

ABSTRACT
The respiratory lineage initiates from the specification of NKX2-1+ progenitor cells that ultimately give rise to a vast gas-exchange surface area. How the size of the progenitor pool is determined and whether this directly impacts final lung size remains poorly understood. Here, we show that epithelium-specific inactivation of Mdm2, which encodes an E3 ubiquitin ligase, led to lethality at birth with a striking reduction of lung size to a single vestigial lobe. Intriguingly, this lobe was patterned and contained all the appropriate epithelial cell types. The reduction of size can be traced to the progenitor stage, when p53, a principal MDM2 protein degradation target, was transiently upregulated. This was followed by a brief increase of apoptosis. Inactivation of the p53 gene in the Mdm2 mutant background effectively reversed the lung size phenotype, allowing survival at birth. Together, these findings demonstrate that p53 protein turnover by MDM2 is essential for the survival of respiratory progenitors. Unlike in the liver, in which genetic reduction of progenitors triggered compensation, in the lung, respiratory progenitor number is a key determinant factor for final lung size.
KEY WORDS: Lung development, Respiratory progenitors, Organ size, E3 ubiquitin ligase, Trp53
Summary: The MDM2-p53 axis determines lung progenitor cell number and subsequent organ size, but not airway patterning and cell differentiation.
INTRODUCTION
The respiratory primordium initiates with the emergence of a small cluster of progenitors in the anterior ventral foregut, marked by their expression of transcription factor NKX2-1. Juxtaposing the NKX2-1 domain, a high level of SOX2 is expressed in the dorsal portion of the common foregut tube, marking the future esophagus. Following the initial specification of the cell fates, morphogenesis separates the common foregut tube into the future trachea and esophagus. Failure to achieve this separation leads to tracheoesophageal fistula (TEF), a relatively common birth defect with a prevalence of ∼1 in 3500 births (Cardoso and Lü, 2006; Jacobs and Que, 2013; Que et al., 2006, 2007; Domyan et al., 2011). Shortly after foregut separation, the posterior portion of the NKX2-1 domain protrudes into the surrounding mesenchyme, forming left and right lung buds. These primary lung buds then elongate and engage in a largely stereotypical branching morphogenesis program to generate the mature lung (Short et al., 2013; Metzger et al., 2008).
Signals such as WNT and BMP are required to promote respiratory specification (Harris-Johnson et al., 2009; Goss et al., 2009). Inactivation of either WNT ligand genes Wnt2 and Wnt2b, or the effector β-catenin, led to agenesis of both the lung and trachea (Harris-Johnson et al., 2009; Goss et al., 2009). Inactivation of Bmpr1a and Bmpr1b led to loss of respiratory identity in the prospective trachea but not in the lung (Domyan et al., 2011). Despite these findings, how the size of the respiratory progenitor population impacts final lung size has not been addressed. A landmark study that investigated this in the pancreas and liver revealed different mechanisms in these two organs (Stanger et al., 2007). Specifically, genetic ablation of pancreatic progenitor cells led to a smaller pancreas. In contrast, genetic ablation of liver progenitor cells triggered compensatory growth, leading to normal organ size.
In this current study, we tested how the size of the respiratory progenitor pool impacts lung size. Mouse double minute 2 (Mdm2) encodes an E3 ubiquitin ligase. E3 ubiquitin ligases add a ubiquitin moiety to proteins, tagging them for proteasome-mediated degradation. Each E3 ubiquitin ligase has a specific, and often limited, repertoire of protein substrates. For MDM2, a primary substrate is p53 (also known as TRP53 in mouse and TP53 in human), a cardinal tumor suppressor (Levine, 1997). Mutations in the p53 gene have been found in a large number of cancer types including lung cancer (Linzer and Levine, 1979; George et al., 2015). Elevated p53 promotes DNA repair, but when the damage is too severe to be repaired, p53 triggers cell apoptosis (Finlay et al., 1989). The p53 pathway also plays a role in regulating progenitor cell proliferation (Michalovitz et al., 1990). In the lung, genetically inactivating p53 in club cells promotes proliferation whereas upregulated p53 signal leads to cell cycle arrest in G2/M phase, and both inactivation and upregulation of p53 altered airway cell composition (McConnell et al., 2016). Therefore, precise control of p53 by factors such as MDM2 is important for maintaining the balance between cell survival and organismal health. Increased Mdm2 expression, leading to low p53 levels, has been seen in a wide variety of human cancers (Levine and Oren, 2009). Mice with germline deletion of Mdm2 are embryonic lethal in a p53-dependent manner (Jones et al., 1995).
