

Summary
Inflammation is a part of the body's immune response for protection against pathogenic infections and other cellular damages; however, chronic inflammation is a major cause of various diseases. One key step in the inflammatory response is the activation of inflammasomes, intracellular protein complexes comprising pattern recognition receptors and other inflammatory molecules. The role of the NLRP3 inflammasome in inflammatory responses has been extensively investigated; however, the caspase‐11 inflammasome has been recently identified and has been classified as a ‘non‐canonical’ inflammasome, and emerging studies have highlighted its role in inflammatory responses. Because the ligands and the mechanisms for the activation of these two inflammasomes are different, studies to date have separately described their roles, although recent studies have reported the functional cooperation between these two inflammasomes during an inflammatory response. This review discusses the studies investigating the functional crosstalk between non‐canonical caspase‐11 and canonical NLRP3 inflammasomes in the context of inflammatory responses; moreover, it provides insight for the development of novel anti‐inflammatory therapeutics to prevent and treat infectious and inflammatory diseases.
Keywords: canonical, caspase‐11, crosstalk, inflammasome, NLRP3, non‐canonical
The functional crosstalk between the non‐canonical caspase‐11 inflammasome and the canonical NLRP3 inflammasome, the most extensively studied canonical inflammasome during inflammatory responses and the pathogenesis of infectious and inflammatory diseases.
Abbreviations
- AIM2
absent in melanoma 2
- ALR
AIM2‐like receptor
- AP‐1
activator protein‐1
- CARD
caspase recruit domain
- DAMPs
danger‐associated molecular patterns
- GBP
guanylate‐binding protein
- GSDMD
gasdermin D
- HMGB1
hepatocyte‐related high‐mobility group box 1
- IL
interleukin
- iLPS
intracellular lipopolysaccharide
- IRF
interferon regulatory factor
- LPG
lipophosphoglycan
- LPS
lipopolysaccharide
- LRRs
leucine‐rich repeats
- METH
methamphetamine
- NACHT
nucleotide‐binding and oligomerization domain
- NLR
NOD‐like receptor
- NLRs
NF‐κB, nuclear factor‐kappa B
- NOD
nucleotide‐binding oligomerization domain
- OMV
outer membrane vesicle
- PAMPs
pathogen‐associated molecular patterns
- PRRs
pattern recognition receptors
- PYD
pyrin domain
- RAGE
receptor for advanced glycation end‐product
- RLR
RIG‐I‐like receptor
- TLR
toll‐like receptor
Introduction
Inflammation is an innate immunity response comprising a series of biological processes that protect the body from invading pathogens and intracellular stress signals. It is characterized by five hallmarks: redness, heat, pain, swelling, and loss of tissue function.1, 2 An inflammatory response is induced by the recognition of pathogen‐associated molecular patterns (PAMPs) and danger‐associated molecular patterns (DAMPs) via pattern recognition receptors (PRRs) that are expressed on the surface or in the cytoplasm of inflammatory cells.1, 2 Activation steps of inflammatory responses are different depending on PRRs. Toll‐like receptor (TLR)‐induced inflammatory responses only activate the priming step, whereas inflammasome‐induced inflammatory responses activate priming and triggering steps. Priming is induced by extracellular PAMPs and DAMPs via cell surface PRRs, such as TLRs, and activates inflammatory signalling pathways, including nuclear factor‐kappa B (NF‐κB), activator protein‐1 (AP‐1) and interferon regulatory factors (IRFs), leading to the production of inflammatory mediators and expression of inflammatory genes.3, 4, 5 Alternatively, triggering is induced by intracellular PAMPs and DAMPs via intracellular PRRs, such as nucleotide‐binding oligomerization domain (NOD)‐like receptors (NLRs), RIG‐I‐like receptors (RLRs), absent in melanoma 2 (AIM2)‐like receptors (ALRs) and caspase‐11, and facilitates the inflammasome activation and pyroptosis.6, 7, 8, 9
Inflammasomes are intracellular protein complexes that comprise intracellular PRRs and inflammatory molecules. Inflammasomes are classified into two main groups: the canonical and non‐canonical inflammasomes. Canonical inflammasomes include the AIM2 inflammasome and NLR family inflammasomes, such as NLRP1, NLRP3, NLRP6, NLRP12 and NLRC4, whereas non‐canonical inflammasomes includes the mouse caspase‐11 and the human homologues of caspase‐11, that is, caspase‐4 and ‐5 inflammasomes.6, 7, 8, 9, 10, 11 These inflammasomes are activated during inflammatory responses and the pathogenesis of infectious and inflammatory diseases. Their activation induces gasdermin D (GSDMD)‐mediated pyroptosis and the proteolytic activation of caspase‐1, leading to subsequent proteolytic maturation of the pro‐inflammatory cytokines interleukin (IL)‐1β and IL‐18 and their secretion by the cell through GSDMD pores.8, 12
A large number of studies exist that have extensively demonstrated the regulatory roles of the canonical inflammasomes during inflammatory responses and the pathogenesis of infectious and inflammatory diseases.13, 14, 15, 16 Mouse caspase‐11 and human caspase‐4/5 inflammasomes have been recently discovered, and emerging studies are demonstrating that caspase‐4/5/11 inflammasomes are also activated during inflammatory responses, ultimately leading to GSDMD‐mediated pyroptosis and caspase‐1‐mediated maturation and secretion of IL‐1β and IL‐18 in a canonical inflammasome‐independent manner. Therefore, caspases‐4/5/11 have been described as ‘non‐canonical’ inflammasomes.17, 18, 19, 20, 21 Canonical and non‐canonical inflammasomes have different PRRs, and are activated by different types of PAMPs and DAMPs that are unique and specific for each inflammasome. Therefore, most studies have focused on the role of inflammasomes individually. Regardless of the inflammasome types, its activation shares similar downstream effector functions, suggesting that inflammasomes, particularly canonical and non‐canonical inflammasomes, functionally cooperate to induce an inflammatory response. Indeed, recent studies have demonstrated the functional crosstalk between inflammasomes. This review aims to summarize and discuss the recent research progress in understanding the functional crosstalk between the non‐canonical caspase‐11 inflammasome and the canonical NLRP3 inflammasome, the most extensively studied canonical inflammasome during inflammatory responses and the pathogenesis of infectious and inflammatory diseases. Moreover, this review highlights the potential pathways that inhibit the activation and functional crosstalk of inflammasomes to provide insight into the future development of effective therapeutics to prevent and treat human infectious and inflammatory diseases.
Non‐canonical caspase‐11 inflammasome and its activators
Activation of caspase‐11 non‐canonical inflammasome by intracellular lipopolysaccharide
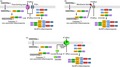
Caspase‐11 consists of three main domains: an N‐terminal caspase recruitment domain (CARD), a p20 domain and a C‐terminal p10 domain (Fig. 1a).10 The non‐canonical caspase‐11 inflammasome is assembled and activated in response to the lipopolysaccharide (LPS) internalized into the cells. LPS is a cell wall component and the most conserved PAMP in Gram‐negative bacteria. Extracellular LPS derived from Gram‐negative bacteria enters cells through TLR4/MD2/CD14 receptor complex‐mediated endocytosis or via bacterial outer membrane vesicle (OMV)‐mediated internalization (Fig. 1b).22 LPS is also released from the internalized or vacuole‐living Gram‐negative bacteria with help from an interferon (IFN)‐inducible GTPase family member, guanylate‐binding protein (GBP). GBPs expressed by the IFN signalling bind with the vacuoles containing Gram‐negative bacteria and disrupt the vacuole membrane integrity, resulting in the cytosolic access of LPS to caspase‐11.23, 24 Extracellular LPS is also internalized by the cells via the cell surface receptor for advanced glycation end‐product (RAGE). Extracellular LPS interacts with hepatocyte‐related high‐mobility group box 1 (HMGB1) to produce the LPS−HMGB1 complex, and this complex then enters the cells via RAGE‐mediated endocytosis (Fig. 1b).25
Figure 1.
Structures and activation of non‐canonical inflammasomes. (a) Domain structures of mouse caspase‐11 and human caspase‐4 and ‐5. Caspase‐4/5/11 consists of three main domains; an N‐terminal CARD, a p20 and a C‐terminal p10, and the amino acid lengths of the caspase‐4/5/11 are 377, 434 and 373, respectively. (b) The mechanisms by which LPS enters the cells. LPS derived from the infected Gram‐negative bacteria binds with TLR4 with the help of CD14 and MD2, and the TLR4‐MD2‐LPS complex is internalized in the cells by the endocytosis. Gram‐negative bacteria also generate the OMVs containing LPS, and the OMVs are internalized in the cells by the endocytosis. In addition, GBPs bind with the vacuoles containing the internalized or vacuole‐living Gram‐negative bacteria and disrupt the vacuoles, resulting in the release of LPS into the cytosol. Moreover, LPS binds to HMGB1, and the HMGB1‐LPS binds to RAGE, leading to the internalization of the RAGE‐HMGB1‐LPS complex by the endocytosis. (c) Structure of the caspase‐11 non‐canonical inflammasome. Caspase‐11 directly binds with iLPS via the caspase‐11 CARD and LPS lipid A moiety, resulting in the formation of the caspase‐11 non‐canonical inflammasome. CARD, caspase recruit domain; GBP, guanylate‐binding protein; HMGB1, hepatocyte‐related high‐mobility group box 1; iLPS, intracellular LPS; LPS, lipopolysaccharide; OMV, outer membrane vesicle; RAGE, receptor for advanced glycation end‐product; TLR4, Toll‐like receptor 4.
