

Abstract
Heart failure (HF) is the leading cause of morbidity and mortality in developed countries, and it is the primary cause of mortality in the elderly worldwide. The processes of inflammatory response activation, production and release of pro-inflammatory cytokines, activation of the complement system, synthesis of autoantibodies, and overexpression of Class II major histocompatibility complex molecules contribute to the HF development and progression. High levels of circulating cytokines correlate with the severity of HF, measured with the use of New York Heart Association’s classification, and prognosis of the disease. In HF, there is an imbalance between pro-inflammatory and anti-inflammatory cytokines. Concentrations of several interleukins are increased in HF, including IL-1β, IL-6, IL-8, IL-9, IL-10, IL-13, IL-17, and IL-18, whereas the levels of IL-5, IL-7, or IL-33 are down-regulated. Concentrations of inflammatory mediators are associated with cardiac function and can be HF markers and predictors of adverse outcomes or mortality. This review presents the role of interleukins, which contribute to the HF initiation and progression, the importance of their pathways in transition from myocardial injury to HF, and the role of interleukins as markers of disease severity and outcome predictors.
Keywords: interleukins, heart failure, interleukin-1, interleukin-6, interleukin-33
Introduction
Heart failure (HF) is an insufficiency of the cardiac output to fulfill the demands of organs and tissues. It can represent the end-stage of several cardiovascular diseases, including ischemic disease, myocardial infarction, or myocarditis, which lead to pathological changes in the myocardium. HF is the leading cause of morbidity and mortality in developed countries, and it is the primary cause of mortality in the elderly worldwide (1). Its typical clinical presentations include shortness of breath, fatigue, and impaired exercise tolerance. Despite several improvements in the management of HF, it is still an incurable and a progressive disease. Its 5-year mortality has been estimated to range between 50% and 70% in the American population (2).
The mechanisms of the HF development are complicated, and several factors have been implicated in its pathophysiology. Many studies confirmed that HF is not only a “pump illness”. In fact, it is a systemic disease, and persistent overactivation of different compensatory systems, such as the renin–angiotensin and β-adrenergic system, is one of the most relevant causes of HF. Nevertheless, the use of inhibitory agents blocking those systems did not influence morbidity and mortality satisfactorily, which may suggest that we still miss some significant pathways. Several trials demonstrated that the process of inflammation might be responsible for the initiation and progression of HF (3-8). Inflammation can be defined as the biological response to tissue injury or irritation. Immune activation may be the result of direct stimulation by foreign antigens, such as in viral myocarditis, or can be indirect, caused by cardiac injury and subsequent exposure to self-antigens, which could be responsible for the response against the heart. Recurrent and sustained immune system activation is implicated in the left ventricular hypertrophy and its progression to HF. Chemical transmitters playing role in mediating the inflammatory signaling from the site of local injury to whole body are known as cytokines. Cytokines recruit cells to the site of inflammation, stimulate cell division, proliferation, and differentiation. In HF there is an activation of the immune system, production and release of autoantibodies, pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α), and complement system recruitment (3-6). High levels of circulating cytokines correlate with the severity of HF, measured with the use of the New York Heart Association (NYHA) classification, and prognosis of the disease (3, 4, 6, 7). They also cause the alterations in the heart function due to the increase of cardiomyocyte apoptosis, cardiac hypertrophy, and matrix metalloproteinase activation (4, 7). The process of T-cell migration to the left ventricle was shown as important in the HF development (8); however, its mechanisms are not well known. An overexpression of TNF-α leads to the development of dilated cardiomyopathy in animal studies (9), which implies that cytokines themselves may cause the initiation and progression of HF.
The processes of the inflammatory response activation, production, and release of pro-inflammatory cytokines, activation of the complement system, production of autoantibodies, and overexpression of Class II major histocompatibility complex molecules contribute to the HF pathogenesis. However, the treatment with several immune pathways inhibitors did not improve the HF outcomes. TNF-α inhibitors had no positive effect on patients’ survival. In fact, high doses of infliximab, a chimeric monoclonal antibody to TNF-α, were found to increase the mortality rate (10, 11). Several trials demonstrated a decreased inflammatory factors concentration after colchicine application. However, it did not improve the clinical state of HF patients (12). The use of nonspecific immunomodulation therapy in HF demonstrated beneficial effects only in some groups of patients (13). On the other side, the levels of inflammatory cytokines did not decrease after traditional therapy was used in acute HF decompensation, even when clinical parameters showed improvement (14). What is more, the use of amiodarone has been associated with up-regulated concentrations of TNF-α in serum (15).
This review presents the role of interleukins, which contribute to the HF initiation and progression, the importance of their pathways in transition from myocardial injury to HF, and the role of interleukins as markers of disease severity and outcome predictors.
Interleukin-1β and its receptor as a therapeutic target in HF and myocardial infarction
Interleukin-1β (IL-1β) is a pro-inflammatory cytokine, which triggers up-regulation of several downstream signaling molecules involved in immune response. After binding to its receptor, multiple adaptor proteins are recruited, including the myeloid differentiation factor 88 (MyD88), which activate the IL-1R associated kinases (IRAKs) and tumor necrosis factor receptor-associated factor 6 (TRAF-6). These all cause the activation of several transcriptional factors. IL-1 triggers transcriptional and posttranslational alterations in sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) and overexpression of nitric oxide synthase (NOS) (16). IL-1 signaling pathway and downstream effector molecules are presented in Figure 1. Myocardial injury induces the synthesis of a macromolecular structure, denoted inflammasome, which is responsible for the cleavage of pro-IL-1β by caspase-1, and therefore activates IL-1β. IL-1β initiates the migration of leukocytes to the site of cardiac insult and triggers the synthesis of other cytokines. The IL-1β concentration was proportional to the ischemia duration in the studies on mice (17).
Figure 1.
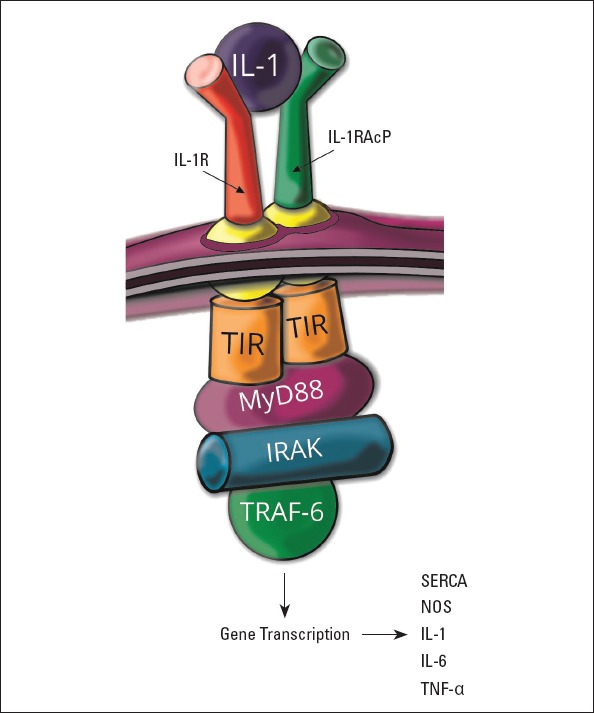
IL-1 Signaling pathway and downstream effector molecules
IL-1R - interleukin-1 receptor; IL-1RAcP - IL-1 receptor accessory protein; TIR - Toll/interleukin-1 receptor kinase; MyD88 - myeloid differentiation factor 88; IRAK - IL-1R associated kinase; TRAF-6 - tumor necrosis factor receptor-associated factor-6; SERCA - sarcoplasmic/endoplasmic reticulum calcium ATPase; NOS - nitric oxide synthase; TNF-α - tumor necrosis factor-α
IL-1β has been implicated in cardiac remodeling after ischemia and in changes in ventricles contractility and relaxation. The level of IL-1β was proportional to the NYHA’s functional class and was regardless of the cause of HF (18). Studies demonstrated that even more patients had elevated concentrations of IL-1 soluble receptor/receptor antagonists than IL-1 (18). An overexpression of IL-1, which caused alterations in myocardium, was noted in sepsis (19) and viral myocarditis (20). Moreover, inhibiting this cytokine suppressed the progression to HF (21).
Treatment with IL-1β promoted contractile dysfunction in cell culture and in vivo studies, and it reduced the LV contractility reserve, evaluated according to the responsiveness to isoproterenol, which is a β agonist. Moreover, the negative influence of IL-1β was reversible (22). IL-1β blocker administration in acute ischemia in animal model prevented from the left ventricle dysfunction, even when used 10 weeks after injury (22, 23). The injection of patients’ plasma, which suffered from acute HF to healthy mice, caused a development of diastolic dysfunction (24, 25). Moreover, treatment with anakinra, which is an IL-1 receptor antagonist, or an IL-1β antibody, protected from the cardiac function depression (23, 25). The administration of plasma from stable systolic HF patients resulted in a lower contractile reserve, whereas the contractile function was unchanged (24).
