

Summary
Immune checkpoint inhibition with monoclonal antibodies is becoming increasingly commonplace in cancer medicine, having contributed to a widening of therapeutic options across oncological indications. Disruption of immune tolerance is the key mechanism of action of checkpoint inhibitors and although immune‐related adverse events are a typical class effect of these compounds, the relationship between toxicity and response is not fully understood. Awareness and vigilance are paramount in recognizing potentially life‐threatening toxicities and managing them in a timely manner. In this review article, we provide an overview of the clinical features, pathological findings and management principles of common immune‐related toxicities, attempting to provide mechanistic insight into an increasingly common complication of cancer therapy.
Keywords: immune checkpoint inhibitors, immunotherapy, toxicity
An overview of the clinical features and management principles of toxicities related to immune checkpoint inhibitors, with a particular focus on current understanding of pathophysiological mechanisms.
Abbreviations
- AIH
autoimmune hepatitis
- CD4, CD8, CD28
cluster of differentiation 4, 8, and 28
- CT
computed tomography
- CTLA‐4
cytotoxic T‐lymphocyte‐associated protein 4
- DILI
drug‐induced liver injury
- GI
gastrointestinal
- IBD
inflammatory bowel disease
- ICI
immune checkpoint inhibitor
- IL‐1
interleukin 1
- iRAE
immune‐related adverse event
- KC
Kupffer cell
- LSEC
liver sinusoidal endothelial cell
- mAb
monoclonal antibody
- NSCLC
non‐small‐cell lung cancer
- PD‐1
programmed cell death protein 1
- PD‐L1 and PD‐L2
programmed cell death protein ligands 1 and 2
- RCC
renal cell cancer
- RGMb
repulsive guidance molecule b
- SCLC
small‐cell lung cancer
Introduction
Anti‐cancer immunotherapy has become an established therapeutic modality for a widening range of malignancies including non‐small‐cell lung cancer (NSCLC), melanoma, urothelial cancer, head and neck and renal cell cancers (RCC). Currently, the most widely used approach is the administration of monoclonal antibodies (mAb) against regulatory immune checkpoint molecules that inhibit T‐cell activation. The use of immune checkpoint inhibitors (ICI) is expected to become more prevalent as new indications for treatment are explored in trials.
Immune checkpoint inhibitors are characterized by an overlapping series of immune‐related adverse events (irAEs), induced by the deregulation of the immune system, which is the basis of their mode of action. Significant attention is required for management, as patterns of toxicity differ from those caused by cytotoxic chemotherapy or molecularly targeted agents. Awareness and early recognition of irAEs is crucial to avert the unnecessary morbidity and mortality associated with the more severe forms of toxicity.
Unlike toxicity from other systemic anti‐cancer treatments, adverse reactions from immunotherapy may occur months to years after the last dose,1, 2 a finding that has prompted the continuation of patient monitoring beyond therapy discontinuation.
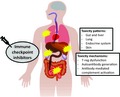
In this review, we provide an overview of the toxicities affected by ICI on key systems, their clinical features and their management principles, and also provide insight into the pathogenesis of these events. Our focus here is the more common or clinically significant toxicities affecting the lung, gut, liver and endocrine system. Detailed clinical guidance on toxicities affecting other systems can be found in published guidelines.3, 4, 5, 6
Pulmonary toxicity
Pulmonary toxicity from ICI involves most typically diffuse inflammation of the lung parenchyma (pneumonitis), whereas other clinicopathological entities such as sarcoid‐like reactions and pleural effusions are rarer.1 Pneumonitis has been described in 2%–5% of patients receiving anti‐programmed cell death protein 1 (PD‐1) monotherapy in clinical trials;2, 7 however, real‐world prevalence might potentially be higher (19%).8 It is the most clinically significant ICI‐associated toxicity, causing death in 1%–2% of cases.1
Association of pneumonitis with PD‐1 axis inhibition is stronger than with that of cytotoxic T‐lymphocyte antigen 4 (CTLA‐4),1 and can be enhanced by combined therapy,2 although this may be explained by the greater number of PD‐1/ programmed cell death protein ligand 1 (PD‐L1) inhibitors in NSCLC trials.9 Rates of pneumonitis with PD‐L1 inhibitors have been found to be lower than with inhibition of PD‐1 in some studies.10, 11 Pneumonitis occurs more frequently in NSCLC and RCC than in melanoma: with precocious onset (2 months) and greater severity in NSCLC,12 where previous radiotherapy and pulmonary co‐morbidities may increase risk.13, 14, 15
Recognition is challenging. Median onset time is approximately 3 months, ranging from a month to 2 years.1, 2, 16 The initial features of dry cough, dyspnoea and hypoxia are non‐specific, being shared with other possible diagnoses, such as disease progression or pseudo‐progression, infection, pulmonary embolus and exacerbation of co‐existing lung disease.2
The key investigation with pneumonitis is high‐resolution computed tomography. Radiographic findings include non‐specific interstitial pneumonia, organizing pneumonia and hypersensitivity pneumonitis, commonly involving the lower lobes.15, 17, 18 Bronchoalveolar lavage aids diagnosis, especially if there is clinical suspicion of infection, or lack of response to immunosuppression.19
Histological findings in pneumonitis include organizing pneumonia, diffuse alveolar damage, and granulomatous inflammation, with a CD4+‐predominant lymphocyte population.2, 15, 20 Although some of these findings are shared with the early stages of lung fibrosis, pneumonitis tends to be reversible. This may be explained, at least in the anti‐PD‐1 setting, by findings pointing to a correlation between lung fibrosis and PD‐1 axis activation. In idiopathic pulmonary fibrosis, anti‐PD‐1 antibody administration in mice leads to reduced fibrosis,21 whereas injection of PD‐L1‐overexpressing fibroblasts into mice promotes fibrosis.22
Understanding of the pathogenesis of ICI pneumonitis is limited and often extrapolated from extrapulmonary findings or inflammatory lung conditions that are idiopathic or related to other aetiologies. Mechanisms probably differ between CTLA‐4 and PD‐1‐targeted therapies: blockade of CTLA‐4 leads to regulatory T‐cell dysfunction and impacts T‐cell priming in draining lymph nodes, whereas PD‐1 blockade has been found to cause regulatory T‐cell dysfunction as well as production of pathological autoantibodies, both in knockout mice and patients.23, 24, 25 Mechanistic insight into anti‐PD‐1‐related pneumonitis is provided by the finding that PD‐L2, found on lung dendritic cells, binds repulsive guidance molecule b (RGMb), found on interstitial macrophages and alveolar epithelium.26 The PD‐L2–RGMb interaction promotes the initial T‐cell expansion required for respiratory immune tolerance in mice.26 Prevention of the interaction of PD‐L2 with PD‐1 is likely to increase the frequency of the PD‐L2–RGMb interaction, so promoting initial T‐cell expansion. However, PD‐1 inhibition may then prevent this T‐cell clone from developing tolerance and lead to immune‐mediated toxicity.26, 27
Treatment of pneumotoxicity is informed by disease severity28 (Table 1) and involves ICI interruption, supportive measures and corticosteroids.4, 29, 30, 31 Subclinical radiographic changes are classified as grade 1 pneumonitis (one‐third of cases) and are treated with treatment cessation until spontaneous resolution. Grade 2 pneumonitis is characterized by mild dyspnoea and cough and can be managed with oral corticosteroids, tapered on symptom resolution for at least 1 month. Grades 3 and 4 (20%–40% of cases) indicate hospitalization. They are defined by severe and life‐threatening symptoms, respectively, and benefit from high‐dose intravenous corticosteroids. Non‐responders may benefit from infliximab, mycophenolate, cyclophosphamide, tocilizumab and interleukin‐1‐targeted agents. Cautious rechallenge with ICI after Grade 1 or 2 pneumonitis is possible, with reported pneumonitis recurrence rates of 25%–33%.2, 32
Table 1.
