

Abstract
Background—
Myocardial metabolic impairment is a major feature in chronic heart failure (HF). As the major coenzyme in fuel oxidation and oxidative phosphorylation and a substrate for enzymes signaling energy stress and oxidative stress response, NAD+ is emerging as a metabolic target in a number of diseases including HF. Little is known on mechanisms regulating homeostasis of NAD+ in the failing heart.
Methods—
To explore possible alterations of NAD+ homeostasis in the failing heart, we quantified expression of NAD+ biosynthetic enzymes in human failing heart and in the heart of a mouse model of dilated cardiomyopathy (DCM) triggered by SRF transcription factor depletion in the heart (SRFHKO) or of cardiac hypertrophy triggered by transverse aorta constriction (TAC). We studied the impact of NAD+ precursor supplementation on cardiac function in both mouse models.
Results—
We observed a 30% loss in levels of NAD+ in the murine failing heart of both DCM and TAC mice that was accompanied by a decrease in expression of the NAMPT enzyme that recycles the nicotinamide (NAM) precursor whereas the nicotinamide riboside kinase NMRK2 that phosphorylates the nicotinamide riboside (NR) precursor is increased, to a higher level in the DCM (40 fold) than in TAC (4 fold). This shift was also observed in human failing heart biopsies compared to non-failing controls. We show that the Nmrk2 gene is an AMPK and PPARalpha responsive gene that is activated by energy stress and NAD+ depletion in isolated rat cardiomyocytes. NR efficiently rescues NAD+ synthesis in response to FK866-mediated inhibition of NAMPT and stimulates glycolysis in cardiomyocytes. Accordingly, we show that NR supplementation in food attenuates the development of HF in mice, more robustly in DCM, and partially after TAC, by stabilizing myocardial NAD+ levels in the failing heart. NR treatment also robustly increases the myocardial levels of three metabolites, nicotinic acid adenine dinucleotide (NAAD), methyl-NAM and N1-Methyl-4-pyridone-5-carboxamide, which can be used as validation biomarkers for the treatment.
Conclusions —
The data show that nicotinamide riboside, the most energy-efficient among NAD precursors, could be useful for treatment of HF notably in the context of DCM, a disease with few therapeutic options.
Keywords: acetyl-coA, dilated cardiomyopathy, energy metabolism, glycolysis, heart failure, NAD, nicotinamide riboside, Serum Response Factor
INTRODUCTION
Despite progress in therapeutic advances, mortality remains high in congestive heart failure (HF), creating a need for new evidence-based and cost-effective treatments. Failure of cardiomyocyte bioenergetics is a key issue in HF that is not well addressed in current therapeutics.1, 2 We focused our attention on pathways that regulate nicotinamide adenine dinucleotide (NAD+) homeostasis in the failing heart, which has emerged as a new avenue for development of metabolic therapy of HF.3, 4 NAD+ is the major hydride transfer coenzyme in fuel oxidation and mitochondrial ATP generation and is the precursor for NADPH, which is required for anabolic pathways and ROS detoxification.5, 6 In addition, NAD+ is an essential substrate of enzymes including sirtuins and poly(ADPribose) polymerases (PARPs), which are important in responding to altered nutritional status and genotoxic stress.6, 7 Sirtuins perform NAD+-dependent deacylation of protein Lys residues modified by Ac-coA derived acyl groups.8 Sirtuins regulate nuclear gene expression as well as mitochondrial enzyme activities and can play a protective role in the heart,9 though chronic over-activation may be deleterious.10 PARP1 is activated by ROS-induced DNA lesions, and could deplete myocardial NAD+ stores in HF.11 In situations of physiological equilibrium, biosynthetic pathways relying on dietary sources of tryptophan (TRP) and NAD+ precursor vitamins counterbalance the consumption of NAD+ by signaling enzymes. However, dietary precursors may become insufficient to maintain the NAD+ metabolome in pathological conditions, thereby necessitating supplementation of NAD+ precursor.5, 12, 13
The homeostasis of NAD+ and regulation of NAD+ biosynthetic enzymes have not been studied in dilated cardiomyopathy (DCM), a leading cause of heart transplant.14 We previously generated a powerful model of non-ischemic DCM in mice triggered by cardiac-specific inducible inactivation of Serum Response Factor (SRFHKO model).15–18 SRF is a major transcriptional regulator of genes and microRNAs involved in contractility as well as in energy flux and calcium handling.15, 17, 18 SRF pathway is altered in human failing heart19, 20 and in animal models of DCM21, 22 The heart of SRFHKO mutant mice display an early down-regulation of myofibrillar creatine kinase (CK) activity encoded by the Ckm gene, which is followed by repression of enzymes of fatty acid β-oxidation (FAO) and loss of ATP, thus recapitulating the transitional steps toward energy failure and cardiac structural remodeling in HF.18
Here we show that NAD+ homeostasis is altered at an early stage in the heart of SRFHKO mice with a striking induction of the NMRK2 pathway for NAD+ synthesis. Nmrk2 gene is one of two mammalian paralogs of the yeast NRK1 gene that was discovered as a eukaryotic route to NAD+ from nicotinamide riboside (NR).23 Given the activity of NR in promoting sirtuin activity and restoring metabolic balance in a variety of model organisms,13, 24–28 we aimed to test whether NR might treat HF in SRFHKO mice and in the pressure overload hypertrophy model induced by transverse aorta constriction (TAC). Here we show that NR greatly protects cardiac function by virtue of improved citrate and Ac-coA metabolism and antioxidant gene expression. Because NR is orally available in people,29 our data rationalize clinical testing of NR in human HF.
METHODS
The data, analytic methods and study materials will be made available from the corresponding author upon request to other researchers for purposes of reproducing the results or replicating the procedure. Detailed Methods are available as supplemental file.
Patients.
LV myocardium was obtained from terminally failing human hearts of 4 patients (mean age 54 years ± 7, S.D.) at the time of transplantation at the “Hôpitaux Universitaires de Strasbourg” (HUS) as previously published with approval of HUS ethics committee.30 All patients gave informed consent. Patients’ characteristics are detailed in Supp. Table S1. The investigations conformed to the principles of the Declaration of Helsinki. Control hearts were obtained from the department of Pharmacology and Pharmacotherapy, University of Szeged, Szeged, Hungary. Experimental protocols were approved by the Ethical Review Board of the Medical Center of the University of Szeged and by the Scientific and Research Ethical Committee of the Medical Scientific Board at the Hungarian Ministry of Health (ETT-TUKEB; No. 51–57/1997 OEj and 4991–0/2010–1018EKU).
Transgenic mice.
All experiments with animals conformed to the Directive 2010/63/EU of the European Parliament and were approved by the ethics committee Charles Darvin #5 (agreement 00369.01). See supplemental information for details on SRF inactivation protocol.
NR supplementation
SRFHKO and control (Sf/Sf) mice were administered A04 rodent maintenance diet (Scientific Animal Food Engineering, Paris, France) supplemented with NR chloride provided by Chromadex (Irvine CA. USA). See supplemental information for details.
Transcriptomic analysis
Transcriptomic data are available on NCBI Gene Expression Database. Accession number: GSE84142.
STATISTICAL ANALYSIS
Animals were randomly assigned into different treatment groups. To assess significance, we performed Student’s t test for independent samples when the experimental design compared only 2 groups, or One-Way ANOVA for multiple group comparisons, or Two Way factorial ANOVA for independent factors when appropriate. Tukey tests were used for comparison of specific experimental groups when One-way ANOVA gave a p value < 0.05 or, in the case of Two Way ANOVA for interaction p value < 0.05. Values are expressed as mean ±SEM.
RESULTS
The nicotinamide riboside kinase 2 pathway is activated in the failing heart of SRFHKO mice
Transcriptome analyses in SRFHKO mice at baseline or after phenylephrine (PE) alpha-adrenergic stimulation16 revealed that the Nmrk2 gene was induced at an early stage in the SRFHKO heart and rose continuously during establishment of HF (Figure 1A and supp. Table S2). The transcript for the ectoenzyme NT5E (CD73) that hydrolyzes extracellular NAD+ and nicotinamide mononucleotide (NMN) to NR, the substrate of NMRK2 kinase31 also increased in SRFHKO hearts (Supp. Table S2). In contrast, the transcripts encoding NAMPT the enzyme converting NAM to NMN, and PNP, which converts NR to NAM,32 were depressed upon PE treatment in SRFHKO hearts (Supp. Table S2). Genes involved in the TRP and nicotinic acid pathways for NAD+ synthesis were not altered (Figure 1A and Supp. Table S2).