Patients with rheumatoid arthritis treated with anakinra had a better systolic and diastolic function of ventricles, greater increase in coronary flow reserve, and improved endothelial function, whereas the use of triamcinolone did not affect the myocardial function (26). Similar results were observed in clinical trials VCU-ART (27) and VCU-ART2 (28), which aimed to verify the effect of anakinra on patients with the ST-segment elevated myocardial infarction (STEMI) treated with primary percutaneous coronary intervention. These studies demonstrated a lower level of inflammatory transmitters, better cardiac function, and decreased extent of pathological remodeling in patients administered with anakinra. However, it did not have an impact on myocardial infarction recurrence or the infarct size. VCU-ART3 is a still continued double-blind randomized clinical trial, which evaluates the effect of high doses of anakinra in comparison to standard doses of anakinra and measures the effects on the acute rise and fall of the plasma C-reactive protein (CRP) levels during the first 14 days after myocardial infarction. Patients with the non-STEMI injected with anakinra had a lower level of CRP, but also, unfortunately, an unchanged infarct size and a higher risk of adverse cardiac events (29). Mice administered with a recombinant human IL-1 receptor antagonist after treatment with doxorubicin (30) or chest irradiation (31) had preserved myocardial systolic and diastolic function. Losartan, which is an angiotensin II receptor antagonist, prevented the depression of the cardiac function after the IL-1 treatment in an animal model (32).
Interleukin-6 and its antagonists as potential therapeutic options in HF
Interleukin-6 (IL-6) is a pleiotropic cytokine implicated in thymocytes, macrophages, and natural killer cells activation; B- and T-cell differentiation; and inducing the acute-phase proteins synthesis (33). This cytokine initiates the leucocyte infiltration; however, a prolonged inflammation can change into destructive response causing tissue fibrosis. IL-6 acts through the IL-6 receptor (IL-6R), which is detected only in some cell types, such as hepatocytes or leukocytes. After IL-6 binds to IL-6R, the ubiquitously expressed glycoprotein 130 receptor subunit (gp130) is recruited to form a complex. However, another pathway known as trans-signaling has been revealed, which uses a soluble form of IL-6R (sIL-6R). This receptor binds IL-6 with a similar affinity. This dimer can associate with gp130 on cells not expressing IL-6R. IL-6 trans-signaling is pro-inflammatory, while classic IL-6 signaling via the membrane-bound IL-6R has anti-inflammatory properties. The Gp130 receptor subunit activates three pathways, including the Janus kinase/signal transducer and activator of the transcription 1/3 (JAK/STAT1/3) pathway, which seems to be the most important in the HF development, Ras/mitogen-activated protein/extracellular signal regulated kinases (Ras/MEK/ERK1/2), and the phosphatidylinositol-3-kinase (PI3K)/Akt pathway (34). Janus kinases are a family of tyrosine kinases linked with the cytoplasmic domain of cytokine and growth factor receptors. After the activation, JAK proteins phosphorylate and recruit STAT proteins whose phosphorylation leads to their homodimerization, and eventually to their translocation to the nucleus or mitochondria (35). STAT3 activates the Survivor Activating Factor Enhancement (SAFE) pathway, which is an intrinsic protective signaling program limiting cell death activated by the heart and triggers ischemic post-conditioning, which prevents the reperfusion injury to occur. The downstream proteins of the JAK/STAT3 pathway are among others cyclin D1, E1, p21, Fas, Bcl-xL, Bcl-2, Mcl-1, or the vascular endothelial growth factor (VEGF). This pathway may also be activated by several factors, including high-density lipoproteins, prostaglandins, bradykinin, leptin, insulin, erythropoietin, adrenoreceptors, cannabinoid agonists, opioids, resveratrol, or biogenic amines present in red wine, including ethanolamine and melatonin (36). The IL-6 signaling pathway and downstream effector molecules are presented in Figure 2. Studies revealed the presence of a decoy receptor, sgp130, which is an endogenous inhibitor of the IL-6 trans-signaling (37). IL-6 inhibited the sarcoplasmic reticulum Ca2+ ATPase (SERCA2) via the transcriptional mechanism (38) and decreased the expression of beta- and alpha-myosin heavy chain and cardiac-alpha-actin in cell culture (39).
Figure 2.
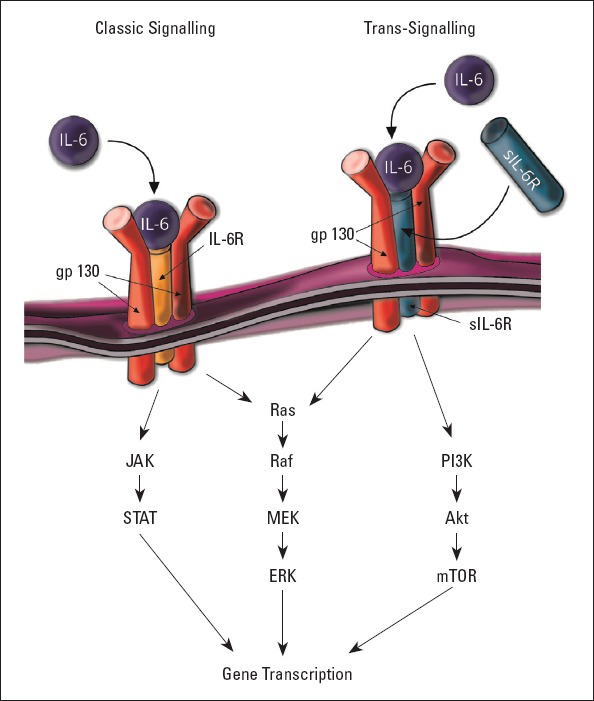
Downstream signaling of IL-6
IL-6R - interleukin-6 receptor; gp130 - glycoprotein 130 receptor subunit; sIL-6R - soluble interleukin-6 receptor; JAK - Janus-activated kinase; STAT - signal transducer and activator of transcription; MEK - mitogen-activated protein kinase; ERK - extracellular signal regulated kinase; PI3K - phosphatidylinositol-3-kinase; mTOR - mammalian target of rapamycin
The levels of IL-6 and soluble IL-6-type receptors were elevated in patients with HF (7) and were proportional to the NYHA functional class (6, 18) and function of either the left or right ventricle (40, 41). A higher number of patients had an increased IL-6 than the sIL-6R level (18). Moreover, studies revealed the overexpression of their genes in ischemic and nonischemic end-stage HF (42). The IL-6 concentration depended on the etiology of HF. It was significantly increased in patients with HF caused by ischemic heart disease and cardiomyopathy than in those affected by valvular heart disease or hypertensive heart disease (43). IL-6 was linked with the sympathetic nervous system activation and correlated with a poor outcome (44). Jug et al. (45) showed that IL-6 was an independent predictor of prognosis, even better than hsCRP in patients with a chronic stable HF. However, the Corona Trial did not demonstrate a correlation between the level of IL-6 and the endpoints, which were cardiovascular mortality, myocardial infarction, or stroke in patients with chronic symptomatic systolic HF (46). Nevertheless, sgp130 was associated with mortality from all causes (46). IL-6 was an independent predictor of systolic pulmonary artery pressure in patients with advanced HF (47). An increased IL-6 level was associated with a higher HF risk in people without previous cardiovascular diseases (48).
In in vitro studies, IL-6 suppressed the systolic function in isolated hamster papillary muscles through the myocardial NOS pathway (49). In animal studies, IL-6 had a negative inotropic effect on myocardium. Its administration resulted in cardiac dilation and the reduction of skeletal muscle mass (50). gp130 played a role in cardiac fibroblast growth, neovascularization, and wound healing (51). The loss of gp130 has been implicated in the cardiac dilation caused by pressure overload (52). Nevertheless, the overexpression of gp130 led to ventricular hypertrophy through the trans-signaling process (53). Activation of the SAFE pathway was required for the cardioprotective effect of ischemic pre- and post-conditioning. Ischemic post-conditioning induced the activation of JAK1, JAK2, STAT1, and STAT3, and the inhibition of this pathway impaired the development of ischemic tolerance (54). The loss of STAT3 resulted in neovascularization alterations, enhanced fibroblast proliferation, HF progression, increased mortality, higher susceptibility to myocardial ischemia/reperfusion injury and infarction, raised apoptotic cell rate, and enhanced infarct size (55). A similar effect was obtained in IL-6-knockout mice, in which the ischemic post-conditioning-induced activation of JAK/STAT was down-regulated, and the infarct size was not reduced (56). Chronic activation of STAT3 through IL-6 and gp130 in subacute infarction was responsible for inflammation, left ventricular rupture, and HF development (57).
The use of an IL-6 and a gp130 antagonist led to a decreased expression of atrial natriuretic peptide (ANP) and beta-myosin heavy chain in isolated cardiomyocytes, reduction of its surface area, and lower proliferation of fibroblast in cardiomyocyte/fibroblast coculture. Fibroblasts, which synthesized angiotensin II (AngII), stimulated the IL-6 production. Losartan, which is an AT-1 receptor antagonist, neutralized the influence of IL-6 and prevented myocyte hypertrophy and fibroblast proliferation (58). Tocilizumab is an anti-human IL-6R monoclonal antibody. It binds to both the soluble and membrane-bound IL-6R (59), and this may be the cause of several serious side effects, including neutropenia and increased susceptibility to infections (60). Another issue that questions the use of tocilizumab in patients with HF is the fact that it leads to increased cholesterol levels and decreased statin effects. However, the concentration of lipoprotein(a), which is a known marker of cardiovascular diseases, was lower after the treatment (60).