Indications for immune checkpoint therapy
| Drug | Target | Combinations | Indication |
|---|---|---|---|
| Ipilimumab (Yervoy™) | CTLA‐4 | Monotherapy | Advanced (unresectable or metastatic) melanoma in adults, and adolescents 12 years of age and older |
| + Nivolumab |
Advanced (unresectable or metastatic) melanoma in adults. First‐line treatment of adult patients with intermediate/poor‐risk advanced renal cell carcinoma (RCC) |
||
| Nivolumab (Opdivo™) | PD‐1 | Monotherapy |
Advanced (unresectable or metastatic) melanoma in adults. Adjuvant treatment of adults with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection |
| + Ipilimumab | Advanced (unresectable or metastatic) melanoma in adults. | ||
| Monotherapy | Treatment of advanced renal cell carcinoma after prior therapy in adults. | ||
| + Ipilimumab | First‐line treatment of adult patients with intermediate/poor‐risk advanced renal RCC | ||
| Monotherapy | Locally advanced or metastatic non‐small cell lung cancer (NSCLC) after prior chemotherapy in adults. | ||
| Adult patients with relapsed or refractory classical Hodgkin lymphoma after autologous stem cell transplant (ASCT) and treatment with brentuximab vedotin | |||
| Recurrent or metastatic squamous cell cancer of the head and neck in adults progressing on or after platinum‐based therapy | |||
| Locally advanced unresectable or metastatic urothelial carcinoma in adults after failure of prior platinum‐containing therapy | |||
| Pembrolizumab (Keytruda™) | PD‐1 | Monotherapy | Advanced (unresectable or metastatic) melanoma in adults |
| Adjuvant treatment of adults with Stage III melanoma and lymph node involvement who have undergone complete resection | |||
| + Axitinib | First‐line treatment of advanced RCC in adults | ||
| Monotherapy | First‐line treatment of metastatic NSCLC in adults whose tumours express PD‐L1 with a ≥50% TPS with no EGFR or ALK positive tumour mutations. | ||
| + Pemetrexed and platinum | First‐line treatment of metastatic non‐squamous NSCLC in adults whose tumours have no EGFR or ALK positive mutations. | ||
| + Carboplatin and paclitaxel or nab‐paclitaxel | First‐line treatment of metastatic squamous NSCLC in adults | ||
| Monotherapy | Treatment of locally advanced or metastatic NSCLC in adults whose tumours express PD‐L1 with a ≥1% TPS and who have received at least one prior chemotherapy regimen (EGFR mutant/ALK positive patients should also have received targeted therapy) | ||
| Adult patients with relapsed or refractory classical Hodgkin lymphoma who have failed ASCT and brentuximab vedotin, or who are transplant‐ineligible and have failed brentuximab vedontin. | |||
| Locally advanced or metastatic urothelial carcinoma in adults who have received prior platinum‐containing chemotherapy | |||
| Locally advanced or metastatic urothelial carcinoma in adults who are not eligible for cisplatin‐containing chemotherapy and whose tumours express PD‐L1 with a CPS ≥10 | |||
| Recurrent or metastatic head and neck squamous cell carcinoma (HNSCC) in adults whose tumours express PD‐L1 with a ≥ 50% TPS and progressing on or after platinum‐containing chemotherapy | |||
| Cemiplimab (Libtayo™) | PD‐1 | Monotherapy | Adults with metastatic or locally advanced cutaneous squamous cell carcinoma who are not candidates for curative surgery or curative radiation |
| Atezolizumab (Tecentriq™) | PD‐L1 | Monotherapy |
Treatment of adult patients with locally advanced or metastatic urothelial carcinoma:
|
| + Bevacizumab, paclitaxel and carboplatin | First‐line treatment of adult patients with metastatic non‐squamous NSCLC (Excluding EGFR mutant/ALK positive patients) | ||
| Monotherapy | Adult patients with locally advanced or metastatic NSCLC after prior chemotherapy (EGFR mutant/ALK positive patients should also have received targeted therapy) | ||
| + Nab‐paclitaxel and carboplatin | First‐line treatment of adult patients with metastatic non‐squamous NSCLC who do not have EGFR mutant or ALK‐positive NSCLC | ||
| + Carboplatin and etoposide | first‐line treatment of adult patients with extensive‐stage small cell lung cancer | ||
| Durvalumab (Imfinzi™) | PD‐L1 | Monotherapy | Locally advanced, unresectable NSCLC in adults whose tumours express PD‐L1 on ≥ 1% of tumour cells and whose disease has not progressed following platinum‐based chemoradiation therapy |
| Avelumab (Bavencio™) | PD‐L1 | Monotherapy | Adults patients with metastatic Merkel cell carcinoma |
ALK, Anaplastic lymphoma kinase; CPS, Combined positive score; EGFR, Epidermal growth factor receptor; TPS, Tumour proportion score.
The table summarises the indications for which immune checkpoint inhibitory antibodies are approved for by the European Medicines Agency, either as monotherapy or as part of a combination of agents.
Unless otherwise stated, treatment continues until progression or unacceptable levels of toxicity. All data here is from the electronic medicines compendium Summary of Product characteristics, and are therefore EMA approved. All FDA indications are not listed here.
Questions remain about the relationship of ICI with radiotherapy in NSCLC. So far, the KEYNOTE‐001 and PACIFIC trials demonstrate that radiotherapy before ICI leads to significantly higher pulmonary toxicity rates than radiotherapy alone. However, the incidence of severe pneumonitis, of Grade 3 or above, remains the same.33 Both trials involved administration of ICI after radiotherapy: prospective data on concomitant immunoradiotherapy will be needed to properly untangle the relationship.
Gastrointestinal toxicity
Gastrointestinal (GI) toxicities are among the most common irAEs. These can be severe, with approximately one‐third of ICI toxicity deaths related to gastrointestinal complications,34 and a 1·1% mortality rate described from ipilimumab‐related colitis.35
The time‐scale of such GI complications is variable: typical onset is at approximately 6–7 weeks after treatment initiation, with onset often later for anti‐PD‐1 than anti‐CTLA‐4;36 however, there are rare reports of GI toxicity occurring as late as 1 year after discontinuation of therapy. Diarrhoea is the most common manifestation of GI toxicity, although the more feared complication is clinically significant colitis. Colitis may present with abdominal pain, haematochezia, fever, or other GI or constitutional symptoms. The risk of GI toxicity is higher for anti‐CTLA‐4 compared with anti‐PD‐1 therapy; recent data have described an incidence of diarrhoea of 12·1%–13·7% after anti‐PD‐1, and 30·2%–35·4% after anti‐CTLA‐4, and incidence of colitis of 0·7%–1·6% after anti‐PD‐1, and 5·7%–9·1% after anti‐CTLA‐4.36 GI toxicity (among other irAEs) is higher with combination ICI therapy,37 with reported rates of all‐grades diarrhoea of 44% and grade 3–4 diarrhoea and colitis of 9% in the initial phase III study in unresectable melanoma. Rates also appear dose‐dependent, with severe diarrhoea and colitis occurring in up to 10% of those treated with ipilimumab 10 mg/kg versus 6% with ipilimumab 3 mg/kg.38
ICI‐related colitis is histologically characterized by mucosal oedema, with infiltration by either neutrophils (including cryptitis and crypt microabscesses), lymphocytes (particularly infiltrating the lamina propria) or both.39 Inflammation may only be microscopic, and collagenous colitis has also been described in association with the use of pembrolizumab.40
The mechanisms underlying GI toxicity in ICI therapy are not well understood. It has been observed that hereditary mutations in the CTLA‐4 gene locus lead to gastrointestinal perturbations (diarrhoea, enteropathy) in the majority of patients.41 Loss of CTLA‐4 expression in regulatory T cells impairs physiological immunosuppression in the periphery, possibly accounting for this effect. Animal models of inflammatory bowel disease (IBD) point to the importance of regulatory T cells in disease pathogenesis, and the essential role of CTLA‐4 therein.42 Moreover, genetic studies in humans have shown that polymorphisms in genes encoding CTLA‐4 and PD‐1 are associated with IBD.43 Given the clinical and pathological similarities, it is unsurprising that several IBD therapies are being used in ICI colitis.