Figure 1. Activation of the Nmrk2 enzyme in dilated cardiomyopathy.
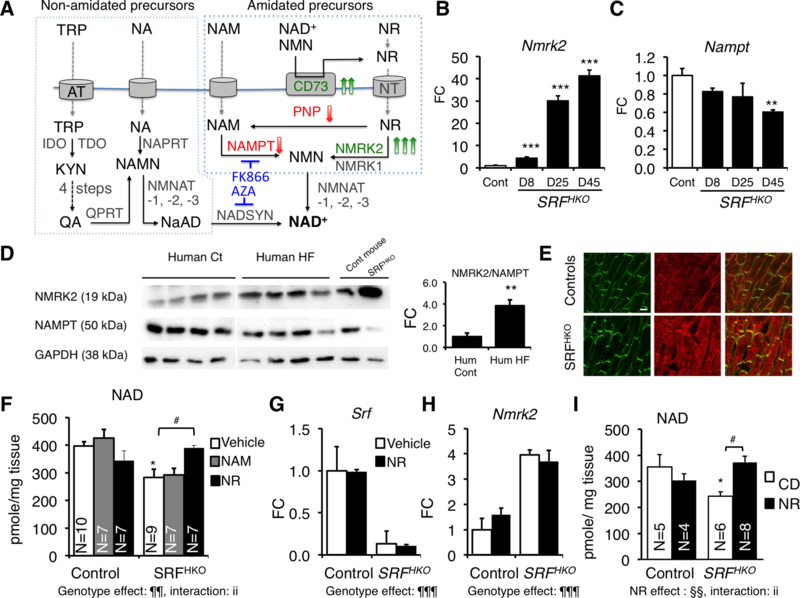
(A) Biosynthetic NAD+ pathways in mammalian cells. NA (Nicotinic acid), NAM, NR and TRP enter cells through specialized transport systems. Extracellular NAD+ and NMN are hydrolyzed to NR by the ectonucleotidase CD73. In the de novo pathway, TRP is converted to kynurenin (KYN) by IDO and TDO enzymes, and then into quinolic acid (QA) in 4 steps (not detailed). QA, NA and NAM are phosphoribosylated by QPRT, NAPRT1, and NAMPT enzymes, respectively, to form mononucleotides NAMN and NMN. NR is phosphorylated by NMRK enzymes to form NMN. NR can be converted to NAM by PNP. NAMN and NMN adenylated by the NMNAT enzymes to form the dinucleotides NAAD+ and NAD+. NAAD+ is amidated by NADSYN1. Genes increased in SRFHKO heart are highlighted in green and those repressed in red (see accompanying Table S2). Inhibitors are shown in blue.
(B, C) RT qPCR analysis of Nmrk2 and Nampt mRNA fold change (FC) in SRFHKO hearts at different days (D) after SRF inactivation. CD, regular chow diet; NR, nicotinamide riboside enriched diet.
(D) Western blot analysis of NMRK2 and NAMPT protein in control and failing human hearts and control and SRFHKO mouse hearts at D45. GAPDH is used as loading control. All human and mouse samples were analyzed on single blot but 2 lanes separating that showed signs of protein degradation and aberrant migration pattern were cut out of the picture
(E) Control and SRFHKO mutant heart sections stained for NMRK2 (red) and vinculin (green) at D45. White bar in upper left panel = 20 μm.
(F-H) NAM or NR (30 μmole) were injected i.p. from D8 to 15 (400 mg/kg/day) to control and SRFHKO mice. Vehicle: saline solution. (F) Myocardial NAD+ levels. (G, H) RT qPCR analysis of Nmrk2 and Nampt mRNA levels.
(I) Myocardial NAD levels in control and SRFHKO mice fed regular chow diet (CD) or NR enriched diet (0.22%. 400–450 mg/kg/day) from D5 to 20.
Throughout the figure data are expressed as mean ± SEM. FC, fold change over control group. Statistical analysis: One-way ANOVA (B, C) or two-way factorial ANOVA for independent samples (F-I). ¶¶ p≤ 0.01, ¶¶¶ p≤ 0.001 for the genotype effect; §§ p≤ 0.01 for the NR treatment effect; ii p≤ 0.01 for the interaction effect. Post-hoc Tukey test: Asterisks indicate statistical significant difference between any group versus the control CD (or vehicle) group: * p≤ 0.05, ** p≤ 0.01, *** p≤ 0.001. # p≤ 0.05 for the effect of NR within the SRFHKO group. A T-test was used for the graph at the right of panel D: ** p≤ 0.01.
Modulation of Nmrk2 and Nampt gene expression detected by transcriptomics were corroborated at the mRNA and protein level (Figure 1B–D). We observed a similar shift in human patients from predominant expression of NAMPT in healthy hearts to increased NMRK2 and depressed NAMPT in failing hearts (Figure 1D, Suppl. Table S2). In mouse heart sections, NMRK2 protein localizes at the proximity of sarcolemma in controls (Figure 1E). The signal was strongly enhanced and spread to cytoplasm in SRFHKO cardiomyocytes.
At day 15 after SRF inactivation, there was a 30% loss of NAD (representing the sum of NAD+ and NADH) (Figure 1F, see supplementary material and method section for details). The gene expression pattern (Nt5e and Nmrk2 up with Pnp and Nampt down) suggested that cardiac tissue is attempting to mobilize and utilize NR as an NAD+ precursor while not increasing NAM usage. We tested this hypothesis by intraperitoneal administration of NR and NAM to SRFHKO mice from D8 to D15. Consistent with this hypothesis, myocardial NAD levels were preserved by NR but not by NAM (Figure 1F). Srf and Nmrk2 expression levels were not changed by these treatments (Figure 1G, H). Because NR is orally available,26, 29 we tested an NR-supplemented diet for 2 weeks. Myocardial NAD levels were reduced in this second series of SRFHKO mice compared to controls and rescued by NR administration (Figure 1I).
NR preserves cardiac function in the SRFHKO heart
We hypothesized that NR supplementation of food might be beneficial for cardiac function in the context of DCM. We fed control and mutant SRFHKO mice a standard chow diet (CD) or NR- supplemented diet to reach a dose of 400 mg/Kg of body weight/day from day 5 to 50, a period in which untreated SRFHKO mice develop DCM and progress toward HF.15–18 NR-diet induced a modest 5 to 7 % increase in body weight in control mice and allowed SRFHKO mice to regain weight after an initial loss while SRFHKO mice on regular diet had lost weight at the end of the protocol (Figure 2A). Cardiac parameters were analyzed by echocardiography between D45–47 (Figure 2B–O). NR did not change heart rate and LV mass index but slightly increased the LV thickness-to-radius ratio (H/R) in control mice (Figure 2N). To assess a potential impact of NR treatment on vascular function, we compared vascular reactivity in isolated mesenteric arteries from NR-fed and control mice. We found no difference between the two groups neither in responses to K+ or agonist-induced contraction nor in the relaxation response to carbachol or a NO donor (Suppl. Figure S1 and Suppl. Table S3).
Figure 2. NR supplementation in diet prevents the onset of heart failure and dilatation.

(A) CD or NR supplemented diet (0.22 %) was given ad libitum to control and SRFHKO mice from D5 after SRF inactivation to the end of the experiment. Body weight was monitored throughout the period. Data are expressed as mean % weight variation ± SEM, compared to weight at D5. A Two-Factor ANOVA with repeated measures on one factor was used for statistical analysis. §§§ p≤ 0.001 for the time effect; iii, p≤ 0.001 for the interaction effect. Tukey test: * p<0.05, ** p<0.01, *** p<0.001 for any time point versus day 5 within a group; #, p≤ 0.05 for the NR treatment in SRFHKO group.
(B-O) Cardiac parameters of control and SRFHKO mutant were analyzed in M-Mode echocardiography between D45 to D47. (B) Heart rate; (C) Left ventricle (LV) mass index; (D) LV ejection fraction; (E) Fractional shortening; (F, G) LV end-systolic diameter (D) and volume (G); (H, I) LV end-diastolic diameter (H) and volume (I); (J, K); LV posterior wall thickness in systole (J) and diastole (K); (L, M) Interventricular septum thickness in systole (L) and diastole (M); (N) H/R: LV thickness (H) to radius (R) ratio; (O) Stroke volume. Dimensions were normalized by the body weight. Data are expressed as mean ± SEM. Statistical analysis: two-way factorial ANOVA for independent samples. ¶¶ p≤ 0.01, ¶¶¶ p≤ 0.001 for the genotype effect; § p≤ 0.05, §§ p≤ 0.01, §§§ p≤ 0.001 for the NR treatment effect; i p≤0.05, ii p≤ 0.01, iii p≤0.001 for the interaction effect. Tukey test: Asterisks indicate statistical significant difference versus the control CD group: * p≤ 0.05, ** p≤ 0.01, *** p≤ 0.001. # p≤ 0.05, ## p≤ 0.01 for the effect of NR within the SRFHKO group.
