

Abstract
Summary
With the advent of fully automated sample preparation robots for Hydrogen–Deuterium eXchange coupled to Mass Spectrometry (HDX-MS), this method has become paramount for ligand binding or epitope mapping screening, both in academic research and biopharmaceutical industries. However, bridging the gap between commercial HDX-MS software (for raw data interpretation) and molecular viewers (to map experiment results onto a 3D structure for biological interpretation) remains laborious and requires simple but sometimes limiting coding skills. We solved this bottleneck by developing HDX-Viewer, an open-source web-based application that facilitates and quickens HDX-MS data analysis. This user-friendly application automatically incorporates HDX-MS data from a custom template or commercial HDX-MS software in PDB files, and uploads them to an online 3D molecular viewer, thereby facilitating their visualization and biological interpretation.
Availability and implementation
The HDX-Viewer web application is released under the CeCILL (http://www.cecill.info) and GNU LGPL licenses and can be found at https://masstools.ipbs.fr/hdx-viewer. The source code is available at https://github.com/david-bouyssie/hdx-viewer.
1 Introduction
By following the isotopic labeling of amide protons, which largely depends on their solvent accessibility and dynamics, Hydrogen–Deuterium eXchange coupled to Mass Spectrometry (HDX-MS) is now an established method for investigating ligand binding, protein–protein interactions, protein dynamics, conformational changes and folding/unfolding. Although considered a low resolution technique compared to other biophysical approaches, HDX-MS has the main advantage of not being limited in size (Sheff et al., 2017), as the proteins of interest are digested before analysis of the labeled peptides. Originating from the 90s, HDX-MS has suffered from its reputation of being poorly reproducible and hard to implement, mostly due to very time-consuming sample preparation and data processing/analysis. Over the past 30 years, academic research successfully developed several strategies to increase the robustness and throughput of the method. Dedicated algorithms were developed to allow the automatic extraction of raw data (Kan et al., 2011; Lou et al., 2010; Pascal et al., 2012; Weis et al., 2006), their statistical analysis (Hourdel et al., 2016; Lau et al., 2019), or both (Rey et al., 2014). However, HDX-MS really gained momentum when MS manufacturers allowed its transfer to biopharma industry, thereby opening a hitherto limited market. From then, commercially available pipelines including automated sample preparation, online proteolytic digestion and dedicated analysis software, such as DynamX (Waters) and HDExaminer (Sierra Analytics), democratized HDX-MS from a handful of specialized teams to structural MS, proteomics laboratories and eventually to industry. These developments coincided with the keen interest of biopharmaceutical companies toward epitope mapping (Wei et al., 2014), monoclonal antibodies and antibody-drug-conjugates structural characterization for quality control assessment (Beck et al., 2015). These all-in-one platforms shifted the methodological bottleneck from sample preparation to data analysis, at least concerning the extraction of the raw data.
The output of an HDX-MS experiment consists in relative deuterium uptakes throughout the amino-acid sequence. Their mapping to 3D structures or models (when available) significantly facilitates biological interpretation. However, in most cases the outputs from the extraction data algorithms are Python scripts that need to be run on a PyMOL console. To our knowledge, MSTools (Kavan and Man, 2011) and MEMHDX (Hourdel et al., 2016) are the only web applications that enable the 3D visualization of HDX-MS data. However, both require manual reformatting of the deuteration data. Furthermore, most recent software such as Deuteros (Lau et al., 2019) or MEMHDX (Hourdel et al., 2016) are restricted to differential analysis only (comparison of two states) and do not allow the visualization of all the measured peptides but only the ones passing a defined statistical threshold. We think that visualization of raw deuteration data for each single condition is a key process in HDX-MS data inspection and interpretation and should be performed alongside visualization of differential deuteration between experimental conditions. To provide a solution that can be readily accessible to most HDX-MS users or neophyte collaborators, we developed a standalone tool to visualize, inspect and compare either individual (single condition) or differential (subtraction between two conditions) data from any HDX-MS software.
2 Materials and methods
HDX-Viewer is an open-source web application leveraging modern web browser features such as the recent WebGL 3D rendering API. Written using HTML5 technologies, it relies on several Javascript libraries: NGL Viewer (v2.0.0-dev.33) (Rose et al., 2018) for the 3D visualization of PDB files, MSAViewer (v1.0.3) (Yachdav et al., 2016) for the visualization of the protein sequences and gif.js (v0.2.0) for the recording of animations in the GIF format.