In parallel with there being an increasing recognition of the influence of the gut microbiota upon the efficacy of ICI therapy, there are also data demonstrating that baseline gut microbiota may be a key risk factor for whether ICI‐related GI toxicities occur or not. In particular, stool microbiota profiling (via 16S rRNA gene sequencing) of 26 patients demonstrated that higher stool relative abundance of the phylum Firmicutes (particularly members of the genus Faecalibacterium) was associated not only with more frequent occurrence of ipilimumab‐induced colitis, but also with longer overall survival.44 In contrast, patients with higher stool relative abundance of the other major bacterial phylum, Bacteroidetes, had lower rates of colitis,44 a finding supported by a further study of 34 patients.45 Bacteroidetes are established as being able to limit inflammation by stimulation of regulatory T‐cell differentiation,46 providing a possible mechanistic link between certain bacterial taxa within the gut microbiota and future immune‐related toxicity.
Regarding treatment of GI toxicity, European Society of Medical Oncology and American Society of Clinical Oncology guidelines recommend that mild cases be treated symptomatically, including measures such as a low‐fibre diet, oral rehydration and loperamide.4, 47 However, if severe symptoms occur from presentation, or if there is no improvement despite symptomatic measures, then flexible sigmoidoscopy or ileocolonoscopy are appropriate,47 as ulceration on colonoscopy makes subsequent steroid‐refractory disease requiring escalation of therapy more likely.4 Abdominal computed tomography is merited because there is a recognized risk of toxic megacolon, intra‐abdominal abscess or colonic perforation after ipilimumab.48
Although it is important to consider other causes of colitis (including cytomegalovirus colitis, Clostridium difficile infection, IBD), investigations should not delay treatment if there is high suspicion of ICI‐induced diarrhoea and colitis. Oral prednisolone or budesonide are suitable for milder cases, but intravenous methylprednisolone is preferred for more severe/non‐resolving cases.4, 47 ICI usage should be postponed until symptomatic resolution. Where there is no significant resolution with corticosteroids, it is generally recommended to use infliximab,4, 47 with case series suggesting that early introduction is associated with better outcomes.49 There is no clinical evidence for the use of prophylactic corticosteroids to prevent GI toxicity, with a trial evaluating prophylactic budesonide in patients receiving ipilimumab demonstrating no benefit.35 Vedolizumab (a mAb blocking the α 4 β 7 integrin) was given to six patients with steroid‐refractory colitis after ipilimumab, and one patient with prior nivolumab;50 although resolution of colitis was observed, the time to steroid‐free remission was 56 days, similar to that observed in spontaneous remission. In addition, clinical improvement was also reported in two patients with steroid‐refractory ICI colitis treated with faecal microbiota transplant,51 further supporting microbiota–immune crosstalk as a contributory factor to the development of GI toxicities.
Liver toxicity
Hepatotoxicity is one of the commonest forms of irAEs from ICI.52 Hepatotoxicity is frequently asymptomatic and identified by elevations in either aspartate or alanine transaminase.47 Markers of synthetic dysfunction, such as bilirubin and clotting indices, can be useful in risk‐stratifying patients in the most severe categories. In patients treated with single‐agent immunotherapy, 5%–10% will develop any hepatitis, and 1%–2% exhibit grade 3–4 injury.4, 47 In dual therapy, hepatitis is more frequently seen, with 25%–30% developing any grade and up to 15% developing grade 3–4 hepatitis.47
In most cases, the hepatocyte is the cellular target of immune‐mediated damage, with lobular hepatitis being the main pattern of injury.53, 54, 55 This is in keeping with the observation that most patients have a hepatocellular pattern of liver function tests, with alanine transaminase/ aspartate transaminase being most significantly deranged.55 Less commonly, ductular injury can be seen, with patients presenting with jaundice and cholestatic liver function tests, and demonstrating poor response to corticosteroids.56
Hepatocyte injury by both necrosis and apoptosis is observed in biopsies of patients with ICI‐related hepatotoxicity. In many patients, lobular inflammation is accompanied by spotty or confluent hepatocyte necrosis and, in a subset of patients, zone 3 necrosis is observed.54 Evidence of multifocal hepatocyte apoptosis and ballooning degeneration can also commonly be seen. Interestingly, anti‐CTLA‐4‐related and anti‐PD‐1‐related hepatotoxicities have been reported to show slightly different histology, though both involve lobular hepatitis.55 In anti‐PD‐1 treatment, granulomatous hepatitis may be observed, with some reports of well‐formed fibrin ring granulomas.57, 58 These are formed of aggregated histiocytes and T cells and are located within the liver lobule. Prominent perivenular injury and endotheliitis have also been reported as a feature of hepatotoxicity in anti‐CTLA‐4‐treated patients.55, 59 Endotheliitis is frequently seen in acute cellular rejection of liver allografts, and suggests an active recruitment of lymphocytes from the sinusoids and venules with endothelial damage.60 The lymphocyte population recruited to the liver may also differ, with one study reporting that the lymphocytic infiltrate in anti‐CTLA‐4‐treated patient was predominantly composed of CD8 T cells, contrasting a more even ratio of CD8 : CD4 cells in anti‐PD‐1 therapy.55 These subtle differences are instructive, hinting at mechanistic distinctions in how liver tolerance is broken and hepatic inflammation is triggered with inhibition of different checkpoint pathways.
Similarly, comparative histological studies between immunotherapy‐related hepatotoxicity and other forms of immune‐mediated liver injury can be valuable. In a direct histopathological comparison between autoimmune hepatitis (AIH), ICI‐related hepatitis and idiosyncratic drug‐induced liver injury (DILI), distinct pathological patterns of injury were demonstrated.53 Patients with ICI‐related hepatitis showed fewer plasma cells and a much lower CD4 : CD8 ratio than AIH. They also demonstrated fewer eosinophils than in cases of idiosyncratic DILI. None of the biopsies from immunotherapy patients demonstrated emperipolesis, the finding of a lymphocyte within the cytoplasm of a hepatocyte, or hepatocyte rosetting, which are hallmarks of AIH. These observations suggest a central role for cytotoxic T cells in the pathogenesis of immunotherapy‐related hepatitis. Neither B‐cell activation with hypergammaglobulinaemia and autoantibody production, nor eosinophilic infiltration and hypersensitivity elements, which are important in AIH and idiosyncratic DILI, respectively, appear to have a significant role.