To examine the role of Mdm2 in lung development, we inactivated Mdm2 in epithelial cells using Shhcre. The Mdm2 mutant lung exhibited one small lobe. Despite this drastic reduction in size, patterning of the airways and alveoli, as well as cell differentiation within this vestigial lobe, was normal. Further analysis suggests that the size reduction is due to a transient increase of p53 and its associated cell death at the early stages of lung development. Our data suggest that progenitor cell number is a determinant factor of lung size.
RESULTS
Inactivation of Mdm2 in the lung epithelium led to lung hypoplasia and TEF
Using RNA in situ hybridization, Mdm2 is primarily detected in the developing lung epithelium (Fig. S1). To determine its role during lung development, we inactivated Mdm2 in lung epithelium using Shhcre, which drives robust cre activity in the endoderm lineage starting at embryonic day (E)8.5 (Shhcre/+;Mdm2f/f, hereafter Shhcre;Mdm2 mutants) (Harris et al., 2006; Mendrysa et al., 2003). All mutants died at birth, likely owing to respiratory distress. At E18.5, unlike normal lung which has five lobes, the Shhcre;Mdm2 mutant exhibited one lobe (Fig. 1A,B). It is also much reduced in size compared with any of the normal lobes. The tube leading onto this single lobe showed a clear loss of cartilage rings (Fig. 1C,D).
Fig. 1.

Inactivation of Mdm2 by Shhcre in the lung epithelium led to lung hypoplasia. (A,B) Whole lung of control (A) and Shhcre;Mdm2 mutant (B) at E18.5. There was variation in the mutant lung size. A representative example was shown. (C,D) Trachea of control (C) and Shhcre;Mdm2 mutant (D) at E18.5. Dashed white lines outline the trachea. (E-H) NKX2-1 and SOX2 co-staining outlined the ventral respiratory and dorsal digestive lineages, respectively, in transverse sections of anterior foregut at E10.5 (E,F) or E11.5 (G,H). (I,J) H&E staining of transverse sections of the foregut indicated that the failure to separate into trachea and esophagus persisted at E18.5 in the Shhcre;Mdm2 mutant (J) compared with control (I). Staining data in this figure are representative from n=3 embryos of each genotype.
We traced the trachea phenotypes to E10.5 when the trachea and esophagus characteristics were first specified, as indicated by juxtaposed NKX2-1 and SOX2 domains before their separation in the control (Fig. 1E). A similar juxtaposition pattern was also found in the Shhcre;Mdm2 mutant common foregut tube, suggesting that the respiratory versus digestive fate specification occurred normally (Fig. 1E,F). However, by E11.5, although the foregut of the control embryos had separated into trachea and esophagus, the foregut of Shhcre;Mdm2 mutants remained as one common tube (Fig. 1G,H). Moreover, in the mutant, the SOX2+ cells occupied about two-thirds of the common foregut compared with the one-third occupied by NKX2-1+ cells, distinct from the more even distribution in the control. The single tube persisted in Shhcre;Mdm2 mutants at E18.5, and was lined by SOX2+ epithelium, and surrounded by a pattern of α-SMA+ smooth muscle cells that mimics the pattern of both the airway and esophagus smooth muscles (Fig. 1I,J; Fig. S2). These phenotypes resemble the symptom of TEF in human (Goyal et al., 2006; Jacobs and Que, 2013). Our previous work showed that a similar defect can be a result of inactivation of BMP signaling (Domyan et al., 2011). To address whether disruption of BMP signaling may underlie the Mdm2 TEF phenotype, we compared the expression pattern of BMP pathway readout pSMAD1/5/8 (note that in mouse SMAD8 is now known as SMAD9) between control and Mdm2 mutant foregut by immunostaining. We found that pSMAD1/5/8 expression remained in the ventral epithelium of the mutant, suggesting that the TEF defect is not due to loss of BMP activity (Fig. S3). These data together indicate that inactivation of Mdm2 led to extreme lung hypoplasia and TEF.
Lung size defect can be traced to reduced primordium size shortly after respiratory fate specification
To address the origin of lung size reduction in the Shhcre;Mdm2 mutant, we traced the phenotype to earlier stages. At E13.5, branching morphogenesis did occur in the Shhcre;Mdm2 mutant as indicated by whole-mount staining with E-cadherin (CDH1) antibody, which outlined the epithelium (Fig. 2A,B). However, the number of branches was severely reduced, constituting the single lobe. Reduced branches were already apparent at E11.5. Compared with the control lung with multiple secondary branches, the mutant lung exhibited one bud on the right side, residual of the right lung which is larger than the left in a normal lung (Fig. 2C,D). Based on Nkx2-1 expression by RNA in situ hybridization, the single bud in the Shhcre;Mdm2 mutant was lung in characteristic (Fig. 2E,F). These data suggest that the size reduction initiated at specification of the lung primordium.
