

Abstract
Summary
Performing highly parallelized preprocessing of methylation array data using Python can accelerate data preparation for downstream methylation analyses, including large scale production-ready machine learning pipelines. We present a highly reproducible, scalable pipeline (PyMethylProcess) that can be quickly set-up and deployed through Docker and PIP.
Availability and implementation
Project Home Page: https://github.com/Christensen-Lab-Dartmouth/PyMethylProcess. Available on PyPI (pymethylprocess), Docker (joshualevy44/pymethylprocess).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Implementation
Studies that measure DNA methylation in large numbers of human biospecimens often use the Illumina Infinium BeadArray platforms known as HumanMethylation27 BeadArray (27K), HumanMethylation450 BeadArray (450K) and HumanMethylationEPIC BeadArray (850K/EPIC) arrays (Bibikova et al., 2009; Moran et al., 2016; Sandoval et al., 2011). However, a straightforward and tractable approach to perform data quality control and normalization in bulk to prepare the data for use in the object-oriented environment is lacking. Here, we introduce a convenient command line interface that makes methylation analyses more object oriented for use in downstream analyses. In addition to traditional differential methylation analyses, machine learning libraries such as scikit-learn, keras and tensorflow (Abadi et al., 2016; Pedregosa et al., 2011) become more accessible in the object oriented environment.
PyMethylProcess is a pip-installable command line interface built using Python 3.6 that interfaces with minfi, ENmix and meffil in R (Aryee et al., 2014; Min et al., 2018; Xu et al., 2016) via rpy2 (Gautier, 2010) to allow users to preprocess and set-up their DNA methylation array data for machine learning, presenting unique methylation datatypes built for the use of python classification, clustering, dimensionality reduction and regression algorithms such as UMAP, random forest, neural networks, k-nearest neighbors and HDBSCAN (Campello et al., 2013; McInnes et al., 2018). Eight Python classes have been introduced to handle the following tasks: package installation (PackageInstaller installs R/bioconductor packages), data acquisition from TCGA and GEO (TCGADownloader) and formatting (PreProcessPhenoData), parallelized quality control (QC), principal component selection via kneedle (Satopaa et al., 2011) and raw, quantile, noob and functional normalization using minfi, meffil, ENmix (PreprocessIDAT), imputation (ImputerObject), feature selection and storage for machine learning applications (MethylationArray[s] store beta and phenotype data), and a basic machine learning class MachineLearning that trains any scikit-learn-like model on MethylationArray objects. These datatypes are abstracted away via a convenient command-line interface. Additional commands are available, such as the removal of non-autosomal and SNP sites (by subsetting CpGs that are not in a list of CpGs supplied by meffil for the respective array platform), and reference-based cell-type estimation (constrained projection/quadratic programming) (Houseman et al., 2012; Jaffe and Irizarry, 2014; Salas et al., 2018). Class methods are available in the help documentation and a wiki details set-up/usage. A visualization module generates interactive 3-D data representations using UMAP and Plotly. We have provided a visual description of the preprocessing workflow (Fig. 1).
Fig. 1.
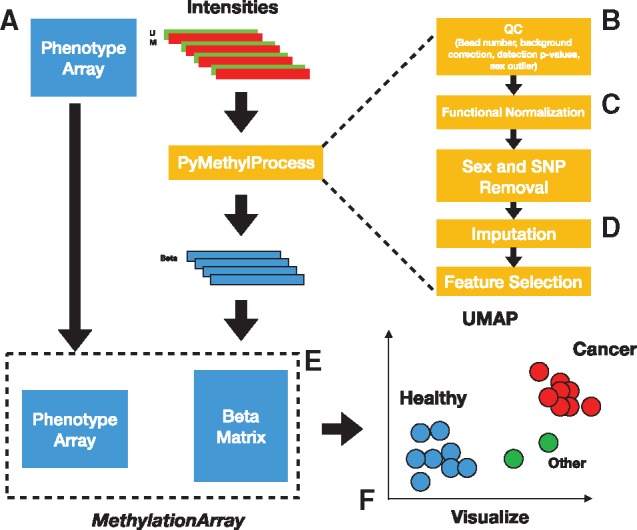
Flow diagram for PyMethylProcess: (A) data from GEO/TCGA, (B, C) QC/normalization, (D, E) storing betas and phenotype data in MethylationArray and (F) interactive visualizations
The beta values and phenotype data can easily be exported to csv format, and the command line interface can reduce the set-up time of standardized methylation data for downstream analyses. The pipeline differs from other python frameworks such as pyMAP (Mahpour, 2016) and GLINT (Rahmani et al., 2017). pyMAP only operates on the 450K framework, relying on a user specific csv annotation file and preprocessed Genome Studio txt file as its input. pyMAP only performs graphical exploration for candidate CpGs, CpG Island feature subsetting, and export to a BED file for downstream analyses. Similarly, GLINT requires a txt phenotype file and either a preprocessed beta values txt file or a R data.frame methylation object (in a RData file) as its inputs. GLINT stores beta values and covariate information as a binary ‘glint’ file. GLINT was designed for epigenome-wide association studies analysis, including reference-based and reference-free estimations, imputed genetic structure and statistical models (linear, logistic and linear mixed effects models). However, it relies on preprocessed data, with some limited quality control options and therefore it could benefit from preprocessed data generated in our pipeline. In addition, GLINT is not designed to export this information to perform user customized downstream machine learning analyses.
