

Enteric viruses initiate infection in the gastrointestinal tract but can disseminate to systemic sites. However, the dynamics of viral dissemination are unclear. In this study, we created a library of 135 barcoded coxsackieviruses to examine viral population diversity across time and space following oral inoculation of mice. Overall, we found that the broad population of viruses disseminates early, followed by monopolization of mouse tissues with three or fewer pool members at later time points. Interestingly, we detected virus in systemic tissues such as pancreas and liver just 20 min after oral inoculation. These results suggest rapid dissemination of diverse viral populations, followed by a major restriction in population diversity and monopolization in all examined tissues.
KEYWORDS: coxsackievirus, dissemination, evolution, viral pathogenesis, viral population dynamics
ABSTRACT
The gastrointestinal tract presents a formidable barrier for pathogens to initiate infection. Despite this barrier, enteroviruses, including coxsackievirus B3 (CVB3), successfully penetrate the intestine to initiate infection and spread systemically prior to shedding in stool. However, the effect of the gastrointestinal barrier on CVB3 population dynamics is relatively unexplored, and the selective pressures acting on CVB3 in the intestine are not well characterized. To examine viral population dynamics in orally infected mice, we produced over 100 CVB3 clones harboring nine unique nucleotide “barcodes.” Using this collection of barcoded viruses, we found diverse viral populations throughout each mouse within the first day postinfection, but by 48 h the viral populations were dominated by fewer than three barcoded viruses in intestinal and extraintestinal tissues. Using light-sensitive viruses to track replication status, we found that diverse viruses had replicated prior to loss of diversity. Sequencing whole viral genomes from samples later in infection did not reveal detectable viral adaptations. Surprisingly, orally inoculated CVB3 was detectable in pancreas and liver as soon as 20 min postinoculation, indicating rapid systemic dissemination. These results suggest rapid dissemination of diverse viral populations, followed by a major restriction in population diversity and monopolization in all examined tissues. These results underscore a complex dynamic between dissemination and clearance for an enteric virus.
IMPORTANCE Enteric viruses initiate infection in the gastrointestinal tract but can disseminate to systemic sites. However, the dynamics of viral dissemination are unclear. In this study, we created a library of 135 barcoded coxsackieviruses to examine viral population diversity across time and space following oral inoculation of mice. Overall, we found that the broad population of viruses disseminates early, followed by monopolization of mouse tissues with three or fewer pool members at later time points. Interestingly, we detected virus in systemic tissues such as pancreas and liver just 20 min after oral inoculation. These results suggest rapid dissemination of diverse viral populations, followed by a major restriction in population diversity and monopolization in all examined tissues.
INTRODUCTION
RNA virus populations are dynamic. Population diversity arises through mutations introduced by the viral RNA-dependent RNA polymerases, which lack proofreading. Although many mutations are detrimental, viruses can benefit from diversity within viral populations since diversity can facilitate replication and dissemination within hosts (1, 2). Viral diversity can be reduced through selective pressures or stochastic events. Because viral diversity is a fundamental parameter in viral evolution and virulence, understanding population dynamics and the factors that influence viral populations is a crucial issue, with consequences for emergence of new pathogens and evolution of new traits and vaccine design.
Previous studies have examined viral population dynamics in a variety of systems and have shown that changes in viral populations can be quantified using genetically marked viruses carrying short unique nucleic acid sequences called “barcodes” (4–8). Several of those previous studies revealed founder effects, representing an extreme reduction in viral population diversity in which viruses in a small subpopulation invade a new tissue or host. This has been described for viruses of plants (9) and of animals, including human immunodeficiency virus (10), hepatitis C virus (11), Zika virus (12, 13), influenza virus (4, 14), and poliovirus (PV) (7).
The gastrointestinal (GI) tract restricts dissemination of lumenal contents, including enteric viruses. Previous work in our laboratory demonstrated that the GI tract limits dissemination of the enteric virus PV, thus reducing the diversity of viral populations in a variety of tissues after oral inoculation (7). However, those studies lacked detailed temporal analysis of viral populations, so how the GI tract influences enteric viral populations over time is unknown.
