small to reach a definite conclusion on the attractive hypothesis that persistent gut permeability and intestinal damage might increase the risk of BSI. Therefore, the role of residual mucosal perturbation and the persistence of intestinal cell damage in the development of n-BSI following CDI should be further investigated in the future.
An additional hallmark of the present study resides in the evaluation of the degree of microbial translocation, inflammation, and intestinal damage biomarkers in patients receiving FMT as a targeted therapy for CDI. Although only 6 patients have been treated with FMT, we have shown that FMT+ subjects tended to have levels of LBP and circulating neutralizing antibodies against LPS endotoxin core antigen similar to those observed in controls. Similarly, circulating IL-6 levels were significantly lower in FMT+ than in FMT- subjects, and that effect was more evident at the end of treatment. In contrast, the degree of intestinal damage did not differ between the 2 groups and remained persistently high. The low levels of microbial translocation and inflammation observed immediately before FMT in subjects belonging to the FMT group might have been influenced by the effect of oral vancomycin therapy, which was administered for 4 days before the procedure and, accordingly, might not reflect the real values of these markers at the beginning of CDI. On the other hand, the results at the end of therapy (10 days after the FMT) might reflect the effect of FMT on these biomarkers. In fact, for unlikely subjects not receiving FMT but receiving oral therapies (ie, vancomycin or fidaxomicin), values of microbial translocation and inflammation 10 days after FMT tended to be more similar to those obtained in controls. This was especially true for microbial translocation and inflammation markers, whereas I-FABP did not differ between the FMT+ and FMT- groups and did not tend to reach values similar to controls. The last result might be easily explained with the described toxicity of oral vancomycin on intestinal cells. Taken together, these findings show that, compared with traditional oral therapies for CDI, the administration of FMT might be able to restore the degree of microbial translocation and inflammation in subjects with CDI.
Overall, our findings highlight the deep and mutual relationship that exists between the extent of inflammation, intestinal damage, and gut mucosal perturbation during the course of CDI. Whether the residual degree of microbial translocation and CD-induced intestinal damage possess a pathogenetic role in the development of n-BSI following CDI or whether FMT has a protective effect on n-BSI has yet to be demonstrated.
As for clinical implications, the results of the present study highlight the concept that, despite clinical resolution of CDI, there is still a non-negligible degree of alteration of gut mucosal integrity, possibly leading to a subsequent passage of microorganisms into the systemic circulation. To this end, markers of microbial translocation, inflammation, and intestinal damage might represent useful tools for the monitoring of mucosal integrity in patients with CDI. Furthermore, clinicians should consider FMT a reliable option for the treatment of CDI, not only due its well-known efficacy in lowering the rate of recurrences [22], but also because the administration of FMT might be able to restore the degree of mucosal permeability and inflammation.
The present investigation has several limitations, such as (i) low number of patients; (ii) evaluation of markers of microbial translocation, inflammation, and intestinal damage before and immediately after CDI-specific therapy, with no information regarding the time to obtaining mucosal integrity restoration after therapy; and (iii) the cohort of subjects with primary n-BSI was not sufficiently powered to demonstrate that the development of n-BSI correlates to any of the observed biomarkers.
Conclusions
In conclusion, it is herein demonstrated for the first time that CDI is associated with an elevation in markers of microbial translocation, inflammation, and epithelial damage, which persists despite the clinical resolution of CDI. Further investigations are needed to test the hypothesis that the development of n-BSI is associated with gut mucosal permeability and intestinal damage following CDI.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
Acknowledgments
The authors thank Silvia Costantini for sample processing and the nursing staff for their contribution to plasma sample collection.
Financial support. This research did not receive any specific grant or funding.
Potential conflicts of interest. All authors: no reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Data availability statement. The data used to support the findings of this study are available from the corresponding author upon request.