Fig. 2.
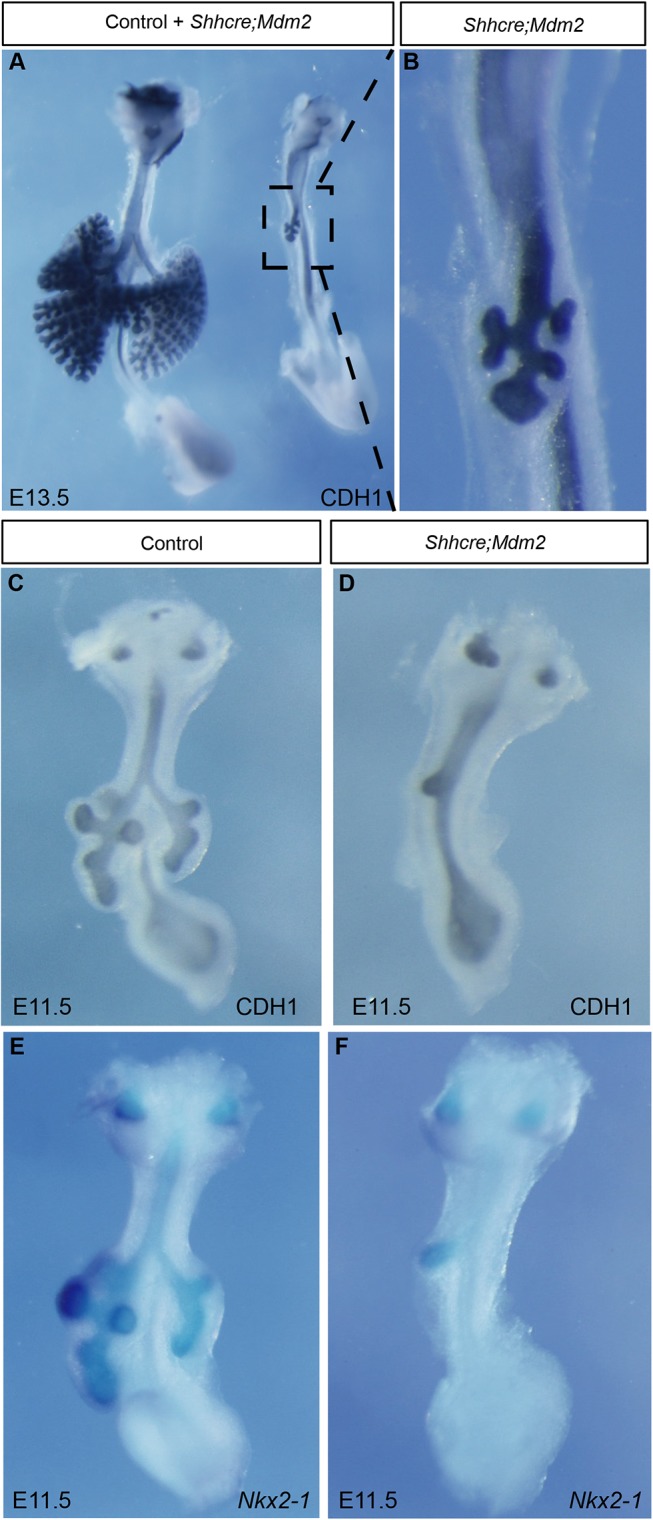
Lung size reduction is apparent at the initiation of branching morphogenesis. (A,B) CDH1 whole-mount antibody staining outlined the epithelium of control and Shhcre;Mdm2 mutant lungs (A) at E13.5. B shows magnification of boxed region in A. (C,D) CDH1 whole-mount antibody staining of lungs at E11.5. (E,F) Nkx2-1 RNA in situ hybridization staining of lungs at E11.5. Staining data in this figure are representative from n=3 embryos of each genotype.
Lung epithelium differentiation continued in the Shhcre;Mdm2 mutant
As a small lung lobe formed in the Shhcre;Mdm2 mutant, we addressed the extent of patterning and differentiation by histological analysis and immunostaining at E18.5. Interestingly, close-up cellular architecture of the mutant lung was comparable with the control lung (Fig. 3A,B). Although minute in size, epithelial cell differentiation took place in the Shhcre;Mdm2 mutant lung, giving rise to ciliated cells, club cells, AT1 and AT2 cells, as shown by FOXJ1, SCGB1A1, HOPX and SFTPC antibody staining, respectively, and quantitative reverse transcription (qRT) PCR quantification of transcripts for the respective cell-type marker genes (Fig. 3C-H). These data suggest that although Mdm2 is crucial for determining the size of progenitor pool, it is not required for the subsequent steps of cell differentiation.