Intracellular LPS (iLPS) is the only known exogenous ligand that directly interacts with caspase‐11, and this interaction is mediated by the caspase‐11 CARD and LPS lipid A moiety (Fig. 1c). Caspase‐11‐iLPS interaction produces the non‐canonical caspase‐11 inflammasome and induces the oligomerization of the caspase‐11−iLPS complex (Fig. 1c). Caspase‐11 inflammasome is in turn activated by auto‐proteolytic cleavage at the aspartic acid residue at the 285th position (Asp285), whereas the cysteine residue at the 254th position (Cys254) is critical for the enzymatic activity of caspase‐11.26 Once the non‐canonical caspase‐11 inflammasome is activated in response to iLPS, it induces the proteolysis of GSDMD at the aspartic acid residue at the 276th position (Asp276), which produces the N‐terminal GSDMD and the C‐terminal GSDMD fragments. The N‐terminal GSDMD fragments then move to the cell surface and create GSDMD pores in the cell membrane, ultimately leading to cell death or pyroptosis. The activation of non‐canonical caspase‐11 inflammasome also facilitates the proteolytic activation of caspase‐1 and caspase‐1‐mediated proteolytic maturation of IL‐1β and IL‐18, leading to the secretion of these cytokines through the GSDMD pores.11, 27 A recent interesting study reported another type of ligand directly interacting with caspase‐11 during the sterile inflammation. Zanoni et al. demonstrated that similar with LPS, the oxidized form of naturally occurring endogenous phospholipids, 1‐palmitoyl‐2‐arachidonoyl‐sn‐glycero‐3‐phosphorylcholine (oxPAPC), directly interacted with caspase‐11 and activated caspase‐11 non‐canonical inflammasome, leading to the NLRP3 inflammasome activation and IL‐1β secretion in dendritic cells.28 However, unlike LPS that interacts with the CARD in caspase‐11, oxPAPC interacted with the caspase domain in caspase‐11, and the downstream activation of NLRP3 inflammasome mediated by oxPAPC‐activated caspase‐11 non‐canonical inflammasome was independent on the caspase‐11 catalytic activity.28 In addition, oxPAPC‐activated caspase‐11 non‐canonical inflammasome did not induce GSDMD‐mediated pyroptosis.28 These results suggest that although oxPAPC is another ligand to directly interact with and activate caspase‐11 non‐canonical inflammasome and downstream NLRP3 inflammasome, caspase‐11 is a unique PRR protease specific for iLPS, and the caspase‐11 non‐canonical inflammasome‐mediated inflammatory responses are induced in a context‐dependent manner.
Chu et al.29 also reported that oxPAPC directly interacts with caspase‐11; however, it competed LPS binding with caspase‐11 and consequently inhibited the activation of caspase‐11 non‐canonical inflammasome, resulting in the suppression of GSDMD‐induced pyroptosis and IL‐1β secretion in macrophages as well as in vivo LPS‐induced septic shock. This finding is in contrast to a previous report proposing that oxPAPC has an agonistic activity for the activation of the caspase‐11 non‐canonical inflammasome in dendritic cells;28 therefore, the role of oxPAPC in the caspase‐11‐mediated inflammatory responses is still unclear and needs to be further investigated.
Although the role of oxPAPC is controversial for the activation of the caspase‐11 non‐canonical inflammasome and the downstream NLRP3 inflammasome in the inflammatory response, these studies provide the critical clue that there might exist other types of ligands than LPS and oxPAPC that directly interact with caspase‐11 and regulate the activation of caspase‐11 non‐canonical inflammasome in the inflammatory responses.
Caspase‐11 is a mouse protein, and efforts have been made to identify the human counterparts of mouse caspase‐11. Shi et al.18 demonstrated that human caspase‐4 and ‐5 directly interact with iLPS and induce pyroptosis. Many studies also demonstrated that the interaction of caspase‐4/5 with iLPS generates non‐canonical caspase‐4/5 inflammasomes, leading to their proteolysis and inducing the GSDMD‐mediated pyroptosis and the proteolytic activation of caspase‐1, leading to the caspase‐1‐mediated secretion of IL‐1β and IL‐18.18, 26, 30, 31, 32, 33, 34 These studies provide a strong evidence that human caspase‐4/5 are homologues of the mouse caspase‐11.
Other activators of caspase‐11 non‐canonical inflammasome
Despite previous studies demonstrating that iLPS can directly activate the non‐canonical caspase‐11 inflammasome during the inflammatory responses induced by Gram‐negative bacterial infections, recent studies have demonstrated that the activation and functional cooperation of the caspase‐11 and NLRP3 inflammasomes during inflammatory responses are also promoted by factors other than iLPS.
As discussed in a previous section, Gram‐negative bacteria strongly activate the caspase‐11 non‐canonical inflammasome during the inflammatory responses by promoting the direct interaction between iLPS and caspase‐11; however, other molecules in the host cells infected with Gram‐negative bacteria have been also reported to play a critical role in the activation of the caspase‐11 non‐canonical inflammasome. Several studies reported that infection of Gram‐negative bacteria, such as Citrobacter rodentium, Escherichia coli, Legionella pneumophila and Yersinia pseudotuberculosis, activated the TIR‐domain‐containing adapter‐inducing interferon β (TRIF), a TLR adaptor and the type I IFN signalling pathway, leading to the caspase‐11 induction and autoactivation and the subsequent NLRP3 inflammasome activation in macrophages.35, 36, 37, 38, 39 These results suggest that the TLR‐TRIF‐IFN signalling axis plays a critical role in the activation of the caspase‐11 non‐canonical inflammasome during the inflammatory responses; however, the molecular mechanism mediating IFN‐induced activation of the caspase‐11 non‐canonical inflammasome is still poorly understood and needs to be demonstrated.
Lupfer et al. reported that NOD and RIP2 are critical factors in the activation of the non‐canonical caspase‐11 inflammasome by modulating reactive oxygen species (ROS) homeostasis during inflammatory responses induced by Gram‐negative bacterial infection. The intracellular NLRs, NOD1 and NOD2 activate the inflammatory NF‐κB and mitogen‐activated protein kinase (MAPK) signalling pathways in response to peptidoglycan fragments, a bacterial cell wall component, by activating their downstream adaptor, RIP2.40, 41 Lupfer et al.42 demonstrated that RIP2 deficiency induced ROS accumulation in macrophages infected with the Gram‐negative bacterium C. rodentium, resulting in the increased expression of caspase‐11 by activation of the c‐Jun N‐terminal kinase signalling pathway and that of the non‐canonical caspase‐11 inflammasome. Moreover, the activated non‐canonical caspase‐11 inflammasome subsequently induced the NLRP3 inflammasome activation in the macrophages. It is noteworthy that ROS generation was induced by RIP2 deficiency because ROS generation is typically increased during inflammatory responses following the activation of NF‐κB and MAPK signalling pathways,3, 4 which are also activated by the NOD2‐RIP2 pathway.40, 41 ROS generation also activates the inflammatory NF‐κB and MAPK signalling pathways,43, 44 suggesting that increased ROS generation by RIP2 deficiency upregulated caspase‐11 expression and that in turn activated the non‐canonical caspase‐11 inflammasome in macrophages. Further studies to elucidate the mechanism of RIP2‐deficiency‐mediated ROS generation and ROS‐induced activation of non‐canonical caspase‐11 inflammasome are required. Cumulatively, these results suggest that the NOD2‐RIP2 axis is a critical player in ROS homeostasis, and that increased ROS level by NOD2‐RIP2 deficiency is another critical factor in the induction and activation of caspase‐11 and caspase‐11‐promoted NLRP3 activation in macrophage‐mediated inflammatory responses.
