

Abstract
Translocases of the AAA+ (ATPases Associated with various cellular Activities) family are powerful molecular machines that use the mechano‐chemical coupling of ATP hydrolysis and conformational changes to thread DNA or protein substrates through their central channel for many important biological processes. These motors comprise hexameric rings of ATPase subunits, in which highly conserved nucleotide‐binding domains form active‐site pockets near the subunit interfaces and aromatic pore‐loop residues extend into the central channel for substrate binding and mechanical pulling. Over the past 2 years, 41 cryo‐EM structures have been solved for substrate‐bound AAA+ translocases that revealed spiral‐staircase arrangements of pore‐loop residues surrounding substrate polypeptides and indicating a conserved hand‐over‐hand mechanism for translocation. The subunits' vertical positions within the spiral arrangements appear to be correlated with their nucleotide states, progressing from ATP‐bound at the top to ADP or apo states at the bottom. Studies describing multiple conformations for a particular motor illustrate the potential coupling between ATP‐hydrolysis steps and subunit movements to propel the substrate. Experiments with double‐ring, Type II AAA+ motors revealed an offset of hydrolysis steps between the two ATPase domains of individual subunits, and the upper ATPase domains lacking aromatic pore loops frequently form planar rings. This review summarizes the critical advances provided by recent studies to our structural and functional understanding of hexameric AAA+ translocases, as well as the important outstanding questions regarding the underlying mechanisms for coordinated ATP‐hydrolysis and mechano‐chemical coupling.
Keywords: AAA+ ATPases, cryo‐EM, spiral staircase, translocation
1. INTRODUCTION
Many cellular processes, including DNA replication,1 protein disaggregation,2 the disassembly of macromolecular complexes,3 and proteolysis,4 require molecular machines of the AAA+ (ATPases Associated with various cellular Activities) ATPase family for the mechanical unfolding of proteins. These ATPases are characterized by conserved Nucleotide Binding Domains (NBDs) that form pentameric or hexameric ring structures and utilize ATP binding and hydrolysis to drive conformational changes for the mechanical “translocation” of substrates through a central channel. In addition, a number of regulatory modules are appended to or inserted into the NBDs of AAA+ ATPases for tailoring the molecular translocation function to specific cellular processes. The mechanical pulling on substrates is important for ATP‐dependent proteolysis, protein retro‐translocation from the endoplasmic reticulum, protein extraction from membranes, and the disassembly of macromolecular complexes, processes that play critical roles in regulating the cell cycle, transcription, stress responses, and general protein quality control.5
1.1. Conserved AAA+‐domain residues
AAA+ proteins have a conserved overall architecture and molecular mechanism of ATP hydrolysis, but can be separated into seven major clades, defined by their shared structural and functional characteristics.6 This review will primarily focus on the Classic Clade (Clade 3) and HCLR Clade (Clade 5) of AAA+ motors, which are involved in protein unfolding and remodeling. For more information on the structural differences observed in other clades, please see these great reviews.5, 6, 7, 8
Conserved across all clades are canonical residues involved in ATP binding and hydrolysis, including Walker‐A, Walker‐B, and Sensor‐1 motifs, as well as Arginine fingers (Figure 1a).5, 8, 9 The nucleotide‐binding site of an AAA+ ATPase is localized between the N‐terminal large AAA+ subdomain with α/β structure and the C‐terminal α‐helical small AAA+ subdomain,10 near the interface to the clockwise‐next neighboring subunit in the pentameric or hexameric rings (Figure 1b). While the Walker and Sensor residues are present in the “cis” subunit, the Arginine fingers are contributed by the adjacent “trans” subunit (Figure 1). The Walker‐A motif, GXXXXGK[T/S] (X can be any amino acid), functions in structuring the nucleotide‐binding pocket, and disrupting these residues by mutagenesis impairs ATP binding. The Walker‐B residues, hhhhD[D/E] (h is a hydrophobic residue), are required for ATP hydrolysis by acting as a catalytic base and activating a water molecule for nucleophilic attack of the γ‐phosphate (Figure 1a). Mutation of Walker‐B residues eliminates or strongly inhibits ATP hydrolysis, and has been used extensively to study AAA+ motor function, investigate the mechanisms of subunit communication and ATP‐hydrolysis‐coupled conformational changes, and to trap AAA+ motors in defined states for structure determination.5, 11, 12, 13, 14, 15, 16, 17
Figure 1.
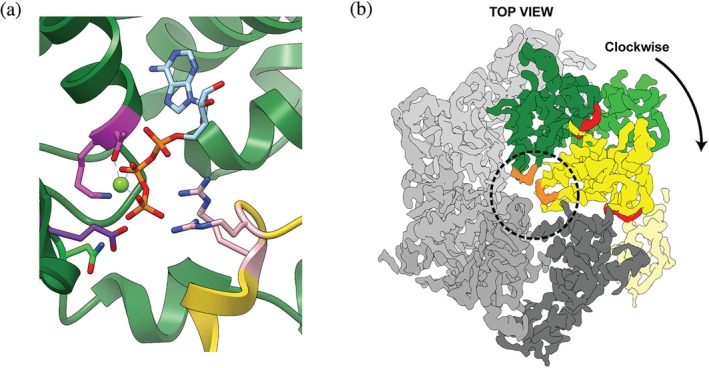
Conserved residues and motifs in AAA+ ATPase motors. (a) Nucleotide‐binding pocket at the interface between the ATPase subunits Rpt1 (green) and Rpt2 (yellow) of the 26S proteasome (PDB:5L4G).74 Highlighted are the intra‐subunit Walker‐A [K/T] (magenta), Walker‐B [E] (purple), and Sensor‐1 [N] (lime green) motifs, as well as the Arginine Finger [R/R] (pink) provided by the clockwise‐next subunit. ATP is shown in cyan and orange, and Mg2+ as a lime‐green sphere. (b) A cryo‐EM density representative “Top” view of the proteasomal AAA+ motor domains (EMDB:4002). The large and small AAA+ subdomains are shown in dark and light green, respectively, for Rpt1, and in dark and light yellow for the neighboring Rpt2. ATP in the binding pocket near the subdomain and subunit interfaces is colored red. Pore‐1 loops are colored orange and the central channel is circled. The remaining Rpt protomers in the hexamer are shown in shades of gray
The Sensor‐1 and Arginine finger motifs18 are critical for ATP hydrolysis (Figure 1a), with the latter spanning the subunit interface and acting in “trans” by supporting the nucleotide pocket of the neighboring subunit. These arginines directly interact with the γ‐phosphate of ATP and stabilize an accumulated negative charge during hydrolysis (Figure 1a). Some of the Classic and HCLR‐clade AAA+ motors also contain an Inter‐Subunit Signaling (ISS) motif, characterized by an α‐helix immediately preceding the sensor‐1 motif and containing a [D/E] residue at its C‐terminus that interacts with the Arg‐finger to sense the nucleotide state of the adjacent subunit.19 Due to the inter‐subunit communication and dependence for catalysis, AAA+ ATPases rely on oligomeric ring structures for proper nucleotide binding and hydrolysis.
A unique structural feature of Classic and HCLR AAA+ motors is a Helix‐2 insertion that extends from every subunit into the central channel of the hexameric ring to contact and propel the substrate polypeptides. Mutational analysis of these “pore‐loops” showed that primarily aromatic residues, such as Tyr or Phe, are required for substrate interactions that appear to be largely of steric nature.11, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 Besides these translocating pore‐1 loops, a second pore loop was found to have substrate‐binding capabilities.11, 19, 23, 24, 31 In this review we will refer to these substrate‐interacting loops as pore‐1 loop and pore‐2 loop.24
Additional domains N‐ or C‐terminal to the AAA+ motor domains provide diversity within the Classic clade, which can thus be further divided into 7 subfamilies: the FtsH‐, katanin‐, TIP49‐, AFG1‐, Proteasomal‐, NSF/Cdc48/Pex‐, and ClpABC‐NBD1 families.6, 8 Similarly, HCLR motors include 4 subfamilies: HsIU/ClpX, ClpABC NBD2, Lon, and RuvB. The type II AAA+ proteins within these clades contain two ATPase domains per subunit that form two stacked rings. These domains, frequently termed NBD1 and NBD2, will in this review be referred to as D1 and D2.