Fig. 3.

Airway and alveolar epithelial cell differentiation appeared normal in Shhcre;Mdm2 mutant. (A,B) H&E staining showed normal alveolar morphology in Shhcre;Mdm2 mutant (B) as compared with control (A) at E18.5. (C,D) Anti-SCGB1A1 staining of club cells and anti-FOXJ1 staining of ciliated cells showed normal airway epithelial cell differentiation in Shhcre;Mdm2 mutant at E18.5. (E) Quantification of qRT-PCR of Scgb1a1 (P=0.9160) and Foxj1 (P=0.7926) expression in control and Shhcre;Mdm2 mutant at E18.5. (F,G) Anti-SFTPC staining of AT2 cells and anti-HOPX staining of AT1 cells showed normal alveolar epithelial cell differentiation in Shhcre;Mdm2 mutant at E18.5. (H) Quantification of qRT-PCR of Sftpc (P=0.8585) and Hopx (P=0.2897) expression in control and Shhcre;Mdm2 mutant at E18.5. Data are mean±s.d. n=3. N.S., not significant. Staining data in this figure are representative from n=3 embryos of each genotype.
Disruption of Mdm2 led to a transient upregulation of p53 protein
To address the molecular mechanism underlying lung primordium reduction in the foregut, we assayed for primary MDM2 target protein p53 expression level using antibody staining. Although no p53+ cells were detected in the control foregut, there was a clear increase of p53 signal in the mutant foregut at E10.5 and E11.5. p53+ cells were restricted to the ventral foregut epithelium where Shhcre was active (Fig. 4A-D). However, no p53 signal was detected in the Shhcre;Mdm2 mutant or control trachea at either E13.5 or E18.5, indicating that p53 upregulation in the mutant was transient (Fig. 4E-H). Increased p53 expression was linked to increased cell death in Shhcre;Mdm2 mutant, as detected at E11.5 by caspase 3 staining on transverse sections as well as by whole-mount Lysotracker Red staining (Fig. 4I-L). Gene expression analysis by qRT-PCR showed that the expression of Mdm2 is lower at the later stage of lung development than at the earlier, providing a plausible explanation for why loss of Mdm2 may have less impact on p53 level at a later stage (Fig. S4). These data suggest that Mdm2 is required in early lung development to degrade p53 protein and promote cell survival.
Fig. 4.
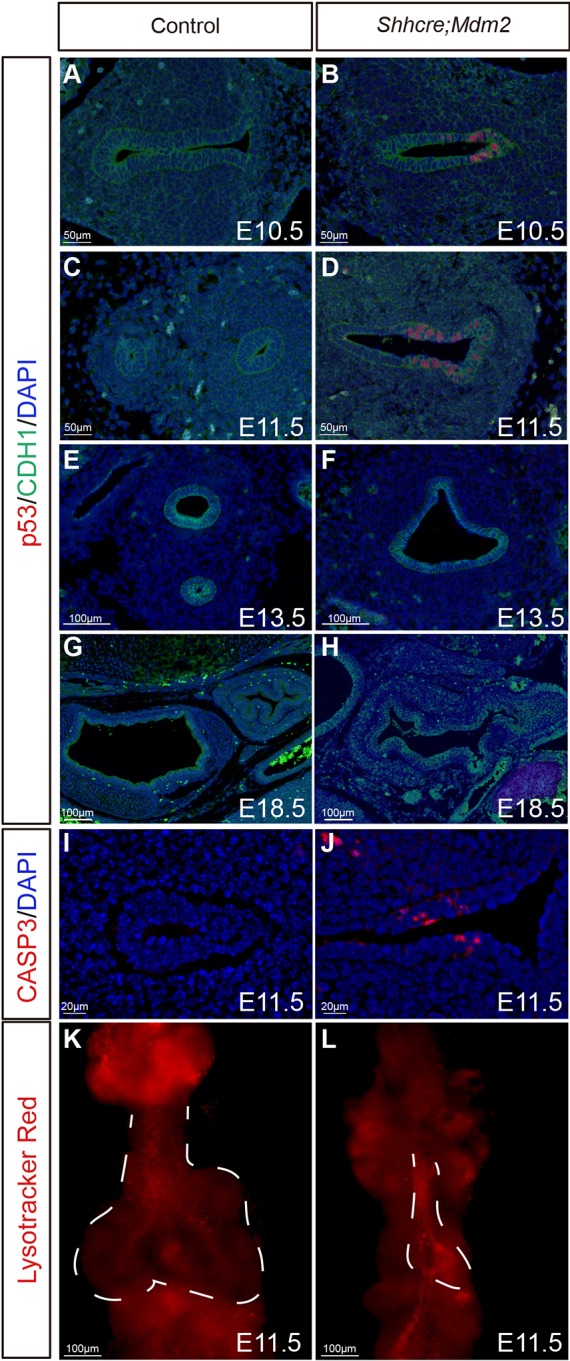
Mdm2 mutant lung exhibited increased p53 signal and cell death. (A-H) Anti-p53 and anti-CDH1 co-staining of transverse sections of foreguts at indicated stages in control and Shhcre;Mdm2 mutants. (I-J) Anti-cleaved caspase 3 staining of transverse sections of control and Shhcre;Mdm2 mutant foreguts at E11.5. (K-L) Whole-mount Lysotracker Red staining of control and Shhcre;Mdm2 mutant lungs at E11.5. Dashed white lines indicate developing lung region. Staining data in this figure are representative from n=3 embryos of each genotype.