Coxsackievirus B3 (CVB3) spreads by the fecal-oral route. After oral infection and passage through the GI tract, CVB3 disseminates to and replicates in extraintestinal tissues prior to shedding in stool. Immune-competent mice are susceptible to oral CVB3 infection but do not succumb to disease. Immune-deficient mice lacking the interferon alpha/beta receptor gene (Ifnar−/−) support high levels of viral replication and succumb to disease, providing a more tractable model to study infection with enteroviruses by the natural oral route (15–18).
In this study, we examined the dissemination patterns of CVB3 over time to assess how viral diversity changes across different tissues. Using a collection of barcoded viruses, we found diverse viral populations across many tissues within the first day after oral inoculation of mice. Surprisingly, dissemination was extremely rapid, with diverse viral populations present in systemic sites at 20 min postinfection. However, by 48 h postinfection (hpi), a small number of viruses monopolized the entire mouse, without detectable virus adaptation. These results reveal dynamic viral dissemination and clearance for an enteric virus.
(This article was submitted to an online preprint archive [3].)
RESULTS
Multiple unique viruses from oral inoculum reach extraintestinal tissues.
We created genetically tagged viruses that allow discrimination of individual members of the population to monitor viral population dynamics. We engineered 135 CVB3 clones carrying unique nine-nucleotide “barcodes” in the 5′ untranslated region (nucleotide 709; Fig. 1A and B). Insertion of a barcode sequence reduced viral replication in HeLa cells only at 5 h postinfection (Fig. 1C), suggesting that the barcoded viruses had sufficient fitness for in vivo experiments. Furthermore, after passaging was performed for 10 viral replication cycles in HeLa cells, the viral barcode was still present, indicating that the barcode insertion was well tolerated (data not shown). Each of the 135 barcoded viruses was generated individually prior to combining equal titers of all 135 to create the barcode virus library. Sequencing replicates of the 135-member barcode virus library confirmed that the pool members were approximately equally represented and that the sequencing procedure was reproducible (Fig. 1D).
FIG 1.

Construction of barcoded collection of CVB3 clones. (A) A random 9-nucleotide sequence was inserted into CVB3 5′ untranslated region (5′UTR) (nucleotide 709) between the internal ribosome entry site (IRES) and the start of coding sequence. (B) Workflow for production and analysis of virus barcode frequencies in a pool of 135 barcoded CVB3 clones. Different colors in plasmids and virions represent uniquely barcoded viruses. (C) Growth curves of a single representative barcoded CVB3 clone (barcode ATCGTACCA) and the wild type (WT) in HeLa cells (n = 4, 2-way analysis of variance [ANOVA], Sidak posttest, standard errors of the means [SEM] shown). (D) Sequencing ratios for each barcode in 4 cDNA replicates of CVB3 barcode virus stock.
To quantify how the viral populations changed over time and space, we infected Ifnar−/− mice orally with 1 × 109 PFU of the 135-member CVB3 library and collected stool and tissues at various time points. At time points within the first day of infection, i.e., 7.5 and 19 h postinfection (hpi), we detected all 135 viruses in the upper GI tract (Fig. 2), indicating relatively rapid dissemination of the broad viral population in the inoculum. Furthermore, these results demonstrate that our sequencing and analysis pipeline was sensitive and unbiased for use with mouse tissues. Interestingly, at 48 and 72 hpi, a small number of pool members, generally three or less, comprised the majority of viral barcodes in each tissue (Fig. 2). Furthermore, nearly all tissues within an animal contained the same small group of viral barcodes, suggesting a relatively unencumbered flow of virus between tissues after 19 hpi. Except for mice 12 and 14, these high-frequency viruses were unique to each mouse, indicating that no barcoded viruses had a selective advantage with respect to replication and dissemination in mice.
FIG 2.

Virus population dynamics in gastrointestinal tract and extraintestinal tissues over time. Ifnar−/− mice were orally infected with 1 × 109 PFU of the CVB3 barcode library and were dissected 7.5 hpi, 19 hpi, 48 hpi, or 72 hpi. The height of the bar corresponds to the titer of virus, and the height of the color bands is proportional to the frequency of a viral barcode within the sample, with total frequencies equaling 1. The horizontal dashed line represents the limit of detection for plaque assay, and bars with slanted lines represent results below the sequencing threshold.