SRFHKO mutant mice fed the standard diet displayed a severe decrease in LV ejection fraction (LVEF) and fractional shortening (FS) (Figure 2D, E). The NR diet clearly protected against this decline in cardiac function. Importantly, NR fully protected SRFHKO mice against the dilatation and thinning of the LV wall (Figure 2F–M), preserving the H/R ratio that normally declines with DCM (Figure 2N). Changes in stroke volume (Figure 2O) and cardiac output (not shown) were not significant.
At the histological level, NR limited the appearance of very long cardiomyocytes (Feret > 100 μm) in SRFHKO mice although the population of large cardiomyocytes (minFeret > 26 μm) remained larger than in controls (Suppl. Figure S2).
NR-enriched diet protects the cardiac NAD+ metabolome in HF
The impact of an NR-enriched diet on the cardiac NAD+ metabolome is not known (Figure 3A). Mice fed with or without NR were sacrificed 3 days after the echocardiography at D50. Myocardial NAD was decreased in the SRFHKO mice at D50 as measured by the NAD cycling assay and the NR diet protected against this drop (Figure 3B). Myocardial NAD+ and NADH varied in the same direction in all conditions leaving the NAD+/NADH ratio unchanged (Supp. Figure S3). LCMS analysis allowed us to provide the levels of NAD+ metabolites on a common scale with NAD+13, 33 (Figure 3C–L). SRFHKO mice hearts did not have higher levels of ADPR or NAM (Figure 3D, E). Levels of NADP+ and NMN showed a similar pattern as NAD+ (Figure 3F, G).
Figure 3. Impact of NR treatment on cardiac NAD+ metabolome.

(A) NR to NAD+ pathway and NAD+ catabolism are depicted. NAD+ is cleaved into NAM and ADP-ribose by NAD+ consuming enzymes. NAM is either recycled by NAMPT or methylated by the Nicotinamide N-Methyl Transferase (NMT) enzyme and oxidized by the aldehyde deoxidase 1 (AOX1) giving rise to degradation products Me-NAM and Me-4PY, respectively.
(B) Myocardial NAD was quantified by colorimetric NAD+ cycling assay at day 50 in controls (N=6), Controls + NR (N=5), SRFHKO (N=6), SRFHKO + NR (N=6). Data are expressed as means ± SEM.
(C-L) Myocardial metabolites were analyzed by LCMS-based metabolomics. See abbreviations in the text. Controls (N=4), Controls + NR (N=5), SRFHKO (N=6), SRFHKO + NR (N=5, except for NAM, MeNAM and Me-4PY, N=4, no peak was identified in one sample). Data are expressed as means ± SEM.
(M-Q) Expression of genes related to oxidative stress signaling (M-O) and cardiac structural and metabolic remodeling (Q). N=5 in each group.
(R) Representative western blot analysis of FOXO1 and acetyl-FOXO1 of 3 independent experiments realized in different duplicates for each group. GAPDH is used as a loading control. Right graphs: Quantification of total and acetyl-FOXO1 ratio on GAPDH and acetyl-FOXO1/Total FOXO1 ratio in N=5 to 6 animals per group.
(S) Acetylation level of mitochondrial Aconitase 2. Aconitase 2 (75 kDa) was immunoprecipitated using a rabbit polyclonal antibody and the immunoprecipitate was analyzed by western blot using a mouse monoclonal anti-Ac(K103) antibody. Inputs were run in a parallel gel and immunoblotted with anti-aconitase 2 and anti-GAPDH for loading control. The ratio of Acetyl-Aconitase2/Total Aconitase2 and Aconitase2/GAPDH are shown on the right. See accompanying Supp. Fig.6A for control immunoprecipitation with preimune rabbit IgG.
T-X) LV cardiac tissue was isolated at 50 days after tamoxifen injection in control and SRFHKO mice fed control diet (CD) or NR-enriched diet. DNA and proteins were extracted from parallel samples to quantify mitochondrial to genomic DNA ratio (T) and enzymatic activities: (U) Complex I, (V) Cytochrome oxidase, (W) Citrate synthase, (X) ATP Citrate Lyase. See associated supplementary figure S7. In (M-S), data are expressed as mean fold change (FC) ± SEM over control group CD.
Statistical analysis: Two-way factorial ANOVA for independent samples was used for all panels. ¶ p≤0.05, ¶¶ p≤0.01, ¶¶¶ p≤ 0.001 for the genotype effect; § p≤ 0.05, §§ p≤ 0.01, §§§ p≤ 0.001 for the NR treatment effect; ii p≤ 0.01 for the interaction effect. Tukey test: Asterisks indicate statistical significant difference versus the control CD group: ** p≤ 0.01.
NR increased the cardiac levels of nicotinic acid adenine dinucleotide (NAAD) a sensitive biomarker of increased NAD+ metabolism29 as well as Methyl-NAM (MeNAM) (Figure 3A, H–J). MeNAM oxidation by AOX1 generates N1-Methyl-4-pyridone-5-carboxamide (Me4PY) and releases hydrogen peroxide (Figure 3A). Changes in unrelated metabolites were not significant (Figure 3K, L). As shown in the nematode,34 NR increased the expression of the Nfel2 gene encoding NRF2 while its target genes Mt2 and Nox4 were already activated as well as G6pdx in the heart of SRFHKO mice (Figure 3M–P). NR treatment in the SRFHK0 hearts limited the level of Myh7 induction, a signature of cardiac stress and metabolic remodeling in the failing heart (Figure 3Q).
The liver is a central organ for the regulation of nutrient metabolism. We assessed the NAD+ metabolome response in the liver of control and SRFHKO mice (Suppl. Table S4). NR increased the Me-NAM and Me-4PY levels similarly in both genotypes. Interestingly, the liver response to NR diet was more robust in the SRFHKO mice with regard to the increase in the steady-state level of NAD, NAAD, ADPR and NAM. Since the liver is not targeted by the Srf deletion, it suggests that the HF condition indirectly modifies the balance of NAD synthesis and signaling in the liver.
NR enriched diet does not result in global cardiac protein deacetylation
The cardiac levels of NAM and ADPR in this HF model are low and show little changes with NR (Figure 3D, E), suggesting that the NR-driven boost in NAD+ synthesis is not necessarily translated into a higher level of NAD+ consumption by sirtuins and PARPs. We performed anti-acetyl-K and anti-poly-ADPribose western blot analyses on cardiac proteins from control and SRFHKO mice fed with the CD or NR-enriched diets. Just as levels of ADPR were not modulated by SRFHKO or NR, neither were levels of PAR (Supp. Figure S4A). There was an increase in the acetylation level of some cardiac proteins in the hearts of NR-fed animals (Supp. Figure S4B). Antibodies specific to the acetylated form of FOXO1 or p53 showed an increase in the acetylation level of these nuclear proteins in the heart in response to NR in controls as in SRFHKO mice (Figure 3R and Supp. Figure S5). By comparison, the acetylation level of the mitochondrial aconitase 2 protein did not change in any of these conditions despite lower protein level in the SRFHKO hearts (Figure 3S, Supp. Figure 6A). These data suggest that the bioenergetic improvement in NR-supplemented hearts may result in greater production of cytosolic Ac-coA and consequent increased nucleocytosolic protein acetylation.
NR improves metabolism of citrate in HF
Several regulators involved in mitochondrial biogenesis and oxidative metabolism as well as lipid metabolism were repressed in SRFHKO hearts (Supp. Figure S7A–B). Simultaneously, the expression of the glucose transporter Glut1 was increased and expression of Pdk2 was decreased which can be seen as a trend of the failing heart to increase glycolysis (Supp. Figure S7C). NR did not alter the expression of these genes.
There was a trend toward a reduced mitochondrial to genomic DNA ratio in the SRFHKO LV (Figure 3T). The NADH-dependent Complex I activity and Cox activity was not changed by the NR diet (Figure 3U, V). Neither the complex I and II maximal respiration rates, nor the apparent Km for ADP, mitochondrial CK coupling and ADP acceptor control ratio showed any alteration at this stage (Day 50) in the SRFHKO LV fibers. These parameters were not altered by NR administration (Supp. Figure S7D–G).