Lung defects in Shhcre;Mdm2 mutants were reversed by introduction of the p53 mutation
To test whether defects in the Mdm2 mutant are dependent on p53 increase, we introduced a p53 mutant allele into the Shhcre;Mdm2 background. At E18.5, the body size of Shhcre/+;Mdm2f/f;p53f/f pups was comparable with that of control pups (Fig. 5A-C). The lung appeared to be grossly normal in size and lobe number, a full reversal of the defect in the Shhcre;Mdm2 mutant (Fig. 5D-F). Inactivation of one copy of p53 did not alleviate the defects (Fig. 5E). Unlike the Shhcre;Mdm2 mutants, which died at birth, the Shhcre/+;Mdm2f/f;p53f/f compound mutants survived birth and lived to at least weaning (Fig. 5G). However, the body size of the compound mutants is smaller, unlike the normal size of the p53 mutants alone. This suggests that in addition to p53, MDM2 may have other substrates in the epithelium. Together, these results indicate that the p53 increase is a primary cause of lung size reduction in the Shhcre;Mdm2 mutant.
Fig. 5.

Introduction of p53 mutation effectively reversed the lung size phenotype in Shhcre;Mdm2 mutant. (A-C) E18.5 pups of indicated genotypes. (D-F) Whole lungs and stomachs of indicated genotypes at E18.5. (G) P18 pups of indicated genotypes. Staining data in this figure are representative from n=3 embryos of each genotype.
DISCUSSION
We and others have shown that signals such as WNTs are essential for the specification of foregut endodermal cells as respiratory primordium progenitors (Goss et al., 2009; Harris-Johnson et al., 2009). Outside of signaling pathways genes, little is known about whether there are other molecular and cellular mechanisms that control respiratory progenitor pool size. Furthermore, as the signaling molecules that impact progenitor cell specification also have later roles in branching morphogenesis and/or cell differentiation, how the initial progenitor pool size impacts later aspects of lung development has not been directly addressed. In this study, we found that epithelial inactivation of Mdm2, an E3 ubiquitin ligase gene, led to a transient upregulation of p53 protein, resulting in respiratory progenitor cell apoptosis. With fewer progenitor cells, the Shhcre;Mdm2 mutant lung was reduced to a single small lobe. This phenotype is unlikely to be because of a defect in left/right asymmetry, as Shhcre is not active in domains involved in left/right asymmetry such as the node or lateral plate mesoderm. Rather, it suggests that lung progenitor cell number is a direct determinant for lobe number and size. Despite the drastic reduction of lung size, branching morphogenesis and epithelial cell differentiation ensued in the remaining lobe. This suggests that Mdm2 is crucial for ensuring that a sufficient number of progenitors are generated to make all lobes of the lung, but it is not required for later aspects of lung development.