To quantify viral population changes, we used two metrics of intrasample diversity: (i) the total number of viral barcodes detected and (ii) the Shannon diversity index (which is sensitive to the number of barcodes present as well to the relative proportions of barcode frequencies). We analyzed viral diversity in GI tract tissues (esophagus, stomach, duodenum, jejunum, ileum, cecum, proximal colon, and stool) versus extraintestinal tissues (mesenteric lymph nodes [MLN], pancreas, and liver) to determine whether populations differed in tissues directly exposed to virus versus tissues exposed following systemic spread. The total number of barcodes and Shannon diversity were high across the GI tract at 7.5 and 19 hpi (Fig. 3). However, by 48 hpi and culminating at 72 hpi, the number of barcodes and Shannon diversity diminished across the GI tract. In contrast, the number of barcodes present and the Shannon diversity were generally lower for extraintestinal tissues regardless of time point. These results suggest broad early dissemination of the viral population in the GI tract and slightly reduced dissemination to extraintestinal sites, with clearance and monopolization by a few pool members at all sites later in infection.
FIG 3.

Viral barcode diversity decreases over time and is lower in extraintestinal tissues. (A and B) Comparing changes in diversity over time, we quantified the total number of barcodes detected (A) and Shannon diversity (B) for the GI tract or for extraintestinal tissues (MLN, pancreas, and liver) combined. Data in panel B were analyzed by Kruskal-Wallis test with Dunn’s correction for multiple comparisons; only statistically significant results are shown (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (C and D) To directly compare GI versus extraintestinal sites, we analyzed the number of barcodes detected (C) and Shannon diversity (D). To obtain the data shown in panel D, we performed mixed-effects analysis with Sidak’s correction for multiple comparisons; only statistically significant results are shown.
The broad “takeover” by a few viruses in nearly all tissues observed at later time points could have been due to adaptive mutations that confer a fitness advantage. To test if the high-frequency viruses harbored viral mutants that were selected for, we obtained CVB3 genome-wide consensus sequence for two tissues, MLN and liver. There were no detectable mutations in 5 of the 8 samples (Fig. 4). The remaining samples had 2 synonymous mutations (T7343C and C3217T) and 1 nonsynonymous mutation (C6506T; 3DH199Y) (Fig. 4). Although consensus sequencing cannot detect low-frequency mutations within a population, these results suggest that a major adaptive mutation did not drive the population monopolization effects that we observed here.
FIG 4.

Monopolization of viral populations occurs in the absence of detectable adaption. Sanger consensus sequencing of CVB3 whole genomes was performed for 48-hpi MLN and liver samples from four representative mice. Each line represents the genome of consensus sequence derived from each sample, and each “X” represents a consensus mutation at that site.
Multiple viruses replicate in all tissues prior to monopolization.
One hypothesis for monopolization by viral subpopulations is that the small number of viruses detectable at later time points had undergone replication whereas the other population members detectable at early time points represented unreplicated viruses from the original inoculum. To test this hypothesis, we used light-sensitive viruses to discriminate between unreplicated inoculum virus and replicated progeny virus. We have previously used light-sensitive, neutral red-labeled barcoded virus to identify which barcoded viruses replicated in mice (19). Viruses propagated in the presence of neutral red incorporate the dye into capsids and lose infectivity when exposed to white light. However, upon replication in the dark, newly replicated viruses are light insensitive, thus facilitating discrimination of inoculum from replicated virus. Using this strategy, we produced a neutral red-labeled stock of barcoded CVB3 clones. Confirming light sensitivity, viral titer from this stock was reduced 250,000-fold after exposure to light. In the dark, we orally infected mice with 1 × 109 PFU and then harvested and processed tissues. Each tissue homogenate was split; one half remained in the dark, and the other half was exposed to light. We then performed experiments to (i) examine viral titers and the percentage of viruses that represented leftover inoculum versus replicated virus and to (ii) quantify viral barcode population diversity in the inoculum versus the replicated virus subsets within each tissue (Fig. 5A).
FIG 5.