In the liver, Kupffer cells (KC) and liver sinusoidal endothelial cells (LSEC) function as antigen‐presenting cells, and are important in the induction of antigen‐specific immune tolerance to soluble and particle‐associated antigen during homeostasis.61 Steady‐state antigen presentation within hepatic sinusoids by LSEC and KC induces T‐cell tolerance and expansion of regulatory T‐cell populations.61 This effect is thought to be mediated by the relatively high expression of PD‐L1 on KC and LSEC. An LSEC PD‐L1Hi phenotype has been shown to be essential for induction of local CD8 T‐cell tolerance.62
Genetic deletion of PD‐L1 in experimental models leads to accumulation of activated CD8+ T cells within the liver, as the pathway is essential for effective deletion of activated cytotoxic T cells by the liver.63 In PD‐L1‐knockout mice, experimentally induced T‐cell‐mediated hepatitis using the Concanavalin A model progressed more rapidly and was more severe than in wild‐type control mice.63 PD‐L1 expression by hepatic stellate cells has also been shown to permit potent T‐cell suppression by these liver‐resident cells and be essential in mediating the T‐cell inhibition that mediates local tolerance in an islet cell transplantation model.64, 65
In murine models of AIH, neonatal thymectomy can deplete the regulatory T‐cell population.66 However, this does not give rise to liver inflammation without genetic deletion of PD‐1 to overcome local T‐cell inhibition.66 This suggests that the combination of functional regulatory T cells with local tolerance maintained by the PD‐1/PD‐L1 checkpoint blockade is necessary for the maintenance of liver tolerance.
The evidence suggests that the PD‐L1/PD‐1 pathway is important for immune regulation within the hepatic environment, contributing to maintenance of tolerance through the regulation of CD8 T‐cell activation and apoptosis. In anti‐PD‐1/PD‐L1 therapy, it can be seen how the threshold for hepatic inflammation may be lowered, permitting local T‐cell activation and unchecked cytotoxicity.
There is also evidence to link CTLA‐4 function with maintenance of tolerance within the hepatic environment. A number of studies have shown associations between CTLA‐4 polymorphisms and spontaneous liver autoimmune conditions, such as primary biliary cholangitis and AIH.67, 68 In animal models of genetic CTLA‐4 deficiency and human studies of dominant CTLA‐4 haplodeficiency, an immune dysregulation syndrome arises with multisystem involvement, including liver infiltration.41 Autoimmune liver diseases have been linked with quantitative and qualitative defects in regulatory T cells,69 a T‐cell subtype that CTLA‐4 is essential in maintaining.26 It is therefore plausible that impairments in regulatory T‐cell function could contribute to hepatic inflammation in the context of CTLA‐4 ICI.
Both pathways have been implicated in the maintenance of hepatic tolerance. Much more research is required to fully understand why only some patients develop liver inflammation in this context.
Treatment of immunotherapy‐related hepatotoxicity involves cessation of checkpoint inhibitors and commencement of high‐dose corticosteroid therapy, sometimes supplemented by a second‐line immunomodulatory agent such as mycophenolate mofetil and/or tacrolimus.70 The spectrum of disease can range from mild grade 1 transaminitis to fulminant liver failure. In severe cases, anti‐thymocyte globulin has been used,71 though this should only be undertaken with advice from an experienced hepatologist. Notably, the recommended first‐line treatment for induction of remission in AIH is azathioprine in addition to corticosteroids,72 as opposed to ICI‐related toxicity.
Endocrine toxicity
Hypophysitis, thyroid disorders, diabetes mellitus and adrenal insufficiency have been widely reported with both PD‐1/PD‐L1 and CTLA‐4 blockade. Endocrine toxicity can be difficult to diagnose due to the non‐specific features of hormone deficiencies.
Thyroid disorders are most common and manifest as hypothyroidism or thyroiditis, presenting initially as thyrotoxicosis, and/or later as hypothyroidism due to inflammatory damage to the gland.73 Primary thyroid dysfunction can occur after both CTLA‐4 and PD‐1/PD‐L1 blockade, unlike other endocrinopathies that are specific for different antibodies. Specifically, after PD‐1 blockade, the rates of hypothyroidism are approximately 5%–8% and of hyperthyroidism they are 3%.74 Rates of hypothyroidism from ipilimumab have been reported as 5·9%.75 In combination therapy, rates of thyroid dysfunction could be up to 50%.76 Fifty per cent of cases of thyrotoxicosis may be self‐limiting; however, if hypothyroidism occurs after PD‐1 blockade, it is more likely to require lifelong thyroxine replacement.77
The underlying molecular mechanism of thyroid toxicity remains unclear. As mentioned above, CTLA‐4 axis targeting is accompanied by regulatory T‐cell dysfunction, as highlighted in a mouse study examining autoimmune thyroiditis.78 Additional to this, PD‐1/PD‐L1 pathway blockade also leads to autoantibody formation: in KEYNOTE‐001, 80% of patients experiencing thyroid dysfunction had positive autoantibodies.73 Finally, PD‐1 axis blockade has also been hypothesized to cause thyroid dysfunction by direct binding of mAb to thyroid cells, which reduces immune tolerance, leading to autoimmune thyroiditis.79
Monitoring involves thyroid function tests, with the addition of thyroid autoantibodies in thyrotoxicosis. Deranged thyroid function tests should prompt a pituitary screen to exclude hypophysitis. If symptomatic, hypothyroidism can be treated with levothyroxine without ICI cessation. Symptomatic hyperthyroidism may require treatment cessation, beta‐blockers and carbimazole.3 ICI may be recommenced once symptoms have resolved, with specialist endocrine input.