The effect of IL-6 may be both positive and negative, depending on the duration of this cytokine activation. IL-6 synthesis restricted to short period after the injury is curative and could protect and preserve the myocardium, while a prolonged synthesis and over-production result in ventricular dilation and HF development. Classical, membrane-bound signaling has been associated with the favorable, anti-inflammatory influence of IL-6, whereas the noxious pro-inflammatory pathway is mediated by trans-signaling. The anti-inflammatory role of IL-6 varies upon the dose, duration, and moment of initiation of its administration and type of receptor stimulated.
Interleukin-8 associated with a worse clinical prognosis in patients with HF
Interleukin-8 (IL-8) is a key mediator associated with inflammation, which is encoded by the CXCL8 gene. IL-8 plays a role in angiogenesis, neutrophil and granulocyte chemotaxis, and phagocytosis stimulation, and it is induced by several stimuli such as shear stress, ischemia, hypoxia, and other factors that activate the nuclear factor kappa–light–chain–enhancer of activated B cells (NF-κB) pathway (61). IL-8 is synthesized by cells with toll-like receptors, including macrophages, epithelial cells, smooth muscle cells, and endothelial cells. In human heart, IL-8 is localized mainly in cardiomyocytes (62).
IL-8 concentration was up-regulated in patients with HF compared to healthy controls (7, 63, 64). Increased IL-8 concentrations have been related with a worse clinical prognosis in patients with HF (65). However, a small study on explanted hearts from patients with end-stage HF showed down-regulated IL-8 mRNA levels in patients with HF compared to healthy donors (62). The IL-8 concentration was demonstrated to be a predictor of the HF development after myocardial infarction (66). High levels of IL-8 were associated with less improvement in left ventricular function during the first 6 weeks after STEMI. The IL-8 level was not proportional to peak troponin T concentration or infarct size after 6 weeks, which suggested that IL-8 might not be implicated in the process of necrosis (67).
Interlekin-10 improved the myocardial function and protected from adverse cardiac remodeling
Interleukin-10 (IL-10) is an anti-inflammatory cytokine produced in macrophages, activated T cells and B cells, or mast cells. IL-10 inhibits pro-inflammatory cytokines and matrix metalloproteinases synthesis. It regulates the growth, differentiation, and function of cytotoxic and helper T cells, B cells, natural killer (NK) cells, granulocytes, dendritic cells, mast cells, keratinocytes, and endothelial cells. IL-10 blocks the infiltration of monocytes and macrophages into the injured site and is a strong repressor of pro-inflammatory cytokines (68).
The IL-10 level was increased in patients with HF compared with healthy people (69) and correlated positively with the NYHA class (70). However, another study demonstrated that the concentration of IL-10 was not altered in patients with HF (7). In studies on mice, IL-10 had a cardioprotective effects. The lack of the IL-10 gene resulted in a worse myocardial function and up-regulated cardiac myocyte apoptosis in animals with isoproterenol- and aortic constriction-induced hypertrophy compared with wild types. IL-10 knockout mice treated with recombinant IL-10 showed an improved systolic function, reversed cardiac remodeling, decreased caspase-3 activation, lowered hypertrophic and inflammatory gene expression, and raised anti-apoptotic Bcl2 concentration. IL-10 acted through STAT3, and it suppressed the nuclear factor-κB activation and blocked the STAT3 inhibition. A persistent STAT3 activation had effects similar to IL-10 administration, a lowered ANP, B-type natriuretic peptide (BNP), and TNF-α gene expression (71).
Several studies demonstrated the important role of IL-10 in pathophysiological pathways after myocardial infarction. The IL-10 level was up-regulated after myocardial ischemia and reperfusion injury in studies on animals, and the highest concentration reached 96–120 hours after the reperfusion (72). IL-10 inhibited infiltration of inflammatory cells and expression of pro-inflammatory cytokines in the myocardium, improved myocardial function, and protected from adverse cardiac remodeling in acute myocardial infarction in animal models (73, 74). IL-10 knockout mice demonstrated a worse left ventricular function, fibrosis, and cardiomyocyte apoptosis (74). The IL-10 administration led to down-regulation of p38 mitogen-activated protein kinase activation, reduced expression of the cytokine mRNA-stabilizing protein Hur, a decreased metalloproteinase-9 (MMP-9) activity, and inhibited fibrosis after myocardial infarction. IL-10 was responsible for increased STAT3 phosphorylation and subsequently up-regulated capillary density in the heart (75). Silencing the Hur gene resulted in an improvement in the systolic function, similarly to IL-10. It reversed cardiac remodeling, down-regulated the TGF-β, MMP-9, and p53 expression, inhibited myocyte apoptosis, and reduced the infarct size (74, 75). Moreover, IL-10 up-regulated the VEGF-A expression in cell culture through the STAT-3 pathway and promoted the mobilization of bone marrow endothelial progenitor cells (75). IL-10 suppressed pathological autophagy in response to Ang II treatment via the PI3K/Akt/mTORC1 pathway (76). Atorvastatin administration enhanced the IL-10 concentration, reduced the TNF/IL-10 ratio, inhibited cardiac remodeling, and improved the systolic function in rats with HF caused by myocardial infarction (77). Regular exercise increased the IL-10 levels in both humans (78) and rats (79).
Interleukin-17 implicated in adverse cardiac remodeling
Interleukin-17 (IL-17) is a pro-inflammatory cytokine, which is synthesized by T helper cells, macrophages, dendritic cells, NK, natural killer T, lymphoid tissue inducer and γδ-T cells. IL-17 stimulates the production of several cytokines, which eventually results in neutrophils and monocytes recruitment to the site of inflammation (80). IL-17 acts via several pathways, including the NF-κB -DNA pathway, mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), and p42/p44 Extracellular signal Regulated Kinases (ERK) pathways (81). IL-17 cooperates with interferon-γ (IFN-γ) in the inflammatory response activation and stimulates several cytokines synthesis, including IL-6, IL-8, C-C motif chemokine ligand 5 (CCL5), C-X-C motif ligand 1 (CXCL1), C-X-C motif ligand 10 (CXCL10), complement component 5a (C5a), and soluble intercellular adhesion molecule-1 (ICAM-1) (82).
The IL-17 concentration was increased in patients with HF compared with healthy controls and correlated with the NYHA class (83). Single nucleotide polymorphisms in the IL-17 gene has been implicated with HF development risk. rs8193037 in IL-17 was an independent risk factor of ischemic and nonischemic HF, whereas rs4819554 in IL17RA was a predictor of cardiovascular mortality in patients with congestive HF (84). Several alterations in the IL-17/gp130-JAK-STAT have been demonstrated in end-stage HF. The expression of non-receptor tyrosine-protein kinase (TYK2), STAT3, and suppressor of cytokine signaling-1 (SOCS3) were reduced, whereas the expression of phospho-glycoprotein 130 (phospho-gp130) and SOCS1 were up-regulated in comparison with healthy people (85). IL-17 has been implicated in adverse cardiac remodeling after myocarditis and its progression to dilated cardiomyopathy in the mouse model (86). IL-17 contributed to the process of cardiac fibrosis, activation of matrix metalloproteinases, and enhanced cardiac cell death. IL-17 led to increased NOS (iNOS) synthesis and subsequently endothelium injury (87). It enhanced the release of pro-inflammatory cytokines, such as IL-1 and IL-6 (88), and decreased intracellular calcium levels (89).
Pro-inflammatory role of interleukin-18
Interleukin-18 (IL-18) is a pro-inflammatory cytokine, which is a member of the IL-1 superfamily. It is produced by macrophages, monocytes, keratinocytes, and other cells. IL-18 cooperates with IL-12 in activating cell-mediated immunity. IL-18 up-regulates the synthesis of several cytokines, including IFN-γ by NK cells and T cells (90). IL-18 has been implicated in fibroblast and smooth muscle cell proliferation and migration through the activation of the JNK and PI3-kinase pathways in in vitro studies (91). Interestingly, acetylsalicylic acid blocks fibroblast migration, reactive oxygen species generation, MMP9 activation, and reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) inhibition (92). IL-18 activates GATA4, which is a member of the GATA family of zinc finger transcription factors, and therefore, it up-regulates the expression of ANP in cardiomyocytes, among others (93). IL-18 also stimulates the activation of NF-κB, p38 MAPK, and ERK. IL-18 plays a role in apoptosis through both the extrinsic and intrinsic pathway (94) and increases pro-apoptotic effects of TNF-α and enhances the caspase-3 activation (95).