2 Results
Some of PyMethylProcess’s preprocessing capabilities are demonstrated on seven datasets (Supplementary Table S1; Capper et al., 2018; Johansson et al., 2013; Li Yim et al., 2016; Pai et al., 2019; Pidsley et al., 2013, 2013; Salas et al., 2017; Soriano-Tárraga et al., 2018) from the 450K and 850K arrays. The preprocessing performance was evaluated for loading, QC and normalization time. After preprocessing, each of these datasets were split into 70% training, 10% validation and 20% test sets.
3 Benefits and future direction
PyMethylProcess streamlines DNA methylation array preprocessing, preserving data accessibility and standardization for the open source Python machine learning community. Additional development based on community needs is welcome through GitHub issues and pull requests. Future development will expand functionality to preprocessing pipelines such as BigMelon (Gorrie-Stone et al., 2019) and feature importance evaluations (e.g. Gini index). This tool is available via Docker (joshualevy44/pymethylprocess) (Boettiger, 2015) and is wrapped using Common Workflow Language (CWL) (Supplementary Figure S1; Amstutz et al., 2016), making the analysis reproducible, operating system agnostic, standardized and sharable.
PyMethylProcess is available on PyPI (pymethylprocess) and GitHub at:https://github.com/Christensen-Lab-Dartmouth/PyMethylProcess.
Funding
This work was supported by NIH grants R01CA216265, R01DE022772 and P20GM104416 to BCC, a Dartmouth College Neukom Institute for Computational Science CompX award to BCC, and training fellowship support for AJT from T32LM012204.
Conflict of Interest: none declared.
Supplementary Material
References
- Abadi M. et al. (2016) Tensorflow: a system for large-scale machine learning. In: OSDI'16 Proceedings of the 12th USENIX Conference on Operating Systems Design and Implementation Savannah, GA, USA, pp. 265–283.
- Amstutz P. et al. (2016) Common Workflow Language, v1.0. Specification, Common Workflow Language Working Group. http://w3id.org/cwl/v1.0/ or 10.6084/m9.figshare.3115156.v2. [DOI]
- Aryee M.J. et al. (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics, 30, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M. et al. (2009) Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics, 1, 177–200. [DOI] [PubMed] [Google Scholar]
- Boettiger C. (2015) An introduction to Docker for reproducible research. SIGOPS Oper. Syst. Rev., 49, 71–79. [Google Scholar]
- Campello R.J.G.B. et al. (2013) Density-based clustering based on hierarchical density estimates In: Pei J.et al. (eds) Advances in Knowledge Discovery and Data Mining, Lecture Notes in Computer Science. Springer, Berlin, Heidelberg, pp. 160–172. [Google Scholar]
- Capper D. et al. (2018) DNA methylation-based classification of central nervous system tumours. Nature, 555, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L. (2010) An intuitive Python interface for Bioconductor libraries demonstrates the utility of language translators. BMC Bioinformatics, 11, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrie-Stone T.J. et al. (2019) Bigmelon: tools for analysing large DNA methylation datasets. Bioinformatics, 6, 981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman E.A. et al. (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics, 13, 86.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe A.E., Irizarry R.A. (2014) Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol., 15, R31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson Å. et al. (2013) Continuous aging of the human DNA methylome throughout the human lifespan. PLoS One, 8, e67378.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Yim A.Y.F. et al. (2016) Peripheral blood methylation profiling of female Crohn’s disease patients. Clin. Epigenet., 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahpour A. (2016) pyMAP: a Python package for small and large scale analysis of Illumina 450k methylation platform. bioRxiv, 078048.
- McInnes L. et al. (2018) UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction.
- Min J.L. et al. (2018) Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics, 34, 3983–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S. et al. (2016) Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics, 8, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai S. et al. (2019) Differential methylation of enhancer at IGF2 is associated with dopamine synthesis in major psychosis. Nature Communications, 10. doi: 10.1038/s41467-019-09786-7. [DOI] [PMC free article] [PubMed]
- Pedregosa F. et al. (2011) Scikit-learn: machine learning in Python. J. Mach. Learn. Res., 12, 2825–2830. [Google Scholar]
- Pidsley R. et al. (2013) A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics, 14, 293.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani E. et al. (2017) GLINT: a user-friendly toolset for the analysis of high-throughput DNA-methylation array data. Bioinformatics, 33, 1870–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas L.A. et al. (2018) An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol., 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas L.A. et al. (2017) Integrative epigenetic and genetic pan-cancer somatic alteration portraits. Epigenetics, 12, 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval J. et al. (2011) Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics, 6, 692–702. [DOI] [PubMed] [Google Scholar]
- Satopaa V. et al. (2011) Finding a ‘Kneedle’ in a haystack: detecting knee points in system behavior. In: 2011 31st International Conference on Distributed Computing Systems Workshops., pp. 166–171. [Google Scholar]
- Soriano-Tárraga C. et al. (2018) Biological age is a predictor of mortality in ischemic stroke. Sci. Rep., 8, 4148.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. et al. (2016) ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res., 44, e20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.