Hypophysitis is a rare but significant complication of ICI. The incidence in patients treated with ipilimumab in initial clinical trials was up to 17%, compared with <1% with anti‐PD1 therapy.80, 81, 82 Pituitary enlargement usually causes headache, but visual disturbance, cranial palsies and cavernous sinus involvement have also been reported.83 In 70% of patients there will be hormone insufficiencies, including thyrotrophin, gonadotrophin and corticotrophin insufficiencies, which manifest as fatigue, nausea, anorexia and temperature intolerance. Adrenocorticotrophic hormone insufficiency associated with hypophysitis is unlikely to recover long term.84
The underlying mechanism is unknown but has been hypothesized. CTLA‐4 protein has been identified in healthy murine pituitary tissue. Infiltrating pituitary gland T cells, activated by inhibiting CTLA‐4, proliferate and secrete inflammatory cytokines, further aggravating the immune response.85 In addition, antibody‐dependent complement activation due to the presence of CTLA‐4 protein in pituitary tissue may result in a mixed type II/IV hypersensitivity reaction.86 This is supported by the lower incidence of hypophysitis with tremelimumab, an IgG4 mAb known to trigger less complement activation. Anti‐PD‐1 therapies are IgG4‐based, potentially explaining the rarity of hypophysitis in this group of patients.86
A visual field assessment and biochemical pituitary screen are advised, whereas brain magnetic resonance imaging will exclude differentials such as cerebral metastases and infection and determine the presence of pituitary enlargement. Using Common Terminology Criteria for Adverse Events, Grade 2 toxicity or above should prompt ICI cessation and consideration of corticosteroids, ideally intravenous if mass effect symptoms or severe hypoadrenalism are present. Options also include hormone replacement if deficient, and endocrinology referral. The majority of patients can recommence ICI once stable, but will probably require long‐term hormone replacement and endocrine input.3
Type 1 diabetes mellitus can be a life‐threatening irAE of anti‐PD‐1/PD‐L1 therapy, with patients requiring permanent insulin replacement.87 The incidence is estimated at around 1%, with half of cases having detectable autoantibodies, most commonly anti‐glutamic acid decarboxylase.88 Detectable antibodies are associated with higher incidence of diabetic ketoacidosis.87 Additionally, PD‐1/PD‐L1 inhibition can lead to worsening of pre‐existing type 2 diabetes.89
This complication is specific to PD‐1/PD‐L1 inhibition, with no reported cases in anti‐CTLA‐4 therapy. Animal models have demonstrated that genetic or pharmacological interruption of the PD‐1/PD‐L1 axis can stimulate the development of diabetes.90 Pancreatic islet cells have been found to express PD‐L1,91 and the interaction of CD80 (an additional PD‐L1 binding site and ligand for CD28) with PD‐1/PD‐L1 could provide an immunoinhibitory effect against self‐reactive effector T cells. Disrupting this interaction may lead to down‐regulation of inhibitory signals from cytotoxic T cells and therefore autoimmune disruption of islet cells.92
Patients should be screened with regular blood glucose testing during therapy. The role of corticosteroid treatment in preventing occurrence is unknown, and steroids are likely to adversely affect outcomes by impacting on glycaemic control. Rechallenge of therapy can be considered once the patient is well and established on insulin.3
Primary adrenal insufficiency or adrenalitis is the rarest endocrine complication, with only a few reported cases. In patients previously treated for ICI toxicity with corticosteroids, secondary adrenal insufficiency should be considered excluded. Other differentials include secondary adrenal insufficiency resulting from adrenocorticotrophic hormone insufficiency from hypophysitis, and bilateral adrenal metastases. Therefore, adrenal and pituitary imaging should be performed in addition to full pituitary axis screening.93
Other toxicities
There are several other irAEs that patients may encounter with ICI treatment that, although not covered extensively in this review, are clinically significant and require diagnostic awareness from the treating clinician. Skin toxicity is the most common irAE, with incidence more common with anti‐CTLA4 compared with PD‐1 therapy, with incidence rates ranging between 34% and 45%.28 Severe cutaneous toxicity is rare, with most cases being managed in the outpatient setting with topical steroids and antihistamines, with continued ICI treatment. A dermatology opinion should be sought in severe or refractory cases.
Nephritis is another clinically significant toxicity that necessitates regular monitoring of renal function, and there should be a low index of clinical suspicion for patients with unexplained rises in urea and creatinine. Haematological toxicity has also been reported, which can range from relatively mild blood dyscrasias to aplastic anaemia94 and acquired haemophilia.95 Further information and detailed guidance on management of these toxicities are available from published guidelines.4, 28 It is also noteworthy that life‐threatening and indeed fatal toxicities have been observed with ICI treatment, principally in the setting of myocarditis or neurotoxicity.96, 97 This has clinical implications in terms of both diagnosis and treatment, but also necessitates thorough, informed consent before commencement of ICI treatment. Such cases should be managed in centres experienced in treating irAEs with early specialist input advised.
The influence of irAEs on efficacy outcomes
The level of immune activation and therefore the anti‐tumour activity achieved by ICI has been hypothesized to be reflected by the intensity of irAEs that occur in each patient. The association of irAEs with outcome of ICI has therefore been an area of debate.9 With ipilimumab, studies looking at irAE occurrence and response have demonstrated both positive correlation in melanoma and RCC,98, 99, 100 and negative correlation in small‐cell lung cancer – although here ICI was given in combination with cytotoxic therapy.101 Larger studies have not shown correlation in melanoma.102, 103 With anti‐PD‐1‐axis therapy, correlation between outcome and cutaneous irAEs has been observed in some studies,104, 105, 106 although the over‐representation of melanoma patients in these cohorts has invited suggestions that toxicities represent lineage‐specific action of therapy and that correlation may not extend to non‐melanoma cases.107 Again, evidence of this correlation has not been consistent, with other series showing no correlation.108 Any positive correlation is hard to disentangle from lead‐time bias, as patients who respond tend to be treated for longer, and so have more time to develop irAEs.107 Another layer of complexity is added by corticosteroids, which are thought to reduce anti‐cancer activity of ICI and have been an exclusion criterion for trials when used to treat premorbid conditions. However, evidence so far has not shown any effect on outcome when these are used to treat irAEs.100, 102
Conclusion
Review of the clinical features, investigation findings, and treatment principles of the major ICI‐related toxicities, as well as of their immunopathology, reveals common patterns. Clinical features can be non‐specific and consistent with a wide range of differentials, with the picture usually complicated by possible progression of primary disease. Some tissues are preferentially affected by anti‐PD‐1 ICI, such as the lung and endocrine pancreas, whereas others are affected more by CTLA‐4 axis inhibition, such as the gut and pituitary gland. Histopathology findings have been varied, and comparisons have been drawn between ICI‐associated toxicity and other autoimmune pathologies, with an ICI‐specific picture emerging in the liver, but not the lung. A common theme across the tissues reviewed has of course been that of regulatory T‐cell dysfunction. In addition to that, PD‐1 axis inhibition is often accompanied by autoantibody generation. A notable mechanism is that of possible anti‐CTLA‐4 antibody‐mediated complement activation in the pituitary. A number of scientific societies have developed guidelines for the management of irAEs,3, 4 which is influenced by toxicity grading, and is based on treatment discontinuation, steroids and further immunosuppression, with additional symptomatic measures. Given that immunotherapy is already widely used across several tumour types and is currently being trialled in more, recognition and prompt treatment of irAEs are essential to minimize patient morbidity. Translational studies aimed at providing a more in‐depth clinical and biological phenotyping of ICI recipients are warranted to increase our understanding of the complex pathophysiology underlying toxicity and response to ICI in patients with cancer.
Disclosure
None declared.
Author contributions
Review concept and design: DJP; drafting of final manuscript: PF; critical revision of the manuscript for important intellectual content: all the authors; administrative, technical, or material support: PF; and study supervision: DJP.
Acknowledgements
The authors would like to acknowledge the infrastructure support provided by Imperial Experimental Cancer Medicine Centre, Cancer Research UK Imperial Centre and the Imperial College Healthcare NHS Trust Tissue Bank. DJP is supported by grant funding from the National Institute for Health Research (NIHR) and the Imperial BRC.