We noted that NR-treatment resulted in a net increase in acetylation of FOXO1 and p53 (Figure 3R and Supp. Figure S5). Availability of cytosolic Ac-coA depends on mitochondrial citrate synthase (CS) production of citrate and conversion to Ac-coA via the cytosolic enzyme, ATP-citrate lyase (ACL). CS activity was reduced to 65% of control levels in the SRFHKO left ventricular myocardium (Figure 3W). Strikingly, NR administration protected against the decline of CS activity in the failing heart. This rescue in activity occurred despite a net reduction in protein level in the SRFHKO myocardium that was not rescued by the NR treatment (Supp. Figure S6B). Interestingly, NR also increased cardiac ATP-citrate lyase (ACL) activity in both control and SRFHKO mice (Figure 3X). Taken together, these data suggest that NR improves CS and ACL activity, resulting in increased nucleocytoplasmic protein acetylation.
Nmrk2 is induced by inhibition of alternative NAD+ biosynthetic pathways in cultured cardiomyocytes
To assess the impact of NMRK2 on NAD+ biosynthesis relative to the other pathways in cardiomyocytes, we used chemical inhibitors to target the alternative pathways in neonatal rat cardiomyocytes (NRC) grown in DMEM medium containing NAM as the vitamin B3. The glutamine analog azaserin (AZA) inhibits the glutamine-dependent NAD synthase NADSYN1,35 which catalyzes the last step of NAD+ synthesis from TRP and NA (Figure 1A). FK866 is a specific inhibitor of Nampt.36
AZA had a slowly repressive effect on NAD pools that resulted in a 50% reduction with respect to non-treated cells at 72 h (Figure 4A–C) while progressively increasing Nmrk2 expression (Figure 4D). FK866 treatment led to a rapid and severe loss of intracellular NAD+ but not NADH (Figure 4A–C). Despite this severe loss, NRC could be maintained up to 72h in continuous layer with FK866 treatment. FK866 treatment strongly increased Nmrk2 expression at 72 h (Figure 4D). Combined AZA and FK866 treatment had a similar impact than FK866 alone (Figure 4A–D). Nmrk1 expression did not change in these conditions (not shown).
Figure 4. Nmrk2 expression is activated by repression of alternative NAD+ biosynthetic pathways.

(A-C) Intracellular levels of NAD+ (A), NADH (B) and NAD+/NADH ratio (C) in NRC after 10 μM FK866 and/or 20 μM Azaserin treatment or no treatment (NT) for 24 h to 72 h as indicated.
(D) Nmrk2 mRNA level in NRC treated as in (A-C)
(E-G) Same as in (A-C) in NRC treated for 72h with 10 μM FK866 or not treated (NT) in normal culture medium (−) or in presence of 250 μM NAD+ or 1 mM NR.
(H) Nmrk2 mRNA level in NRC treated as in (E-G)
(I) NAD+ levels in NRCs treated with 10 μM FK866 for 72 h, or not treated (NT) in the presence of increasing concentration of NR in culture medium.
(J) NAD+ content in non-treated (NT) or following 24 h NR treatment (1mM) in NRC infected with Ad-GFP or HA-Nmrk2. Bottom: western blot detection of HA-Nmrk2 with anti-HA antibody.
(K-M) Mitochondrial stress test in Seahorse analyzer. NRC grown on Seahorse 96 well plates were analyzed for oxygen consumption rate (OCR) at day 8 after 5 days of treatment. (K) Basal mitochondrial respiration is calculated from total cellular respiration minus non-mitochondrial respiration. (L) ATP production is calculated from basal mitochondrial respiration minus respiration after oligomycin injection. (M) Maximal respiration is measured after FCCP injection. See accompanying Suppl. Figure S9 for other respiration parameters.
(N-P) Glycolysis stress test in Seahorse analyzer. NRC grown on Seahorse 96 well plates were analyzed at day 8 after 5 days of treatment. (N) Glycolysis was measured as a function of extracellular acidification rate (ECAR) after injection of glucose 10 mM. (O) Glycolytic capacity as the maximum ECAR following injection of oligomycin. (P) Glycolytic reserve as the difference between glycolysis and maximal glycolytic capacity.
Throughout the figure the data are expressed as mean fold change (FC) ± SEM over the control group, except when indicated. Statistical analysis: a two-way factorial ANOVA for independent samples was used for panels A to J. ¶ and § symbols as indicated in the panels; i p≤0.05, ii p≤ 0.01, iii p≤0.001 for the interaction effect. One-Way ANOVA was used for panels K-P: ¶ p< 0.05, ¶¶ p< 0.01, ¶¶¶ p< 0.001. Tukey test: *, p< 0.05, ** p< 0.01, *** p< 0.001 between any group versus NT control cells; ## p< 0.01, ### p< 0.001 for indicated comparisons.
We tested whether NR could preserve NAD+ levels in the presence of FK866. NR slightly increased the baseline level of intracellular NAD+ in non-treated NRC while exogenous NAD+ had no impact (Figure 4E). Both compounds increased the NAD+/NADH ratio (Figure 4F–G). NR fully protected NAD+ levels in NRC treated with FK866 while the protection by exogenous NAD+ was partial (Figure 4E), though both treatments blocked Nmrk2 induction (Figure 4E). At least 100 μM of NR was required to rescue the NAD+ loss induced by 10 μM FK866 (Figure 4I). To assess the functionality of NMRK2 in absence of a stress like the FK866, we infected NRC with a recombinant adenovirus expressing HA-tagged Nmrk2 cDNA (Figure 4J). Overexpression of NMRK2 did not modulate the NAD+ levels in NRC. In presence of NR, Nmrk2 overexpression robustly increased the NAD+ level by a factor of 6. NR alone slightly increased the NAD+ level in control NRC infected with Ad-GFP (Figure 4J). In isolated primary cultures of adult rat cardiomyocytes (ARC), NR increased intracellular NAD nearly 3 fold showing that the NMRK pathway is more active in ARC than in NRC (Suppl. Figure S8A). FK866 reduced NAD levels by a factor 2 (Suppl. Figure S8A) in ARC and increased Nmrk2 expression by a factor 4.5 while AZA had no impact (Suppl. Figure S8B). We assessed the impact of NR on oxygen consumption rates (OCR) in NRCs following a mitochondrial stress protocol. NR had no impact on any mitochondrial respiration parameter in NRC at baseline (Figure 4K–M, Suppl. Figure S9). NR increased glycolysis at baseline in a dose-dependent manner (Figure 4N), but not the maximal glycolytic capacity (Figure 4O, P). We assessed the capacity of NR to rescue any impact of FK866 on metabolism. FK866 reduced basal OCR and maximal respiration and trended to reduce ATP production (adjusted p value = 0.0547) (Figure 4K–M). Addition of NR at doses that we showed to fully rescue the drop in NAD+ levels triggered by the FK866, did not correct these alterations and even further reduced maximal respiration at high doses (Figure 4K–M, Suppl. Figure S9). FK866 strongly reduced basal glycolysis and glycolytic capacities in NRC. NR strikingly preserved NRC glycolytic capacities to 100% control levels and more in FK866-treated cells and stimulated glycolysis at baseline in non-treated cells in a dose-dependent manner (Figure 4N–P).
SRF deficiency-induced depression of the Nmrk2 promoter is counterbalanced by the sensitivity of Nmrk2 gene to NAD+ depletion and energetic stress
Because Nmrk2 expression is activated in vivo upon Srf gene inactivation, we addressed the role of the SRF transcription factor in Nmrk2 gene regulation. We identified an SRF binding site with 9/10 bp match to the consensus CC(A/T)6GG (CArG) sequence in a conserved 600 bp region between the murine and human genes (Figure 5A). We cloned the upstream region and 5’UTR of the murine Nmrk2 gene (−581/+61) into a luciferase reporter vector and mutated the CArG-like sequence to a motif matching the consensus sequence (5’-CC) or a motif unable to bind SRF (5’-GG) (Figure 5A). FK866 increased the expression of the WT construct 8-fold while a consensus CArG reduced the extent of activation (Figure 5B). The 5’-GG mutation reduced baseline expression by 75% and abolished the response to FK866. The full intergenic 3009 bp sequence upstream of Nmrk2 had a much higher activity, especially in response to FK866 (Figure 5C). SiRNA-mediated inhibition of Srf reduced, but did not abolish, the induction of endogenous Nmrk2 by FK866 in NRCs suggesting that FK866-triggered activation of the Nmrk2 promoter is partially independent of SRF (Figure 5D–E). FK866 treatment reduces ATP to 50% of the control level in NRC (Figure 5F). SRF is major upstream regulator of Ckm gene involved in high-energy phosphate compounds balance. A siRNA-mediated 5-fold knock-down of Ckm gene expression lowered the cellular ATP levels down to 70% control level and triggered a 6.8 fold increase in Nmrk2 expression (Figure 5G–H). A 2-fold reduction in Srf expression resulted in a 2-fold reduction of Ckm expression leaving Nmrk2 expression intact. Enforcing a more profound depression of Ckm expression in the context of Srf repression in NRC by co-transfection with both Srf and Ckm siRNAs resulted in a 2.3 fold increase in Nmrk2 expression (Figure 5H).