Gabrielli et al. reported that secreted aspartyl proteinases (Saps) of Candida albicans play a pivotal role in the activation of the non‐canonical caspase‐11 inflammasome. Candida species are one of the most common causes of systemic infection, with up to 50% mortality rate.45 Candida albicans is a pathogenic yeast found in the human gut flora and the main Candida species that causes most of the reported candidiasis cases. Saps are the key virulence determinants in C. albicans,46 with Sap2 and Sap6 reported to induce activation of the NLRP3 inflammasome, resulting in caspase‐1 activation and secretion of IL‐1β and IL‐18 in both monocytes and macrophages.47 Gabrielli et al. reported that Sap2 and Sap6 are also critical activators of the non‐canonical caspase‐11 inflammasome in macrophages. They demonstrated that Sap2 and Sap6 internalized by endocytosis activated the caspase‐11 inflammasome in a type I IFN‐dependent manner.48 Activated caspase‐11 inflammasome cooperated with the NLRP3 inflammasome to activate caspase‐1, leading to the subsequent IL‐1β secretion.48 It was previously believed that LPS is the only activator of the caspase‐11 non‐canonical inflammasome; therefore, Gram‐negative bacteria but not Gram‐positive bacteria and other types of pathogens are able to induce the non‐canonical caspase‐11 inflammasome during the inflammatory responses. Unlike previous observations, this study identified Saps of Candida species as critical factors other than LPS to activate the caspase‐11 non‐canonical inflammasome, leading to the subsequent activation of the NLRP3 inflammasome in macrophages during inflammatory responses. Moreover, this study provided critical evidence that not only Gram‐negative bacteria but also yeast is able to activate the non‐canonical caspase‐11 inflammasome in macrophage‐mediated inflammatory responses. However, further studies demonstrating the mechanisms of how yeast Saps activate the non‐canonical caspase‐11 inflammasome and whether Saps, similar to LPS, interact with caspase‐11 are urgently required. Cumulatively, these results suggest that similar to LPS of Gram‐negative bacteria, other virulence factors from pathogenic yeasts can also induce the caspase‐11 inflammasome activation and facilitate the interplay between caspase‐11 and NLRP3 inflammasome pathways.
Another molecule has been recently reported to activate the non‐canonical caspase‐11 inflammasome, and to promote the functional crosstalk between the caspase‐11 and NLRP3 inflammasomes in the neuroinflammatory responses. Du et al. reported that methamphetamine (METH), an amphetamine‐type drug, acts as a stimulator of caspase‐11 and that functional cooperation between the METH‐induced activation of non‐canonical caspase‐11 inflammasome and the NLRP3 inflammasome function occurs during METH‐mediated neuroinflammation. They demonstrated that METH increased caspase‐11 expression and the non‐canonical caspase‐11 inflammasome in vitro and in vivo.49 Inhibition of the METH‐induced caspase‐11 expression reduced the expression of NLRP3, ASC, caspase‐1, IL‐1β and IL‐18, as well as suppressed the NLRP3 inflammasome activation in astrocytes.49 This study is interesting because not only pathogenic microbes but also an exogenous drug can activate the non‐canonical caspase‐11 inflammasome and induce the functional cooperation between the caspase‐11 and NLRP3 inflammasomes during the inflammatory responses, indicating that the caspase‐11 could be a promising target to alleviate the toxicity induced by drug‐mediated inflammation. However, molecular interaction between exogenous drugs and caspase‐11 and how these drugs can induce the inflammatory responses by activating the caspase‐11 and NLRP3 inflammasomes remains unclear. Therefore, further studies solving these questions need to be conducted. Taken together, these results suggest that unlike pathogenic microbes, exogenous drug METH is the activator of the non‐canonical caspase‐11 inflammasome and that the caspase‐11 inflammasome plays a pivotal role in the METH‐induced neuroinflammation by increasing the expression of NLRP3 inflammasome components as well as by activating the NLRP3 inflammasome pathway.
Lipopolysaccharide is the major glycolipid in Gram‐negative bacterial cell walls, with lipophosphoglycan (LPG) being another type of glycolipid present on the wall surfaces of Leishmania parasites, a group of single‐cell protozoan parasites causing Leishmaniasis in humans and other mammals.50 LPG interacts with TLR2 and enters the macrophages via endocytosis,51 resulting in the production of pro‐inflammatory cytokines.52, 53 Recently, de Carvalho et al. reported that Leishmania parasites‐derived LPG is a critical agonist for the activation of caspase‐11 and NLRP3 inflammasomes in macrophages. They demonstrated that cytoplasmic delivery of LPG in the infected macrophages promoted the non‐canonical caspase‐11 inflammasome activation, leading to pyroptosis, caspase‐1 activation and IL‐1β secretion in macrophages.54 These pathways were impaired in Lpg1−/−‐Leishmania‐infected macrophages.54 Moreover, the LPG‐activated caspase‐11 inflammasome subsequently triggered the activation of NLRP3 inflammasome.54 This study provides critical evidence that caspase‐11 non‐canonical inflammasome could be activated by another type of glycolipid, LPG derived from Leishmania protozoan parasites, indicating that similar to Gram‐negative bacteria, protozoan parasite infection also induces the inflammatory responses by activating the non‐canonical caspase‐11 and NLRP3 inflammasomes in macrophages and that selective targeting of LPG during Leishmania parasite infection could be a promising strategy to ameliorate the Leishmania parasite‐induced inflammation and Leishmaniasis. Although LPG was identified as an activator of the caspase‐11 non‐canonical inflammasome in macrophages, the molecular mechanisms of this event need to be further elucidated. Cumulatively, these results suggest that LPG is another critical agonist to induce the caspase‐11 inflammasome activation, and to trigger the functional cooperation between caspase‐11 and NLRP3 inflammasomes via the caspase‐11‐dependent NLRP3 inflammasome activation in the protozoan parasite‐infected inflammatory responses.
In summary, the non‐canonical caspase‐11 inflammasome is activated by not only iLPS derived from Gram‐negative bacteria, but also other factors derived from Gram‐negative bacteria, yeast, protozoan parasites and even by exogenous drugs, such as METH. Interestingly, unlike iLPS, all these factors activate the caspase‐11 non‐canonical inflammasome by inducing the caspase‐11 expression rather than by directly binding with caspase‐11 during the inflammatory responses. The activation of non‐canonical caspase‐11 inflammasome by any of these factors induces its cooperation with the NLRP3 inflammasome to ultimately trigger an inflammatory response. However, further efforts in identifying and validating more molecular ligands that activate these pathways as well as investigating the molecular mechanisms by which these ligands activate the non‐canonical caspase‐11 and NLRP3 inflammasomes during the inflammatory responses are required.
Canonical and non‐canonical NLRP3 inflammasomes
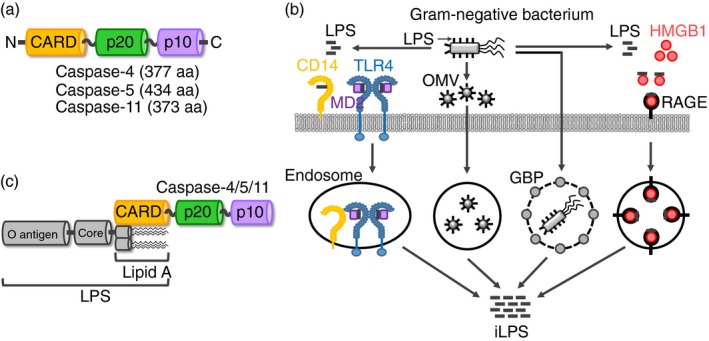
NLRP3 belongs to the NLR family of PRRs and consists of an N‐terminal pyrin domain (PYD), a nucleotide‐binding and oligomerization domain (NACHT), and leucine‐rich repeats (LRRs; Fig. 2a). Under the stimulation, NLRP3 inflammasome is activated and assembled by interaction of NLRP3 with other inflammatory molecules, such as the ASC, a bipartite adapter through their PYDs and subsequent recruitment and interaction with inactive pro‐caspase‐1 through the CARDs of ASC and pro‐caspase‐1 (Fig. 2b).6, 8, 9
Figure 2.
Structure and activation of canonical and non‐canonical NLRP3 inflammasomes (a) Domain structures of NLRP3, ASC and pro‐caspase‐1. NLRP3 consists of three main domains; an N‐terminal PYD, a NACHT and C‐terminal LRRs. ASC consists of two main domains; an N‐terminal PYD and a C‐terminal CARD. Pro‐caspase‐1 consists of three main domains; an N‐terminal CARD, a p20 and a C‐terminal p10. (b) Structure of the NLRP3 inflammasome. NLRP3 binds with ASC via their PYDs, and then pro‐caspase‐1 is recruited and binds with ASC via their CARDs. (c) Two‐signal model for the activation of canonical and non‐canonical NLRP3 inflammasomes. In the priming response, interaction between extracellular PAMPs and PRRs, such as TLRs, induces the transduction cascades of the inflammatory NF‐κB and IRF3 signalling pathways by activating TRIF, resulting in the upregulation of the expression of inflammatory molecules, such as NLRP3, pro‐caspase‐1, caspase‐11 and type I IFN necessary for the subsequent activation of the canonical and non‐canonical NLRP3 inflammasome pathways. The produced type I IFN in turn interacts with extracellular IFNAR in an autocrine manner, and the IFN signalling upregulates the expression of caspase‐11 and GBP. In the triggering response, extracellular and intracellular NLRP3 ligands interact with NLRP3, resulting in canonical NLRP3 inflammasome activation. LPS internalized from the Gram‐negative bacteria directly interacts with caspase‐11, leading to caspase‐11 non‐canonical inflammasome activation, and the activated caspase‐11 non‐canonical inflammasome, in turn, induces non‐canonical NLRP3 inflammasome activation. Activation of canonical and non‐canonical NLRP3 inflammasomes activates caspase‐1, and the activated caspase‐1 induces IL‐1β and IL‐18 secretion and GSDMD‐mediated pyroptosis. CARD, caspase recruit domain; GBP, guanylate‐binding protein; GSDMD, gasdermin D; IFN, interferon; IFNAR, type I IFN receptor; IL, interleukin; IRF3, interferon regulatory factor 3; LRRs, leucine‐rich repeats; NACHT, nucleotide‐binding and oligomerization domain; NF‐κB, nuclear factor‐kappa B; NLR, NOD‐like receptor; PAMP, pathogen‐associated molecular pattern; PRR, pattern recognition receptor; PYD, pyrin domain; TRIF, TIR‐domain‐containing adapter‐inducing interferon β.