References
- 1. Shannon VR. Pneumotoxicity associated with immune checkpoint inhibitor therapies. Curr Opin Pulm Med 2017; 23:305–16. [DOI] [PubMed] [Google Scholar]
- 2. Naidoo J, Wang X, Woo KM, Iyriboz T, Halpenny D, Cunningham J et al Pneumonitis in patients treated with anti‐programmed death‐1/programmed death ligand 1 therapy. J Clin Oncol 2017; 35:709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haanen JBAG, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J et al Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2018;29(Suppl 4):iv264–6. [DOI] [PubMed] [Google Scholar]
- 4. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM et al Management of immune‐related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2018; 36:1714–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thompson JA, Schneider BJ, Brahmer J, Andrews S, Armand P, Bhatia S et al Management of immunotherapy‐related toxicities, version 1.2019. J Natl Compr Canc Netw 2019; 17:255–89. [DOI] [PubMed] [Google Scholar]
- 6. Puzanov I, Diab A, Abdallah K, Bingham CO, Brogdon C, Dadu R et al Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer 2017;5:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson DB, Chandra S, Sosman JA. Immune checkpoint inhibitor toxicity in 2018. JAMA 2018; 320:1702–3. [DOI] [PubMed] [Google Scholar]
- 8. Suresh K, Voong, Shankar B, KR Forde PM, Ettinger DS, Marrone KA et al Pneumonitis in non‐small cell lung cancer patients receiving immune checkpoint immunotherapy: incidence and risk factors. J Thorac Oncol 2018; 13:1930–9. [DOI] [PubMed] [Google Scholar]
- 9. Inno A, Metro G, Bironzo P, Grimaldi AM, Grego E, Di Nunno V et al Pathogenesis, clinical manifestations and management of immune checkpoint inhibitors toxicity. Tumori 2017; 103:405–21. [DOI] [PubMed] [Google Scholar]
- 10. Khunger M, Rakshit S, Pasupuleti V, Hernandez AV, Mazzone P, Stevenson J et al Incidence of pneumonitis with use of programmed death 1 and programmed death‐ligand 1 inhibitors in non‐small cell lung cancer: a systematic review and meta‐analysis of trials. Chest 2017; 152:271–81. [DOI] [PubMed] [Google Scholar]
- 11. Pillai RN, Behera M, Owonikoko TK, Kamphorst AO, Pakkala S, Belani CP et al Comparison of the toxicity profile of PD‐1 versus PD‐L1 inhibitors in non–small cell lung cancer: a systematic analysis of the literature. Cancer 2018; 124:271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nishino M, Ramaiya NH, Awad MM, Sholl LM, Maattala JA, Taibi M et al PD‐1 inhibitor‐related pneumonitis in advanced cancer patients: radiographic patterns and clinical course. Clin Cancer Res 2016; 22:6051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Howell M, Lee R, Bowyer S, Fusi A, Lorigan P. Optimal management of immune‐related toxicities associated with checkpoint inhibitors in lung cancer. Lung Cancer 2015; 88:117–23. [DOI] [PubMed] [Google Scholar]
- 14. Winer A, Nicholas Bodor J, Borghaei H. Identifying and managing the adverse effects of immune checkpoint blockade. J Thorac Dis 2018; 10:S480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delaunay M, Cadranel J, Lusque A, Meyer N, Gounaut V, Moro‐Sibilot D et al Immune‐checkpoint inhibitors associated with interstitial lung disease in cancer patients. Eur Respir J 2017; 50:1700050. [DOI] [PubMed] [Google Scholar]
- 16. Wu J, Hong D, Zhang X, Lu X, Miao J. PD‐1 inhibitors increase the incidence and risk of pneumonitis in cancer patients in a dose‐independent manner: a meta‐analysis. Sci Rep 2017; 7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Postow MA, Sidlow R, Hellmann MD. Immune‐related adverse events associated with immune checkpoint blockade. N Engl J Med 2018; 378:158–68. [DOI] [PubMed] [Google Scholar]
- 18. Nishino M, Sholl LM, Hodi FS, Hatabu H, Ramaiya NH. Anti‐PD‐1‐related pneumonitis during cancer immunotherapy. N Engl J Med 2015; 373:288–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chuzi S, Tavora F, Cruz M, Costa R, Chae YK, Carneiro BA et al Clinical features, diagnostic challenges, and management strategies in checkpoint inhibitor‐related pneumonitis. Cancer Manag Res 2017; 9:207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berthod G, Lazor R, Letovanec I, Romano E, Noirez L, Mazza Stalder J et al Pulmonary sarcoid‐like granulomatosis induced by ipilimumab. J Clin Oncol 2012; 30:e156–9. [DOI] [PubMed] [Google Scholar]
- 21. Celada LJ, Kropski JA, Herazo‐Maya JD, Luo W, Creecy A, Abad AT et al PD‐1 up‐regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3‐mediated IL‐17A and TGF‐β 1 production. Sci Transl Med 2018; 10 10.1126/scitranslmed.aar8356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Geng Y, Liu X, Liang J, Habiel DM, Kulur V, Coelho AL et al PD‐L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight 2019; 4:e125326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J et al Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD‐1‐deficient mice. Nat Med 2003; 9:1477–83. [DOI] [PubMed] [Google Scholar]
- 24. Kanameishi S, Otsuka A, Nonomura Y, Fujisawa A, Endo Y, Kabashima K. Idiopathic thrombocytopenic purpura induced by nivolumab in a metastatic melanoma patient with elevated PD‐1 expression on B cells. Ann Oncol 2016; 27:546–7. [DOI] [PubMed] [Google Scholar]
- 25. Kong YM, Flynn JC. Opportunistic autoimmune disorders potentiated by immune‐checkpoint inhibitors anti‐CTLA‐4 and anti‐PD‐1. Front Immunol 2014; 5:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM et al RGMb is a novel binding partner for PD‐L2 and its engagement with PD‐L2 promotes respiratory tolerance. J Exp Med 2014; 211:943–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tabchi S, Messier C, Blais N. Immune‐mediated respiratory adverse events of checkpoint inhibitors. Curr Opin Oncol 2016; 28:269–77. [DOI] [PubMed] [Google Scholar]
- 28. Haanen JBAG, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J et al Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2017; 28: iv119–iv142. [DOI] [PubMed] [Google Scholar]
- 29. Eigentler TK, Hassel JC, Berking C, Aberle J, Bachmann O, Grünwald V et al Diagnosis, monitoring and management of immune‐related adverse drug reactions of anti‐PD‐1 antibody therapy. Cancer Treat Rev 2016; 45:7–18. [DOI] [PubMed] [Google Scholar]
- 30. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel‐Vinay S et al Immune‐related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016; 54:139–48. [DOI] [PubMed] [Google Scholar]
- 31. Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME et al Toxicities of the anti‐PD‐1 and anti‐PD‐L1 immune checkpoint antibodies. Ann Oncol 2015; 26:mdv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Santini FC, Rizvi H, Plodkowski AJ, Ni A, Lacouture ME, Gambarin‐Gelwan M et al Safety and efficacy of re‐treating with immunotherapy after immune‐related adverse events in patients with NSCLC. Cancer Immunol Res 2018; 6:1093–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wirsdörfer F, De Leve S, Jendrossek V. Combining radiotherapy and immunotherapy in lung cancer: can we expect limitations due to altered normal tissue toxicity? Int J Mol Sci 2019; 20:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khoja L, Day D, Wei‐Wu Chen T, Siu LL, Hansen AR. Tumour‐ and class‐specific patterns of immune‐related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol 2017; 28:2377–85. [DOI] [PubMed] [Google Scholar]
- 35. Weber J, Thompson JA, Hamid O, Minor D, Amin A, Ron I et al A Randomized, double‐blind, placebo‐controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res 2009; 15:5591–8. [DOI] [PubMed] [Google Scholar]
- 36. Soularue E, Lepage P, Colombel JF, Coutzac C, Faleck D, Marthey L et al Enterocolitis due to immune checkpoint inhibitors: a systematic review. Gut 2018; 67:2056–67. [DOI] [PubMed] [Google Scholar]