CVB3 in vivo replication dynamics. (A) Mice were orally infected with neutral red-labeled CVB3 barcoded viruses in the dark. Mice were dissected in the dark, and tissue samples were homogenized and either kept in the dark or exposed to light. Virus was quantified by plaque assay and then amplified for a single cycle in HeLa cells, and deep sequencing was performed on barcodes of progeny. In the example here, the blue virus had replicated in vivo, and therefore it was light insensitive and amplified in HeLa cells. (B) Viral titers in samples kept in the dark, representing both input/inoculum and replicated viruses. Whiskers show maximums to minimums; points labeled with numbers indicate mouse number. (C) Percentages of replicated virus (PFU from light-exposed samples divided by PFU of dark-exposed samples multiplied by 100). The lower dotted line indicates the limit of detection derived from plaque assay for light-exposed samples, and the upper dotted line indicates 100% replication within a population.
First, to determine total tissue titers and the percentages of virus that had undergone replication, samples were quantified by plaque assay. Titers from samples kept in the dark, representing total tissue titers, are presented in Fig. 5B. Trends were similar to the data shown in Fig. 2, with low early extraintestinal tissue titers that increased with time. To reveal the percentage of viruses that had undergone replication, we divided the light titers by the dark titers for each tissue and multiplied by 100. As shown in Fig. 5C, a fraction of virus in stool and colon was light insensitive at 7.5 hpi, indicating that some viral replication had occurred in vivo even at that early time point. The amount of light-insensitive virus was 10-fold higher than the background of light-insensitive/unlabeled virions in the inoculum, arguing that the majority of light-insensitive virus in these samples had replicated in vivo (Fig. 6). The majority of viruses in extraintestinal tissues were still light sensitive, indicating that little viral replication had occurred by 7.5 hpi (Fig. 5C). At 19 hpi, two mice had substantial viral replication in the MLN and pancreas, but nearly all viruses in the liver had replicated in vivo (Fig. 5C). By 48 hpi, nearly all viruses in all tissues were light insensitive (Fig. 5C).
FIG 6.
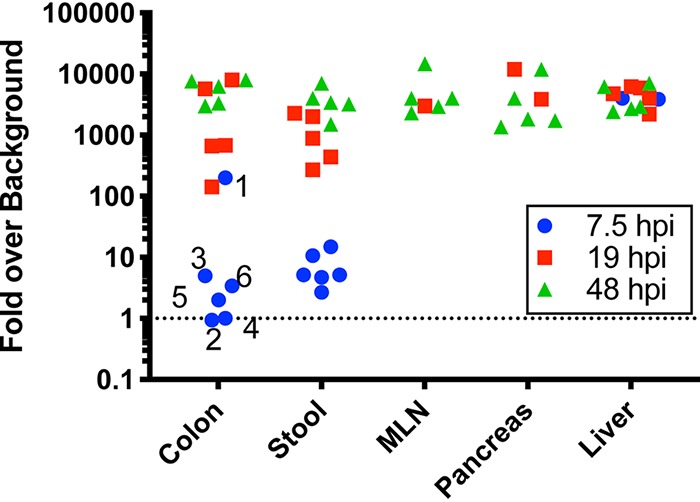
The level of nearly all light-insensitive virus clones recovered from mice is higher than background in neutral red-labeled virus stock. A small fraction of viruses within neutral red-labeled stock are light insensitive, and their presence can confound interpretation of results from subsequent mouse experiments. To rule out this effect, we quantified the fraction of light-insensitive viruses in our stock and compared it with the data from mouse experiments. We found that 1/4,000 PFU in neutral red-labeled virus inoculum was light insensitive. Assuming that (i) neutral red-labeled and unlabeled virions equally penetrated each tissue and (ii) no viral replication occurred, then 1/4,000 PFU from dark-treated tissues would represent light-insensitive inoculum PFU. This sets the background level for each sample. The light-treated virus titer was divided by this background level to obtain the fold signal over background. Values over 1 indicate that virus replication had occurred; the probability of bona fide viral replication increased exponentially as indicated on the y axis. Numbers indicate mouse identifiers.