An inflammatory response induced by inflammasomes commonly comprises two main consecutive steps: priming and triggering. Priming is the preparatory phase for inflammatory responses, which includes upregulation of the expression of inflammatory molecules, such as PRRs, inflammatory enzymes and pro‐inflammatory cytokines, whereas triggering is the process that boosts the inflammatory responses by inducing the activation of inflammasome responses and inflammatory cell death known as pyroptosis.7, 8 The activation of NLRP3 inflammasome also follows this two‐signal model. The priming response is initiated by the interaction of extracellular PRRs, such as TLR, with PAMPs and subsequently activates the inflammatory signalling pathways, such as NF‐κB and IRF3 via TRIF activation, which induces the expression of NLRP3, pro‐caspase‐1, caspase‐11 and IFN. The produced IFN in turn interacts with its extracellular receptor, IFNAR in an autocrine manner, resulting in the IFN signalling‐induced expression of caspase‐11 and GBP (Fig. 2c). The triggering response is initiated by the activation of NLRP3 in response to various extracellular and intracellular upstream activators, such as ATP, cholesterol, crystals (endogenous and exogenous), alum, asbestos, hyaluronan, β‐amyloids, bacterial pore‐generating toxins, monosodium urate, silica and pathogen‐originated nucleic acid hybrids as well as the downstream cellular mechanisms, such as K+ efflux, Ca2+ influx, phagosomal rupture, oxidized mitochondrial DNA, mitochondrial damage and ROS, leading to the formation of the active NLRP3 inflammasome.6, 7, 8, 9, 55, 56 The activation of NLRP3 inflammasome subsequently induces the proteolytic activation of the inactive pro‐caspase‐1, generating the active caspase‐1 dimers, and the active caspase‐1 in turn induces the GSDMD‐mediated pyroptosis as well as the proteolytic maturation and secretion of IL‐1β and IL‐18 through the GSDMD pores. This process is called the ‘canonical’ NLRP3 inflammasome activation and caspase‐11 is not required for its activation (Fig. 2c).6, 8, 9, 58
Intracellular LPS found in Gram‐negative bacteria, such as E. coli, Haemophilus influenzae, Klebsiella pneumoniae, Neisseria gonorrhea, Shigella flexneri, Enterobacter cloacae, Proteus mirabilis, L. pneumophila and Legionella gratiana, is internalized by several different ways (Fig. 1b), directly interacts with caspase‐11 (Fig. 1c), and activates the non‐canonical caspase‐11 inflammasome, which in turn activates the NLRP3 inflammasome.35, 59 The caspase‐11 non‐canonical inflammasome‐induced NLRP3 inflammasome activation also induces proteolytic activation of caspase‐1, leading to the caspase‐1‐mediated maturation and secretion of IL‐1β and IL‐18 in a manner similar to that of the activation of canonical NLRP3 inflammasome. Moreover, the non‐canonical caspase‐11 inflammasome along with active caspase‐1 also induces the proteolytic processing of GSDMD and the consequent GSDMD‐mediated pyroptosis via generating GSDMD pores in the membrane.6, 8, 9, 58 This kind of NLRP3 inflammasome activation is distinguished from the manner of canonical NLRP3 inflammasome activation, which is caspase‐11‐dependent, leading to the identification of the ‘non‐canonical’ NLRP3 inflammasome activation (Fig. 2c).9, 58
Although the canonical and non‐canonical NLRP3 inflammasomes are activated in different ways, they functionally cooperate to induce the inflammatory responses. NLRP3 inflammasome is activated by canonical NLRP3 stimulants; however, caspase‐11 non‐canonical inflammasome activated by a different stimulant, iLPS, can also activate the NLRP3 inflammasome. The activation signals and mechanisms are different and independent of each other; however, the canonical and non‐canonical NLRP3 inflammasomes have a crosstalk and work together to induce the inflammatory responses. Moreover, both share the common downstream effector functions that induce the inflammatory response via the GSDMD‐mediated pyroptosis and caspase‐1‐mediated maturation and secretion of pro‐inflammatory cytokines, IL‐1β and IL‐18 through the GSDMD pores. Taken together, given the evidence, the activation of non‐canonical NLRP3 inflammasome during an inflammatory response strongly indicates that the canonical NLRP3 inflammasome and non‐canonical caspase‐11 inflammasomes can functionally cooperate to induce the inflammatory response. The molecular mechanisms mediating the crosstalk between these two inflammasomes will be discussed next.
Functional crosstalk of caspase‐11 and NLRP3 inflammasomes during inflammatory responses
As discussed earlier, the regulatory roles of the canonical inflammasomes, particularly NLR family inflammasomes, have long been described in inflammatory responses and the pathogenesis of infectious and inflammatory diseases. Interestingly, a large number of studies have focused on the NLRP3 inflammasome, resulting in the remarkable understanding of its regulatory roles during the inflammatory response and its role in the exacerbation of various human infectious and inflammatory diseases.15, 16, 60, 61, 62, 63, 64, 65, 66, 67, 68 Recently, the caspase‐11 inflammasome was identified as a novel non‐canonical inflammasome to induce inflammatory responses in response to iLPS from Gram‐negative bacteria, and the emerging studies have been successfully demonstrating that similar to the canonical inflammasomes, the caspase‐11 non‐canonical inflammasome also induces inflammatory responses and that the activation of the caspase‐11 non‐canonical inflammasome might play critical roles in the pathogenesis of infectious, inflammatory and autoimmune diseases.8, 27, 69, 70 Despite many studies regarding the regulatory roles of both the canonical NLRP3 and non‐canonical caspase‐11 inflammasomes in inflammatory responses, they have been separately studied, which might be due to their different ligands and activation mechanisms. However, many efforts have been made to demonstrate the functional relationship and cooperation between the two inflammasomes.
Rathinam et al. demonstrated the caspase‐11‐dependent activation of the NLRP3 inflammasome following infection of the Gram‐negative bacteria C. rodentium and E. coli. Gram‐negative bacterial infection activated the TLR4‐TRIF and the type I IFN signalling pathways, leading to the IFN‐mediated caspase‐11 induction and autoactivation in macrophages.35 The caspase‐11 inflammasome activation subsequently synergized the assembly and activation of the NLRP3 inflammasome, which in turn induced caspase‐1 activation and caspase‐1‐mediated pyroptosis.35 Interestingly, these events were not induced by Gram‐positive bacterial infection.35 This study identified TRIF as a critical modulator of the TLR4 pathway in the activation of the non‐canonical caspase‐11 inflammasome during the pathogenesis of Gram‐negative bacterial infection, and suggested that solely Gram‐negative bacteria induce the caspase‐11‐dependent activation of the NLRP3 inflammasome. Another study by Gurung et al.36 demonstrated that infection by the Gram‐negative bacteria C. rodentium and E. coli induced the synthesis and activation of the caspase‐11 inflammasome via the TLR4‐TRIF axis, and that caspase‐11 inflammasome activation in turn activated the NLRP3 inflammasome, leading to caspase‐1 activation and secretion of IL‐1β and IL‐18. This study further confirmed that the TLR4‐TRIF pathway plays a pivotal role in the activation of the caspase‐11 non‐canonical inflammasome, subsequently leading to the activation of the canonical NLRP3 inflammasome in macrophages during infection by enteric Gram‐negative bacteria, but not Gram‐positive bacteria. Although these two studies successfully demonstrated the Gram‐negative bacterium‐induced activation of the non‐canonical caspase‐11 inflammasome and caspase‐11 inflammasome‐dependent activation of the canonical NLRP3 inflammasome, further studies would be necessary to confirm that of all Gram‐negative bacterial PAMPs, LPS is a key determinant for the activation and functional cooperation of these two inflammasomes.