Figure 5. SRF is a component of Nmrk2 gene transcription.

(A) CArG-like binding site for SRF in the conserved region between the murine (M.m) Nmrk2 promoter and human (H.s) NMRK2 intron 1. The CArG-like motif was mutated to a CArG consensus sequence (cons) and a CArG mutant (mut) site unable to bind SRF.
(B) Activity of p586-Firefly Luciferase construct bearing wild-type (WT), consensus (CONS) or mutated (Mut) CArG motif (H) or longer fragments (I), without (NT) or with FK866 treatment (10 μM). SV40-renilla luciferase was co-transfected with the Nmrk2-Firefly Luc constructs for normalization of transfection efficiency.
(C) Various lengths of the murine Nmrk2 regulatory region were inserted into pGL4 vector and transfected in non-treated or FK866 treated cells.
(D, E) Srf and Nmrk2 mRNA levels in NRC transfected with control scrambled siRNA (siScr) or Srf si-RNA, without (NT) or with FK866 treatment.
(F, G) ATP levels in cells treated for 72h with FK866 (F) or siCkm (G).
(H) Srf, Ckm and Nmrk2 mRNA level in NRCs transfected for 72h with siRNAs as indicated.
Throughout the figure, data are expressed as mean ± SEM over control group, Fc, fold change over control group except for panel B and C, fold change over promoterless pGL4 plasmid and H FC over siCtneg. Two-way factorial ANOVA for independent factors was used for panels B to E. ¶ and § symbols as indicated in the panels; iii p≤0.001 for the interaction effect. A T-test was used for panels F and G; * p< 0.05, ** p< 0.01,between FK treated cells or siCkm transfected cells versus control cells or siCtneg transfected cells, respectively. A one-way ANOVA was used in panel H. §§§ p≤ 0.001. Post-hoc Tukey test: ** p< 0.01, between any group versus siCtneg transfected control cells.
The Nmrk2 gene is induced by the AMPK-PPARα axis
In line with the previous results, we hypothesized that pathways related to energy failure might activate Nmrk2. The level of phosphorylated AMPKα, the energy stress sensing AMP-activated kinase, and its target Ac-CoA Carboxylase (ACC) was increased at an early stage when Nmrk2 induction begins in the SRFHKO hearts (Figure 6A–B). 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) treatment stimulated AMPK phosphorylation in NRC and increased NMRK2 protein level in NRC (Figure 6C–D). The energetic stress induced by glucose-deprivation for 24 h or FK866 for 48h also increased the NMRK2 protein.
Figure 6. Nmrk2 expression is increased by AMPK and PPARα pathways.

(A) Representative western blot analysis of cardiac proteins in control and SRFHKO mice at D9 using antibodies directed against ACC, phospho-ACC (Ser79), AMPKα and phospho-AMPKα (Thr172). Phosphorylated and total proteins were analyzed on 2 separate gels (gel 1 and 2) and GAPDH antibody was used for loading control.
(B) Quantification of total and phospho-protein signal from western blot analyses. Data are normalized on GAPDH signal. The Phos/Total ratio is calculated from GAPDH normalized levels for each individual. N= 6 for each group. Data are expressed as mean fold change (FC) ± SEM over control group. T-test: * p≤ 0.05, ** p ≤0.01, *** p≤ 0.001 over control group.
(C) NRC were treated with FK866 (10 μM, 72h), AICAR (500 μM, 48h) or grown in absence of glucose in the medium (Glc 0) for 48h and proteins were extracted for western blot analyses. Representative western blot.
(D) Quantification on n=3 samples for each condition of NRC treated as in (C). Data are expressed as mean FC ± SEM over NT group. * p≤ 0.05 over control group.
(E-G) Intracellular NAD+ content (E), NADH (F) and NAD+/NADH ratio (G) in NRC after 24h of treatment with AICAR [500 μM]. NT; non-treated cells.
(H) RT qPCR analysis of Nmrk2 mRNA level in NRC treated with AICAR. T-test: *** p≤ 0.001 over non-treated control group.
(I, J) NRC were co-transfected with Nmrk2-luciferase constructs containing 586 or 3009 base pairs of upstream Nmrk2 regulatory region and a dominant negative (DN) AMPK expression vector. NRC were transfected at D3 after plating, followed by AICAR treatment (500 μM) at D4. Luciferase levels were analyzed at D5. Normalized Fireflyl/Renilla values are expressed as in Figure 5B as FC ± SEM over the promoterless pGL4 vector. ¶¶ p≤0.01, ¶¶¶ p≤ 0.001 for treatment effect. . ** p≤ 0.01, *** p≤ 0.001 over non-treated control group. ## p< 0.01 for AICAR vs. AICAR+AMPK-DN.
(K) Nmrk2 promoter deletion analysis by luciferase assay. Rectangular boxes show the position of the putative PPAR binding sites. Data are expressed as mean FC ± SEM over the promoterless pGL4 plasmid. One way ANOVA: ¶¶¶ p≤ 0.001 for promoter length effect. Post-hoc Tukey test: **p≤ 0.01, *** p≤ 0.001 over the promoter less pGL4 vector. . ## p< 0.01 for over the immediately shorter construct
(L) Neonatal rat cardiac fibroblasts and cardiomyocyte-enriched fractions were separated on a discontinuous percoll gradient. Cardiac fibroblasts were transfected with the p-228, p-581 and p-3009-FLuc constructs. Cardiomyocytes were transfected with the p-3009-FLuc. SV40-RLuc construct was cotransfected for normalization. Data are expressed as mean FC ± SEM over the mean p228-FLuc activity in cardiac fibroblasts. One-way ANOVA: ¶ p≤ 0.05. Post-hoc Tukey test: * p≤ 0.05 Cardio-3009 over Fibro-30089.
(M) NRC were co-transfected with the p3009-FLuc construct and the RXR expression vector and with either PPARα, PPAR β/δ or PPARγ expression vectors. NRC were treated 24h later with the agonists GW7647 [0.6 μM], GW501516 [0.6 μM], and G1929 [0.6 μM], for PPARα, PPARβ/δ and PPARγ, respectively, or with their respective antagonists, GW6471 [10 μM], GSK3787 [2 μM] or GW9662 [2 μM]. Data are expressed as mean FC ± SEM over normalized luciferase levels of NRC transfected with the p3009-FLuc construct alone (dashed line) in the same experiment. A one-way ANOVA was used since each group is independent of the other (different agonists and antagonists): ¶¶¶ p≤ 0.001. Post-hoc Tukey test: ** p≤ 0.01 versus p3009-FLuc alone. ## p≤ 0.01 for comparison between agonist and antagonist.
(N) NRC were transfected with the p3009-FLuc construct. Transfected NRC were treated 24h later with the antagonists G6471, GSK3787, or G9662, 30 minutes before adding AICAR for a further 24 h period. All concentrations were as in L. Data are expressed as mean FC ± SEM over the p3009-luc construct treated with AICAR alone. One-way ANOVA: ¶ p≤ 0.05. Post-hoc Tukey: * p≤ 0.05 vs AICAR treated cells.
AICAR treatment did not modulate NAD+/NADH but robustly induced Nmrk2 expression (Figure 6E–H). To assess whether the effect of AICAR took place at the level of the Nmrk2 promoter, we transfected Nmrk2-luciferase constructs into NRC and treated the cells with AICAR. The p586-Luc and moreover the p3009-Luc reporter was highly responsive to AICAR treatment (Figure 6I–J). Co-transfection of a plasmid overexpressing a dominant negative (DN) isoform of AMPK efficiently blunted induction by AICAR (Figure 6I–J).