References
- 1. Lessa FC, Winston LG, McDonald LC; Emerging Infections Program C. difficile Surveillance Team. Burden of Clostridium difficile infection in the United States. N Engl J Med 2015; 372:825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Russo A, Falcone M, Fantoni M, et al. Risk factors and clinical outcomes of candidaemia in patients treated for Clostridium difficile infection. Clin Microbiol Infect 2015; 21:493.e1–4. [DOI] [PubMed] [Google Scholar]
- 3. Falcone M, Russo A, Iraci F, et al. Risk factors and outcomes for bloodstream infections secondary to Clostridium difficile infection. Antimicrob Agents Chemother 2016; 60:252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guastalegname M, Russo A, Falcone M, et al. Candidemia subsequent to severe infection due to Clostridium difficile: is there a link? Clin Infect Dis 2013; 57:772–4. [DOI] [PubMed] [Google Scholar]
- 5. Tsay S, Williams SR, Benedict K, et al. A tale of two healthcare-associated infections: Clostridium difficile coinfection among patients with candidemia. Clin Infect Dis 2019; 68:676–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vallabhaneni S, Almendares O, Farley MM, et al. Epidemiology and factors associated with candidaemia following Clostridium difficile infection in adults within metropolitan Atlanta, 2009-2013. Epidemiol Infect 2016; 144:1440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giuliano S, Guastalegname M, Jenco M, et al. Severe community onset healthcare-associated Clostridium difficile infection complicated by carbapenemase producing Klebsiella pneumoniae bloodstream infection. BMC Infect Dis 2014; 14:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amit S, Mishali H, Kotlovsky T, et al. Bloodstream infections among carriers of carbapenem-resistant Klebsiella pneumoniae: etiology, incidence and predictors. Clin Microbiol Infect 2015; 21:30–4. [DOI] [PubMed] [Google Scholar]
- 9. Taur Y, Xavier JB, Lipuma L, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis 2012; 55:905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roghmann MC, McCarter RJ Jr, Brewrink J, et al. Clostridium difficile infection is a risk factor for bacteremia due to vancomycin-resistant enterococci (VRE) in VRE-colonized patients with acute leukemia. Clin Infect Dis 1997; 25:1056–9. [DOI] [PubMed] [Google Scholar]
- 11. Thomas JA, Newman KC, Doshi S, et al. Bacteraemia from an unrecognized source (occult bacteraemia) occurring during Clostridium difficile infection. Scand J Infect Dis 2011; 43:269–74. [DOI] [PubMed] [Google Scholar]
- 12. Ulrich RJ, Santhosh K, Mogle JA, et al. Is Clostridium difficile infection a risk factor for subsequent bloodstream infection? Anaerobe 2017; 48:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol 2012; 30:149–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mainous MR, Tso P, Berg RD, Deitch EA. Studies of the route, magnitude, and time course of bacterial translocation in a model of systemic inflammation. Arch Surg 1991; 126:33–7. [DOI] [PubMed] [Google Scholar]
- 15. Czepiel J, Biesiada G, Brzozowski T, et al. The role of local and systemic cytokines in patients infected with Clostridium difficile. J Physiol Pharmacol 2014; 65:695–703. [PubMed] [Google Scholar]
- 16. Wang Y, Wang S, Kelly CP, et al. TPL2 is a key regulator of intestinal inflammation in Clostridium difficile infection. Infect Immun 2018; 86:e00095-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aktories K, Barbieri JT. Bacterial cytotoxins: targeting eukaryotic switches. Nat Rev Microbiol 2005; 3:397–410. [DOI] [PubMed] [Google Scholar]
- 18. March DS, Marchbank T, Playford RJ, et al. Intestinal fatty acid-binding protein and gut permeability responses to exercise. Eur J Appl Physiol 2017; 117:931–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gajda AM, Storch J. Enterocyte fatty acid-binding proteins (FABPs): different functions of liver and intestinal FABPs in the intestine. Prostaglandins Leukot Essent Fatty Acids 2015; 93:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McGeough MD, Pena CA, Mueller JL, et al. Cutting edge: IL-6 is a marker of inflammation with no direct role in inflammasome-mediated mouse models. J Immunol 2012; 189:2707–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perkins MR, Bartha I, Timmer JK, et al. ; Swiss HIV Cohort Study The interplay between host genetic variation, viral replication, and microbial translocation in untreated HIV-infected individuals. J Infect Dis 2015; 212:578–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vindigni SM, Surawicz CM. Fecal microbiota transplantation. Gastroenterol Clin North Am 2017; 46:171–85. [DOI] [PubMed] [Google Scholar]