1.2. Previous structural analyses
Most of the early structures for AAA+ proteins of the Classic and HCLR clades were solved by X‐ray crystallography in the presence of various nucleotides. These structures gave important insights into the overall architecture of AAA+ motors, but often revealed hexameric open lock‐washer or spiral‐shaped assemblies that were largely thought to represent inaccurate or functionally irrelevant conformations.6, 32, 33 Moreover, these structures lacked any bound substrate, which complicated an assessment regarding their relevance for active translocation. Due to the lack of high‐resolution information on the ATPase hexamers and their substrate‐bound states, major functional questions remained: how are subunits in AAA+ rings occupied by nucleotide, how do they interact with substrate, and how does ATP hydrolysis drive mechanical translocation?
The first substrate‐bound structures were revealed for motors in other clades, including crystal structures of DNA helicases and clamp loaders.34, 35, 36, 37, 38 In the structure of the DNA‐bound E1 helicase, a representative of the superfamily III helicase clade, the AAA+ domains form a right‐handed spiral staircase with a vertical rise of 1 nucleotide/subunit and a 60° turn between neighboring subunits in the ring. The nucleotide states observed for individual subunits progress around the ring and along the spiral arrangement, from ATP‐bound at the top to ADP‐bound or apo state at the bottom, suggesting that ATP binding and hydrolysis occur sequentially in the hexamer and lead to uniform step sizes for DNA threading. The combination of this spiral architecture and potentially sequential hydrolysis led to the first hypothesis of a “hand‐over‐hand” mechanism for substrate translocation, in which subunits upon ATP hydrolysis and nucleotide exchange transition from the bottom to the top position in the spiral staircase.34
Due to their high conservation, it was postulated that all AAA+ motors may operate by similar mechanisms.39 However, it was harder to imagine how the protein‐processing AAA+ motors interact with their substrates, considering that polypeptide chains do not show the same helical regularity as their nucleic acid counterparts in helicases. Furthermore, biochemical data for ClpX, one of the most well‐characterized HCLR clade AAA+ motors, contradicted a strictly sequential ATP‐hydrolysis model. Numerous studies suggested that ClpX hydrolyzes ATP in a more stochastic manner, and that subunits unable to hydrolyze can be skipped with no major effect on neighboring subunits.40, 41, 42, 43 Furthermore, single‐molecule measurements of substrate processing suggested that ClpX operates in “dwell” and “burst” phases,44, 45, 46 and thereby translocates its substrates with variable step sizes of 1–4 nm, depending on how many subunits hydrolyze during a given dwell‐burst cycle.40, 44, 45, 47 These findings thus contradict a more regular stepping with uniform step sizes expected for strictly sequential ATP hydrolysis.48 Solving high‐resolution substrate‐bound structures of AAA+ motors was consequently important to reconcile previous structural knowledge, elucidate the motor interactions with substrate polypeptides, and reveal the principles underlying ATP‐hydrolysis‐coupled protein translocation.
Recent technological advancements in the cryo‐EM field, including the development of direct electron detectors, made high‐resolution structure determination feasible, especially for larger macromolecular complexes such as AAA+ hexamers. As we now know, AAA+ ATPases undergo large conformational changes and transition through highly asymmetric hexameric ring states during substrate translocation, which explains the previous challenges in crystallographic studies. In the past 2 years, 41 substrate‐bound structures of AAA+ motors have been solved by cryo‐EM, with resolutions ranging from 2.9–4.8 Å, and including several datasets with multiple conformations and nucleotide states for a particular motor (Table 1). Here we highlight the structures of these substrate‐engaged motors, which greatly expand our knowledge of how AAA+ motors couple ATP hydrolysis to mechanical translocation.
Table 1.
Summary of substrate‐bound AAA+ motor structures determined by cryo‐EM. Abbreviations are as follows: N‐terminal domain (NTD), Walker B (WB), and not applicable (N/A)
| Protein | Species | Nucleotide state | Mutation | Substrate | Resolution (Å) | PDB | EMDB | Citation |
|---|---|---|---|---|---|---|---|---|
| Yme1 | S. cerevisiae | ATP | WB (E381Q), CCHex | No added substrate | 3.4 | 6AZ0 | 7,023 | Puchades 201750 |
| AFG3L2 | H. sapiens | AMPPNP | WB (E408Q), protease inactive (E575Q) | No added substrate | 3.1 | 6NYY | 552 | Puchades 201951 |
| Vps4 | S. cerevisiae | ADP•BeFx | N/A | Peptide | 4.3 | 5UIE | 8,549 | Monroe54 |
| Vps4 | S. cerevisiae | ADP•BeFx | N/A | Peptide | 3.2 | 6BMF, 6AP1 | 8,887 | Han 201752 |
| Vps4 | S. cerevisiae | ADP•BeFx | N/A | Circular peptide | 3.6 | 6NDY | 443 | Han 201953 |
| Spastin | D. melanogaster | ATP | WB (E583Q) | Poly‐glutamate peptide | 3.2 | 6P07 | 20,226 | Sandate55 |
| 26S | S. cerevisiae | ATP | N/A | Poly‐Ub'ed titin(PY) | 4.1–4.7 | 6EF0, 6EF1, 6EF2, 6EF3 | 9,042–9,045 | de la Peña56 |
| 26S | H. sapiens | ATPγS | N/A | Poly‐Ub'ed Sic1(PY) | 2.8–3.6 | 6MSB, 6MSD, 6MSE, 6MSG, 6MSH, 6MSJ, 6MSK | 9,215–9,229 | Dong57 |
| VAT | T. acidophilum | ATPγS | ΔNTD | No added substrate | 4.8 | 5VCA | 8,658, 8,659 | Ripstein58 |
| NSF | C. griseus | ATP, EDTA, no Mg2+ | N/A | SNARE protein, SNAP‐25A | 3.8, 3.9 | 6MDO, 6MDP | 9,102, 9,103 | White62 |
| Cdc48 | S. cerevisiae | ATP | D2 WB (E588Q) | Poly‐Ub'ed mEOS | 3.9 | 60A9 | 0665 | Twomey60 |
| Cdc48 | S. cerevisiae | ADP•BeFx | N/A | Poly‐Ub'ed mEOS | 4.1, 3.6 | 60AA, 60AB | 0666, 20,000 | Twomey60 |
| Cdc48 | S. cerevisiae | ADP•BeFx | N/A | Endogenous substrate | 3.7 | 60 MB, 60PC | 20,124 | Cooney59 |
| Rix7 | S. cerevisiae | No added nucleotide | Double WB (E303Q, E602Q) | No added substrate | 4.5 | 6MAT | 9,063 | Lo61 |
| Hsp104 | S. cerevisiae | ATPγS | N/A | Casein | 3.9, 4.1 | 5VJH, 5VYA | 8,697, 8,746 | Gates63 |
| ClpB | E. coli | ATPγS | BAP variant, double WB (E279A/E678A) | Casein | 4.5–5 | 5OFO | 3,776, 3,777 | Deville 201765 |
| ClpB | M. Tuberculosis | ATPγS | N/A | Casein | 3.8–3.9 | 6DJU, 6DJV | 7,492, 7,493 | Yu64 |
| ClpB | E. coli | ATPγS | De‐repressed (K476C) | Casein | 2.9 | 6OAX, 6OAY | 20,004–20,005 | Rizo66 |
| ClpB | E. coli | ATPγS | De‐repressed (K476C), double WB (E279A/E678A) | Casein | 3.6–4.1 | 6qs6, 6qs7, 6qs8, 6qs4 | 4,621, 4,624–4,627 | Deville 201969 |
| ClpB | E. coli | ATPγS | Double WB (E279A/E678A) | Casein | 4–6 | 6rn2, 6rn3, 6rn4 | 4,640–4,642 | Deville 201969 |
| Hsp101 | P. malariae | ATPγS | N/A | PTEX cargo polypeptide | 3–3.6 | X6E10 | 8,951 | Ho67 |
| TRIP13 | H. sapiens | ATPγS | WB (E253Q) | p31comet‐C‐MAD2‐CDC20 complex | 4.3–4.5 | 6F0X | 4,166 | Alfieri68 |
1.3. Spiral‐staircase arrangement of pore‐1 loops
The first substrate‐bound Classic AAA+ motor structure was solved for the 26S proteasome from S. cerevisiae,49 albeit with low resolution and substrate density not visible. The details of substrate interactions thus remained elusive, but this structure revealed a defined spiral‐staircase arrangement of the distinct proteasomal ATPase subunits, Rpt1–Rpt6, in the hexameric ring. Since that early discovery, many structures of AAA+ motors have been solved at higher resolution and with visible substrate bound in the central channel, including: Yme1,50 AFG3L2,51 Vps4,52, 53, 54 spastin,55 proteasome,56, 57 VAT,58 Cdc48,59, 60 Rix7,61 NSF,62 Hsp104,63 ClpB,64, 65, 66 Hsp101,67 and Trip13.68 The majority of studies took advantage of hydrolysis‐inhibiting Walker‐B mutations or non‐hydrolyzable ATP analogs to stabilize substrate‐bound complexes (Table 1). Despite the large variability in sample preparations, utilized substrates, and regulatory domains present in these motor complexes, all structures revealed highly similar spiral‐staircase arrangements of ATPase subunits and pore loops that directly interact with protein substrates in the central channel (Figure 2).