Analysis of the 5’ regulatory sequences of Nmrk2 revealed enrichment in putative peroxisome proliferator-activated receptor (PPAR) binding sites (Figure 6K). The fragments −3009/−2552 and −1028/−228 contain most of the activity. By comparison, the activity of the Nmrk2 promoter was very low in cardiac fibroblasts (Figure 6L). To assess the role of PPAR factors in Nmrk2 gene regulation, we co-transfected NRC with the p3009-Luc reporter and PPAR expression vectors. Only PPARα increased the activity of the Nmrk2 promoter in presence of its agonist GW7647 (Figure 6M). The PPARα antagonist G6471 repressed the Nmrk2 promoter (Figure 6M) and blunted the activating effect of AICAR (Figure 6N). Neither the PPARβ nor the PPARγ antagonists had any effect on Nmrk2 promoter induction by AICAR. These results establish that Nmrk2 is an AMPK-PPARα responsive gene that is induced by energy stress in cardiac cells.
NR preserves cardiac function in the TAC model
To determine whether NR treatment could be useful in other forms of HF, we assessed its efficiency in the TAC model of pressure overload cardiac hypertrophy. NR was administrated starting 2 days after the TAC or SHAM surgery. Kaplan-Meier survival curve analysis showed no significant difference, mainly because of a similar early mortality between the two groups within one week after TAC (Figure 7A). NR treatment reduced the final drop in LVEF that occurred between the 4rth and 6th week after TAC (Figure 7B and Supp. Table S5). NR did not attenuate the dilatation of the LV chamber and had only a transient effect on the thinning of the IVS (Figure 7C, D). At sacrifice, the cardiac hypertrophy index was similar between the CD and NR groups (Figure 7E).
Figure 7. NR treatment preserves myocardial NAD levels and limits the drop in ejection fraction in the pressure overload induced hypertrophy model.

2-month old Control and SRFHKO male mice were subjected to TAC or SHAM surgery and fed with control chow diet (CD) or NR enriched diet from day 2 after surgery to day 42.
(A) Kaplan-Meier survival curve analysis. Log-rank statistic: p> 0.05. Color code shown on the right for each group is valid for panels A to E.
(B-D) Echocardiography follow-up analysis from base line (2 days before surgery) to 6 weeks after. IVSThD, interventricular septum thickness in diastole; LVEDD, Left ventricle end-diastolic diameter; LVEF, left ventricle ejection fraction.
(E) Heart weight to body weight ratio after sacrifice at 6 weeks.
(F-H) Cardiac NAD+ and NADH levels and redox state assessed by the NAD+ cycling assay.
(I-Q) Cardiac mRNA levels of the indicated genes assessed by RT-qPCR.
Data are expressed as mean ± SEM. Statistical analysis: Two-Way ANOVA for independent factors statistical analysis is shown at 6 week and was used for panels B to Q, followed by post-hoc Tukey test. ¶¶ p≤ 0.01, ¶¶¶ p≤ 0.001 for the TAC effect; §§§ p≤ 0.001 for the NR treatment effect; i p≤0.05 for the interaction effect. Asterisks indicate statistical significant difference for the indicated comparisons: * p≤ 0.05, ** p≤ 0.01. # p≤ 0.05 for the effect of NR within the TAC group. See accompanying Table S5 for other echocardiography data at 6 weeks and previous stages.
The TAC induced HF resulted in a decrease of the myocardial NAD+ pool, which was partially preserved by NR, with little impact on the NAD+/NADH ratio (Figure 7F–H). TAC increased Nmrk2 expression level in the non-treated group and NR-treated group while Nmrk1 expression remained unchanged and Nampt was repressed (Figure 7I–K). TAC repressed Sirt3 expression, Sirt1 and Ckm expression while increasing the expression of Myh7, Anf and Bnp (Figure 7L–Q) but NR treatment had no impact on gene expression.
DISCUSSION
We report that depressed NAD+ homeostasis and activation of NMRK2 kinase are early and persistent events in a mouse model of DCM leading to heart failure and that administration of NR, the substrate of NMRK2 and most energetically favorable NAD+ precursor, prevents deterioration of cardiac function and adverse remodeling (Figure 8).
Figure 8. Activation of the NMRK2 path to NAD+ synthesis as an adaptive energy sparing mechanism in the failing heart.

DCM is a form of pathological cardiac remodeling that is associated with severe energy depletion leading to HF. The energy stress sensor AMP-kinase is an adaptive signaling pathway aiming to preserve energy in the cells. The NAMPT enzyme is a major, rate-limiting step for NAD+ synthesis whose energetic cost equals four ATP molecules, including one converted to AMP, for the synthesis of one NAD+. NAMPT is repressed in most forms of HF through unknown regulatory mechanism. Opposite to this repression, Nmrk2 gene is an AMPK responsive gene that is activated in several models of DCM and other forms of pathological remodeling such as the pressure-overload cardiac hypertrophy. NMR-Kinase 2 allows the synthesis of NAD+ at a lower cost of 2 ATP per molecule. Although NR content may be limited in rodent and human diet, NR can easily be provided as a nutraceutical to help the failing heart to maintain NAD+ levels.
Altered NAD+ homeostasis has been reported in several models of HF including pressure overload, myocardial infarction and angiotensin 2 infusion.3, 37, 38 In this context, much attention has focused on the NAMPT enzyme that is repressed in several models of cardiac injuries.38 On the other hand, we found a robust upregulation of Nmrk2 expression in the heart of SRFHKO developing DCM and a more modest but significant increase following TAC-induced HF. Interestingly, we observed a similar shift from repressed NAMPT to increased NMRK2 in human failing heart. Interrogation of the Gene Expression Omnibus (GEO) database reveals that Nmrk2 is induced in several models of cardiomyopathy related to mutations in the lamin-A (GEO dataset GDS2746),39 Idh2 (GDS4893),40 PGC1α (GDS4776),41 or Tfr1 (GSE68745)42 genes. Considering the relative heterogeneity of molecular functions encoded by these genes, we propose that the activation of the NMRK2 pathway represents a common adaptive mechanism in the failing heart. Here we show that Nmrk2 gene can be activated in response to NAMPT inhibition and activation of the energy stress sensor AMPK. In NRC, we show this activation to depend on PPARα. PPARα is often expressed at lower levels in the adult failing heart43 while on the other hand AMPK was shown to activate PPARα in various context.44 Hence AMPK activation could partially compensate for the lower levels of PPARα to activate Nmrk2. The precise contribution of these actors in the adult failing heart remains an open question. In the case of SRFHKO hearts, it is interesting to note that despite the fact that SRF is an upstream regulator of Nrmk2 gene that we would expect to be repressed following Srf gene inactivation, SRF is also a major upstream regulator of Ckm gene, which is important to maintain PCr/ADP/ATP balance15, 18 and AMPK is activated early on in the SRFHKO heart (this study). We show in vitro that Ckm depression alone is sufficient to induce Nmrk2 expression. In addition Srf inactivation alters the expression of numerous sarcomeric genes and triggers a severe eccentric remodeling15 that is likely to further increase the energetic cost of contraction in vivo. A similar phenomenon could occur for instance in the lamin-A mutants, that display severe alterations in cardiomyocyte cytoskeletal integrity and in which SRF activity is repressed and Nmrk2 expression induced.22, 39 In addition to its well-known roles in stimulating fuel oxidation to restore ATP levels, we show that AMPK stimulation induces Nmrk2 expression. NMN synthesis from NR by NMRK enzymes requires a single ATP while synthesis from NAM by NAMPT requires more than 3 ATP equivalents: one for the autophosphorylation of the enzyme, and 2 (plus a carbohydrate) in formation of PRPP (Figure 8). Both pathways consume an additional ATP for fusion with NMN to generate the dinucleotide. Hence, the shift from NAMPT to NMRK2 for NAD+ synthesis is an energy-sparing mechanism that may be favored in HF. In turn, we show that one of the beneficial actions of NR, at least in vitro, may be to increase glycolysis in cardiomyocytes. These different mechanisms of action may contribute to increase cardiac efficiency, which should be assessed in future studies to establish whether metabolic substrate preference and potentially glucose oxidation which is associated with a better respiratory quotient and ATP availability is improved upon NR treatment.