Second, we quantified viral barcode diversity in each tissue, differentiating diversity in inoculum versus replicated populations. Because RNA from neutral red-inactivated virus could still be amplified by reverse transcription-PCR (RT-PCR) (data not shown), we enriched for viruses that had replicated in vivo by the use of light exposure and amplification of light-insensitive viruses in HeLa cells for a single viral replication cycle (Fig. 5A). Viruses that had replicated in vivo would be insensitive to light exposure and would undergo viral RNA amplification in HeLa cells, whereas inoculum viruses would not undergo RNA amplification. In parallel, the same procedure was used on samples kept in the dark, to amplify both inoculum and replicated viral RNAs. We then subjected the viral barcodes to deep sequencing to assess the diversity of viral barcodes from inoculum (“dark” samples) or replicated viruses (“light” samples) in each tissue. Curiously, at 7.5 hpi, the viruses in stool were diverse and had undergone replication whereas the replicated viruses in colon had minimal diversity (Fig. 7 and 8). Since viral diversity in stool reflects viral replication/shedding in multiple sites along the GI tract and since we examined only colon tissue in these experiments, it is likely that viral replication in tissues other than colon seeded the diverse replicated viruses observed in feces. In extraintestinal tissues at 7.5 hpi, we sporadically detected replicated viral barcodes, and these had low diversity (Fig. 7 and 8). At 19 hpi in stool, all 135 barcoded viruses had replicated (Fig. 7 and 8) in most mice. Viral barcode diversity in colon samples did not change from 7.5 hpi to 19 hpi, although several samples had distinctly higher diversity at 19 hpi. At 48 hpi, we could detect replicated viral barcodes in all samples, and diversity was low in nearly all tissues. Consistent with Fig. 2 data, populations across all mice were very similar, supporting a model of whole-mouse monopolization by a subpopulation of virus. Taking the results together, diversity among replicated viruses in the intestinal tract was high at 7.5 and 19 hpi, but was low at 48 hpi, and diversity among replicated viruses was low at all time points in extraintestinal tissues. Therefore, we conclude that the loss of viral diversity at 48 hpi was not due to replication of only a small fraction of virus in vivo.
FIG 7.

Diverse viruses replicated prior to population monopolization. The ratio of viral barcodes within each sample is shown, with viral barcodes derived from virus that had replicated in vivo (light exposed) and total replicated-plus-unreplicated inoculum viral barcodes (dark samples). The crosshatch (#) symbol represents samples with results below the sequencing depth threshold.
FIG 8.
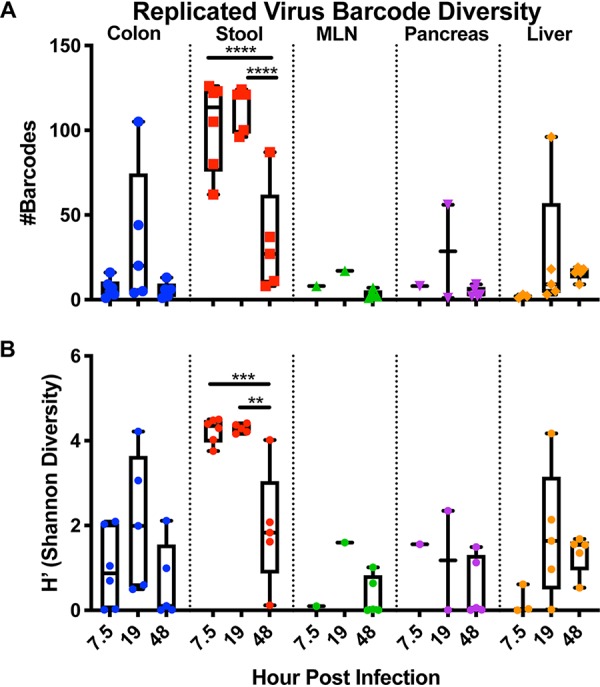
Viral barcode diversity of viruses replicated in vivo. Data represent the number of viral barcodes in each tissue at each time point from samples exposed to light and amplified for a single cycle in HeLa cells. (A) Number of barcodes present of replicated virus in each sample. (B) Shannon diversity (H’) of replicated viral barcodes. Analyses were performed using 2-way ANOVA and Tukey multiple comparisons; only statistically significant results are shown (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Orally inoculated CVB3 rapidly spreads to extraintestinal tissues in mice.