In the Shhcre;Mdm2 mutant, the p53 level is increased at E10.5 and E11.5, but not at E13.5 or E18.5, suggesting a temporally restricted role of MDM2-p53 relationship during lung development. A similar result is found in the auditory supporting cells and hair cells of the Mdm2 mutant, in which cell apoptosis triggered by p53 upregulation was detected in embryonic and early postnatal stages, but not in the adult (Laos et al., 2017). These findings suggest the possibility that the p53-dependent DNA repair and cell survival system is more active in early progenitors and recently differentiated cells rather than in terminally differentiated cells. Consistent with this, embryonic stem cells are hypersensitive to DNA damage-induced apoptosis (Liu et al., 2014; Li and Huang, 2010; Sabapathy et al., 1997). In comparison, when embryonic stem cells initiated differentiation, the p53 level declined significantly. These findings highlight the importance of p53 at the center of a heightened surveillance machinery to detect and eliminate damaged cells, with the goal to maintain a healthy pool of progenitors.
In the adult model of pneumonectomy, resection of one lung lobe would trigger compensatory regrowth of the remainder lobes through renewed cell proliferation and differentiation (Liu et al., 2016; Lechner et al., 2017). From this setting, it has been concluded that lung regrowth is regulated by available space in the chest cavity, and changes in the associated mechanical force. In comparison, it is interesting that in the Shhcre;Mdm2 mutant, the much-reduced lung only occupied a small portion of the chest cavity. Nevertheless, all the rest of the lung development program was deployed. A recent study elegantly demonstrated that changes in intra-luminal fluid pressure alters the ratio of AT1 versus AT2 cells near birth (Li et al., 2018). As embryonic lung cells produce a large portion of the fluid within the fetal lung lumen, it is possible that in the Shhcre;Mdm2 mutant, the amount of fluid produced is reduced proportionally to the smaller lung size. As a consequence, the fluid pressure may have remained normal, allowing proper differentiation of AT1 and AT2 cells. Overall, our findings suggest that, during development, prenatal lung size is largely dependent on progenitor cell number, and less influenced by available chest cavity space.
This dependence of organ size on progenitor cell number is also observed in the pancreas, but not in the liver (Stanger et al., 2007). In the liver, genetically engineered reduction of progenitor cell number did not change organ size. This is achieved by compensatory growth from remaining cells. Given that the liver is an exceptionally regenerative tissue, we postulate that the pre-determination of organ size by progenitor cell number occurs in organs with less regenerative potential, such as the lung and pancreas. In conclusion, our findings demonstrate a cardinal role of the MDM2-p53 axis in the determination of lung progenitor cell number and organ size.
MATERIALS AND METHODS
Mice
Embryos were harvested at the stages as indicated, counting noon on the day when the vaginal plug was found as E0.5. The alleles used in this study have been described previously: Mdm2fl (Mendrysa et al., 2003), p53fl (Marino et al., 2000) and Shhcre (Harfe et al., 2004). Mdm2fl mice were obtained from National Cancer Institute Mouse Repository under a material transfer agreement. To generate the conditional mutants, males were heterozygous for both cre and the conditional allele. They were mated to females homozygous for the conditional allele. Littermate embryos carrying cre with heterozygous conditional allele were used as controls. All animal experimental procedures were approved by the University of California-San Diego and University of Wisconsin-Madison Animal Care and Use Committee.
Histological and immunofluorescence staining
Embryonic tracheas and lungs were dissected in PBS solution, fixed in 4% paraformaldehyde overnight at 4°C, processed for paraffin embedding and sectioned at 5 μm. Hematoxylin and Eosin (H&E) staining was carried out using a standard protocol. Immunofluorescence was carried out using standard protocols and citric acid antigen retrieval. Primary antibodies used were: anti-SCGB1A1 (1:200, Seven Hills Bioreagents, WRAB-3950), anti-FOXJ1 (1:100, eBioscience, 14-9965-80), anti-HOPX (1:100, Santa Cruz Biotechnology, sc-398703), anti-SFTPC (1:100, Millipore, AB3786), anti-p53 (1:100, Abcam, ab26-100 µl), anti-CDH1 (1:100, Cell Signaling Technology, 3195S), anti-caspase 3 (1:100, Cell Signaling Technology, 9661S), anti-SOX2 (1:100, Abcam, ab97959), anti-α-SMA (1:400, Sigma-Aldrich, C6198-.2ML) and anti-NKX2-1 (1:100, Fisher, MS-699-P0).
qRT-PCR
Lungs from a minimum of three animals per genotype were individually homogenized in TRIzol, and RNA was extracted using an RNeasy Plus Micro Kit (Qiagen). cDNA was prepared with a SuperScript-III First-Strand Synthesis System (Invitrogen) and PCR-quantified using SYBR green (Bio-Rad) and in a Bio-Rad sequence detection system. Three technical replicates were performed per biological sample, and three biological replicates were performed per genotype. The transcript level was normalized to Actb (β-actin) transcript level and compared using a two-tailed Student's t-test. Results are reported as transcript quantity relative to control±s.d. and considered statistically significant if P<0.05. qRT-PCR primers are listed in Table S1.