Figure 2.
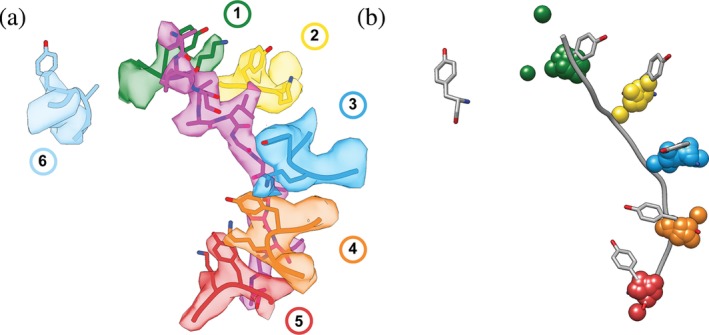
Spiral‐staircase arrangement of pore‐1‐loop residues in contact with substrate. (a) Atomic model (PDB:6EF2) and EM density (EMDB 9440) for the spiral staircase of pore‐1‐loop residues in the 5T state of the 26S proteasome. Shown are the substrate in magenta as well as the conserved Tyr and flanking Lys residues for each protomer numbered by the position in the “spiral staircase,” with Rpt1 in green, Rpt2 in yellow, Rpt6 in blue, Rpt3 in orange, Rpt4 in red, and Rpt5 in cyan. (b) The pore‐1‐loop Tyr's and the substrate polypeptide from the 5T model of the 26S proteasome (56) are displayed in gray and overlaid with 1‐Å centroids indicating the Cα positions for the aromatic “paddle” residues in the pore‐1 loops of substrate‐bound YME1 (6AZ0), AFG3L2 (6NYY), VPS4 (5UIE, 6BMF, 6AP1), the 26S proteasome (1D*‐state (6EF0), 5D‐state (6EF1), 5T‐state (6EF2), 4D‐state (6EF3), ED1‐state (6MSJ), ED2‐state (6MSK)), D2 domains of VAT/p97 (5VCA), D2 domains of NSF (6MDO, 6MDP), D2 domains of Rix7 (6MAT), D2 domains of Hsp104 in the closed (5VJH) and extended states (5VYA), D2 domains of ClpB D2 (ATPγS‐bound double‐WB mutant (5OFO), KC‐1 state (6qs6), KC‐2A state (6qs7), KC‐2B state (6qs8), KC‐3 state (6qs4), WT‐1 (6rn2), WT‐2A (6rn3), WT‐2B (6rn4), conformer1 (6DJU), conformer2 (6DJV), Pre (6OAX), Post (6OAY)), TRIP13 (6F0X), D2 domains of Hsp101 (6E10), and Spastin (6P07). The Cα's for the aromatic pore‐1‐loop residue of protomers 1–5 in each motor were aligned to the proteasomal 5T state as a reference model (6EF2), using Matchmaker in Chimera.75 RMSD values for this alignment ranged from 0.403–1.526 Å. Centroids for the substrate‐disengaged subunit were omitted, as they have the largest variance in position. The two green centroids that do not overlay are from AFG3L2 (6NYY) and 1D* (6EF0), which both have two substrate‐disengaged subunits
Most of these substrate‐bound structures show five subunits in contact with the substrate polypeptide (with exceptions having 4 or 6), and one disengaged “seam” subunit that lies between the “top”‐most and “bottom”‐most subunits in the spiral staircase. This subunit arrangement and substrate binding can be clearly seen in the example staircase of Rpts1‐6 of the proteasome (Figure 2a). The “spiral staircase” described in these studies is largely defined by the arrangement of the substrate‐engaged pore‐1 loops (Figure 2a), which usually contain an aromatic residue (Tyr/Phe/Trp) that comes into close proximity (∼4 Å) of the polypeptide backbone and intercalates the side chains. These direct interactions with the backbone are non‐specific, which explains why these AAA+ motors can accommodate and efficiently translocate a large variety of substrate proteins.
The substrate contacts of translocating pore‐1 loops are vertically separated by a rise of ∼6 Å, or two amino acids, and a ∼60° rotation between neighboring subunits, corresponding to the hexameric arrangement of NBDs. This spiral distribution is highly conserved among the different structures and can be overlaid extremely well, with an RMSD of <1.6 Å when aligning the pore‐1 loop residues (Figure 2b). Importantly, this spiral‐staircase arrangement of pore loops and ATPase subunits in general is also strikingly similar to the previously described structures of DNA helicases and clamp loaders, hinting to a common mechanism of ATP‐hydrolysis‐driven substrate translocation.
1.4. Additional substrate contacts ‐ a complex interaction network
Many structures of AAA+ motors, including Vps4,52, 53, 54 Rix7,61 Rpt1‐6 of the proteasome,56, 57 ClpB,65 and NSF62 indicate a secondary spiral of pore‐2 loops that form a similar staircase below the pore‐1 loops, but do not directly interact with substrate or lack bulky side chains. In other structures, however, the pore‐2 loop residues make direct contact with substrate through aromatic residues. Both, YME150 and AFG3L251 contain pore‐2 loop residues, Y396 and F421, respectively, that form a second spiral staircase of substrate contacts. Mutation of these residues to alanine impairs substrate translocation and degradation by the protease domains that reside C‐terminal of the AAA+ domain in those motors.50, 51 In AFG3L2, the pore‐2 loops of the fourth and fifth subunits appear to reach into the interior of the proteolytic cavity to handoff substrate from the ATPase ring to the protease for degradation.51
Spastin,55 TRIP13,68 and ClpB66 also have pore‐2 loop residues that form direct interactions with the substrate. In spastin, residues H596 and R601 form a second spiral staircase of electrostatic interactions with every other sidechain in a polyglutamate peptide substrate.55 Similarly, in ClpB, the pore‐2‐loop residues E639, K640 and H641 project into the central channel and appear to interact directly with the substrate.66 In the TRIP13 structure, seven N‐terminal residues of the MAD2 substrate extend into the pore, where one of them, R7, directly interacts with E269 and D272 in the pore‐2 loop of the “top” subunit. Mutating these pore‐2‐loop residues to Ala or Arg made TRIP13 completely defective in substrate remodeling.