Case et al.37 reported that infection by the Gram‐negative bacterium L. pneumophila in addition to LPS treatment could also activate the caspase‐11 inflammasome in macrophages, resulting in the induction of the caspase‐1 proteolytic activation and caspase‐1‐mediated pyroptosis by activation of the ASC and NLRP3 inflammasome, but not the NLRC4 inflammasome, indicating that caspase‐11‐mediated canonical inflammasome activation is not universal but an inflammasome‐specific event in macrophages during the inflammatory responses. Predictively, L. pneumophila‐mediated caspase‐11 inflammasome activation and the caspase‐11‐induced pyroptosis were deficient in the TRIF/MyD88 double knockout (KO) or type I IFN receptor KO macrophages.37 Interestingly, the caspase‐11 inflammasome activation and caspase‐11‐dependent responses were induced in the macrophages deficient for either the TRIF or MyD88 intracellular adaptors of TLR4.37 This observation differs from that of the studies discussed earlier in this review,35, 36 and the reasons for the discrepancy in these results remain unclear. A possible explanation is that the molecular mechanisms by which Gram‐negative bacteria induce the non‐canonical caspase‐11 inflammasome and the caspase‐11‐dependent NLRP3 inflammasome activation could be different depending on the bacterial subtype, and further studies in this regard need to be conducted. Cumulatively, these studies suggest that Gram‐negative bacteria‐induced caspase‐11 inflammasome activation and caspase‐11‐dependent NLRP3 inflammasome activation require the TLR4‐TRIF‐MyD88 axis and the type I IFN signalling in macrophage‐mediated inflammatory responses.
Another study reported by Casson et al.38 demonstrated that caspase‐11 activation by the Gram‐negative bacteria L. pneumophila and Y. pseudotuberculosis requires TLR4 and TRIF‐dependent type I IFN signalling, and caspase‐11 inflammasome activation consequently induced the caspase‐1 activation and the secretion of both IL‐1β and IL‐1α. Consistent with previous studies discussed earlier in this review, caspase‐11 inflammasome activation induced IL‐1β secretion by activating the NLRP3 inflammasome. However, unlike those previous studies, caspase‐11 inflammasome‐induced IL‐1α secretion was independent from NLRP3 and NLRC4 inflammasomes as well as the TLR4‐TRIF axis and type I IFN signalling.38 These results suggest that IL‐1α secretion is not process‐specific for inflammasome activation, and that caspase‐11 and NLRP3 inflammasomes do or do not cooperate, thereby resulting in different outcomes during macrophage‐mediated inflammatory responses.
Lacey et al.39 recently investigated the role of caspase‐11 and NLRP3 inflammasomes in the pathogenesis of joint inflammation induced by Gram‐negative bacterial infection. They demonstrated that Brucella melitensis infection induced inflammatory joint arthritis by activating the caspase‐11 inflammasome and caspase‐1, subsequently inducing pyroptosis and caspase‐1‐mediated secretion of IL‐1β and IL‐18. Interestingly, the caspase‐11 inflammasome activation did not induce the NLRP3 inflammasome activation, and the NLRP3 inflammasome was dispensable for the caspase‐11 inflammasome‐dependent restriction of Brucella burden in inflammatory joint arthritis.39 It is known that activated macrophages play a detrimental role in the pathogenesis of inflammatory joint arthritis,71, 72, 73, 74 and that caspase‐11 and NLRP3 inflammasomes are highly activated in the activated macrophages during inflammatory responses. However, it remains unclear why Gram‐negative bacterial infection activated the caspase‐11 inflammasome but not NLRP3 inflammasome in this study, and one possible explanation is that not all Gram‐negative bacteria induce inflammatory joint arthritis or that the molecular mechanisms of Gram‐negative bacterium‐induced pathogenesis of inflammatory joint arthritis might not be universal. This study indicates that despite using a similar Gram‐negative bacterial group, the inflammatory response depends on the bacterial species and strain and perhaps their different cell wall structure, which can include different PAMPs. Further studies demonstrating the mechanisms of the pathogenesis of inflammatory joint arthritis and other inflammatory and infectious diseases are required.
Despite previous studies demonstrating the functional cooperation between caspase‐11 and NLRP3 inflammasomes, the underlying molecular mechanisms were poorly understood. Elucidating the mechanisms of their functional crosstalk in the inflammatory responses is essential not only to improve our knowledge of how caspase‐11 and NLRP3 inflammasomes work together to trigger an inflammatory response but also to identify and validate critical molecules that could act as molecular targets for the development of anti‐inflammatory drugs. Recent studies have investigated the molecular and cellular mechanisms of the functional cooperation of the caspase‐11 and NLRP3 inflammasomes in the inflammatory responses.
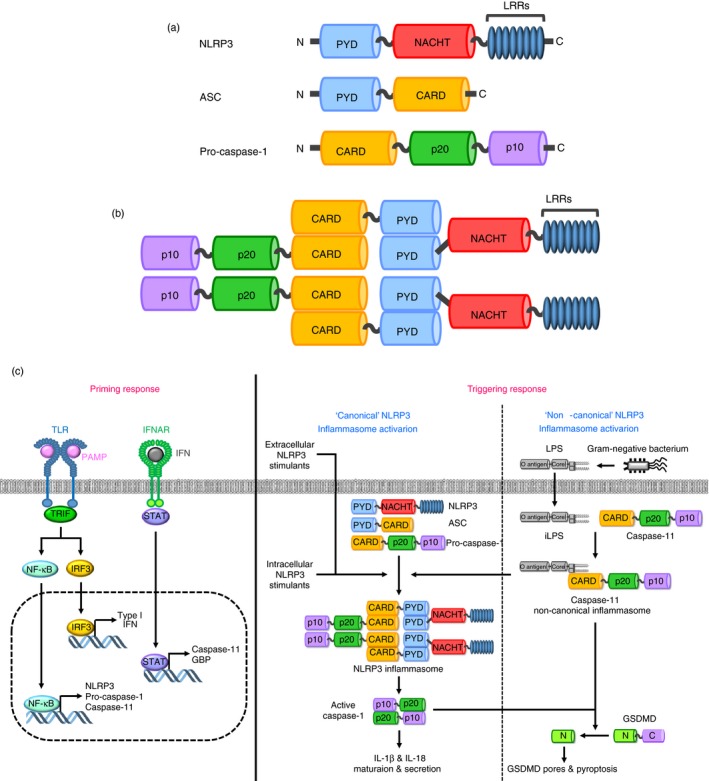
Rühl and Broz investigated the mechanism by which the caspase‐11 inflammasome controls the NLRP3 inflammasome activation and IL‐1β secretion in macrophages. They demonstrated that LPS transfection activated the non‐canonical caspase‐11 inflammasome, subsequently activating NLRP3 inflammasome in macrophages,75 indicating that non‐canonical caspase‐11 inflammasome functions upstream of the canonical NLRP3 inflammasome. They further found that the activation of the non‐canonical caspase‐11 inflammasome promoted K+ efflux, which is essential for the NLRP3 inflammasome activation,6, 9 consequently resulting in the NLRP3 inflammasome activation in macrophages.75 Moreover, caspase‐11‐induced activation of NLRP3 inflammasome induced caspase‐1 activation and caspase‐1‐mediated IL‐1β secretion in macrophages.75 Despite the observation of caspase‐11 non‐canonical inflammasome‐induced K+ efflux, this study did not specify which molecules mediate K+ efflux. Recent studies, however, reported that bacterial pore‐forming toxins and P2X7 promote K+ efflux;76, 77 therefore, caspase‐11 non‐canonical inflammasome‐induced K+ efflux was probably promoted by these molecules. This study clearly outlined the mechanism of non‐canonical caspase‐11 inflammasome‐induced activation of NLRP3 inflammasome by promoting intracellular K+ efflux (Fig. 3a). Studies demonstrating how activation of the non‐canonical caspase‐11 inflammasome induces K+ efflux during the inflammatory responses as well as identifying and validating the critical factors in this event are required.
Figure 3.
Mechanisms of caspase‐11 non‐canonical inflammasome‐mediated activation of NLRP3 inflammasome (a) Caspase‐11 non‐canonical inflammasome activated by the interaction between iLPS and caspase‐11 induces the K+ efflux through the bacterial pore‐forming toxins and P2X7, leading to the activation of NLRP3 canonical inflammasome by assembling NLRP3, ASC and pro‐caspase‐1. (b) Activation of caspase‐11 non‐canonical inflammasome induces the membrane damage of the cells, and the membrane damage consequently triggers the K+ efflux, leading to the activation of NLRP3 canonical inflammasome by assembling NLRP3, ASC and pro‐caspase‐1. (c) Activation of caspase‐11 non‐canonical inflammasome produces the N‐terminal GSDMD fragments by proteolytic cleavage of the full‐length GSDMD, and the N‐terminal GSDMD fragments generate GSDMD pores in the cell membrane, leading to the pyroptosis. K+ efflux is induced through the GSDMD pores, resulting in the activation of NLRP3 canonical inflammasome by assembling NLRP3, ASC and pro‐caspase‐1. C, C‐terminal fragment of GSDMD; CARD, caspase recruit domain; GSDMD, gasdermin D; iLPS, intracellular LPS; LRRs, leucine‐rich repeats; N, N‐terminal fragment of GSDMD; NACHT, nucleotide‐binding and oligomerization domain; NLR, NOD‐like receptor; PYD, pyrin domain.