Although the NMRK2 pathway is activated in the failing heart of SRFHKO mice, the myocardial NAD+ level is depressed, which suggests that circulating and tissue levels of NAD+ precursors are insufficient to sustain cardiac NAD+ synthesis in mice on a regular rodent diet, stimulating an interest in NR supplementation to correct this defect. A few studies have shown that short-term NAD+ supplementation following chronic Angiotensin-2 administration37 or NMN supplementation following ischemia-reperfusion injury45 or TAC surgery46 can normalize alterations in NAD+/NADH ratio in the mouse failing heart. Interestingly, cells do not take up NMN intact and, instead, NMN is extracellularly dephosphorylated to NR, such that its cellular conversion to NAD+ depends on the NR kinase pathway.47, 48
We discovered a strongly beneficial effect of NR in the SRFHKO model of HF in preservation of cardiac function and limitation of cardiac remodeling that was associated with maintenance of NAD+ levels in the heart. NAD+ metabolomics and expression analysis showed no evidence for increased levels of PAR, ADPR, Nam or PARylated proteins, which would indicate a storm of NAD+ consuming activities in this model of HF. Instead, a progressive loss of Nampt expression, coupled with the known depression of ATP and high-energy phosphates, is postulated to make it challenging for damaged hearts to maintain NAD+ homeostasis.
Beyond the repletion in myocardial NAD+ levels, the most striking NR-reversible deficit we found in HF was depression of CS and ACL activity. Many regulatory processes from gene expression to enzyme activity are controlled by reversible protein Lys acetylation. Though there are a variety of acetylases and deacetylases that are NAD+-dependent and NAD+-independent, production of citrate in mitochondria and its conversion to cytosolic Ac-coA by the ACL is required to drive changes in substrate fuel selection2 and protein acetylation level.8, 49 NR had no impact on the acetylation level of a mitochondrial protein like Aco2 but robustly increased the acetylation level of FOXO1 or p53 transcription factors in SRFHKO hearts and control hearts, suggesting a tight link between NR-derived NAD+ synthesis and Ac-coA availability for post-translational acetylation of nucleo-cytosolic proteins. This apparently overrides the ability of SIRT1 to deacetylate these factors. At the present stage, it is difficult to assess the consequences of these modifications. Moreover, there is presently no reason to think that FOXO1 and p53 are the only targets whose acetylation level is modified by long term NR treatment. Rather, our data suggest that the acetylation levels of FOXO1 and p53 can be considered as surrogate markers of an increase in nucleo-cytosolic Ac-coA metabolism.
Finally, we observed that NR led to heightened production of Me4PY, a terminal NAM metabolite associated with local production of H2O2. This pathway was shown to promote life extension in the C. elegans through a hormetic response to a moderate rise in ROS, which activates the cytoprotective NRF-2 transcription factor involved in oxidative stress response.34 In the SRFHKO heart, NR was shown to result in Me4PY simultaneous with induction of NRF-2.
Here, we also show a modest though significant protective effect of NR treatment on the drop of LVEF observed in the TAC model. The effect of NR in the TAC model seems to be less important than in the DCM model, which may be linked to the different nature of the cardiac stress in these models, i.e. pressure overload in the TAC model that increases afterload versus volume overload in DCM that increases preload, and the fact that the Nmrk2 gene is less strongly induced in the TAC model than in the SRFHKO model. We note also that the early mortality induced by the TAC led to an early dropout in the untreated and NR-treated TAC groups that may have introduced a bias by removing from the analysis the animals with the worst outcome.
In conclusion, our work shows that oral NR supplementation is a powerful approach to preserve cardiac function and limit remodeling in DCM, a devastating disease currently lacking effective therapies. Recently, mutations in the de novo NAD+ biosynthetic pathway were shown to lead to a new category of genetic diseases called congenital NAD deficiency disorders, which are associated with cardiac and skeletal defects but correctible by niacin supplementation in mouse models of the disease.50 Our study and previous finding by others13, 26–28 suggest that NR has the potential not only to improve the cardiac symptoms in congenital syndromes and acquired HF but also to act at a systemic level notably on liver metabolism, insulin-resistance and skeletal muscle performances that are altered concomitantly with cardiac defects. However, a limitation of our study is the fact that we did not study the impact of NR on the survival rate of SRFHKO mice and occurrence of terminal congestive HF. Further work will be required to determine the pharmacokinetics and pharmacodynamics of therapeutic NR administration in animal models and people suffering HF to enable safe and effective human translation.
Supplementary Material
Clinical Perspective.
What Is New?
Myocardial NAD coenzyme levels are depressed in mouse models of heart failure.
An expression shift occurs in murine and human failing hearts in which the normally predominant NAMPT enzyme using nicotinamide as a precursor for NAD+ synthesis is repressed whereas the NMRK2 enzyme using the nucleoside nicotinamide riboside (NR) is strongly upregulated.
NMRK2 pathway is activated by energy stress in an AMPKα/PPARα-dependent manner in cardiomyocytes.
NR-supplemented diet administrated to murine models of dilated cardiomyopathy or pressure overload-induced heart failure restores myocardial NAD levels and preserves cardiac function.
NR increases glycolysis as well as citrate and acetyl-coA metabolism in cardiomyocytes.
What Are the Clinical Implications?
NR is the most energetically favorable NAD precursor and was recently shown to be uniquely and orally bioavailable in humans in a phase I clinical trial.
NR-supplemented diet could help patients suffering heart failure to cope with limited myocardial ATP supply by restoring NAD coenzyme levels and associated signaling.
NR-supplemented diet has been shown in several studies to enhance metabolic flexibility in the context of obesity and diabetes and to improve muscle function justifying that NR is put in the front line for metabolic therapy of heart failure syndrome in which these functions are altered.
ACKNOWLEDGMENTS
We thank V. Veksler and R. Ventura-Clapier (Inserm UMR-S 1180) for expert advises on respiration assays, Luc Bertrand (UCL, Belgium) for expert advises on AMPK detection, A. Angelini (UMR 8256) for technical help in these assays, A. Grosfeld (UMR 8256) for preliminary set-up experiments for Nmrk2 cloning, Florence Lefebvre for ARC isolation.
SOURCES OF FUNDING
This study was supported by the Association Française contre les Myopathies (AFM N 16282), the Agence Nationale de la Recherche (ANR) grant NAD-Heart ANR-17-CE17-0015-01 and ANR-08-GENOPAT-038, Fondation de France grant 00075811 and fundings from Institut National pour la Santé Et la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), Université Pierre et Marie Curie Paris 6 and University Paris Sud. N.D, C.T., R.D. were supported by a PhD fellowship of French Ministère de la Recherche et de l’Enseignement Supérieur. N.D. was further supported by a one year PhD fellowship from AFM. C.B. is supported by the Roy J. Carver Trust.
Footnotes
DISCLOSURES
C.B. discloses an intellectual property interest in uses of NR. He serves on the Scientific Advisory Board of ChromaDex, which distributes NR and is cofounder and Chief Scientific Advisor of Prohealthspan, which sells NR supplements. The other authors declare no competing financial interest.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Methods, 6 supplemental figures, and 4 supplemental tables.