In addition, these studies identified many other inter‐protomer contacts that form a complex substrate‐interaction network beyond the classic pore‐loop spiral staircases. As obvious from a sequence alignment of pore‐1 loops from all structures discussed here, the position preceding the conserved aromatic amino acid is often charged (Lys/Arg) and can form electrostatic interactions with residues of neighboring pore loops, including Glu or Asp frequently found three amino acids downstream of the aromatic residue (Figure 3). ClpB has two charged amino acids, K250 and R252, flanking the aromatic pore‐1‐loop residue Y251. In two ClpB studies,66, 69 K250 and R252 were observed to form a salt bridge or hydrogen bond with E254 and E256 of the clockwise‐ and counterclockwise neighboring pore‐1 loops, respectively, and thus create a clamp around the substrate polypeptide. Furthermore, the ClpB study by Yu et al.64 showed R252 oriented toward Y251 of the neighboring subunit and hydrogen‐bonding with S249. Mutational analyses in each study revealed that eliminating the charged residues flanking the conserved aromatic “paddle” in the pore‐1 loop leads to defects in substrate processing, while charge‐reversal mutations are active.
Figure 3.
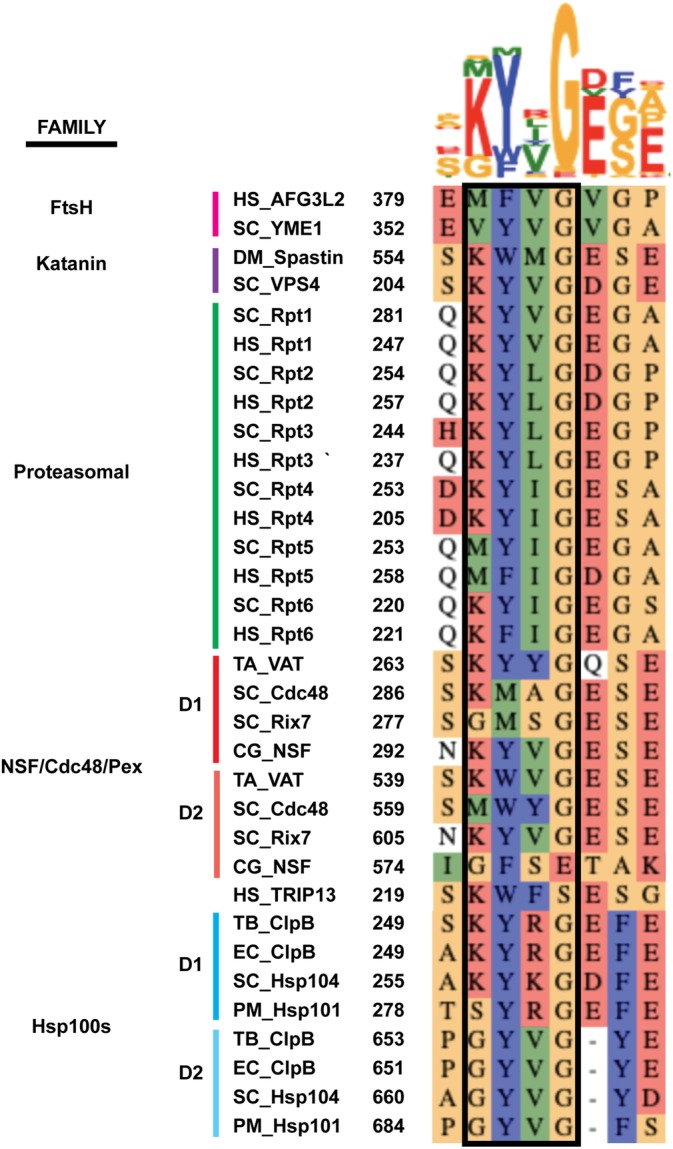
Sequence alignment for the pore‐1‐loop residues (boxed) and flanking amino acids in the AAA+ motor domains of protein translocases, including both the D1 and D2 domains for the double‐ring Type II AAA + 's. Abbreviations are as follows: Homo sapiens (HS), Saccharomyces cerevisiae (SC), Drosophila melanogaster (DM), Thermoplasma acidophilum (TA), Cricetulus griseus (CG), Mycobacterium tuberculosis (TB), Escherichia coli (EC), and Plasmodium malariae (PM). Alignments were done in MUSCLE76 and illustrated with AlignmentViewer 77
Similarly, in the pore‐1 loop of spastin, the flanking Lysine, K555, forms a salt bridge with E462 in the alpha‐1 helix of a neighboring subunit, which is a unique structural feature to severing enzymes.70, 71 Spastin contains a series of other Arg and Glu residues that appear to form a network of salt bridges and hydrogen bonds between subunits, including interactions between the pore‐2 and a newly identified pore‐3 loop. Mutations of any of these Arg or Glu residues impair microtubule severing. The AFG3L251 pore‐1‐loop residue F381 is flanked by a hydrophobic residue, M380 that contacts F381 of the counterclockwise neighboring subunit and thereby surrounds the polypeptide with tight interactions. Mutating M380 to a Val, as present in YME1, or Lys, as present in many other motors (Figure 3), decreased or eliminated substrate degradation, respectively, showing how specific inter‐pore‐loop interactions are tuned for each system.
These studies thus revealed many new interactions between ATPase protomers that are highly conserved and specific to each individual system and include both salt bridges and hydrogen‐bonding networks to facilitate effective substrate binding and translocation.
1.5. Role of non‐aromatic pore‐loops
Although most of the spiral staircases for substrate‐bound AAA+ motors can be overlaid, there are two major outliers, Rix7 and Cdc48,60, 61 which are both Type II AAA+ motors containing a Met as a non‐aromatic residue in the pore‐1 loop of the D1 ATPase domain (Figure 3). The Rix7 structure was determined using Walker‐B mutations in both the D1 and D2 domains, and revealed a planar D1 ring that makes no pore‐1‐loop contacts with substrate and is vertically much more compact (∼15 Å) than the D2 spiral staircase contacting the substrate (∼28 Å). The D1 ring was found to contain ATP in all six nucleotide‐binding pockets, suggesting that the D1 domain may hydrolyze ATP in a different manner than the sequential mechanism assumed for D2 or other AAA+ motors. Mutational analyses in Rix7 revealed that replacing the D1 pore‐1 loop (Gly/Met/Ser) with the D2 pore‐1 loop (Lys/Tyr/Val) causes a growth defect. While the Met to Tyr mutation alone was viable, the replacement of Ser by Tyr was lethal, owing to the importance of this subtle change in pore loop spacing.61
One of the substrate‐bound structures of Cdc48 solved by Twomey et al60 is similar to Rix7 with respect to a spiral staircase in the D2 ring and a near‐planar D1 ring that contains ATP in all NBD pockets and contacts substrate through only two pore‐1 loop Met's. This structure was determined with a D2 Walker‐B mutant in the presence of ATP, whereas wild‐type Cdc48 with bound ADP/BeFx resulted in two alternative structures that exhibit the D1 ring with four substrate contacts in a similar spiral as D2. These ADP/BeFx structures thus resemble the state observed in the Cdc48 study by Cooney et al.,59 in which ADP/BeFx was used as well and five subunits were found in contact with substrate for both the D1 and D2 ATPase rings. According to Twomey et al.,60 the angle between the D1 and D2 domains in the ATP‐bound structure increases for individual Cdc48 protomers with their increasingly lower position in the D2 spiral staircase, whereas this trend is not observed in the ADP/BeFx‐bound states. It therefore appears that ADP/BeFx fixes the angle of the hinge segment between the D1 and D2 domains, favoring a spiral‐staircase arrangement in the D1 ring that mirrors the spiral in D2. Interestingly, similar to Rix7, introducing aromatic residues in the pore‐1 loop of Cdc48's D1 domain leads to a lethal phenotype, likely through aberrant engagement and processing of substrates.72
Multiple studies thus demonstrated that non‐aromatic pore‐1 loops in the D1 rings of Type II AAA+ motors can form spiral staircases as well as more planar and compact arrangements. It is unclear whether both D1‐ring states are relevant for substrate translocation, as they may have been induced by the ATP analogs used for structure determination, yet mutational studies clearly indicated that the pore‐1 loop residues in the D1 domains are highly conserved for proper motor function in the cell. While the bottom D2 ring in Type II AAA+ motors appears responsible for the majority of substrate translocation, the top D1 ring with its non‐aromatic pore‐1 loops may play a role for initial substrate engagement and/or in a ratchet‐like manner to prevent the backsliding of substrates between power strokes of the D2 ring. The latter model is supported by recent single‐molecule studies of ClpA, in which a mutant with ATPase‐deficient D1 ring showed significant defects in substrate unfolding and translocation, as well as back slips through the enzyme.73
1.6. ATP hydrolysis drives translocation
As the spiral staircase arrangements are very similar among the substrate‐bound AAA+ motor structures, so are the nucleotide states observed for particular positions in these staircases. A few of the structural studies did not assign nucleotide states and are therefore not discussed in this section: VAT,58 ClpB,65 and Hsp101.67
Assigning nucleotide states was often highly challenging due to intermediate resolution, especially for the nucleotide pockets, and the averaging of different nucleotides in any given pocket during the single‐particle averaging inherent to cryo‐EM data analysis. For high‐enough resolution structures, the authors determined the nucleotide state (ATP/ADP/apo) based on the observed densities. Otherwise they performed a variety of measurements to assess the openness of ATPase pockets or the distances between hydrolysis‐relevant motifs that report on nucleotide identity, including the trans‐acting Arg‐fingers and the intersubunit signaling (ISS) motif. In an active ATP‐bound pocket, the Arg‐finger and ISS are in close proximity to the γ‐phosphate (∼3‐4 Å). Following hydrolysis and as the γ‐phosphate is released, the Arg‐finger and ISS retract from the nucleotide (∼15–20 Å), and these pockets are assigned either ADP‐bound or apo states. Furthermore, the inter‐protomer contact area greatly decreases in a post‐hydrolysis pocket, providing an additional important parameter for the assignment of nucleotide states.