The concentrations of IL-18 mRNA and protein were increased in myocardium in patients with ischemic cardiomyopathy. The expression of IL-18-binding protein (IL-18BP) mRNA was reduced in end-stage HF, whereas the IL-18 receptor α chain (IL-18Rα) level was up-regulated in both ischemic and dilated cardiomyopathy (96). The IL-18 concentration was an independent risk factor of mortality in elderly patients with HF (97). The expression of IL-18, IL-18Rα, and IL-18 mRNA were up-regulated in pressure overload-induced hypertrophy and subsequent HF in animals, whereas the IL-18BP concentration was unchanged in hypertrophied hearts and reduced in HF. Also in in vitro pressure overload models, the expression of IL-18 and IL-18Rα were increased. The IL-18 inhibition attenuated hypertrophy (97). Mice treated with IL-18 presented with cardiac hypertrophy, reduced left ventricular ejection fraction, enhanced end-diastolic pressure, up-regulated expression of ANP mRNA, and decreased β-adrenergic responsiveness to isoproterenol (98). Inhibiting the IL-18 pathway prevented the HF development (25). Moreover, cardiomyocytes incubated with recombinant IL-18 demonstrated an enhanced cell surface area, and increased ANP and BNP concentration and Akt activation (97). IL-18 knockout mice, which underwent pressure overload, demonstrated attenuated ventricular dilatation (99).
Cardioprotective effect of interleukin-33 in HF development and progression
Interleukin-33 (IL-33) is a member of the IL-1 superfamily. This protein is constitutively expressed in the nuclei of endothelial cells in both small and large blood vessels, in epithelial cells, smooth muscle cells, fibroblasts, and keratinocytes (100). IL-33 acts through IL-1 receptor-like 1 (IL1RL1, ST2) and IL-1 receptor accessory protein (IL-1RAcP) dimeric receptor complex, which promotes the activation of NF-κB and MAPK pathways. Two isoforms of the IL-33 receptor have been discovered, denoted a soluble (sST2) receptor and membrane-bound (ST2L) receptor, which are present in cardiomyocytes and fibroblasts. sST2 is a soluble decoy receptor, which is a mechanically induced cardiomyocyte protein and inhibits anti-hypertrophic effects of IL-33 (101). IL-33 by signaling via the MyD88, IRAK-1/4, and TRAF activates the Th2 type immune responses. In endothelial cells, IL-33 stimulates the synthesis of IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1/CCL2), vascular cell adhesion molecule-1 (VCAM-1), ICAM-1 and endothelial selectin (E-selectin), increases the vascular permeability and promotes angiogenesis (102). The IL-33 signaling pathway and downstream effector molecules are presented in Figure 3 IL-33 inhibits the atherosclerotic plaques formation via the induction of IL-5 and ox-LDL antibodies (103) and decreases adiposity, lowers fasting glucose, and improves glucose and insulin tolerance in obesity as revealed by studies on animals (104). In myocardium, it is produced mainly in cardiac fibroblasts in response to mechanical stress (105). IL-33 prevents angiotensin II- and phenylephrine-induced hypertrophy. Moreover, IL-33 activates NF-κB and blocks the phosphorylation of inhibitor of NF-κB caused by angiotensin II or phenylephrine (105). IL-33 plays a dual role, and under certain circumstances, it can act both as a pro-inflammatory or anti-inflammatory protein.
Figure 3.
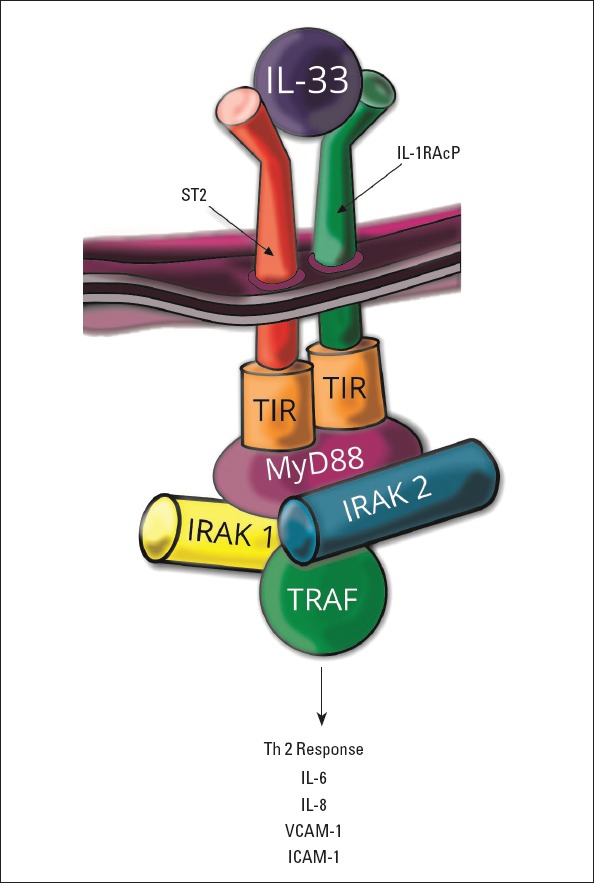
IL-33 signaling pathway and downstream effector molecules
ST2 - interleukin-1 receptor-like 1; IL-1RAcP - IL-1 receptor accessory protein; TIR - Toll/interleukin-1 receptor kinase; MyD88 - myeloid differentiation factor 88; IRAK - IL-1R associated kinase; TRAF - tumor necrosis factor receptor-associated factor; VCAM-1 - vascular cell adhesion molecule-1; ICAM - intercellular adhesion molecule-1
A previous study demonstrated a decreased concentration of IL-33 in patients with HF in comparison with healthy controls (106). Another study indicated that both IL-33 and ST2 were down-regulated in patients with unstable HF submitted to Left Ventricular Assist Device (LVAD) Support compared to those with stable HF. The ST2 expression was proportional to markers of inflammation. Treatment with LVAD resulted in an increase of IL-33 and ST2 level, and therefore, it was hypothesized that the IL-33/ST2 pathway was implicated in mechanical unloading and probably in inhibiting adverse remodeling (107). However, a recent study showed that the serum level of IL-33 was increased in patients with HF, whereas its bioactivity was reduced (108). IL-33 was proportional to serum concentration of oxidative stress markers, whereas the IL-33/sST2 ratio was inversely proportional to the malondialdehyde (MDA) level. IL-33 directly down-regulated the MDA and reactive oxygen species production and enhanced the superoxide dismutase activity in cultured cells stimulated by angiotensin II (108).
Mice administered with IL-33 demonstrated decreased hypertrophy, cardiac remodeling, fibrosis, and a better survival rate after the pressure overload (105). Under hypoxic conditions, cardiomyocytes incubated with IL-33 expressed a higher level of antiapoptotic proteins, including XIAP, cIAP1, and survivin. IL-33 used in rats subjected to the ischemia–reperfusion injury led to a decreased caspase synthesis and therefore prevented apoptosis. It reduced the infarct size and fibrosis, and improved the systolic function. These effects were not seen in ST2 knockouts animals, and therefore, positive IL-33 effects could be mediated through the ST2 pathway (101).
IL-33 knockout mice subjected to pressure overload presented with more severe hypertrophy, reduced fractional shortening, adverse fibrosis, inflammation, poorer prognosis, and an increased level of natriuretic peptides RNA, including BNP and C-Myc. They also showed an up-regulated level of Th1 cytokines mRNA expression and infiltration of inflammatory cells in myocardium (109). Animals treated with IL-33 demonstrated lower expression of natriuretic peptides RNA (105). Studies on animals showed that the mRNA levels of IL-33 were enhanced in the heart up to 12 weeks after the infarction, whereas the sST2 expression was up-regulated in the first week and reduced at 4 weeks after the infarction. The level of sST2 mRNA was correlated with the expression of inflammatory, including IL-6, TNF-α, TGF-β and MCP-1, and fibrosis markers (110).
Studies on animals revealed that the lack of ST2 led to a more pronounced systolic dysfunction, left ventricular hypertrophy, adverse remodeling, myocardial fibrosis, and worse survival in both ischemic and nonischemic HF (105). sST2 competed with ST2L for IL-33 binding, inhibiting the interaction of IL-33 and ST2L. Clinical trials revealed that serum sST2 levels predicted an adverse outcome and death in patients with myocardial infarction or chronic HF (111). ST2L mediated the effect of IL-33 on Th2-dependent inflammatory processes, whereas sST2 has been implicated in the attenuation of Th2 inflammatory responses. Moreover, an increased concentration of sST2 in healthy subjects preceded adverse outcomes (112). The change in the ST2 level in patients with acute HF was predictive of 90-day mortality and was independent of BNP or N-terminal pro-brain natriuretic peptide (NT-proBNP) concentration (113).
Other interleukins playing an important role in HF
Interleukin-5 (IL-5) is a cytokine, implicated in the growth and differentiation of eosinophils and B cells. It plays a role in the immunoglobulin production and eosinophil activation. It is synthesized by Type 2 T helper cells and mast cells (114). The IL-5 plasma concentration was reduced in patients with HF and inversely proportional to disease duration in the NYHA Class III and IV in patients with ischemic cardiomyopathy (64). The expression of IL-5 in heart was down-regulated in mice after myocardial infarction (115).