Our data indicate that orally inoculated virus disseminates broadly within a few hours, but how quickly can virus disseminate to GI tract tissues and extraintestinal sites? To assess how quickly and where oral virus inoculum disseminates before viral replication occurs, we infected Ifnar−/− and Ifnar+/+ mice with 1 × 109 PFU CVB3 and assessed viral titers at 20 min postinfection. Surprisingly, we detected virus in the pancreas, liver, and MLN at this extremely early time point (Fig. 9A). Additionally, early dissemination of CVB3 to extraintestinal sites was observed in Ifnar−/− mice, but not in Ifnar+/+ mice. In Ifnar−/− mice, viral barcode diversity was high in extraintestinal tissues, indicating rapid dissemination of a relatively diverse population (Fig. 9B). To determine whether the rapid systemic dissemination was specific to CVB3, we quantified dissemination of a related enteric virus, poliovirus. Poliovirus did not disseminate to extraintestinal tissues at the same frequency as CVB3 by 20 min postinfection in Ifnar−/− mice expressing the poliovirus receptor (PVR) (Fig. 9C and D), indicating that rapid viral spread is not shared among all enteroviruses. To test if rapid dissemination differed for nonviral molecules in Ifnar−/− and Ifnar+/+ mice, we orally administered 35S-labeled cysteine and methionine and examined radioactive counts in a variety of tissues. We detected radioactivity in extraintestinal tissues at 20 min postadministration, and the levels were equivalent in Ifnar−/− and Ifnar+/+ mice. Taken together, these data indicate that CVB3 and other molecules can disseminate systemically very soon after oral delivery in Ifnar−/− mice.
FIG 9.

Orally inoculated CVB3 rapidly spreads to extraintestinal tissues in mice. (A) Ifnar+/+ and Ifnar−/− mice were orally inoculated with 1 × 109 PFU CVB3, and tissues were collected at 20 min postinfection (mpi). Virus was quantified in homogenized tissues by plaque assay (n = 4). (B) Dissemination of CVB3 barcode collection at 20 mpi (n = 5). The horizontal dashed line represents the limit of detection for the plaque assay, and bars with slanted lines represent results below the sequencing threshold. (C) PVR Ifnar−/− mice were orally inoculated with 1 × 109 PFU poliovirus, and tissues were collected at 20 mpi. Virus was quantified in homogenized tissues by plaque assay (n = 13). (D) Comparison of CVB3 and PV extraintestinal tissue titers represented in panels A and C. (E) Dissemination of 35S radiolabeled cysteine and methionine at 20 min after oral feeding.
DISCUSSION
In this work, we engineered a barcoded collection of CVB3 clones to monitor viral populations across time and space in orally infected mice. Consistent with our previous observation that the GI tract limits poliovirus diversity (7), we detected lower viral diversity in extraintestinal tissues than in the GI tract (Fig. 2). We also observed monopolization of viral populations by a small viral subpopulation, and this viral subpopulation was shared among virtually all tissues. In our earlier work with poliovirus, tissues were harvested upon disease onset—generally at 72 hpi or later. We observed low poliovirus population diversity at those late time points, and we concluded that this loss of diversity was due to combined bottlenecks interfering with passage from mouth to gut and from gut to blood (7). However, here we found that replication of diverse CVB3 populations in the GI tract preceded the loss of diversity (Fig. 7 and 8). Thus, loss of CVB3 diversity was not due only to a small subset of viruses from the inoculum replicating in vivo. Strikingly, nearly all tissues in each mouse were monopolized by a small subpopulation of viral barcodes (Fig. 2 and 7). These results suggest either that (i) all viral populations derived from 1 or 2 infectious events or that (ii) viral populations in separate tissues disseminated and intermingled and that the viral population in a given site could be subsumed by an invading viral population. While we focused on Ifnar−/− mice in this study, in the future we aim to explore the nature of viral population dynamics in wild-type mice as well as other immunodeficient mouse strains.