Comparing the nucleotide occupancies for numerous structures of substrate‐bound single‐ring AAA+ motors reveals a highly consistent continuum of ATP‐hydrolysis states around the spiral staircase (Figure 4a). Subunits 1–3 at the top of the spiral are always ATP‐bound, while the “seam” subunit bridging the lowest and highest positions is generally in a post‐hydrolysis state (ADP/apo). This is not surprising, given that its large distance to the clockwise‐next neighbor does not support a closed nucleotide pocket (Figure 4a). There is some discrepancy between structures regarding the nucleotide state of subunits 4 and 5, varying between ATP‐bound, ADP‐bound, or apo (Figure 4a). For most structures of single‐ring motors, subunit 4 is assigned to be ATP‐bound, except for Vps452 and the 5T state of the 26S proteasome,56 in which it appears to contain ADP. Conversely, subunit 5 is mostly ADP‐bound or apo, except for TRIP1368 and spastin.55 In agreement with these assignments, the nucleotide pockets also display a pattern of opening up as subunits progress down the spiral and from ATP‐bound to ADP‐bound states.50, 53, 54, 57, 60, 62 For example, the angle between the large and small AAA subdomains (Figure 1b) in NSF is larger for ADP‐bound or apo pockets (∼10°) than for ATP‐bound pockets (∼1–2°).62 Based on these structural snapshots, we can start to understand how ATP binding and hydrolysis may occur sequentially around the ring, leading to the observed continuum of nucleotide states that depend on the vertical position within the spiral staircase (Figure 4a).
Figure 4.
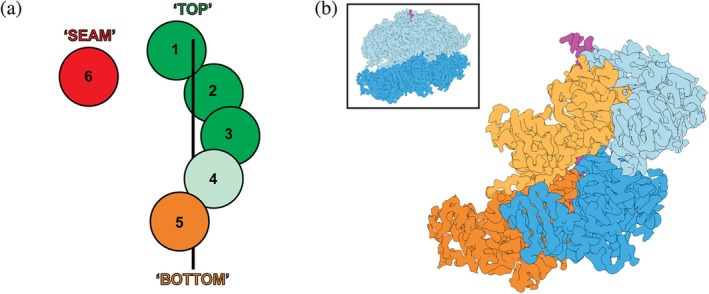
The nucleotide state of a AAA+ subunit depends on its position within the spiral staircase. (a) Cartoon representation of the spiral staircase arrangement with five substrate‐engaged and one dis‐engaged subunit, depicting their nucleotide states. ATP‐bound protomers are colored green, with a lighter green in position 4 indicating some accounts of ADP and potential hydrolysis in this subunit. The substrate‐disengaged “seam” subunit is colored red to represent an ADP‐bound or apo state, while the “Bottom” subunit is shown in orange to indicate a mostly ADP‐bound state, with some accounts of ATP in this position. (b) EM density for the “KC‐2” state of ClpB (EMDB:4625, complete hexamer shown in the inset with D1 domains in light blue and D2 domains in dark blue) reveals the slanted conformation observed in double ring AAA+ motors. The D1 and D2 domains are shown in light and dark blue for protomer 3, and in light and dark orange for protomer 4, highlighting how the D1 domain of a subunit overlaps with the D2 domain of the counterclockwise‐next neighbor
During the ATPase cycle, the substrate‐engaged subunits at the top of the spiral remain ATP‐bound, and ATP hydrolysis likely initiates in subunit 4 or 5. Phosphate release and opening of the ATPase pocket destabilize interactions between AAA+ subdomains within the hydrolyzing subunit and towards the clockwise‐next neighbor, triggering disengagement of the bottom subunit from the substrate, and its transition towards the top of the spiral. At the same time, the structural compaction between subunits required to rebind ATP to the disengaged subunit favors its movement to the top of the spiral and provides directionality for translocation. Hence, disengaged subunits move upwards, while substrate‐engaged subunits progress down in the spiral, leading to a vertical downward translocation of substrate through the central pore. Important information about the coordination between these movements and the ATPase cycle was provided by structural studies that revealed multiple states for the same AAA+ motor, as described below.
1.7. Multiple motor states of the 26S proteasome temporally detail translocation
Most of the studies discussed here determined a single substrate‐bound structure with subunits in different nucleotide states, which were then used to interpolate how the ATP‐hydrolysis events and conformational changes may propagate around the ring for processive translocation.50, 51, 52, 53, 54, 55, 58, 59, 61, 65, 67, 68 Fortunately, several studies revealed multiple substrate‐engaged states and corresponding differences in nucleotide occupancies for the 26S proteasome,56, 57 ClpB,64, 66, 69 and Hsp104,56 providing insight into the coordination between ATP‐hydrolysis events and conformational changes.
Of particular importance was thereby the 26S proteasome with its heterohexameric motor that is a component of the 19S regulatory particle sitting atop the 20S core peptidase. Visualizing multiple states of this motor provided insight into the progression of individual subunits through the various stages of the ATPase cycle and indicated a potential temporal order of events and conformations. The study by Dong et al.57 revealed seven conformations, with two of them, E D1 and E D2, likely representing actively translocating motor states. E D1 and E D2 show five substrate‐bound subunits consistent with the spiral staircase described above (Figure 2), and the identities of the top, bottom, and seam subunits are rotated by one position in the heterohexamer, suggesting that these two structures represent consecutive motor states during sequential ATP hydrolysis.