Whole-mount RNA in situ hybridization
Lungs were dissected in PBS, fixed in 4% paraformaldehyde overnight at 4°C, and then dehydrated to 100% methanol. Whole-mount in situ hybridization was carried out using established protocols (Herriges et al., 2015).
Whole-mount immunohistochemical staining
The staining was performed following a previously published protocol (Zhang et al., 2016). The anti-CDH1 antibody (Cell Signaling Technology) was diluted by 1 to 200.
Lysotracker staining
Embryos were incubated in Lysotracker staining medium (Abcam) for 30 min at 37°C. They were then fixed in 4% paraformaldehyde with minimal Hanks’ Balanced Salt solution at 4°C overnight, and dehydrated through 25%, 50%, 75% and 100% methanol solution before imaging.
Supplementary Material
Acknowledgements
We thank members of the Sun lab for constructive discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.M.V., X.S.; Methodology: J.M.V., X.S.; Formal analysis: P.S., J.M.V., X.S.; Investigation: P.S., R.L., Y.Z., A.G., C.T.; Data curation: P.S., R.L., Y.Z., A.G., C.T.; Writing - original draft: P.S., J.M.V., X.S.; Writing - review & editing: P.S., R.L., J.M.V., X.S.; Visualization: P.S., R.L., Y.Z., A.G., C.T.; Supervision: J.M.V., X.S.; Project administration: J.M.V., X.S.; Funding acquisition: X.S.
Funding
This work was supported by the National Heart, Lung, and Blood Institute (R01 HL142215, HL143256, HL122406, HL119946). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.179820.supplemental
References
- Cardoso W. V. and Lü J. (2006). Regulation of early lung morphogenesis: questions, facts and controversies. Development 133, 1611-1624. 10.1242/dev.02310 [DOI] [PubMed] [Google Scholar]
- Domyan E. T., Ferretti E., Throckmorton K., Mishina Y., Nicolis S. K. and Sun X. (2011). Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development 138, 971-981. 10.1242/dev.053694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay C. A., Hinds P. W. and Levine A. J. (1989). The p53 proto-oncogene can act as a suppressor of transformation. Cell 57, 1083-1093. 10.1016/0092-8674(89)90045-7 [DOI] [PubMed] [Google Scholar]
- George J., Lim J. S., Jang S. J., Cun Y., Ozretić L., Kong G., Leenders F., Lu X., Fernández-Cuesta L., Bosco G. et al. (2015). Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47-53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss A. M., Tian Y., Tsukiyama T., Cohen E. D., Zhou D., Lu M. M., Yamaguchi T. P. and Morrisey E. E. (2009). Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev. Cell 17, 290-298. 10.1016/j.devcel.2009.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A., Jones M. O., Couriel J. M. and Losty P. D. (2006). Oesophageal atresia and tracheo-oesophageal fistula. Arch. Dis. Child. Fetal Neonatal Ed. 91, F381-F384. 10.1136/adc.2005.086157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe B. D., Scherz P. J., Nissim S., Tian H., McMahon A. P. and Tabin C. J. (2004). Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118, 517-528. 10.1016/j.cell.2004.07.024 [DOI] [PubMed] [Google Scholar]
- Harris K. S., Zhang Z., McManus M. T., Harfe B. D. and Sun X. (2006). Dicer function is essential for lung epithelium morphogenesis. Proc. Natl. Acad. Sci. USA 103, 2208-2213. 10.1073/pnas.0510839103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris-Johnson K. S., Domyan E. T., Vezina C. M. and Sun X. (2009). beta-Catenin promotes respiratory progenitor identity in mouse foregut. Proc. Natl. Acad. Sci. USA 106, 16287-16292. 10.1073/pnas.0902274106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herriges J. C., Verheyden J. M., Zhang Z., Sui P., Zhang Y., Anderson M. J., Swing D. A., Zhang Y., Lewandoski M. and Sun X. (2015). FGF-regulated etv transcription factors control fgf-shh feedback loop in lung branching. Dev. Cell 35, 322-332. 10.1016/j.devcel.2015.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs I. J. and Que J. (2013). Genetic and cellular mechanisms of the formation of esophageal atresia and tracheoesophageal fistula. Dis. Esophagus 26, 356-358. 10.1111/dote.12055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. N., Roe A. E., Donehower L. A. and Bradley A. (1995). Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206-208. 10.1038/378206a0 [DOI] [PubMed] [Google Scholar]