Due to the propensity for RNA viruses to rapidly develop diverse populations, it was possible the monopolized populations that we had observed were the consequence of mutations that were selected for and enriched. Indeed, others have reported selection for poliovirus mutations uniquely adapted for specific tissues (20). However, we found that nearly all viral populations within a mouse had been overwhelmed by the same small viral subpopulation by 48 hpi. In that time frame, we did not find detectable mutations within the whole-genome consensus sequences that could explain the adaptive advantage of this subpopulation (Fig. 4). In the absence of major adaptive mutations, key future objectives are to determine (i) how viral subpopulations dominate within a tissue and whole animal, (ii) the anatomic source of monopolizing viral population, and (iii) whether clearance enables monopolization.
In order to delineate the population dynamics of an enteric virus over time, we set out to measure kinetics of CVB3 dissemination after oral administration of virus. We found that virus could reach extraintestinal tissues as soon as 20 min postinfection in Ifnar−/− mice, but not in Ifnar+/+ mice (Fig. 9). Previous work has shown that interferon gamma can influence GI barrier permeability, but roles for interferon alpha/beta in rapid viral dissemination have not been described (21). Furthermore, poliovirus did not disseminate to extraintestinal sites by 20 min postinfection, indicating a unique rapid dissemination mechanism for CVB3 that is not shared for all enteroviruses. What mechanisms could explain this rapid dissemination? Passage either across the intestinal epithelia or via M cells can lead to viral access to the lymphatic system and then to the portal vein and subsequently the bloodstream, or other mechanisms may contribute. Because we found that radioactive amino acids were also present in extraintestinal sites at 20 min postfeeding (Fig. 9E), the early dissemination mechanism may not be restricted to viruses. Future experiments will explore the physiological routes of rapid enterovirus dissemination and the contribution of type 1 interferon signaling to GI permeability.
In conclusion, we found that diverse viral populations disseminate and replicate in GI and extraintestinal sites but that viral population diversity is severely restricted postreplication. These results support a model of early broad dissemination and subsequent clearance for an enteric virus.
MATERIALS AND METHODS
Cells and media.
293T and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with either 10% normal calf serum (HeLa) or fetal calf serum (293T), 2 mM l-glutamine, and 10 mM HEPES. Cells were maintained at 37°C and 5% CO2. All transfections with viral plasmids were performed with Lipofectamine 2000 (Thermo Fisher). Our HeLa cells (originally from Karla Kirkegaard’s laboratory) are highly permissive for replication of enteroviruses compared with other HeLa cell lines, including the ATCC line.
Viruses.
In this work, we used the CVB3 H3 strain (Woodruff U57056.1, with 16-nucleotide [nt] differences of unknown origin) and poliovirus Mahoney serotype 1. All poliovirus work was done under WHO-approved elevated biosafety level 2 (BSL2+)/poliovirus conditions. Stocks were generated by transfecting 293T cells with (i) plasmids containing viral genomes under T7 promoter and (ii) plasmid expressing T7 polymerase. For the CVB3 barcode clones, each individual stock was propagated separately to control the ratio of each virus in the final mixture. After 2.5 days, cells were subjected to freezing-thawing 3 times, and clarified supernatant was added to HeLa cells. Cells were incubated until cytopathic effects were evident and were then subjected to freezing-thawing 3 times. These initial viral stocks were quantified by plaque assay and then amplified in HeLa cells at multiplicity of infection (MOI) of >10. When cytopathic effects were observed, cells were collected by scraping, subjected to freezing-thawing 3 times, and resuspended in a small volume of phosphate-buffered saline (PBS) containing 0.1 mg/ml MgCl2 and CaCl2. Virus within clarified supernatant was quantified and used for experiments.
Plaque assay.
Samples were serially diluted, and 200 μl was plated onto HeLa cells in 6-well plates (70% confluence). Cells were incubated at room temperature for >20 min, media with 1% agar were added to wells, and the reaction mixtures were incubated at 37°C for 2 to 3 days. Wells were stained with 1% crystal violet–20% ethanol and rinsed with water, and plaques were counted.
Neutral red/light-sensitive viruses.
Virus stocks were amplified as described above except that the amplification was performed in the presence of 50 μg/ml neutral red (7). Neutral red-labeled viruses were handled in the presence of a red light to maintain infectivity. To quantify percent light-sensitive (inoculum/unreplicated) virus, an aliquot of virus was exposed to white light from a desk lamp for 20 min. For tissue homogenates, samples were exposed to light for 1 h with occasional mixing.