Of the four motor states described by de la Peña et al. for the wild‐type proteasome in the presence of ATP,56 three appear to be consecutive based on the movement of subunits within the spiral‐staircases arrangement and the changes in their nucleotide‐binding pockets. During the transition from the 5D to the 5T state, subunit 4 hydrolyzes ATP, while the substrate‐disengaged “seam” subunit 6 exchanges ADP for ATP. Importantly, the spiral staircase remains unchanged during this transition, and there is no translocation of substrate. These hydrolysis and nucleotide‐exchange events appear to be prerequisites for the subsequent large conformational arrangement and translocation step occurring during the transition from the 5T to the 4D state. Subunit 5 at the bottom of the spiral disengages from substrate and separates from the neighboring subunit 4, likely as a consequence of phosphate release from the ATPase pocket at this interface, while the “seam” subunit 6 takes on the top position, 6 Å above subunit 1, and engages the substrate. Consequently, the substrate‐bound subunits 1–4 move downward as a rigid body, pulling the substrate polypeptide along. These protomer movements can be directly observed relative to the 20S core particle and confirm that the ATP‐hydrolysis driven conformational changes are linked to substrate translocation. Taken together, these structures demonstrate a compelling argument for sequential ATP hydrolysis and coupled conformational changes that progress around the hexameric ring and lead to substrate translocation by a hand‐over‐hand mechanism.
1.8. Double ring motors show an offset in hydrolysis events
Structures for double‐ring, Type II AAA+ motors with assigned nucleotide states are available for Rix7, Cdc48, ClpB, and Hsp104. As previously discussed, the D1 ring of Rix7 and Cdc48 contains non‐aromatic pore‐1 loops, is in a planar conformation, and appears to be occupied by ATP in all subunits. Their D2 rings, however, adopt spiral‐staircase arrangements with patterns of nucleotide states that resemble those for the single‐ring AAA+ translocases, with ATP bound to subunits at the top and ADP‐bound or apo‐state subunits near the bottom and in the seam (Figure 4a). ClpB and Hsp104 form spiral staircases in both their D1 and D2 rings, with the same consequent patterns of nucleotide states. Multiple motor states from these studies reveal likely successive hydrolysis events that present a more complex model, involving offset cycling of hydrolysis between D1 and D2.
There are three ClpB studies64, 66, 69 that detail multiple states with differences in nucleotide occupancies for the D1 and D2 domains of individual ClpB protomers. While Yu et al.64 used a ClpB variant from Mycobacterium tuberculosis, the studies by Deville et al.69 and Rizo et al.66 focus on bacterial ClpB with a de‐repressing K476C mutation that causes faster ATP hydrolysis and substrate processing. All structures were determined in the presence of casein as a substrate and ATPyS as an ATP analog. In addition, Deville et al.69 introduced Walker‐B mutations into both the D1 and D2 domains. Described states include KC1, KC2, KC3,69 “Pre” and “Post” states,66 and “conf1” and “conf2”,64 which are thought to be consecutive states in the ATP‐hydrolysis and substrate‐translocation cycles.
The overall consensus of these studies is that the nucleotide‐hydrolysis and ‐exchange events for a particular protomer in D2 ring often precede the same events in the D1 ring, suggesting an offset coordination between the two ATPase domains. Due to their slanted arrangement, the D1 domain of one subunit is positioned partially on top of the D2 domain for the counter‐clockwise next subunit (Figure 4b). Although direct interactions between these domains have not been implicated, the ATP hydrolysis cycles appear to be correlated between these stacked domains, potentially due to steric reasons when a particular staircase arrangement in the bottom D2 ring has to be accommodated by a corresponding arrangement in the D1 ring above.69 For example, in the transition between the KC‐2 and KC‐3 states,69 and “Pre” and “Post” states,66 the D1 domain of subunit 5 and D2 domain of subunit 4 hydrolyze ATP in a coordinated manner, potentially driving the conformational changes occurring between those states (Figure 4b). A similar offset is observed in the study by Yu et al.,64 where the D2 domain of the “seam” subunit is already occupied with ATP in the “conf1” state, whereas the D1 domain exchanges ADP for ATP during the transition to “conf2,” and both the D1 and D2 domains then bind the substrate.
Although the results for mutant ClpB are consistent with each other, this offset cycling was not observed in the structures of wild‐type ClpB (WT‐1, WT‐2A, and WT‐2B) described in Deville et al69 or in the structures of the yeast homolog, Hsp104, presented by Gates et al.63 This Hsp104 study describes both “closed” and “extended” states of the hexamer, with either five or six substrate‐bound subunits. During the presumed transition between these states, the sixth subunit moves from an “off” position to the top of the spiral staircase and engages the substrate about 6 Å above the next subunit, similar to the arrangement observed for the substrate‐bound proteasome. Furthermore, the “seam” subunit exchanges ADP for ATP in both its D1 and D2 domains, reinforcing that ATP binding is a pre‐requisite for substrate interactions at the top of the spiral staircase.
In each of these studies, the use of non‐hydrolyzable ATP analogs or hydrolysis‐eliminating mutations, together with the homohexameric architecture of the ATPase rings, limits our ability to determine whether the offset cycling of the D1 and D2 ATPase domains is a true occurrence or a consequence of the experimental design. Larger datasets may reveal additional states, helping to convolute this mechanism even more, and biochemical experiments provide important functional information to verify these structure‐based models.69
In summary, the multiple, likely consecutive states observed for some of the AAA+ motors support the concept that translocation originates from ATP‐hydrolysis‐induced conformational changes occurring sequentially around the ring. These detailed structural insights thus significantly furthered our understanding of the hand‐over‐hand mechanism for ATP‐hydrolysis‐driven polypeptide translocation.
1.9. Substrate engagement and translocation through mechano‐chemical coupling
The multitude of substrate‐bound structures solved over the past couple of years provided a major advance to our understanding of AAA+ motor function, yet there still remain numerous open questions, for instance about initial substrate engagement, and whether spiral staircase arrangements of ATPase subunits are induced by the substrate or represent an intrinsic feature of to the actively translocating hexamer. A few of the studies describe substrate‐free motor states that may give us insight into the mechanisms of substrate engagement. Strikingly, these substrate‐free motors show very different conformations, which could be in part due to variations in extra domains and insertions. Both VAT and Cdc4858, 59 adopt symmetric substrate‐free states that may be important for cofactor binding atop the hexameric ring. Hsp104 and ClpB56, 64 have left‐handed spirals with a large open cleft, potentially representing a primed state for lateral substrate entry into the ring during handoff from Hsp70/DnaK chaperones. The substrate‐free states for other motors, including Hsp101,67 TRIP13,68 and another ClpB,65 are more similar to substrate‐engaged conformations. These structures indicate that substrate engagement generally affects the overall ring conformation and might trigger the switch to the canonical staircase‐arrangement, yet the complex networks of interactions with the substrate as well as between neighboring subunits point to the AAA+ motor dictating the spiral conformation during translocation. Since the substrate‐free states appear to vary dramatically for different AAA+ motors, more detailed studies will be necessary to determine how substrates are initially engaged in each individual case.
Following an engagement step, AAA+ motors form substrate contacts in a spiral staircase arrangement of pore‐1 and pore‐2 loops, with other nearby elements contributing additional steric interactions. For example, charged residues flanking the aromatic “paddle” in the pore‐1 loop form salt bridges or hydrogen‐bonds with residues in the neighboring subunit to stabilize a network of substrate contacts. The variability in these residues provides substrate‐gripping abilities that are appropriate for each individual motor and cellular function. Similarly, non‐aromatic pore‐1‐loop residues, as found in the D1 rings of Type II AAA+ motors, seem correlated with a planar pore‐loop arrangement rather than a spiral staircase, and apparently do not bind substrate as well as the aromatic pore loops. Mutational analyses revealed that altering pore loops and salt bridges often reduces substrate degradation or compromises cell viability, indicating that these substrate interactions are finely tuned for each cellular process.