Interleukin-7 (IL-7) acts as a hematopoietic growth factor, promoting the maturation of multipotent hematopoietic stem cells into lymphoid progenitor cells. It is produced by stromal cells in the thymus and bone marrow, fibroblastic reticular cells in T-cell zones of lymph nodes, dendritic cells, epithelial cells, hepatocytes, intestinal epithelial and epithelial goblet cells, and neurons. IL-7 is responsible for the development, homeostasis, and survival of B cells, T cells, and NK cells (116). This interleukin was down-regulated in patients with HF (64). Local IL-7 synthesis played an important role in growth and maintenance of CD8+ T cells in heart, which have been implicated in myocardial damage in chronic Chagas’ disease cardiomyopathy (117).
Interleukin-9 (IL-9) is a cytokine that plays a role in regulating the differentiation of hematopoietic cells, their proliferation, and prevention of cell death. IL-9 stimulates T, B, and NK cells and the mast cell growth and function, and it induces the Th2 cytokine production. Its secretion by mast cells, NK cells, Th2, Th17, Treg, ILC2, and Th9 cells is stimulated by IL-2, IL-4, IL-25, IL-33, and TGF-β, among others. IL-9 acts through the STAT pathway (118). This cytokine was increased in patients with HF and was negatively proportional to the left ventricular ejection fraction (64). The presence of IL-9 in blood was found to be a risk factor of adverse outcome in patients with HF, and it was correlated with an impaired cardiopulmonary functional capacity (119). IL-9 acted through activating the Gp130/JAK/STAT pathway protecting from progression to HF (120).
Interleukin-13 (IL-13) is produced by CD4 cells, eosinophils, basophils, mast cells, NK cells, NK T cells, and lymphoid tissue associated with the gastrointestinal tract. IL-13 stimulates the immunoglobulin E (IgE) class switching, mediates synthesis of IgE in B cells, up-regulates the CD23 and MHC Class II expression, and modulates the resistance to intracellular parasites. It has been implicated in B-cell maturation and differentiation, reduction of the macrophage activity, and therefore inhibiting the production of pro-inflammatory cytokines (121). Plasma concentration of IL-13 in HF patients was up-regulated in comparison to healthy people. It was positively correlated with the NYHA functional class, brain natriuretic peptide and CRP levels, and negatively correlated with the left ventricular ejection fraction (122).
Conclusion
HF is a systemic disease with a multifactorial etiology. A great amount of studies demonstrated that the activation of inflammation is implicated as an important factor in the initiation and progression of HF. The immune system plays an important role in myocardial remodeling, hypertrophy, apoptosis, and fibrosis. In HF, there is an imbalance between pro-inflammatory and anti-inflammatory cytokines. Concentrations of several interleukins are increased in HF, including IL-1β, IL-6, IL-8, IL-9, IL-10, IL-13, IL-17, and IL-18, whereas the levels of IL-5, IL-7, or IL-33 are down-regulated. Concentrations of inflammatory mediators are associated with cardiac function and can be HF markers and predictors of adverse outcomes or mortality. IL-6 is an independent predictor of prognosis in chronic stable HF and is associated with a higher risk of HF in people without previous cardiovascular diseases. The IL-8 concentration was demonstrated to be a predictor of HF development after myocardial infarction. IL-18 was an independent risk factor of mortality in elderly patients with HF, whereas the presence of IL-9 in blood was found to be a risk factor of adverse outcomes in HF. The comparison of interleukins regarding their effects in cell culture, animal and clinical studies, and their role as a predictor of prognosis is demonstrated in Table 1. However, although a great number of trials have been conducted, the exact role of interleukins and pathophysiological pathways are not fully understood. Unfortunately, all attempts to use anti-inflammatory treatment in HF appeared to be ineffective. Moreover, several interleukins play a dual role and can act as both pro- or anti-inflammatory factors and have either cardioprotective or deleterious properties. This could be dependent on their concentration, period of their synthesis, and types of receptors they activate. A moderate activation of the SAFE pathway has a protective effect. Nevertheless, the chronic activation of STAT3 is responsible for inflammation, left ventricular rupture, and HF development. Therefore, further studies regarding the assessment of inflammatory pathways in HF in view of identifying new opportunities to improve the quality of life, slow disease progression, and improve the survival need to be carried out.
Table 1.
Comparison of interleukins regarding their effects in cell culture, animal and clinical studies, and their role as a predictor of prognosis. ↑ - increased concentration, ↓ - decreased concentration, ↔ - concentration not changed
| Interleukin | Characteristic features | Effect of treatment in cell culture | Effect of treatment in animal studies | Effect of inhibition in in vivo studies | Effect of antagonist treatment in patients | Concentration in patients with heart failure | Independent predictor of poor prognosis |
|---|---|---|---|---|---|---|---|
| Interleukin-1 | Involved in cardiac remodeling after ischemia | Promoted contractile dysfunction and reduced LV contractility reserve | Decreased contractile function and reduced LV contractility reserve | Prevented from LV dysfunction | Protected from depression of cardiac function | ↑ proportional to NYHA functional class | + |
| Interleukin-6 | If restricted to short period after the injury- cardioprotective; when prolonged- ventricular dilation and HF development | Negative inotropic effect | Negative inotropic effect; cardiac dilation | Prevented from cardiac dilation | Cardioprotective effect of ischemic pre- and post-conditioning | ↑ proportional to NYHA functional class and function of either left and right ventricle | + |
| Interleukin-8 | Key mediator associated with inflammation | ? | ? | Worse myocardial function and up-regulated cardiac myocyte apoptosis | ↑ | + | |
| Interleukin-10 | Anti-inflammatory; cardioprotective effects | Suppressed pathological autophagy | Inhibited infiltration of inflammatory cells; improved myocardial function; protected from adverse cardiac remodeling | Worse left ventricular function, increased fibrosis, and cardiomyocyte apoptosis | ↑ or ↔; proportional to NYHA functional class | ||
| Interleukin-17 | Pro-inflammatory | ? | Adverse cardiac remodeling; activation of matrix metalloproteinases; enhanced cardiac apoptosis | ↑ proportional to NYHA functional class | + | ||
| Interleukin-18 | Pro-inflammatory | Enhanced cell surface area, increased ANP and BNP concentration and Akt activation | Cardiac hypertrophy, reduced left ventricular ejection fraction, enhanced end-diastolic pressure, up-regulated expression of natriuretic peptides | Prevented from HF development; attenuated ventricular dilatation | ↑ | + | |
| Interleukin-33 | Dual role, pro-inflammatory or anti-inflammatory | Prevented from hypertrophy; increased level of antiapoptotic proteins | Decreased hypertrophy, reduced infarct size and fibrosis, improved systolic function after ischemia–reperfusion injury | More severe hypertrophy, reduced fractional shortening, adverse fibrosis, inflammation, poorer prognosis, and increased level of natriuretic peptides | ↓; especially in patients with unstable heart failure | Serum soluble decoy receptor (sST2) levels predicted adverse outcome and death |
HF - heart failure; LV – left ventricular; NYHA - New York Heart Association
Footnotes
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – O.A.S.; Design – O.A.S.; Supervision – O.A.S., E.N.K.; Funding – O.A.S., A.P., M.M., E.N.K.; Materials – O.A.S., A.P., Ł.M.; Data collection &/or processing – O.A.S., A.P., Ł.M., M.M.; Analysis &/or interpretation – O.A.S., A.P.; Literature search – O.A.S., A.P.; Writing – O.A.S., Ł.M., M.M.; Critical review – O.A.S., Ł.M., E.N.K.