- 37. Larkin J, Chiarion‐Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD et al Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ascierto PA, Del Vecchio M, Robert C, Mackiewicz A, Chiarion‐Sileni V, Arance A et al Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double‐blind, multicentre, phase 3 trial. Lancet Oncol 2017; 18:611–22. [DOI] [PubMed] [Google Scholar]
- 39. Karamchandani DM, Chetty R. Immune checkpoint inhibitor‐induced gastrointestinal and hepatic injury: pathologists’ perspective. J Clin Pathol 2018; 71:665–71. [DOI] [PubMed] [Google Scholar]
- 40. Baroudjian B, Lourenco N, Pagès C, Chami I, Maillet M, Bertheau P et al Anti‐PD1‐induced collagenous colitis in a melanoma patient. Melanoma Res 2016; 26:308–11. [DOI] [PubMed] [Google Scholar]
- 41. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A et al Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 2014; 20:1410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte‐associated antigen 4 plays an essential role in the function of CD25+ CD4+ regulatory cells that control intestinal inflammation. J Exp Med 2000; 192:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu JZ, Van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A et al Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015; 47:979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C et al Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol 2017; 28:1368–79. [DOI] [PubMed] [Google Scholar]
- 45. Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L et al Intestinal microbiome analyses identify melanoma patients at risk for checkpoint‐blockade‐induced colitis. Nat Commun 2016; 7:10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe–host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci Transl Med 2014; 6:220ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haanen JBAG, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J et al Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up†. Ann Oncol 2017; 28(suppl_4): iv119–42. [DOI] [PubMed] [Google Scholar]
- 48. Mitchell KA, Kluger H, Sznol M, Hartman DJ. Ipilimumab‐induced perforating colitis. J Clin Gastroenterol 2013; 47:781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Abu‐Sbeih H, Ali FS, Wang X, Mallepally N, Chen E, Altan M et al Early introduction of selective immunosuppressive therapy associated with favorable clinical outcomes in patients with immune checkpoint inhibitor‐induced colitis. J Immunother Cancer 2019; 7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bergqvist V, Hertervig E, Gedeon P, Kopljar M, Griph H, Kinhult S et al Vedolizumab treatment for immune checkpoint inhibitor‐induced enterocolitis. Cancer Immunol Immunother 2017; 66:581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL et al Fecal microbiota transplantation for refractory immune checkpoint inhibitor‐associated colitis. Nat Med 2018; 24:1804–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cramer P, Bresalier RS. Gastrointestinal and hepatic complications of immune checkpoint inhibitors. Curr Gastroenterol Rep 2017; 19:3. [DOI] [PubMed] [Google Scholar]
- 53. Zen Y, Yeh MM. Hepatotoxicity of immune checkpoint inhibitors: a histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug‐induced liver injury. Mod Pathol 2018; 31:965–73. [DOI] [PubMed] [Google Scholar]
- 54. Johncilla M, Misdraji J, Pratt DS, Agoston AT, Lauwers GY, Srivastava A et al Ipilimumab‐associated hepatitis: clinicopathologic characterization in a series of 11 cases. Am J Surg Pathol 2015; 39:1075–84. [DOI] [PubMed] [Google Scholar]
- 55. De Martin E, Michot JM, Papouin B, Champiat S, Mateus C, Lambotte O et al Characterization of liver injury induced by cancer immunotherapy using immune checkpoint inhibitors. J Hepatol 2018; 68:1181–90. [DOI] [PubMed] [Google Scholar]
- 56. Doherty GJ, Duckworth AM, Davies SE, Mells GF, Brais R, Harden SV et al Severe steroid‐resistant anti‐PD1 T‐cell checkpoint inhibitor‐induced hepatotoxicity driven by biliary injury. ESMO Open 2017; 2:e000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Black JR, Goldin RD, Foxton M, Marafioti T, Akarca AU, Pria AD et al PD‐L1 expressing granulomatous reaction as an on‐target mechanism of steroid‐refractory immune hepatotoxicity. Immunotherapy 2019; 11:585–90. [DOI] [PubMed] [Google Scholar]
- 58. Everett J, Srivastava A, Misdraji J. Fibrin ring granulomas in checkpoint inhibitor‐induced hepatitis. Am J Surg Pathol 2016; 41:134–7. [DOI] [PubMed] [Google Scholar]
- 59. Kim KW, Ramaiya NH, Krajewski KM, Jagannathan JP, Tirumani SH, Srivastava A et al Ipilimumab associated hepatitis: imaging and clinicopathologic findings. Invest New Drugs 2013; 31:1071–7. [DOI] [PubMed] [Google Scholar]
- 60. Shi Y, Dong K, Zhang YG, Michel RP, Marcus V, Wang YY et al Sinusoidal endotheliitis as a histological parameter for diagnosing acute liver allograft rejection. World J Gastroenterol 2017; 23:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Heymann F, Peusquens J, Ludwig‐Portugall I, Kohlhepp M, Ergen C, Niemietz P et al Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 2015; 62:279–91. [DOI] [PubMed] [Google Scholar]
- 62. Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7‐homolog 1‐dependent CD8+ T cell tolerance. Hepatology 2008; 47:296–305. [DOI] [PubMed] [Google Scholar]
- 63. Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7–H1 determines accumulation and deletion of intrahepatic CD8+ T lymphocytes. Immunity 2004; 20:327–36. [DOI] [PubMed] [Google Scholar]
- 64. Charles R, Chou HS, Wang L, Fung JJ, Lu L, Qian S. Human hepatic stellate cells inhibit T‐cell response through B7–H1 pathway. Transplantation 2013; 96:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen CH, Kuo LM, Chang Y, Wu W, Goldbach C, Ross MA et al In vivo immune modulatory activity of hepatic stellate cells in mice. Hepatology 2006; 44:1171–81. [DOI] [PubMed] [Google Scholar]
- 66. Kido M, Watanabe N, Okazaki T, Akamatsu T, Tanaka J, Saga K et al Fatal autoimmune hepatitis induced by concurrent loss of naturally arising regulatory T cells and PD‐1‐mediated signaling. Gastroenterology 2008; 135:1333–43. [DOI] [PubMed] [Google Scholar]
- 67. Eskandari‐Nasab E, Tahmasebi A, Hashemi M. Meta‐analysis: the relationship between CTLA‐4 +49 A/G polymorphism and primary biliary cirrhosis and type I autoimmune hepatitis. Immunol Invest 2015; 44:331–48. [DOI] [PubMed] [Google Scholar]
- 68. Fan LY, Tu XQ, Cheng QB, Zhu Y, Feltens R, Pfeiffer T et al Cytotoxic T lymphocyte associated antigen‐4 gene polymorphisms confer susceptibility to primary biliary cirrhosis and autoimmune hepatitis in Chinese population. World J Gastroenterol 2004; 10:3056–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grant CR, Liberal R, Holder BS, Cardone J, Ma Y, Robson SC et al Dysfunctional CD39(POS) regulatory T cells and aberrant control of T‐helper type 17 cells in autoimmune hepatitis. Hepatology 2014; 59:1007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Champiat S, Lambotte O, Barreau E, Belkhir R, Berdelou A, Carbonnel F et al Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol 2016; 27:559–74. [DOI] [PubMed] [Google Scholar]
- 71. McGuire HM, Shklovskaya E, Edwards J, Trevillian PR, McCaughan GW, Bertolino P et al Anti‐PD‐1‐induced high‐grade hepatitis associated with corticosteroid‐resistant T cells: a case report. Cancer Immunol Immunother 2018; 30:563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lowe D, John S. Autoimmune hepatitis: appraisal of current treatment guidelines. World J Hepatol 2018; 10:911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Osorio JC, Ni A, Chaft JE, Pollina R, Kasler MK, Stephens D et al Antibody‐mediated thyroid dysfunction during T‐cell checkpoint blockade in patients with non‐small‐cell lung cancer. Ann Oncol 2017; 28:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Byun DJ, Wolchok JD, Rosenberg LM, Girotra M. Cancer immunotherapy – immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol 2017; 13:195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ryder M, Callahan M, Postow MA, Wolchok J, Fagin JA. Endocrine‐related adverse events following ipilimumab in patients with advanced melanoma: a comprehensive retrospective review from a single institution. Endocr Relat Cancer 2014; 21:371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Morganstein DL, Lai Z, Spain L, Diem S, Levine D, Mace C et al Thyroid abnormalities following the use of cytotoxic T‐lymphocyte antigen‐4 and programmed death receptor protein‐1 inhibitors in the treatment of melanoma. Clin Endocrinol 2017; 86:614–20. [DOI] [PubMed] [Google Scholar]