Analyzing the nucleotide state for individual subunits in the hexamer suggests that ATP hydrolysis causes an overall downward movement of substrate‐engaged subunits within the vertical spiral‐staircase arrangement to thread the substrate through the central channel. Hydrolysis likely occurs in the fourth position of the spiral, destabilizing the contacts with the clockwise‐next neighbor and forcing that “bottom” subunit to dissociate from substrate. The previously disengaged “off” subunit exchanges ADP for ATP, before becoming the next “top” subunit in contact with the substrate 6 Å above the previous “top” pore loop. Substrate interactions are likely controlled by hinge movements between the AAA+ subdomains of each subunit, which in turn depend on a subunit's nucleotide state and position within the spiral staircase. Four subunits are consistently found to be substrate‐bound and thus, in addition to efficiently propelling the substrate forward, may prevent backsliding or substrate escape. Overall, rebinding of ATP after hydrolysis favors a conformation that swings up the respective subunit to bind substrate at the top of the spiral, and this process seems to occur sequentially around the ring.
This structure‐based translocation model thus encompasses a very regular stepping or inch‐worming along the substrate. Substrate interactions are moved from the bottom to the top of the spiral staircase when a post‐hydrolysis subunit exchanges ADP for ATP, while four ATP‐bound, substrate‐engaged subunits move downward as a rigid body, leading to the translocation of two amino acids per hydrolyzed ATP, or 1/6 of the vertical spiral extension. This mechanism is in contrast to the “power‐stroke” model, in which a single subunit hydrolyzes ATP, moves downwards, and pulls the substrate along. The power stroke model was proposed primarily based on extensive biochemical and single‐molecule studies of ClpX from E. coli, which indicated non‐strictly‐sequential ATP hydrolysis and larger translocation step sizes that originate from coordinated “firing” of several subunits during a burst phase. Interestingly, recent cryo‐EM studies of ClpX revealed a spiral staircase arrangement and substrate interactions of subunits that strongly resemble the scenario for other AAA+ motors described above.48 The model for translocation being driven by sequential ATP hydrolysis around the ring and substrate‐disengaged subunits moving upwards in the spiral staircase appears quite compelling, as 41 substrate‐bound structures of AAA+ protein translocases spanning many different families, functions, and experimental design have yielded similar results regarding substrate contacts and nucleotide states. However, these structures represent snapshots, with numerous questions remaining about their order and temporal transitions. Resolving the apparent discrepancy between translocation models based on structural information versus functional data is an important goal for the AAA+ field and will require biochemical and biophysical measurements on several Classic and HCLR AAA+ motors to better clarify the hand‐over‐hand mechanisms.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
ACKNOWLEDGMENTS
We thank Ken Dong and Adam Yokom for helpful discussions. A.M. is an HHMI investigator and S.N.G. is a Howard Hughes Medical Institute Fellow of the Damon Runyon Cancer Research Foundation, DRG‐2342‐18. This work was funded by HHMI and the National Institutes of Health (R01‐GM094497 to A.M.).
Gates SN, Martin A. Stairway to translocation: AAA+ motor structures reveal the mechanisms of ATP‐dependent substrate translocation. Protein Science. 2020;29:407–419. 10.1002/pro.3743
Funding information Damon Runyon Cancer Research Foundation, Grant/Award Number: DRG‐2342‐18; Howard Hughes Medical Institute; National Institute of General Medical Sciences, Grant/Award Number: R01‐GM094497
REFERENCES
- 1. Duderstadt KE, Berger JM. AAA+ ATPases in the initiation of DNA replication. Crit Rev Biochem Mol Biol. 2008;43:163–187. [DOI] [PubMed] [Google Scholar]
- 2. Duran EC, Weaver CL, Lucius AL. Comparative analysis of the structure and function of AAA+ motors ClpA, ClpB, and Hsp104: Common threads and disparate functions. Front Mol Biosci. 2017;4:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ryu J‐K, Jahn R, Yoon T‐Y. Progresses in understanding N‐ethylmaleimide sensitive factor (NSF) mediated disassembly of SNARE complexes. Biopolymers. 2016;105:518–531. [DOI] [PubMed] [Google Scholar]
- 4. Yedidi RS, Wendler P, Enenkel C. AAA‐ATPases in protein degradation. Front Mol Biosci. 2017;4:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hanson PI, Whiteheart SW. AAA+ proteins: Have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. [DOI] [PubMed] [Google Scholar]
- 6. Miller JM, Enemark EJ. Fundamental characteristics of AAA+ protein family structure and function. Archaea Vanc BC. 2016;2016:9294307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of aaa+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. [DOI] [PubMed] [Google Scholar]
- 8. Iyer LM, Leipe DD, Koonin EV, Aravind L. Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol. 2004;146:11–31. [DOI] [PubMed] [Google Scholar]
- 9. Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha‐ and beta‐subunits of ATP synthase, myosin, kinases and other ATP‐requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ammelburg M, Frickey T, Lupas AN. Classification of AAA+ proteins. J Struct Biol. 2006;156:2–11. [DOI] [PubMed] [Google Scholar]
- 11. Beckwith R, Estrin E, Worden EJ, Martin A. Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat Struct Mol Biol. 2013;20:1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doyle SM, Shorter J, Zolkiewski M, Hoskins JR, Lindquist S, Wickner S. Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein‐remodeling activity. Nat Struct Mol Biol. 2007;14:114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Franzmann TM, Czekalla A, Walter SG. Regulatory circuits of the AAA+ disaggregase Hsp104. J Biol Chem. 2011;286:17992–18001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: Allosteric control of a protein machine. Cell. 2005;121:1017–1027. [DOI] [PubMed] [Google Scholar]
- 15. Mogk A, Schlieker C, Strub C, Rist W, Weibezahn J, Bukau B. Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J Biol Chem. 2003;278:17615–17624. [DOI] [PubMed] [Google Scholar]
- 16. Weibezahn J, Schlieker C, Bukau B, Mogk A. Characterization of a trap mutant of the AAA+ chaperone ClpB. J Biol Chem. 2003;278:32608–32617. [DOI] [PubMed] [Google Scholar]
- 17. Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE. N‐ethylmaleimide‐sensitive fusion protein: A trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol. 1994;126:945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ogura T, Whiteheart SW, Wilkinson AJ. Conserved arginine residues implicated in ATP hydrolysis, nucleotide‐sensing, and inter‐subunit interactions in AAA and AAA+ ATPases. J Struct Biol. 2004;146:106–112. [DOI] [PubMed] [Google Scholar]
- 19. Augustin S, Gerdes F, Lee S, Tsai FTF, Langer T, Tatsuta T. An intersubunit signaling network coordinates ATP hydrolysis by m‐AAA proteases. Mol Cell. 2009;35:574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DeLaBarre B, Christianson JC, Kopito RR, Brunger AT. Central pore residues mediate the p97/VCP activity required for ERAD. Mol Cell. 2006;22:451–462. [DOI] [PubMed] [Google Scholar]
- 21. Gerega A, Rockel B, Peters J, Tamura T, Baumeister W, Zwickl P. VAT, the thermoplasma homolog of mammalian p97/VCP, is an N domain‐regulated protein unfoldase. J Biol Chem. 2005;280:42856–42862. [DOI] [PubMed] [Google Scholar]
- 22. Graef M, Langer T. Substrate specific consequences of central pore mutations in the i‐AAA protease Yme1 on substrate engagement. J Struct Biol. 2006;156:101–108. [DOI] [PubMed] [Google Scholar]
- 23. Hinnerwisch J, Fenton WA, Furtak KJ, Farr GW, Horwich AL. Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell. 2005;121:1029–1041. [DOI] [PubMed] [Google Scholar]
- 24. Martin A, Baker TA, Sauer RT. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor‐dependent recognition of ssrA‐tagged substrates. Mol Cell. 2008;29:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martin A, Baker TA, Sauer RT. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol. 2008;15:1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park E, Rho YM, Koh O‐J, et al. Role of the GYVG pore motif of HslU ATPase in protein unfolding and translocation for degradation by HslV peptidase. J Biol Chem. 2005;280:22892–22898. [DOI] [PubMed] [Google Scholar]
- 27. Siddiqui SM, Sauer RT, Baker TA. Role of the processing pore of the ClpX AAA+ ATPase in the recognition and engagement of specific protein substrates. Genes Dev. 2004;18:369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song HK, Hartmann C, Ramachandran R, et al. Mutational studies on HslU and its docking mode with HslV. Proc Natl Acad Sci U S A. 2000;97:14103–14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamada‐Inagawa T, Okuno T, Karata K, Yamanaka K, Ogura T. Conserved pore residues in the AAA protease FtsH are important for proteolysis and its coupling to ATP hydrolysis. J Biol Chem. 2003;278:50182–50187. [DOI] [PubMed] [Google Scholar]