References
- 1.Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2018 Update:A Report From the American Heart Association. Circulation. (e67-) 2018;137:e492. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 3.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure:an analysis of the cytokine database from the Vesnarinone trial (VEST) Circulation. 2001;103:2055–9. doi: 10.1161/01.cir.103.16.2055. [DOI] [PubMed] [Google Scholar]
- 4.Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000;102:3060–7. doi: 10.1161/01.cir.102.25.3060. [DOI] [PubMed] [Google Scholar]
- 5.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 6.Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction:a report from the Studies of Left Ventricular Dysfunction (SOLVD) J Am Coll Cardiol. 1996;27:1201–6. doi: 10.1016/0735-1097(95)00589-7. [DOI] [PubMed] [Google Scholar]
- 7.Gullestad L, Ueland T, Brunsvig A, Kjekshus J, Simonsen S, Frøland SS, et al. Effect of metoprolol on cytokine levels in chronic heart failure--a substudy in the Metoprolol Controlled-Release Randomised Intervention Trial in Heart Failure (MERIT-HF) Am Heart J. 2001;141:418–21. doi: 10.1067/mhj.2001.112785. [DOI] [PubMed] [Google Scholar]
- 8.Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velázquez F, Aronovitz M, et al. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ Heart Fail. 2015;8:776–87. doi: 10.1161/CIRCHEARTFAILURE.115.002225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res. 1997;81:627–35. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- 10.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure:results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–40. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 11.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure:results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004;109:1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 12.Deftereos S, Giannopoulos G, Panagopoulou V, Bouras G, Raisakis K, Kossyvakis C, et al. Anti-inflammatory treatment with colchicine in stable chronic heart failure:a prospective, randomized study. JACC Heart Fail. 2014;2:131–7. doi: 10.1016/j.jchf.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Torre-Amione G, Anker SD, Bourge RC, Colucci WS, Greenberg BH, Hildebrandt P, et al. Advanced Chronic Heart Failure CLinical Assessment of Immune Modulation Therapy Investigators. Results of a non-specific immunomodulation therapy in chronic heart failure (ACCLAIM trial):a placebo-controlled randomised trial. Lancet. 2008;371:228–36. doi: 10.1016/S0140-6736(08)60134-8. [DOI] [PubMed] [Google Scholar]
- 14.Milani RV, Mehra MR, Endres S, Eigler A, Cooper ES, Lavie CJ, Jr, et al. The clinical relevance of circulating tumor necrosis factor-alpha in acute decompensated chronic heart failure without cachexia. Chest. 1996;110:992–5. doi: 10.1378/chest.110.4.992. [DOI] [PubMed] [Google Scholar]
- 15.Oral H, Fisher SG, Fay WP, Singh SN, Fletcher RD, Morady F. Effects of amiodarone on tumor necrosis factor-alpha levels in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1999;83:388–91. doi: 10.1016/s0002-9149(98)00874-1. [DOI] [PubMed] [Google Scholar]
- 16.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–32. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011;108:19725–30. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Testa M, Yeh M, Lee P, Fanelli R, Loperfido F, Berman JW, LeJemtel TH. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol. 1996;28:964–71. doi: 10.1016/s0735-1097(96)00268-9. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med. 1996;183:949–58. doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim BK, Choe SC, Shin JO, Ho SH, Kim JM, Yu SS, et al. Local expression of interleukin-1 receptor antagonist by plasmid DNA improves mortality and decreases myocardial inflammation in experimental coxsackieviral myocarditis. Circulation. 2002;105:1278–81. [PubMed] [Google Scholar]
- 21.Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A. Interleukin-1βinduces a reversible cardiomyopathy in the mouse. Inflamm Res. 2013;62:637–40. doi: 10.1007/s00011-013-0625-0. [DOI] [PubMed] [Google Scholar]
- 22.Toldo S, Mezzaroma E, Van Tassell BW, Farkas D, Marchetti C, Voelkel NF, et al. Interleukin-1βblockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol. 2013;98:734–45. doi: 10.1113/expphysiol.2012.069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toldo S, Mezzaroma E, Bressi E, Marchetti C, Carbone S, Sonnino C, et al. Interleukin-1βblockade improves left ventricular systolic/diastolic function and restores contractility reserve in severe ischemic cardiomyopathy in the mouse. J Cardiovasc Pharmacol. 2014;64:1–6. doi: 10.1097/FJC.0000000000000106. [DOI] [PubMed] [Google Scholar]
- 24.Van Tassell BW, Arena RA, Toldo S, Mezzaroma E, Azam T, Seropian IM, et al. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS One. 2012;7:e33438. doi: 10.1371/journal.pone.0033438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toldo S, Mezzaroma E, O'Brien L, Marchetti C, Seropian IM, Voelkel NF, et al. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014;306:H1025–31. doi: 10.1152/ajpheart.00795.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikonomidis I, Lekakis JP, Nikolaou M, Paraskevaidis I, Andreadou I, Kaplanoglou T, et al. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117:2662–9. doi: 10.1161/CIRCULATIONAHA.107.731877. [DOI] [PubMed] [Google Scholar]
- 27.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, et al. VCU-ART Investigators. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010;105:1371–7. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 28.Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study] Am J Cardiol. 2013;111:1394–400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes:the MRC-ILA Heart Study. Eur Heart J. 2015;36:377–84. doi: 10.1093/eurheartj/ehu272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu J, Zhang J, Xiang D, Zhang Z, Zhang L, Wu M, et al. Recombinant human interleukin-1 receptor antagonist protects mice against acute doxorubicin-induced cardiotoxicity. Eur J Pharmacol. 2010;643:247–53. doi: 10.1016/j.ejphar.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 31.Mezzaroma E, Mikkelsen RB, Toldo S, Mauro AG, Sharma K, Marchetti C, et al. Role of Interleukin-1 in Radiation-Induced Cardiomyopathy. Mol Med. 2015;21:210–8. doi: 10.2119/molmed.2014.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Q, Wang T, Yu H, Liu B, Jia R. Interaction between interleukin-1 beta and angiotensin II receptor 1 in hypothalamic paraventricular nucleus contributes to progression of heart failure. J Interferon Cytokine Res. 2014;34:870–5. doi: 10.1089/jir.2013.0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kishimoto T, Akira S, Taga T. Interleukin-6 and its receptor:a paradigm for cytokines. Science. 1992;258:593–7. doi: 10.1126/science.1411569. [DOI] [PubMed] [Google Scholar]
- 34.Fischer P, Hilfiker-Kleiner D. Role of gp130-mediated signalling pathways in the heart and its impact on potential therapeutic aspects. Br J Pharmacol. 2008;153(Suppl 1):S414–27. doi: 10.1038/bjp.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway:from protection to failure. Pharmacol Ther. 2008;120:172–85. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Lecour S, James RW. When are pro-inflammatory cytokines SAFE in heart failure? Eur Heart J. 2011;32:680–5. doi: 10.1093/eurheartj/ehq484. [DOI] [PubMed] [Google Scholar]
- 37.Narazaki M, Yasukawa K, Saito T, Ohsugi Y, Fukui H, Koishihara Y, et al. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood. 1993;82:1120–6. [PubMed] [Google Scholar]
- 38.Villegas S, Villarreal FJ, Dillmann WH. Leukemia Inhibitory Factor and Interleukin-6 downregulate sarcoplasmic reticulum Ca2+ ATPase (SERCA2) in cardiac myocytes. Basic Res Cardiol. 2000;95:47–54. doi: 10.1007/s003950050007. [DOI] [PubMed] [Google Scholar]
- 39.Patten M, Krämer E, Bünemann J, Wenck C, Thoenes M, Wieland T, et al. Endotoxin and cytokines alter contractile protein expression in cardiac myocytes in vivo. Pflugers Arch. 2001;442:920–7. doi: 10.1007/s004240100612. [DOI] [PubMed] [Google Scholar]
- 40.Yan AT, Yan RT, Cushman M, Redheuil A, Tracy RP, Arnett DK, et al. Relationship of interleukin-6 with regional and global left-ventricular function in asymptomatic individuals without clinical cardiovascular disease:insights from the Multi-Ethnic Study of Atherosclerosis. Eur Heart J. 2010;31:875–82. doi: 10.1093/eurheartj/ehp454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harhay MO, Tracy RP, Bagiella E, Barr RG, Pinder D, Hundley WG, et al. Relationship of CRP, IL-6, and fibrinogen with right ventricular structure and function:The MESA-Right Ventricle Study. Int J Cardiol. 2013;168:3818–24. doi: 10.1016/j.ijcard.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eiken HG, Øie E, Damås JK, Yndestad A, Bjerkeli V, Aass H, et al. Myocardial gene expression of leukaemia inhibitory factor, interleukin-6 and glycoprotein 130 in end-stage human heart failure. Eur J Clin Invest. 2001;31:389–97. doi: 10.1046/j.1365-2362.2001.00795.x. [DOI] [PubMed] [Google Scholar]
- 43.Fedacko J, Singh RB, Gupta A, Hristova K, Toda E, Kumar A, et al. Inflammatory mediators in chronic heart failure in North India. Acta Cardiol. 2014;69:391–8. doi: 10.1080/ac.69.4.3036655. [DOI] [PubMed] [Google Scholar]