- Laos M., Sulg M., Herranen A., Anttonen T. and Pirvola U. (2017). Indispensable role of Mdm2/p53 interaction during the embryonic and postnatal inner ear development. Sci. Rep. 7, 42216 10.1038/srep42216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner A. J., Driver I. H., Lee J., Conroy C. M., Nagle A., Locksley R. M. and Rock J. R. (2017). Recruited monocytes and type 2 immunity promote lung regeneration following pneumonectomy, Cell Stem Cell 21, pp. 120-134.e7. 10.1016/j.stem.2017.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine A. J. (1997). p53, the cellular gatekeeper for growth and division. Cell 88, 323-331. 10.1016/S0092-8674(00)81871-1 [DOI] [PubMed] [Google Scholar]
- Levine A. J. and Oren M. (2009). The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer 9, 749-758. 10.1038/nrc2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. and Huang J. (2010). A new puzzling role of p53 in mouse embryonic stem cells. Cell Cycle 9, 1669-1670. 10.4161/cc.9.9.11596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Wang Z., Chu Q., Jiang K., Li J. and Tang N. (2018). The strength of mechanical forces determines the differentiation of alveolar epithelial cells, Dev. Cell 44, pp. 297-312.e5. 10.1016/j.devcel.2018.01.008 [DOI] [PubMed] [Google Scholar]
- Linzer D. I. H. and Levine A. J. (1979). Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43-52. 10.1016/0092-8674(79)90293-9 [DOI] [PubMed] [Google Scholar]
- Liu J. C., Lerou P. H. and Lahav G. (2014). Stem cells: balancing resistance and sensitivity to DNA damage. Trends Cell Biol. 24, 268-274. 10.1016/j.tcb.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Wu H., Jiang K., Wang Y., Zhang W., Chu Q., Li J., Huang H., Cai T., Ji H. et al. (2016). MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep. 16, 1810-1819. 10.1016/j.celrep.2016.07.020 [DOI] [PubMed] [Google Scholar]
- Marino S., Vooijs M., van Der Gulden H., Jonkers J. and Berns A. (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14, 994-1004. [PMC free article] [PubMed] [Google Scholar]
- McConnell A. M., Yao C., Yeckes A. R., Wang Y., Selvaggio A. S., Tang J., Kirsch D. G. and Stripp B. R. (2016). p53 regulates progenitor cell quiescence and differentiation in the airway. Cell Rep. 17, 2173-2182. 10.1016/j.celrep.2016.11.007 [DOI] [PubMed] [Google Scholar]
- Mendrysa S. M., McElwee M. K., Michalowski J., O'Leary K. A., Young K. M. and Perry M. E. (2003). mdm2 is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol. Cell. Biol. 23, 462-472. 10.1128/MCB.23.2.462-473.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger R. J., Klein O. D., Martin G. R. and Krasnow M. A. (2008). The branching programme of mouse lung development. Nature 453, 745-750. 10.1038/nature07005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalovitz D., Halevy O. and Oren M. (1990). Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell 62, 671-680. 10.1016/0092-8674(90)90113-S [DOI] [PubMed] [Google Scholar]
- Que J., Choi M., Ziel J. W., Klingensmith J. and Hogan B. L. M. (2006). Morphogenesis of the trachea and esophagus: current players and new roles for noggin and Bmps. Differentiation 74, 422-437. 10.1111/j.1432-0436.2006.00096.x [DOI] [PubMed] [Google Scholar]
- Que J., Okubo T., Goldenring J. R., Nam K.-T., Kurotani R., Morrisey E. E., Taranova O., Pevny L. H. and Hogan B. L. (2007). Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development 134, 2521-2531. 10.1242/dev.003855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K., Klemm M., Jaenisch R. and Wagner E. F. (1997). Regulation of ES cell differentiation by functional and conformational modulation of p53. EMBO J. 16, pp. 6217-6229. 10.1093/emboj/16.20.6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short K., Hodson M. and Smyth I. (2013). Spatial mapping and quantification of developmental branching morphogenesis. Development 140, 471-478. 10.1242/dev.088500 [DOI] [PubMed] [Google Scholar]
- Stanger B. Z., Tanaka A. J. and Melton D. A. (2007). Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature 445, 886-891. 10.1038/nature05537 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Yokoyama S., Herriges J. C., Zhang Z., Young R. E., Verheyden J. M. and Sun X. (2016). E3 ubiquitin ligase RFWD2 controls lung branching through protein-level regulation of ETV transcription factors. Proc. Natl. Acad. Sci. USA 113, 7557-7562. 10.1073/pnas.1603310113 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.