Cunha et al. also reported the mechanism of caspase‐11‐dependent NLRP3 inflammasome activation by K+ efflux in the Gram‐negative bacterium, L. pneumophila‐induced inflammatory responses. They demonstrated that L. pneumophila infection activated the non‐canonical caspase‐11 inflammasome in macrophages and that this activation induced cell membrane damage in cooperation with the AIM2 inflammasome.79 Cell membrane damage promoted K+ efflux, an essential and sufficient process to activate the NLRP3 inflammasome,75, 80, 81 consequently activating the NLRP3 inflammasome in macrophages.79 Activation of non‐canonical caspase‐11 inflammasome induces pyroptosis, an inflammatory form of cell death by forming GSDMD‐mediated membrane pores, and the mature pro‐inflammatory cytokines, IL‐1β and IL‐18 were then secreted through these pores.10, 11, 82 This study also did not figure out the mechanism by which the activation of caspase‐11 non‐canonical inflammasome‐induced K+ efflux in macrophages. Although it is believed that K+ efflux occurs through the damaged cell membrane, the studies examining whether the caspase‐11 non‐canonical inflammasome‐induced membrane damage in this study is indeed the GSDMD‐mediated pyroptosis or differs from the GSDMD‐mediated pyroptosis. Additionally, apart from the GSDMD‐mediated pyroptosis, other mechanisms of the non‐canonical caspase‐11 inflammasome‐induced cell membrane damage need to be further elucidated. These results suggest that the activation of the non‐canonical caspase‐11 inflammasome by Gram‐negative bacteria promotes NLRP3 activation by inducing membrane damage‐mediated K+ efflux (Fig. 3b). This is another study that adds to the understanding of the mechanisms by which activation of the non‐canonical caspase‐11 inflammasome induces the NLRP3 inflammasome by triggering cell membrane damage and consequent K+ efflux in macrophages.
Kayagaki et al. also reported GSDMD as an activator of NLRP3 inflammasome in macrophage‐mediated inflammatory responses. They demonstrated that the activation of non‐canonical caspase‐11 inflammasome by the delivery of LPS and Gram‐negative bacterial infection processed GSDMD, and the resulting N‐terminal GSDMD fragments promoted the pyroptosis by generating GSDMD pores in the cell membranes and the NLRP3 inflammasome activation, leading to the NLRP3‐dependent caspase‐1 activation and the maturation and secretion of IL‐1β in macrophages.83 Moreover, activation of the NLRP3 inflammasome and caspase‐1‐induced IL‐1β secretion were defective in the GSDMD KO macrophages.83 Despite the clear evidence of GSDMD‐induced activation of NLRP3 inflammasome, the underlying molecular mechanism by which N‐terminal GSDMD fragments produced by caspase‐11 non‐canonical inflammasome activate the NLRP3 inflammasome was unknown. However, recent studies reported that K+ efflux occurs through the GSDMD pores generated by caspase‐11 non‐canonical inflammasome‐processed N‐terminal GSDMD fragments, resulting in the activation of NLRP3 inflammasome.76, 77, 78 The purpose of this study was to demonstrate a novel function of GSDMD as an agonist of the NLRP3 inflammasome, and to elucidate another mechanism by which iLPS‐induced activation of the non‐canonical caspase‐11 inflammasome triggers the NLRP3 canonical inflammasome in the inflammatory responses. These results strongly suggest that the caspase‐11 inflammasome activates NLRP3 inflammasome by inducing K+ efflux through GSDMD pores, resulting in the NLRP3 inflammasome‐mediated downstream inflammatory events, and that GSDMD is a key molecule in the activation of the NLRP3 inflammasome in the inflammatory responses (Fig. 3c).
Despite the small number of studies, these are critical for understanding the mechanisms of the functional crosstalk between the caspase‐11 and NLRP3 inflammasomes. Activation of the non‐canonical caspase‐11 inflammasome triggers K+ efflux by cellular molecules, such as bacterial pore‐forming toxins and P2X7 and also by cell membrane damage, resulting in the canonical NLRP3 inflammasome activation in an inflammatory response. Activation of the non‐canonical caspase‐11 inflammasome produces N‐terminal fragments of GSDMD by proteolytic processing, an effector molecule that promotes the assembly and activation of NLRP3 by inducing K+ efflux through GSDMD pores. Cell membrane damage facilitating K+ efflux might be the GSDMD‐mediated pyroptosis and, if so, GSDMD plays a pivotal role in the functional crosstalk between the caspase‐11 and NLRP3 inflammasomes by inducing the GSDMD pore‐mediated K+ efflux and the activation of NLRP3 canonical inflammasome during the inflammatory responses. Although these studies provided some evidence for the mechanisms of the non‐canonical caspase‐11 inflammasome‐induced activation of the canonical NLRP3 inflammasome, further mechanistic studies are still required for a complete understanding of these pathways.
Conclusion and perspectives
Inflammation is an innate immune response and protective mechanism by which the body eliminates infected pathogens and cellular damages. Chronic inflammation has been considered a critical determinant of the infectious and inflammatory diseases. Therefore, a large number of studies have analysed the mechanisms of the inflammatory response and the pathogenesis of infectious and inflammatory diseases in an attempt to identify and validate the molecular targets critical in the regulation of the inflammatory response to develop effective anti‐inflammatory drugs.
One of the key steps in an inflammatory response is the activation of intracellular inflammasomes in response to unique ligands, leading to the pyroptosis and the caspase‐1‐induced proteolytic maturation and secretion of pro‐inflammatory cytokines IL‐1β and IL‐18 by the inflamed cells. Many studies have mostly focused on the NLRP3 canonical inflammasome because it has been demonstrated to play critical roles in the inflammatory responses and pathogenesis of various infectious, inflammatory and autoimmune diseases.15, 16, 76, 84, 85, 86 However, a novel caspase‐11 inflammasome has been recently identified and demonstrated to induce an inflammatory response by activating downstream effectors, such as caspase‐1 and GSDMD, leading to the GSDMD‐mediated pyroptosis and caspase‐1‐induced secretion of the pro‐inflammatory cytokines IL‐1β and IL‐18. The downstream effector events are similar to those of NLRP3 inflammasome activation; therefore, caspase‐11 inflammasome was considered as the non‐canonical caspase‐11 inflammasome.17 Unlike NLRP3 inflammasome, the non‐canonical caspase‐11 inflammasome was recently identified, and the studies demonstrating the roles of the caspase‐11 non‐canonical inflammasome in the inflammatory responses are limited. However, emerging studies have successfully demonstrated some of the functions of the non‐canonical caspase‐11 inflammasome and the pathogenesis of the infectious and inflammatory diseases. Additionally, they have demonstrated the functional crosstalk between the non‐canonical caspase‐11 and the canonical NLRP3 inflammasomes and the underlying mechanisms in inflammatory responses. These were summarized in Table 1. These studies are still insufficient for a complete and in‐depth understanding of these pathways. Specifically, studies investigating the molecular and cellular mechanisms by which the non‐canonical caspase‐11 inflammasome activates the canonical NLRP3 inflammasome in an inflammatory response are still required. In addition, the functional crosstalk between the non‐canonical caspase‐11 inflammasome and the other types of canonical inflammasomes, such as NLRP1, NLRP6, NLRP12, NLRC4 and AIM2 inflammasomes, in the inflammatory responses and the pathogenesis of the infectious and inflammatory diseases also needs to be further investigated. The identification and validation of novel ligands that activate non‐canonical caspase‐11 inflammasome will expand our understanding of the mechanisms by which it can then subsequently activate the canonical NLRP3 inflammasome.
Table 1.