- 44.Tsutamoto T, Hisanaga T, Wada A, Maeda K, Ohnishi M, Fukai D, et al. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol. 1998;31:391–8. doi: 10.1016/s0735-1097(97)00494-4. [DOI] [PubMed] [Google Scholar]
- 45.Jug B, Salobir BG, Vene N, Sebestjen M, Sabovic M, Keber I. Interleukin-6 is a stronger prognostic predictor than high-sensitive C-reactive protein in patients with chronic stable heart failure. Heart Vessels. 2009;24:271–6. doi: 10.1007/s00380-008-1111-4. [DOI] [PubMed] [Google Scholar]
- 46.Rogers JK, Jhund PS, Perez AC, Böhm M, Cleland JG, Gullestad L, et al. Effect of rosuvastatin on repeat heart failure hospitalizations:the CORONA Trial (Controlled Rosuvastatin Multinational Trial in Heart Failure) JACC Heart Fail. 2014;2:289–97. doi: 10.1016/j.jchf.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 47.Dolenc J, Šebeštjen M, Vrtovec B, Koželj M, Haddad F. Pulmonary hypertension in patients with advanced heart failure is associated with increased levels of interleukin-6. Biomarkers. 2014;19:385–90. doi: 10.3109/1354750X.2014.918654. [DOI] [PubMed] [Google Scholar]
- 48.Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, et al. Framingham Heart Study. Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction:the Framingham Heart Study. Circulation. 2003;107:1486–91. doi: 10.1161/01.cir.0000057810.48709.f6. [DOI] [PubMed] [Google Scholar]
- 49.Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–9. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- 50.Janssen SP, Gayan-Ramirez G, Van den Bergh A, Herijgers P, Maes K, Verbeken E, et al. Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation. 2005;111:996–1005. doi: 10.1161/01.CIR.0000156469.96135.0D. [DOI] [PubMed] [Google Scholar]
- 51.Tsuruda T, Jougasaki M, Boerrigter G, Huntley BK, Chen HH, D'Assoro AB, et al. Cardiotrophin-1 stimulation of cardiac fibroblast growth:roles for glycoprotein 130/leukemia inhibitory factor receptor and the endothelin type A receptor. Circ Res. 2002;90:128–34. doi: 10.1161/hh0202.103613. [DOI] [PubMed] [Google Scholar]
- 52.Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J, Jr, et al. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–98. doi: 10.1016/s0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- 53.Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA. 1995;92:4862–6. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci U S A. 2001;98:9050–5. doi: 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–95. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 56.Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein AB, Hunt G, et al. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res. 2004;64:61–71. doi: 10.1016/j.cardiores.2004.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hilfiker-Kleiner D, Shukla P, Klein G, Schaefer A, Stapel B, Hoch M, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122:145–55. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- 58.Fredj S, Bescond J, Louault C, Delwail A, Lecron JC, Potreau D. Role of interleukin-6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J Cell Physiol. 2005;204:428–36. doi: 10.1002/jcp.20307. [DOI] [PubMed] [Google Scholar]
- 59.Smolen JS, Schoels MM, Nishimoto N, Breedveld FC, Burmester GR, Dougados M, et al. Consensus statement on blocking the effects of interleukin-6 and in particular by interleukin-6 receptor inhibition in rheumatoid arthritis and other inflammatory conditions. Ann Rheum Dis. 2013;72:482–92. doi: 10.1136/annrheumdis-2012-202469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rubbert-Roth A. Assessing the safety of biologic agents in patients with rheumatoid arthritis. Rheumatology (Oxford) 2012;51(Suppl 5):38–47. doi: 10.1093/rheumatology/kes114. [DOI] [PubMed] [Google Scholar]
- 61.Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol. 1994;56:559–64. [PubMed] [Google Scholar]
- 62.Damås JK, Eiken HG, Oie E, Bjerkeli V, Yndestad A, Ueland T, et al. Myocardial expression of CC- and CXC-chemokines and their receptors in human end-stage heart failure. Cardiovasc Res. 2000;47:778–87. doi: 10.1016/s0008-6363(00)00142-5. [DOI] [PubMed] [Google Scholar]
- 63.Damås JK, Gullestad L, Ueland T, Solum NO, Simonsen S, Frøland SS, et al. CXC-chemokines, a new group of cytokines in congestive heart failure—possible role of platelets and monocytes. Cardiovasc Res. 2000;45:428–36. doi: 10.1016/s0008-6363(99)00262-x. [DOI] [PubMed] [Google Scholar]
- 64.Cappuzzello C, Di Vito L, Melchionna R, Melillo G, Silvestri L, Cesareo E, et al. Increase of plasma IL-9 and decrease of plasma IL-5, IL-7, and IFN-gamma in patients with chronic heart failure. J Transl Med. 2011;9:28. doi: 10.1186/1479-5876-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nymo SH, Hulthe J, Ueland T, McMurray J, Wikstrand J, Askevold ET, et al. Inflammatory cytokines in chronic heart failure:interleukin-8 is associated with adverse outcome. Results from CORONA. Eur J Heart Fail. 2014;16:68–75. doi: 10.1093/eurjhf/hft125. [DOI] [PubMed] [Google Scholar]
- 66.De Gennaro L, Brunetti ND, Montrone D, De Rosa F, Cuculo A, Di Biase M. Subacute inflammatory activation in subjects with acute coronary syndrome and left ventricular dysfunction. Inflammation. 2012;35:363–70. doi: 10.1007/s10753-011-9326-4. [DOI] [PubMed] [Google Scholar]
- 67.Husebye T, Eritsland J, Arnesen H, Bjørnerheim R, Mangschau A, Seljeflot I, et al. Association of interleukin 8 and myocardial recovery in patients with ST-elevation myocardial infarction complicated by acute heart failure. PLoS One. 2014;9:e112359. doi: 10.1371/journal.pone.0112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 69.Silvestre JS, Mallat Z, Tamarat R, Duriez M, Tedgui A, Levy BI. Regulation of matrix metalloproteinase activity in ischemic tissue by interleukin-10:role in ischemia-induced angiogenesis. Circ Res. 2001;89:259–64. doi: 10.1161/hh1501.094269. [DOI] [PubMed] [Google Scholar]
- 70.Dixon DL, Griggs KM, Bersten AD, De Pasquale CG. Systemic inflammation and cell activation reflects morbidity in chronic heart failure. Cytokine. 2011;56:593–9. doi: 10.1016/j.cyto.2011.08.029. [DOI] [PubMed] [Google Scholar]
- 71.Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E, et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-κB. Circulation. 2012;126:418–29. doi: 10.1161/CIRCULATIONAHA.112.112185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frangogiannis NG, Mendoza LH, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, et al. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000;165:2798–808. doi: 10.4049/jimmunol.165.5.2798. [DOI] [PubMed] [Google Scholar]
- 73.Stumpf C, Seybold K, Petzi S, Wasmeier G, Raaz D, Yilmaz A, et al. Interleukin-10 improves left ventricular function in rats with heart failure subsequent to myocardial infarction. Eur J Heart Fail. 2008;10:733–9. doi: 10.1016/j.ejheart.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 74.Krishnamurthy P, Lambers E, Verma S, Thorne T, Qin G, Losordo DW, et al. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB J. 2010;24:2484–94. doi: 10.1096/fj.09-149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res. 2009;104:e9–18. doi: 10.1161/CIRCRESAHA.108.188243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kishore R, Krishnamurthy P, Garikipati VN, Benedict C, Nickoloff E, Khan M, et al. Interleukin-10 inhibits chronic angiotensin II-induced pathological autophagy. J Mol Cell Cardiol. 2015;89:203–13. doi: 10.1016/j.yjmcc.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stumpf C, Petzi S, Seybold K, Wasmeier G, Arnold M, Raaz D, et al. Atorvastatin enhances interleukin-10 levels and improves cardiac function in rats after acute myocardial infarction. Clin Sci (Lond) 2009;116:45–52. doi: 10.1042/CS20080042. [DOI] [PubMed] [Google Scholar]
- 78.Davies EJ, Moxham T, Rees K, Singh S, Coats AJ, Ebrahim S, et al. Exercise training for systolic heart failure:Cochrane systematic review and meta-analysis. Eur J Heart Fail. 2010;12:706–15. doi: 10.1093/eurjhf/hfq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nunes RB, Tonetto M, Machado N, Chazan M, Heck TG, Veiga AB, et al. Physical exercise improves plasmatic levels of IL-10, left ventricular end-diastolic pressure, and muscle lipid peroxidation in chronic heart failure rats. J Appl Physiol (1985) 2008;104:1641–7. doi: 10.1152/japplphysiol.00062.2008. [DOI] [PubMed] [Google Scholar]
- 80.Onishi RM, Gaffen SL. Interleukin-17 and its target genes:mechanisms of interleukin-17 function in disease. Immunology. 2010;129:311–21. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wuyts WA, Vanaudenaerde BM, Dupont LJ, Van Raemdonck DE, Demedts MG, Verleden GM. Interleukin-17-induced interleukin-8 release in human airway smooth muscle cells:role for mitogen-activated kinases and nuclear factor-kappaB. J Heart Lung Transplant. 2005;24:875–81. doi: 10.1016/j.healun.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 82.Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. 2009;119:1424–32. doi: 10.1161/CIRCULATIONAHA.108.827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li XF, Pan D, Zhang WL, Zhou J, Liang JJ. Association of NT-proBNP and interleukin-17 levels with heart failure in elderly patients. Genet Mol Res. 2016;15 doi: 10.4238/gmr.15028014. doi:10.4238/gmr.15028014. [DOI] [PubMed] [Google Scholar]