REFERENCES
- 1.Ventura-Clapier R, Garnier A, Veksler V and Joubert F. Bioenergetics of the failing heart. Biochim Biophys Acta. 2011;1813:1360–72. [DOI] [PubMed] [Google Scholar]
- 2.Des Rosiers C, Labarthe F, Lloyd SG and Chatham JC. Cardiac anaplerosis in health and disease: food for thought. Cardiovascular research. 2011;90:210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mericskay M Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential. Arch Cardiovasc Dis. 2016;109:207–15. [DOI] [PubMed] [Google Scholar]
- 4.Lee CF and Tian R. Mitochondrion as a Target for Heart Failure Therapy- Role of Protein Lysine Acetylation. Circulation journal : official journal of the Japanese Circulation Society. 2015;79:1863–70. [DOI] [PubMed] [Google Scholar]
- 5.Belenky P, Bogan KL and Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–9. [DOI] [PubMed] [Google Scholar]
- 6.Canto C, Menzies KJ and Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell metabolism. 2015;22:31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fouquerel E and Sobol RW. ARTD1 (PARP1) activation and NAD(+) in DNA repair and cell death. DNA Repair (Amst). 2014;23:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghanta S, Grossmann RE and Brenner C. Mitochondrial protein acetylation as a cell-intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl-lysine modifications. Crit Rev Biochem Mol Biol. 2013;48:561–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bindu S, Pillai VB and Gupta MP. Role of Sirtuins in Regulating Pathophysiology of the Heart. Trends in endocrinology and metabolism: TEM. 2016;27:563–73. [DOI] [PubMed] [Google Scholar]
- 10.Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B and Sadoshima J. PPARalpha-Sirt1 Complex Mediates Cardiac Hypertrophy and Failure through Suppression of the ERR Transcriptional Pathway. Cell metabolism. 2011;14:598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillai JB, Isbatan A, Imai S and Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. The Journal of biological chemistry. 2005;280:43121–43130. [DOI] [PubMed] [Google Scholar]
- 12.Bogan KL and Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annual review of nutrition. 2008;28:115–30. [DOI] [PubMed] [Google Scholar]
- 13.Trammell SA, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, Obrosov A, Kardon RH, Yorek MA and Brenner C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci Rep. 2016;6:26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lund LH, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb S, Levvey BJ, Meiser B, Rossano JW, Yusen RD and Stehlik J. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Heart Transplantation Report−-2015; Focus Theme: Early Graft Failure. J Heart Lung Transplant. 2015;34:1244–54. [DOI] [PubMed] [Google Scholar]
- 15.Parlakian A, Charvet C, Escoubet B, Mericskay M, Molkentin JD, Gary-Bobo G, De Windt LJ, Ludosky MA, Paulin D, Daegelen D, Tuil D and Li Z. Temporally controlled onset of dilated cardiomyopathy through disruption of the SRF gene in adult heart. Circulation. 2005;112:2930–9. [DOI] [PubMed] [Google Scholar]
- 16.Tritsch E, Mallat Y, Lefebvre F, Diguet N, Escoubet B, Blanc J, De Windt LJ, Catalucci D, Vandecasteele G, Li Z and Mericskay M. An SRF/miR-1 axis regulates NCX1 and annexin A5 protein levels in the normal and failing heart. Cardiovascular research. 2013;98:372–80. [DOI] [PubMed] [Google Scholar]
- 17.Touvron M, Escoubet B, Mericskay M, Angelini A, Lamotte L, Santini MP, Rosenthal N, Daegelen D, Tuil D and Decaux JF. Locally expressed IGF1 propeptide improves mouse heart function in induced dilated cardiomyopathy by blocking myocardial fibrosis and SRF-dependent CTGF induction. Dis Model Mech. 2012;5:481–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diguet N, Mallat Y, Ladouce R, Clodic G, Prola A, Tritsch E, Blanc J, Larcher JC, Delcayre C, Samuel JL, Friguet B, Bolbach G, Li Z and Mericskay M. Muscle creatine kinase deficiency triggers both actin depolymerization and desmin disorganization by advanced glycation end-products in dilated cardiomyopathy. The Journal of biological chemistry. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang J, Wei L, Otani T, Youker KA, Entman ML and Schwartz RJ. Inhibitory cardiac transcription factor, SRF-N, is generated by caspase 3 cleavage in human heart failure and attenuated by ventricular unloading. Circulation. 2003;108:407–13. [DOI] [PubMed] [Google Scholar]
- 20.Kuwahara K, Teg Pipes GC, McAnally J, Richardson JA, Hill JA, Bassel-Duby R and Olson EN. Modulation of adverse cardiac remodeling by STARS, a mediator of MEF2 signaling and SRF activity. The Journal of clinical investigation. 2007;117:1324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frank D, Rangrez AY, Poyanmehr R, Seeger TS, Kuhn C, Eden M, Stiebeling K, Bernt A, Grund C, Franke WW and Frey N. Mice with cardiac-restricted overexpression of Myozap are sensitized to biomechanical stress and develop a protein-aggregate-associated cardiomyopathy. J Mol Cell Cardiol. 2014;72:196–207. [DOI] [PubMed] [Google Scholar]
- 22.Ho CY, Jaalouk DE, Vartiainen MK and Lammerding J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature. 2013;497:507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bieganowski P and Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. [DOI] [PubMed] [Google Scholar]
- 24.Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS and Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell. 2007;129:473–84. [DOI] [PubMed] [Google Scholar]
- 25.Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L and Auwerx J. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154:430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA and Auwerx J. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell metabolism. 2012;15:838–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, Viscomi C and Zeviani M. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell metabolism. 2014;19:1042–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsstrom S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J and Suomalainen A. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO molecular medicine. 2014;6:721–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME and Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nature communications. 2016;7:12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garnier A, Zoll J, Fortin D, N’Guessan B, Lefebvre F, Geny B, Mettauer B, Veksler V and Ventura-Clapier R. Control by circulating factors of mitochondrial function and transcription cascade in heart failure: a role for endothelin-1 and angiotensin II. Circulation Heart failure. 2009;2:342–350. [DOI] [PubMed] [Google Scholar]
- 31.Grozio A, Sociali G, Sturla L, Caffa I, Soncini D, Salis A, Raffaelli N, De Flora A, Nencioni A and Bruzzone S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. The Journal of biological chemistry. 2013;288:25938–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belenky P, Christensen KC, Gazzaniga F, Pletnev AA and Brenner C. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals. Quantitative basis for Urh1 and purine nucleoside phosphorylase function in NAD+ metabolism. The Journal of biological chemistry. 2009;284:158–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trammell SA and Brenner C. Targeted, LCMS-based Metabolomics for Quantitative Measurement of NAD(+) Metabolites. Computational and structural biotechnology journal. 2013;4:e201301012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmeisser K, Mansfeld J, Kuhlow D, Weimer S, Priebe S, Heiland I, Birringer M, Groth M, Segref A, Kanfi Y, Price NL, Schmeisser S, Schuster S, Pfeiffer AF, Guthke R, Platzer M, Hoppe T, Cohen HY, Zarse K, Sinclair DA and Ristow M. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol. 2013;9:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Preiss J and Handler P. Biosynthesis of diphosphopyridine nucleotide. II. Enzymatic aspects. The Journal of biological chemistry. 1958;233:493–500. [PubMed] [Google Scholar]
- 36.Galli U, Travelli C, Massarotti A, Fakhfouri G, Rahimian R, Tron GC and Genazzani AA. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J Med Chem. 2013;56:6279–96. [DOI] [PubMed] [Google Scholar]
- 37.Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A and Gupta MP. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. The Journal of biological chemistry. 2010;285:3133–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu CP, Yamamoto T, Oka S and Sadoshima J. The function of nicotinamide phosphoribosyltransferase in the heart. DNA Repair (Amst). 2014;23:64–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G and Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. The Journal of clinical investigation. 2007;117:1282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akbay EA, Moslehi J, Christensen CL, Saha S, Tchaicha JH, Ramkissoon SH, Stewart KM, Carretero J, Kikuchi E, Zhang H, Cohoon TJ, Murray S, Liu W, Uno K, Fisch S, Jones K, Gurumurthy S, Gliser C, Choe S, Keenan M, Son J, Stanley I, Losman JA, Padera R, Bronson RT, Asara JM, Abdel-Wahab O, Amrein PC, Fathi AT, Danial NN, Kimmelman AC, Kung AL, Ligon KL, Yen KE, Kaelin WG Jr., Bardeesy N and Wong KK D-2-hydroxyglutarate produced by mutant IDH2 causes cardiomyopathy and neurodegeneration in mice. Genes Dev. 2014;28:479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, Attie AD, Muoio DM and Kelly DP. A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res. 2014;114:626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu W, Barrientos T, Mao L, Rockman HA, Sauve AA and Andrews NC. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015;13:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vega RB and Kelly DP. Cardiac nuclear receptors: architects of mitochondrial structure and function. The Journal of clinical investigation. 2017;127:1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pol CJ, Lieu M and Drosatos K. PPARs: Protectors or Opponents of Myocardial Function? PPAR Res. 2015;2015:835985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S and Sadoshima J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PloS one. 2014;9:e98972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE and Tian R. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation. 2016;134:883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, Kulkarni SS, Rodrigues M, Redpath P, Migaud ME, Auwerx J, Yanes O, Brenner C and Canto C. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nature communications. 2016;7:13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fletcher RS, Ratajczak J, Doig CL, Oakey LA, Callingham R, Da Silva Xavier G, Garten A, Elhassan YS, Redpath P, Migaud ME, Philp A, Brenner C, Canto C and Lavery GG. Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol Metab. 2017;6:819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR and Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi H, Enriquez A, Rapadas M, Martin E, Wang R, Moreau J, Lim CK, Szot JO, Ip E, Hughes JN, Sugimoto K, Humphreys DT, McInerney-Leo AM, Leo PJ, Maghzal GJ, Halliday J, Smith J, Colley A, Mark PR, Collins F, Sillence DO, Winlaw DS, Ho JWK, Guillemin GJ, Brown MA, Kikuchi K, Thomas PQ, Stocker R, Giannoulatou E, Chapman G, Duncan EL, Sparrow DB and Dunwoodie SL. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. The New England journal of medicine. 2017;377:544–552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.