- 23. Crobach MJT, Planche T, Eckert C. et al. European Society of Clinical Microbiology and Infectious Diseases: update of the diagnostic guidance document for Clostridium difficile infection. Clin Microbiol Infect 2016; 22(Suppl 4):S63–81. [DOI] [PubMed] [Google Scholar]
- 24. Wei Y, Yang J, Wang J, et al. Successful treatment with fecal microbiota transplantation in patients with multiple organ dysfunction syndrome and diarrhea following severe sepsis. Crit Care 2016; 20:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cohen SH, Gerding DN, Johnson S, et al. ; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 2010; 31:431–55. [DOI] [PubMed] [Google Scholar]
- 26. Horan TC, Andrus M, Dudeck MA. CDC/NHSN surveillance definition of health care-associated infection and criteria for specific types of infections in the acute care setting. Am J Infect Control 2008; 36:309–32. [DOI] [PubMed] [Google Scholar]
- 27. Perez F, Pultz MJ, Endimiani A, et al. Effect of antibiotic treatment on establishment and elimination of intestinal colonization by KPC-producing Klebsiella pneumoniae in mice. Antimicrob Agents Chemother 2011; 55:2585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steffen EK, Berg RD, Deitch EA. Comparison of translocation rates of various indigenous bacteria from the gastrointestinal tract to the mesenteric lymph node. J Infect Dis 1988; 157:1032–8. [DOI] [PubMed] [Google Scholar]
- 29. Eaves-Pyles T, Alexander JW. Comparison of translocation of different types of microorganisms from the intestinal tract of burned mice. Shock 2001; 16:148–52. [DOI] [PubMed] [Google Scholar]
- 30. Adrie C, Parlato M, Salmi L, et al. Bacterial translocation and plasma cytokines during transcatheter and open-heart aortic valve implantation. Shock 2015; 43:62–7. [DOI] [PubMed] [Google Scholar]
- 31. Al-Fares A, Pettenuzzo T, Del Sorbo L. Extracorporeal life support and systemic inflammation. Intensive Care Med Exp 2019; 7(Suppl 1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maselli R, Oliva A, Badalamenti M, et al. Risk of microbial translocation in per-oral endoscopic miotomy (POEM) for achalasia: antibiotic prophylaxis or short therapy? An interim analysis of a prospective randomized clinical trial. Endoscopy 2019; 51:S82. [Google Scholar]
- 33. Hasegawa M, Yamazaki T, Kamada N, et al. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol 2011; 186:4872–80. [DOI] [PubMed] [Google Scholar]
- 34. Johansson ME, Gustafsson JK, Holmén-Larsson J, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014; 63:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hasegawa M, Yada S, Liu MZ, et al. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity 2014; 41:620–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hasegawa M, Kamada N, Jiao Y, et al. Protective role of commensals against Clostridium difficile infection via an IL-1β-mediated positive-feedback loop. J Immunol 2012; 189:3085–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cox ER, Nayak SU, Kuruppu JC. Klebsiella oxytoca bacteremia-causal relationship to symptomatic colitis? Int J Infect Dis 2013; 17:e472–3. [DOI] [PubMed] [Google Scholar]
- 38. Ngo JT, Parkins MD, Gregson DB, et al. Population-based assessment of the incidence, risk factors, and outcomes of anaerobic bloodstream infections. Infection 2013; 41:41–8. [DOI] [PubMed] [Google Scholar]
- 39. Santos-Antunes J, Magro F, Macedo G. Listeria monocytogenes bacteremia and CMV colitis in a patient with ulcerative colitis. J Crohns Colitis 2014; 8:254–5. [DOI] [PubMed] [Google Scholar]
- 40. Sun X, Hirota SA. The roles of host and pathogen factors and the innate immune response in the pathogenesis of Clostridium difficile infection. Mol Immunol 2015; 63:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Warren CA, van Opstal EJ, Riggins MS, et al. Vancomycin treatment’s association with delayed intestinal tissue injury, clostridial overgrowth, and recurrence of Clostridium difficile infection in mice. Antimicrob Agents Chemother 2013; 57:689–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van Opstal E, Kolling GL, Moore JH 2nd, et al. Vancomycin treatment alters humoral immunity and intestinal microbiota in an aged mouse model of Clostridium difficile infection. J Infect Dis 2016; 214:130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lewis BB, Buffie CG, Carter RA, et al. Loss of microbiota-mediated colonization resistance to Clostridium difficile infection with oral vancomycin compared with metronidazole. J Infect Dis 2015; 212:1656–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.