3 Results
HDX-Viewer is a user-friendly online tool that does not require any programming skills to use. It generates PDB files containing the HDX-MS results for each time-point/experimental condition, and their 3D visualization allows to dynamically inspect and spot surfaces of interest. DynamX (PML) or HDExaminer (CSV) outputs as well as a custom template (CSV) can be uploaded with the associated PDB structure in the ‘Input Data’ fields (see the help document for a detailed description of the supported input formats). HDX-MS results will then be plotted to the 3D structure on the right panel (Fig. 1). Clicking on the ‘Load Demo Files’ button displays the binding interface of the ChAD peptide to the SecB tetrameric chaperone (Guillet et al., 2019) and can be used to test the application. The different time points can be visualized independently with the embedded 3D viewer. The user can rotate the structure, tune the color scale to enhance the rendering and define the undetected regions and background colors, as well as the mode of structural representation. Animation options are also available. Finally, the protein sequence(s) appear in the bottom of the window, with the same coloring scale as the 3D display. The resulting image/video can be exported with a simple click on the button ‘Export as PNG’ or ‘Record GIF’. We believe HDX-Viewer bridges the gap between manual curation of raw HDX data and their biological interpretation based on structural knowledge. Together, it facilitates and significantly quickens protein analysis using HDX-MS.
Fig. 1.
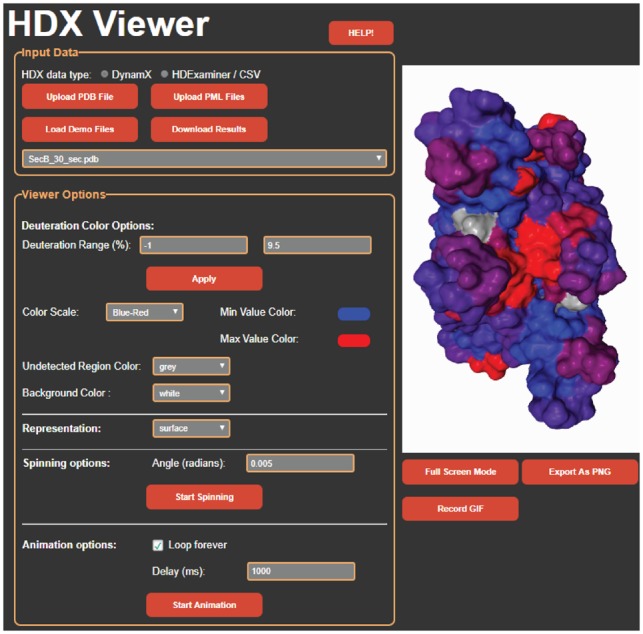
Screenshot of the HDX-Viewer web application
Funding
This project was supported by the Région Occitanie and the French Ministry of Research: Investissement d'Avenir Infrastructures Nationales en Biologie et Santé program (ProFI, Proteomics French Infrastructure project) [ANR‐10‐INBS‐08]. Novo Nordisk Foundation Center for Protein Research was supported financially by the Novo Nordisk Foundation [Grant Agreement NNF14CC0001].
Conflict of Interest: none declared.
References
- Beck A. et al. (2015) Cutting-edge mass spectrometry characterization of originator, biosimilar and biobetter antibodies. J. Mass Spectrom., 50, 285–297. [DOI] [PubMed] [Google Scholar]
- Guillet V. et al. (2019) Structural insights into chaperone addiction of toxin-antitoxin systems. Nat. Commun., 10, 1187.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourdel V. et al. (2016) MEMHDX: an interactive tool to expedite the statistical validation and visualization of large HDX-MS datasets. Bioinformatics, 32, 3413–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z.Y. et al. (2011) ExMS: data analysis for HX-MS experiments. J. Am. Soc. Mass Spectrom., 22, 1906–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavan D., Man P. (2011) MSTools—web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom., 302, 53–58. [Google Scholar]
- Lau A.M. et al. (2019) Deuteros: software for rapid analysis and visualization of data from differential hydrogen deuterium exchange-mass spectrometry. Bioinformatics. doi: 10.1093/bioinformatics/btz022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X. et al. (2010) Deuteration distribution estimation with improved sequence coverage for HX/MS experiments. Bioinformatics, 26, 1535–1541. [DOI] [PubMed] [Google Scholar]
- Pascal B.D. et al. (2012) Hdx workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom., 23, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey M. et al. (2014) Mass spec studio for integrative structural biology. Structure, 22, 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose A.S. et al. (2018) NGL viewer: web-based molecular graphics for large complexes. Bioinformatics, 34, 3755–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff J.G. et al. (2017) Nanospray HX-MS configuration for structural interrogation of large protein systems. Analyst, 142, 904–910. [DOI] [PubMed] [Google Scholar]
- Weis D.D. et al. (2006) Semi-automated analysis of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom., 17, 1700–1703. [DOI] [PubMed] [Google Scholar]
- Wei H. et al. (2014) Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discov. Today, 19, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachdav G. et al. (2016) MSAViewer: interactive JavaScript visualization of multiple sequence alignments. Bioinformatics, 32, 3501–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]