Summary of functional cooperation between caspase‐11 non‐canonical and NLRP3 canonical inflammasomes
| Study results | Exp. models | Ref. |
|---|---|---|
| Gram‐negative bacterial infection activated TRIF and type I IFN signalling | Gram‐negative bacterium‐infected BMDMs (Citrobacter rodentium and Escherichia coli) | 35 |
| Gram‐negative bacterial infection activated caspase‐11 inflammasome | ||
| Caspase‐11 inflammasome activation subsequently activated NLRP3 inflammasome, resulting in caspase‐1 activation and caspase‐1‐mediated pyroptosis | ||
| Gram‐negative bacterial infection activated TLR4‐TRIF axis | Gram‐negative bacterium‐infected BMDMs (C. rodentium and E. coli) | 36 |
| Gram‐negative bacterial infection activated caspase‐11 inflammasome | ||
| Caspase‐11 inflammasome activation subsequently activated NLRP3 inflammasome, resulting in caspase‐1 activation and caspase‐1‐mediated IL‐1β and IL‐18 secretion | ||
| Gram‐negative bacterial infection activated caspase‐11 inflammasome | Gram‐negative bacterium‐infected BMDMs (Legionella pneumophila) | 37 |
| Caspase‐11 inflammasome activation subsequently activated NLRP3 inflammasome, resulting in caspase‐1 activation and caspase‐1‐mediated pyroptosis | ||
| Caspase‐11 inflammasome activation did not occur in TRIF/MyD88 double KO or type I IFN receptor KO macrophages | ||
| Gram‐negative bacterial infection activated caspase‐11, NLRP3 inflammasomes and caspase‐1, and induced IL‐1β and IL‐1α secretion | Gram‐negative bacterium‐infected BMDMs (L. pneumophila and Yersinia pseudotuberculosis) | 38 |
| Caspase‐11‐mediated IL‐1β secretion was dependent on NLRP3 inflammasome and caspase‐1, | ||
| but caspase‐11‐mediated IL‐1α secretion was NLRP3 inflammasome/caspase‐1‐independent | ||
| Gram‐negative bacterial infection activated caspase‐1 and caspase‐11, resulting in pyroptosis and caspase‐1‐mediated IL‐1β and IL‐18 secretion | Gram‐negative bacterium‐infected arthritic mice (Brucella melitensis) | 39 |
| Caspase‐11 inflammasome activation did not activate NLRP3 inflammasome | ||
| NLRP3 inflammasome was dispensable for caspase‐11‐mediated restriction of bacterial burdens | ||
| RIP2‐deficiency increased ROS production | Nod2‐ or Rip2‐deficient BMDMs | 42 |
| Increased ROS‐induced caspase‐11 inflammasome expression and activation, resulting in NRLP3 inflammasome activation | ||
| Sap2 and Sap6 secreted from pathogenic yeast (Candida albicans) activated caspase‐11 inflammasome | Sap2 or Sap6‐treated Raw264·7 and murine peritoneal macrophages | 48 |
| Caspase‐11 inflammasome activation subsequently activated NLRP3 inflammasome, resulting in caspase‐1 activation and caspase‐1‐mediated IL‐1β secretion | ||
| METH‐induced caspase‐11 expression and activated caspase‐11 non‐canonical inflammasome in vitro and in vivo | METH‐treated astrocytes | 49 |
| Inhibition of METH‐induced caspase‐11 expression decreased expression of NLRP3, ASC, caspase‐11, IL‐1β and IL‐18, as well as suppressed NLRP3 inflammasome activation in astrocytes | ||
|
Leishmania parasite infection and LPG transfection activated caspase‐11 inflammasome Caspase‐11 inflammasome activation subsequently activated NLRP3 inflammasome, resulting in pyroptosis, caspase‐1 activation and IL‐1β secretion |
Leishmania parasite‐infected BMDMs (L. major, L. braziliensis, and L. amazonensis) LPG‐transfected BMDMs | 54 |
| iLPS‐induced caspase‐11 inflammasome activation decreased intracellular K+ level | LPS‐transfected BMDMs | 75 |
| Activated caspase‐11 inflammasome subsequently activated NLRP3 inflammasome by promoting K+ efflux, resulting in caspase‐1 activation and caspase‐1‐mediated IL‐1β secretion | ||
| Gram‐negative bacterial infection activated caspase‐11 | Gram‐negative bacterium‐infected BMDMs (L. pneumophila) | 79 |
| Caspase‐11 inflammasome activation promoted cell membrane damage and K+ efflux in cooperation with AIM2 inflammasome | ||
| Membrane damage‐triggered K+ efflux induced NLRP3 inflammasome activation | ||
| iLPS‐induced caspase‐11 inflammasome activation cleaved GSDMD | LPS‐transfected BMDMs | 83 |
| Resulting N‐terminal GSDMD fragments induced NLRP3 inflammasome activation and pyroptosis, resulting in caspase‐1 activation and caspase‐1‐mediated IL‐1β secretion |
In conclusion, the studies outlined in this review discussed the functional crosstalk between the non‐canonical caspase‐11 and canonical NLRP3 inflammasomes during inflammatory responses. Selectively targeting these inflammasomes or intervention of their functional crosstalk using pharmacological agents may provide promising strategies to prevent and treat various infectious and inflammatory diseases as well as other inflammation‐induced diseases, such as cancer.
Disclosures
The author declares no conflict of interests.
References
- 1. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002; 20:197–216. [DOI] [PubMed] [Google Scholar]
- 2. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140:805–20. [DOI] [PubMed] [Google Scholar]
- 3. Yi YS, Son YJ, Ryou C, Sung GH, Kim JH, Cho JY. Functional roles of Syk in macrophage‐mediated inflammatory responses. Mediators Inflamm 2014; 2014:270 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang Y, Kim SC, Yu T, Yi YS, Rhee MH, Sung GH et al Functional roles of p38 mitogen‐activated protein kinase in macrophage‐mediated inflammatory responses. Mediators Inflamm 2014; 2014:352 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu T, Yi YS, Yang Y, Oh J, Jeong D, Cho JY. The pivotal role of TBK1 in inflammatory responses mediated by macrophages. Mediators Inflamm 2012; 2012:979 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016; 16:407–20. [DOI] [PubMed] [Google Scholar]
- 7. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014; 157:1013–22. [DOI] [PubMed] [Google Scholar]
- 8. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 2017; 277:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mathur A, Hayward JA, Man SM. Molecular mechanisms of inflammasome signaling. J Leukoc Biol 2018; 103:233–57. [DOI] [PubMed] [Google Scholar]
- 10. Yi YS. Caspase‐11 non‐canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage‐mediated inflammatory responses. Immunology 2017; 152:207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ding J, Shao F. SnapShot: The noncanonical inflammasome. Cell 2017; 168:544–e1. [DOI] [PubMed] [Google Scholar]
- 12. Shi J, Gao W, Shao F. Pyroptosis: gasdermin‐mediated programmed necrotic cell death. Trends Biochem Sci 2017; 42:245–54. [DOI] [PubMed] [Google Scholar]
- 13. Satoh T, Otsuka A, Contassot E, French LE. The inflammasome and IL‐1beta: implications for the treatment of inflammatory diseases. Immunotherapy 2015; 7:243–54. [DOI] [PubMed] [Google Scholar]
- 14. Yang CS, Shin DM, Jo EK. The role of NLR‐related protein 3 inflammasome in host defense and inflammatory diseases. Int Neurourol J 2012; 16:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res 2015; 8:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yi YS. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J Physiol Pharmacol 2018; 22:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J et al Non‐canonical inflammasome activation targets caspase‐11. Nature 2011; 479:117–21. [DOI] [PubMed] [Google Scholar]
- 18. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P et al Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014; 514:187–92. [DOI] [PubMed] [Google Scholar]
- 19. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM et al Caspase‐11 increases susceptibility to Salmonella infection in the absence of caspase‐1. Nature 2012; 490:288–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 2013; 341:1250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi‐Takamura S et al Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013; 341:1246–9. [DOI] [PubMed] [Google Scholar]
- 22. Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD et al Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase‐11 activation. Cell 2016; 165:1106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S et al Caspase‐11 activation requires lysis of pathogen‐containing vacuoles by IFN‐induced GTPases. Nature 2014; 509:366–70. [DOI] [PubMed] [Google Scholar]
- 24. Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K et al Guanylate binding proteins promote caspase‐11‐dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci USA 2014; 111:6046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X et al The Endotoxin delivery protein HMGB1 mediates caspase‐11‐dependent lethality in sepsis. Immunity 2018; 49:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose‐Girma M, Anderson K et al Caspase‐11 auto‐proteolysis is crucial for noncanonical inflammasome activation. J Exp Med 2018; 215:2279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yi YS. Regulatory roles of the caspase‐11 non‐canonical inflammasome in inflammatory diseases. Immune Netw 2018; 18:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J et al An endogenous caspase‐11 ligand elicits interleukin‐1 release from living dendritic cells. Science 2016; 352:1232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM et al The oxidized phospholipid oxPAPC protects from septic shock by targeting the non‐canonical inflammasome in macrophages. Nat Commun 2018; 9:996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM et al Human caspase‐4 mediates noncanonical inflammasome activation against gram‐negative bacterial pathogens. Proc Natl Acad Sci USA 2015; 112:6688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, Bozdagi O et al A critical role for human caspase‐4 in endotoxin sensitivity. J Immunol 2014; 193:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goddard PJ, Sanchez‐Garrido J, Slater SL, Kalyan M, Ruano‐Gallego D, Marches O et al Enteropathogenic Escherichia coli stimulates effector‐driven rapid caspase‐4 activation in human macrophages. Cell Rep 2019; 27:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bitto NJ, Baker PJ, Dowling JK, Wray‐McCann G, De Paoli A, Tran LS et al Membrane vesicles from Pseudomonas aeruginosa activate the noncanonical inflammasome through caspase‐5 in human monocytes. Immunol Cell Biol 2018; 96:1120–30. [DOI] [PubMed] [Google Scholar]