- 84.Sandip C, Tan L, Huang J, Li Q, Ni L, Cianflone K, et al. Common variants in IL-17A/IL-17RA axis contribute to predisposition to and progression of congestive heart failure. Medicine (Baltimore) 2016;95:e4105. doi: 10.1097/MD.0000000000004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Podewski EK, Hilfiker-Kleiner D, Hilfiker A, Morawietz H, Lichtenberg A, Wollert KC, et al. Alterations in Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation. 2003;107:798–802. doi: 10.1161/01.cir.0000057545.82749.ff. [DOI] [PubMed] [Google Scholar]
- 86.Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. 2010;106:1646–55. doi: 10.1161/CIRCRESAHA.109.213157. [DOI] [PubMed] [Google Scholar]
- 87.Krstić J, Jauković A, Mojsilović S, Ðorđević IO, Trivanović D, Ilić V, et al. In vitro effects of IL-17 on angiogenic properties of endothelial cells in relation to oxygen levels. Cell Biol Int. 2013;37:1162–70. doi: 10.1002/cbin.10144. [DOI] [PubMed] [Google Scholar]
- 88.Lee JH, Cho ML, Kim JI, Moon YM, Oh HJ, Kim GT, et al. Interleukin 17 (IL-17) increases the expression of Toll-like receptor-2, 4, and 9 by increasing IL-1beta and IL-6 production in autoimmune arthritis. J Rheumatol. 2009;36:684–92. doi: 10.3899/jrheum.080169. [DOI] [PubMed] [Google Scholar]
- 89.Hu MH, Zheng QF, Jia XZ, Li Y, Dong YC, Wang CY, et al. Neuroprotection effect of interleukin (IL)-17 secreted by reactive astrocytes is emerged from a high-level IL-17-containing environment during acute neuroinflammation. Clin Exp Immunol. 2014;175:268–84. doi: 10.1111/cei.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dinarello CA. Interleukin-18. Methods. 1999;19:121–32. doi: 10.1006/meth.1999.0837. [DOI] [PubMed] [Google Scholar]
- 91.Fix C, Bingham K, Carver W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine. 2011;53:19–28. doi: 10.1016/j.cyto.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Siddesha JM, Valente AJ, Sakamuri SS, Gardner JD, Delafontaine P, Noda M, et al. Acetylsalicylic acid inhibits IL-18-induced cardiac fibroblast migration through the induction of RECK. J Cell Physiol. 2014;229:845–55. doi: 10.1002/jcp.24511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chandrasekar B, Mummidi S, Claycomb WC, Mestril R, Nemer M. Interleukin-18 is a pro-hypertrophic cytokine that acts through a phosphatidylinositol 3-kinase-phosphoinositide-dependent kinase-1-Akt-GATA4 signaling pathway in cardiomyocytes. J Biol Chem. 2005;280:4553–67. doi: 10.1074/jbc.M411787200. [DOI] [PubMed] [Google Scholar]
- 94.Chandrasekar B, Vemula K, Surabhi RM, Li-Weber M, Owen-Schaub LB, Jensen LE, et al. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem. 2004;279:20221–33. doi: 10.1074/jbc.M313980200. [DOI] [PubMed] [Google Scholar]
- 95.Yoshida T, Friehs I, Mummidi S, del Nido PJ, Addulnour-Nakhoul S, Delafontaine P, et al. Pressure overload induces IL-18 and IL-18R expression, but markedly suppresses IL-18BP expression in a rabbit model. IL-18 potentiates TNF-α-induced cardiomyocyte death. J Mol Cell Cardiol. 2014;75:141–51. doi: 10.1016/j.yjmcc.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mallat Z, Heymes C, Corbaz A, Logeart D, Alouani S, Cohen-Solal A, et al. Evidence for altered interleukin 18 (IL)-18 pathway in human heart failure. FASEB J. 2004;18:1752–4. doi: 10.1096/fj.04-2426fje. [DOI] [PubMed] [Google Scholar]
- 97.Sanchez I, Santana S, Escobar C, Santiago JL, Gonzalez A, Ribas N, et al. Clinical implications of different biomarkers in elderly patients with heart failure. Biomark Med. 2014;8:535–41. doi: 10.2217/bmm.14.24. [DOI] [PubMed] [Google Scholar]
- 98.Woldbaek PR, Sande JB, Strømme TA, Lunde PK, Djurovic S, Lyberg T, et al. Daily administration of interleukin-18 causes myocardial dysfunction in healthy mice. Am J Physiol Heart Circ Physiol. 2005;289:H708–14. doi: 10.1152/ajpheart.01179.2004. [DOI] [PubMed] [Google Scholar]
- 99.Colston JT, Boylston WH, Feldman MD, Jenkinson CP, de la Rosa SD, Barton A, et al. Interleukin-18 knockout mice display maladaptive cardiac hypertrophy in response to pressure overload. Biochem Biophys Res Commun. 2007;354:552–8. doi: 10.1016/j.bbrc.2007.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roussel L, Erard M, Cayrol C, Girard JP. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep. 2008;9:1006–12. doi: 10.1038/embor.2008.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seki K, Sanada S, Kudinova AY, Steinhauser ML, Handa V, Gannon J, et al. Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ Heart Fail. 2009;2:684–91. doi: 10.1161/CIRCHEARTFAILURE.109.873240. [DOI] [PubMed] [Google Scholar]
- 102.Choi YS, Choi HJ, Min JK, Pyun BJ, Maeng YS, Park H, et al. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood. 2009;114:3117–26. doi: 10.1182/blood-2009-02-203372. [DOI] [PubMed] [Google Scholar]
- 103.Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339–46. doi: 10.1084/jem.20071868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Miller AM, Asquith DL, Hueber AJ, Anderson LA, Holmes WM, McKenzie AN, et al. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res. 2010;107:650–8. doi: 10.1161/CIRCRESAHA.110.218867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest. 2007;117:1538–49. doi: 10.1172/JCI30634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Segiet OA, Romuk E, Nowalany-Kozielska E, Wojciechowska C, Piecuch A, Wojnicz R. The concentration of interleukin-33 in heart failure with reduced ejection fraction. Anatol J Cardiol. 2019;21:305–13. doi: 10.14744/AnatolJCardiol.2019.64614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Caselli C, D'Amico A, Ragusa R, Caruso R, Prescimone T, Cabiati M, et al. IL-33/ST2 pathway and classical cytokines in end-stage heart failure patients submitted to left ventricular assist device support:a paradoxic role for inflammatory mediators? Mediators Inflamm. 2013;2013:498703. doi: 10.1155/2013/498703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang HF, Xie SL, Chen YX, Mai JT, Wang JF, Zhu WL, et al. Altered serum levels of IL-33 in patients with advanced systolic chronic heart failure:correlation with oxidative stress. J Transl Med. 2012;10:120. doi: 10.1186/1479-5876-10-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Veeraveedu PT, Sanada S, Okuda K, Fu HY, Matsuzaki T, Araki R, et al. Ablation of IL-33 gene exacerbate myocardial remodeling in mice with heart failure induced by mechanical stress. Biochem Pharmacol. 2017;138:73–80. doi: 10.1016/j.bcp.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 110.Sánchez-Más J, Lax A, Asensio-López Mdel C, Fernandez-Del Palacio MJ, Caballero L, Santarelli G, et al. Modulation of IL-33/ST2 system in postinfarction heart failure:correlation with cardiac remodelling markers. Eur J Clin Invest. 2014;44:643–51. doi: 10.1111/eci.12282. [DOI] [PubMed] [Google Scholar]
- 111.Weinberg EO, Shimpo M, Hurwitz S, Tominaga S, Rouleau JL, Lee RT. Identification of serum soluble ST2 receptor as a novel heart failure biomarker. Circulation. 2003;107:721–6. doi: 10.1161/01.cir.0000047274.66749.fe. [DOI] [PubMed] [Google Scholar]
- 112.Wang TJ, Wollert KC, Larson MG, Coglianese E, McCabe EL, Cheng S, et al. Prognostic utility of novel biomarkers of cardiovascular stress:the Framingham Heart Study. Circulation. 2012;126:1596–604. doi: 10.1161/CIRCULATIONAHA.112.129437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Boisot S, Beede J, Isakson S, Chiu A, Clopton P, Januzzi J, et al. Serial sampling of ST2 predicts 90-day mortality following destabilized heart failure. J Card Fail. 2008;14:732–8. doi: 10.1016/j.cardfail.2008.06.415. [DOI] [PubMed] [Google Scholar]
- 114.Sanderson CJ. Interleukin-5, eosinophils, and disease. Blood. 1992;79:3101–9. [PubMed] [Google Scholar]
- 115.Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102:257–64. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 116.ElKassar N, Gress RE. An overview of IL-7 biology and its use in immunotherapy. J Immunotoxicol. 2010;7:1–7. doi: 10.3109/15476910903453296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fonseca SG, Reis MM, Coelho V, Nogueira LG, Monteiro SM, Mairena EC, et al. Locally produced survival cytokines IL-15 and IL-7 may be associated to the predominance of CD8+ T cells at heart lesions of human chronic Chagas disease cardiomyopathy. Scand J Immunol. 2007;66:362–71. doi: 10.1111/j.1365-3083.2007.01987.x. [DOI] [PubMed] [Google Scholar]
- 118.Goswami R, Kaplan MH. A brief history of IL-9. J Immunol. 2011;186:3283–8. doi: 10.4049/jimmunol.1003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marra AM, Arcopinto M, Salzano A, Bobbio E, Milano S, Misiano G, et al. Detectable interleukin-9 plasma levels are associated with impaired cardiopulmonary functional capacity and all-cause mortality in patients with chronic heart failure. Int J Cardiol. 2016;209:114–7. doi: 10.1016/j.ijcard.2016.02.017. [DOI] [PubMed] [Google Scholar]
- 120.Knoops L, Renauld JC. IL-9 and its receptor:from signal transduction to tumorigenesis. Growth Factors. 2004;22:207–15. doi: 10.1080/08977190410001720879. [DOI] [PubMed] [Google Scholar]
- 121.Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–56. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 122.Nishimura Y, Inoue T, Nitto T, Morooka T, Node K. Increased interleukin-13 levels in patients with chronic heart failure. Int J Cardiol. 2009;131:421–3. doi: 10.1016/j.ijcard.2007.07.128. [DOI] [PubMed] [Google Scholar]