- 30. Zhang F, Wu Z, Zhang P, Tian G, Finley D, Shi Y. Mechanism of substrate unfolding and translocation by the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009;34:485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weibezahn J, Tessarz P, Schlieker C, et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–665. [DOI] [PubMed] [Google Scholar]
- 32. Chang C‐W, Lee S, Tsai FTF. Structural elements regulating AAA+ protein quality control machines. Front Mol Biosci. 2017;4:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wendler P, Ciniawsky S, Kock M, Kube S. Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta Mol Cell Res. 2012;1823:2–14. [DOI] [PubMed] [Google Scholar]
- 34. Enemark EJ, Joshua‐Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. [DOI] [PubMed] [Google Scholar]
- 35. Itsathitphaisarn O, Wing RA, Eliason WK, Wang J, Steitz TA. The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell. 2012;151:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kelch BA, Makino DL, O'Donnell M, Kuriyan J. How a DNA polymerase clamp loader opens a sliding clamp. Science. 2011;334:1675–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simonetta KR, Kazmirski SL, Goedken ER, et al. The mechanism of ATP‐dependent primer‐template recognition by a clamp loader complex. Cell. 2009;137:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomsen ND, Berger JM. Running in reverse: The structural basis for translocation polarity in hexameric helicases. Cell. 2009;139:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nyquist K, Martin A. Marching to the beat of the ring: Polypeptide translocation by AAA+ proteases. Trends Biochem Sci. 2014;39:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cordova JC, Olivares AO, Shin Y, et al. Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell. 2014;158:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin A, Baker TA, Sauer RT. Rebuilt AAA + motors reveal operating principles for ATP‐fuelled machines. Nature. 2005;437:1115–1120. [DOI] [PubMed] [Google Scholar]
- 42. Stinson BM, Nager AR, Glynn SE, Schmitz KR, Baker TA, Sauer RT. Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine. Cell. 2013;153:628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stinson BM, Baytshtok V, Schmitz KR, Baker TA, Sauer RT. Subunit asymmetry and roles of conformational switching in the hexameric AAA+ ring of ClpX. Nat Struct Mol Biol. 2015;22:411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aubin‐Tam M‐E, Olivares AO, Sauer RT, Baker TA, Lang MJ. Single‐molecule protein unfolding and translocation by an ATP‐fueled proteolytic machine. Cell. 2011;145:257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maillard RA, Chistol G, Sen M, et al. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell. 2011;145:459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rodriguez‐Aliaga P, Ramirez L, Kim F, Bustamante C, Martin A. Substrate‐translocating loops regulate the mechanochemical coupling and power production in a AAA+ protease. Nat Struct Mol Biol. 2016;23:974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sen M, Maillard RA, Nyquist K, et al. The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell. 2013;155:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fei X, Bell TA, Jenni S, et al. Structures of the ATP‐fueled ClpXP proteolytic machine bound to protein substrate. Biophysics. 2019. 10.1101/704999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matyskiela ME, Lander GC, Martin A. Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol. 2013;20:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Puchades C, Rampello AJ, Shin M, et al. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science. 2017;358:eaao0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Puchades C, Ding B, Song A, Wiseman RL, Lander GC, Glynn SE. Unique structural features of the mitochondrial AAA+ protease AFG3L2 reveal the molecular basis for activity in health and disease. Mol Cell. 2019;75:1073–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han H, Monroe N, Sundquist WI, Shen PS, Hill CP. The AAA ATPase Vps4 binds ESCRT‐III substrates through a repeating array of dipeptide‐binding pockets. Elife. 2017;6:e31324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Han H, Fulcher JM, Dandey VP, et al. Structure of Vps4 with circular peptides and implications for translocation of two polypeptide chains by AAA+ ATPases. Elife. 2019;8:e44071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Monroe N, Han H, Shen PS, Sundquist WI, Hill CP. Structural basis of protein translocation by the Vps4‐Vta1 AAA ATPase. Elife. 2017;6:e24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sandate CR, Szyk A, Zehr EA, Lander GC, Roll‐Mecak A. An allosteric network in spastin couples multiple activities required for microtubule severing. Nat Struct Mol Biol. 2019;26:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. de la Peña AH, Goodall EA, Gates SN, Lander GC, Martin A. Substrate‐engaged 26S proteasome structures reveal mechanisms for ATP‐hydrolysis‐driven translocation. Science. 2018;362:eaav0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dong Y, Zhang S, Wu Z, et al. Cryo‐EM structures and dynamics of substrate‐engaged human 26S proteasome. Nature. 2019;565:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ripstein ZA, Huang R, Augustyniak R, Kay LE, Rubinstein JL. Structure of a AAA+ unfoldase in the process of unfolding substrate. Elife. 2017;6:e25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cooney I, Han H, Stewart MG, et al. Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science. 2019;365:502–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Twomey EC, Ji Z, Wales TE, et al. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science. 2019;365:eaax1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lo Y‐H, Sobhany M, Hsu AL, et al. Cryo‐EM structure of the essential ribosome assembly AAA‐ATPase Rix7. Nat Commun. 2019;10:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. White KI, Zhao M, Choi UB, Pfuetzner RA, Brunger AT. Structural principles of SNARE complex recognition by the AAA+ protein NSF. Elife. 2018;7:e38888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gates SN, Yokom AL, Lin J, et al. Ratchet‐like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science. 2017;357:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yu H, Lupoli TJ, Kovach A, et al. ATP hydrolysis‐coupled peptide translocation mechanism of Mycobacterium tuberculosis ClpB. Proc Natl Acad Sci U S A. 2018;115:E9560–E9569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Deville C, Carroni M, Franke KB, et al. Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. Sci Adv. 2017;3:e1701726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rizo AN, Lin J, Gates SN, et al. Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase. Nat Commun. 2019;10:2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ho C‐M, Beck JR, Lai M, et al. Malaria parasite translocon structure and mechanism of effector export. Nature. 2018;561:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alfieri C, Chang L, Barford D. Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature. 2018;559:274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Deville C, Franke K, Mogk A, Bukau B, Saibil HR. Two‐step activation mechanism of the ClpB disaggregase for sequential substrate threading by the main ATPase motor. Cell Rep. 2019;27:3433–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roll‐Mecak A, Vale RD. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature. 2008;451:363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zehr E, Szyk A, Piszczek G, Szczesna E, Zuo X, Roll‐Mecak A. Katanin spiral and ring structures shed light on power stroke for microtubule severing. Nat Struct Mol Biol. 2017;24:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Esaki M, Islam MT, Tani N, Ogura T. Deviation of the typical AAA substrate‐threading pore prevents fatal protein degradation in yeast Cdc48. Sci Rep. 2017;7:5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kotamarthi HC, Sauer RT, Baker TA. The non‐dominant AAA+ ring in the ClpAP protease functions as an anti‐stalling motor to accelerate protein unfolding and translocation. Biophysics. 2019. 10.1101/585554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schweitzer A, Aufderheide A, Rudack T, et al. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc Natl Acad Sci U S A. 2016;113:7816–7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 76. Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zimmermann L, Stephens A, Nam S‐Z, et al. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol. 2018;430:2237–2243. [DOI] [PubMed] [Google Scholar]