- 34. Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human caspase‐4 and caspase‐5 regulate the one‐step non‐canonical inflammasome activation in monocytes. Nat Commun 2015; 6:8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM et al TRIF licenses caspase‐11‐dependent NLRP3 inflammasome activation by gram‐negative bacteria. Cell 2012; 150:606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z et al Toll or interleukin‐1 receptor (TIR) domain‐containing adaptor inducing interferon‐beta (TRIF)‐mediated caspase‐11 protease production integrates Toll‐like receptor 4 (TLR4) protein‐ and Nlrp3 inflammasome‐mediated host defense against enteropathogens. J Biol Chem 2012; 287:34 474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA et al Caspase‐11 stimulates rapid flagellin‐independent pyroptosis in response to Legionella pneumophila . Proc Natl Acad Sci USA 2013; 110:1851–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B et al Caspase‐11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog 2013; 9:e1003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lacey CA, Mitchell WJ, Dadelahi AS, Skyberg JA. Caspase‐1 and Caspase‐11 mediate pyroptosis, inflammation, and control of Brucella joint infection. Infect Immun 2018; 86: e00361‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Girardin SE, Tournebize R, Mavris M, Page AL, Li X, Stark GR et al CARD4/Nod1 mediates NF‐kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep 2001; 2:736–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body‐Malapel M et al RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol 2007; 178:2380–6. [DOI] [PubMed] [Google Scholar]
- 42. Lupfer CR, Anand PK, Liu Z, Stokes KL, Vogel P, Lamkanfi M et al Reactive oxygen species regulate caspase‐11 expression and activation of the non‐canonical NLRP3 inflammasome during enteric pathogen infection. PLoS Pathog 2014; 10:e1004410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y et al ROS and ROS‐mediated cellular signaling. Oxid Med Cell Longev 2016; 2016:4 350 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu Q, Nie SP, Wang JQ, Yin PF, Huang DF, Li WJ et al Toll‐like receptor 4‐mediated ROS signaling pathway involved in Ganoderma atrum polysaccharide‐induced tumor necrosis factor‐alpha secretion during macrophage activation. Food Chem Toxicol 2014; 66:14–22. [DOI] [PubMed] [Google Scholar]
- 45. Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 2010; 36:1–53. [DOI] [PubMed] [Google Scholar]
- 46. Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev 2003; 67:400–28, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pietrella D, Pandey N, Gabrielli E, Pericolini E, Perito S, Kasper L et al Secreted aspartic proteases of Candida albicans activate the NLRP3 inflammasome. Eur J Immunol 2013; 43:679–92. [DOI] [PubMed] [Google Scholar]
- 48. Gabrielli E, Pericolini E, Luciano E, Sabbatini S, Roselletti E, Perito S et al Induction of caspase‐11 by aspartyl proteinases of Candida albicans and implication in promoting inflammatory response. Infect Immun 2015; 83:1940–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Du SH, Qiao DF, Chen CX, Chen S, Liu C, Lin Z et al Toll‐Like receptor 4 mediates methamphetamine‐induced neuroinflammation through caspase‐11 signaling pathway in astrocytes. Front Mol Neurosci 2017; 10:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Turco SJ, Descoteaux A. The lipophosphoglycan of Leishmania parasites. Annu Rev Microbiol 1992; 46:65–94. [DOI] [PubMed] [Google Scholar]
- 51. Srivastava S, Pandey SP, Jha MK, Chandel HS, Saha B. Leishmania expressed lipophosphoglycan interacts with Toll‐like receptor (TLR)‐2 to decrease TLR‐9 expression and reduce anti‐leishmanial responses. Clin Exp Immunol 2013; 172:403–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de Assis RR, Ibraim IC, Nogueira PM, Soares RP, Turco SJ. Glycoconjugates in New World species of Leishmania: polymorphisms in lipophosphoglycan and glycoinositolphospholipids and interaction with hosts. Biochim Biophys Acta 2012; 1820:1354–65. [DOI] [PubMed] [Google Scholar]
- 53. Franco LH, Beverley SM, Zamboni DS. Innate immune activation and subversion of Mammalian functions by leishmania lipophosphoglycan. J Parasitol Res 2012; 2012:165 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. de Carvalho RVH, Andrade WA, Lima‐Junior DS, Dilucca M, de Oliveira CV, Wang K et al Leishmania lipophosphoglycan triggers caspase‐11 and the non‐canonical activation of the NLRP3 inflammasome. Cell Rep 2019; 26:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tozser J, Benko S. Natural compounds as regulators of NLRP3 inflammasome‐mediated IL‐1beta production. Mediators Inflamm 2016; 2016:5 460 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sarvestani ST, McAuley JL. The role of the NLRP3 inflammasome in regulation of antiviral responses to influenza A virus infection. Antiviral Res 2017; 148:32–42. [DOI] [PubMed] [Google Scholar]
- 57. Sharma D, Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol 2016; 213:617–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rivers‐Auty J, Brough D. Potassium efflux fires the canon: Potassium efflux as a common trigger for canonical and noncanonical NLRP3 pathways. Eur J Immunol 2015; 45:2758–61. [DOI] [PubMed] [Google Scholar]
- 59. Gomes MTR, Cerqueira DM, Guimaraes ES, Campos PC, Oliveira SC. Guanylate‐binding proteins at the crossroad of noncanonical inflammasome activation during bacterial infections. J Leukoc Biol 2019; 106:553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mao L, Kitani A, Strober W, Fuss IJ. The role of NLRP3 and IL‐1beta in the pathogenesis of inflammatory bowel disease. Front Immunol 2018; 9:2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D et al The ketone metabolite beta‐hydroxybutyrate blocks NLRP3 inflammasome‐mediated inflammatory disease. Nat Med 2015; 21:263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto‐inflammatory disease CAPS mutations. Nat Immunol 2016; 17:1176–86. [DOI] [PubMed] [Google Scholar]
- 63. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL et al The NLRP3 inflammasome instigates obesity‐induced inflammation and insulin resistance. Nat Med 2011; 17:179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT et al Fatty acid‐induced NLRP3‐ASC inflammasome activation interferes with insulin signaling. Nat Immunol 2011; 12:408–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira‐Saecker A et al NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013; 493:674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010; 464:1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang S, Yuan YH, Chen NH, Wang HB. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson's disease. Int Immunopharmacol 2019; 67:458–64. [DOI] [PubMed] [Google Scholar]
- 68. Shaw PJ, Lukens JR, Burns S, Chi H, McGargill MA, Kanneganti TD. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol 2010; 184:4610–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Krause K, Caution K, Badr A, Hamilton K, Saleh A, Patel K et al CASP4/caspase‐11 promotes autophagosome formation in response to bacterial infection. Autophagy 2018; 14:1928–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hayward JA, Mathur A, Ngo C, Man SM Cytosolic recognition of microbes and pathogens: inflammasomes in action. Microbiol Mol Biol Rev, 2018; 82: e00015‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yi YS, Ayala‐Lopez W, Kularatne SA, Low PS. Folate‐targeted hapten immunotherapy of adjuvant‐induced arthritis: comparison of hapten potencies. Mol Pharm 2009; 6:1228–36. [DOI] [PubMed] [Google Scholar]
- 72. Hu Y, Wang B, Shen J, Low SA, Putt KS, Niessen HWM et al Depletion of activated macrophages with a folate receptor‐beta‐specific antibody improves symptoms in mouse models of rheumatoid arthritis. Arthritis Res Ther 2019; 21:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Feng Y, Shen J, Streaker ED, Lockwood M, Zhu Z, Low PS et al A folate receptor beta‐specific human monoclonal antibody recognizes activated macrophage of rheumatoid patients and mediates antibody‐dependent cell‐mediated cytotoxicity. Arthritis Res Ther 2011; 13:R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yi YS. Folate receptor‐targeted diagnostics and therapeutics for inflammatory diseases. Immune Netw 2016; 16:337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rühl S, Broz P. Caspase‐11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol 2015; 45:2927–36. [DOI] [PubMed] [Google Scholar]
- 76. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019; 19:477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xia S, Hollingsworth LR, Wu H Mechanism and regulation of gasdermin‐mediated cell death. Cold Spring Harb Perspect Biol. 2019. 10.1101/cshperspect.a036400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 2019; 20: E3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cunha LD, Silva ALN, Ribeiro JM, Mascarenhas DPA, Quirino GFS, Santos LL et al AIM2 engages active but unprocessed Caspase‐1 to induce noncanonical activation of the nlrp3 inflammasome. Cell Rep 2017; 20:794–805. [DOI] [PubMed] [Google Scholar]
- 80. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D'Silva DB et al NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase‐4 and caspase‐5. Eur J Immunol 2015; 45:2918–26. [DOI] [PubMed] [Google Scholar]
- 81. Schmid‐Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase‐4 mediates non‐canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol 2015; 45:2911–7. [DOI] [PubMed] [Google Scholar]
- 82. Rathinam VAK, Zhao Y, Shao F. Innate immunity to intracellular LPS. Nat Immunol 2019; 20:527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S et al Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 2015; 526:666–71. [DOI] [PubMed] [Google Scholar]
- 84. Yi YS. Roles of ginsenosides in inflammasome activation. J Ginseng Res 2019; 43:172–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 2018; 17:588–606. [DOI] [PubMed] [Google Scholar]
- 86. Xu C, Lu Z, Luo Y, Liu Y, Cao Z, Shen S et al Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat Commun 2018; 9:4092. [DOI] [PMC free article] [PubMed] [Google Scholar]