- 77. Delivanis DA, Gustafson MP, Bornschlegl S, Merten MM, Kottschade L, Withers S et al Pembrolizumab‐induced thyroiditis: comprehensive clinical review and insights into underlying involved mechanisms. J Clin Endocrinol Metab 2017; 102:2770–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Morris GP, Brown NK, Kong YM. Naturally‐existing CD4+CD25+Foxp3+ regulatory T cells are required for tolerance to experimental autoimmune thyroiditis induced by either exogenous or endogenous autoantigen. J Autoimmun 2009; 33:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yamauchi I, Sakane Y, Fukuda Y, Fujii T, Taura D, Hirata M et al Clinical features of nivolumab‐induced thyroiditis: a case series study. Thyroid 2017; 27:894–901. [DOI] [PubMed] [Google Scholar]
- 80. Torino F, Barnabei A, De Vecchis L, Salvatori R, Corsello SM. Hypophysitis induced by monoclonal antibodies to cytotoxic T lymphocyte antigen 4: challenges from a new cause of a rare disease. Oncologist 2012; 17:525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH et al Survival, durable tumor remission, and long‐term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014; 32:1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R et al Anti‐programmed‐death‐receptor‐1 treatment with pembrolizumab in ipilimumab‐refractory advanced melanoma: a randomised dose‐comparison cohort of a phase 1 trial. Lancet 2014; 384:1109–17. [DOI] [PubMed] [Google Scholar]
- 83. Honegger J, Schlaffer S, Menzel C, Droste M, Werner S, Elbelt U et al Diagnosis of primary hypophysitis in Germany. J Clin Endocrinol Metab 2015; 100:3841–9. [DOI] [PubMed] [Google Scholar]
- 84. Albarel F, Gaudy C, Castinetti F, Carré T, Morange I, Conte‐Devolx B et al Long‐term follow‐up of ipilimumab‐induced hypophysitis, a common adverse event of the anti‐CTLA‐4 antibody in melanoma. Eur J Endocrinol 2015; 172:195–204. [DOI] [PubMed] [Google Scholar]
- 85. Lin H‐H, Gutenberg A, Chen T‐Y, Tsai N‐M, Lee C‐J, Cheng Y‐C et al In situ activation of pituitary‐infiltrating T lymphocytes in autoimmune hypophysitis. Sci Rep 2017; 7:43492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Iwama S, De Remigis A, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary expression of CTLA‐4 mediates hypophysitis secondary to administration of CTLA‐4 blocking antibody. Sci Transl Med 2014; 6:230ra45. [DOI] [PubMed] [Google Scholar]
- 87. Akturk HK, Kahramangil D, Sarwal A, Hoffecker L, Murad MH, Michels AW. Immune checkpoint inhibitor‐induced type 1 diabetes: a systematic review and meta‐analysis. Diabet Med 2019; 36:1075–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA et al Collateral damage: insulin‐dependent diabetes induced with checkpoint inhibitors. Diabetes 2018; 67:1471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kotwal A, Haddox C, Block M, Kudva YC. Immune checkpoint inhibitors: an emerging cause of insulin‐dependent diabetes. BMJ Open Diabetes Res Care 2019; 7:e000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yadav D, Hill N, Yagita H, Azuma M, Sarvetnick N. Altered availability of PD‐1/PD ligands is associated with the failure to control autoimmunity in NOD mice. Cell Immunol 2009; 258:161–71. [DOI] [PubMed] [Google Scholar]
- 91. Rajasalu T, Brosi H, Schuster C, Spyrantis A, Boehm BO, Chen L et al Deficiency in B7–H1 (PD‐L1)/PD‐1 coinhibition triggers pancreatic beta‐cell destruction by insulin‐specific, murine CD8 T‐cells. Diabetes 2010; 59:1966–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kapke J, Shaheen Z, Kilari D, Knudson P, Wong S. Immune checkpoint inhibitor‐associated type 1 diabetes mellitus: case series, review of the literature, and optimal management. Case Rep Oncol 2017; 10:897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Illouz F, Briet C, Cloix L, Le Corre Y, Baize N, Urban T et al Endocrine toxicity of immune checkpoint inhibitors: essential crosstalk between endocrinologists and oncologists. Cancer Med 2017; 6:1923–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Meyers DE, Hill WF, Suo A, Jimenez‐Zepeda V, Cheng T, Nixon NA. Aplastic anemia secondary to nivolumab and ipilimumab in a patient with metastatic melanoma: a case report. Exp Hematol Oncol 2018; 7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Delyon J, Mateus C, Lambert T. Hemophilia A induced by ipilimumab. N Engl J Med 2011; 365:1747–8. [DOI] [PubMed] [Google Scholar]
- 96. Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM et al Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol 2018; 71:1755–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Johnson DB, Manouchehri A, Haugh AM, Quach HT, Balko JM, Lebrun‐Vignes B et al Neurologic toxicity associated with immune checkpoint inhibitors: a pharmacovigilance study. J Immunother Cancer 2019; 7:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE et al Enterocolitis in patients with cancer after antibody blockade of cytotoxic T‐lymphocyte–associated antigen 4. J Clin Oncol 2006; 24:2283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL et al Ipilimumab (anti‐CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother 2007; 30:825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Downey SG, Klapper JA, Smith FO, Yang JC, Sherry RM, Royal RE et al Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL‐associated antigen‐4 blockade. Clin Cancer Res 2007; 13(22 Pt 1):6681–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Arriola E, Wheater M, Galea I, Cross N, Maishman T, Hamid D et al Outcome and biomarker analysis from a multicenter phase 2 study of ipilimumab in combination with carboplatin and etoposide as first‐line therapy for extensive‐stage SCLC. J Thorac Oncol 2016; 11:1511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Horvat TZ, Adel NG, Dang TO, Momtaz P, Postow MA, Callahan MK et al Immune‐related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol 2015; 33:3193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Di Giacomo AM, Grimaldi AM, Ascierto PA, Queirolo P, Del Vecchio M, Ridolfi R et al Correlation between efficacy and toxicity in pts with pretreated advanced melanoma treated within the Italian cohort of the ipilimumab expanded access programme (EAP). J Clin Oncol 2013; 31(15_suppl);9065. [Google Scholar]
- 104. Hua C, Boussemart L, Mateus C, Routier E, Boutros C, Cazenave H et al Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol 2016; 152:45. [DOI] [PubMed] [Google Scholar]
- 105. Sanlorenzo M, Vujic I, Daud A, Algazi A, Gubens M, Luna SA et al Pembrolizumab cutaneous adverse events and their association with disease progression. JAMA Dermatol 2015; 151:1206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Freeman‐Keller M, Kim Y, Cronin H, Richards A, Gibney G, Weber JS. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune‐related adverse events and association with outcomes. Clin Cancer Res 2016; 22:886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lo JA, Fisher DE, Flaherty KT. Prognostic significance of cutaneous adverse events associated with pembrolizumab therapy. JAMA Oncol 2015; 1:1340–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Grimaldi A, Simeone E, Festino L, Giannarelli D, Palla M, Caracò C et al Correlation between immune‐related adverse events and response to pembrolizumab in advanced melanoma patients. J Immunother Cancer 2015; 3(Suppl 2):P186. [Google Scholar]