Virus growth curve.
HeLa cells were infected in suspension at an MOI of 10 for 30 min on ice and washed 3 times, and 50,000 cells were plated per well in a 96-well plate. At the indicated time points, plates were frozen. Three replicates were performed for each condition at each time point, and the experiment was repeated 3 times. For the growth curve analyses, virus stock from a single representative barcoded virus clone was used.
Construction of virus barcode library.
To insert the 9-nt barcode, primers (IDT) were designed (NEBaseChanger) with random overhangs on the 5′ end to be inserted between nucleotides 708 and 709 of the CVB3 genome. PCR was performed using Q5 high-fidelity DNA polymerase (NEB; M0491L). The PCR product was treated using KLD enzyme mix (NEB; M0554S), and transformed into competent bacteria (Zymo; T3007). Individual colonies were screened for the presence of the 9-nt barcode by colony PCR and then verified by Sanger sequencing. Individual stocks of virus were prepared as described above, mixed at equal PFU levels, and then concentrated by ultracentrifugation (66,549 × g overnight through the use of 10 ml 30% sucrose). The characteristics of barcode collection were as follows. At a distance of 1 nt (1 deletion or 1 mutation), a few barcodes had “near neighbors.” For example, 18 barcodes had at least one other library member barcode within a distance of one nucleotide and 4 had at least two other library member barcodes within one nucleotide. Thus, 13% of the entire library was within 1 nt of other barcodes. Additionally, 13 barcodes contained an ATG start codon, and 5 of those were in a different frame from the downstream canonical start codon. However, all of them were well represented in the virus stock.
Mice.
All mice experiments were approved by the University of Texas Southwestern (UTSW) Institutional Animal Care and Use Committee. C57BL/6J, C57BL/6-Ifnar−/−, C57BL/6-PVR (transgenic for the human poliovirus receptor), and C57BL/6-PVR-Ifnar−/− (16) mouse strains were maintained using standard animal husbandry practice in UTSW animal facilities. Mice were orally infected with a total of 1 × 109 PFU of the 135-virus mixture (7.4 × 106 PFU of each individual stock). All mice were infected or given radiolabel mix in 25 μl by pipetting the volume into the mouse’s mouth. For radiolabeled amino acid dissemination, we diluted Express protein labeling mix, 35S (Perkin Elmer, neg072007mc) and administered 131,000 to 246,000 cpm/mouse. Tissues were collected and homogenized as described below, and the radioactivity in homogenates was measured on a scintillation counter.
Dissections and tissue processing.
Tissues were collected and snap-frozen on liquid nitrogen. After weighing, beads (Fisher Scientific, NC9862662) and PBS with 0.1 mg/ml MgCl2 and CaCl2 were added and were homogenized (Next Advance Bullet Blender Storm). Samples were divided into portions for RNA isolation and viral quantification. Fractions destined for plaque assay were subjected to freezing-thawing 3 times and spun at 16,000 × g, and virus was quantified within the supernatant. RNA was isolated from intestinal contents and stool using a viral RNA extraction kit (Zymo, R1035) and from tissues using TRI LS (Sigma-Aldrich, T3934) per the manufacturer’s protocol.
Sequencing and analysis of barcode library.
cDNA was synthesized using primers and Superscript II enzyme (Fisher Scientific, 18064071). Libraries were produced by amplifying cDNA using primers targeting the barcoded viral locus in PCR. Primers contained Illumina adaptor sequences, Illumina indices within reverse primers for demultiplexing by the sequencing core, and variable-length custom indices within forward primers to increase multiplexing potential and to phase sequencing reactions. Samples were gel purified and pooled by amplicon band intensity. These pools were combined together, normalizing by nucleic acid concentration. Sequencing was performed on a NextSeq Illumina sequencer with 75 cycles at the UTSW McDermott Sequencing Core. FASTQ files were demultiplexed using the reverse index and then demultiplexed using the forward index. Barcodes were extracted from the reads plus the surrounding 30 nucleotides of viral genomic sequence to ensure that the barcodes were associated with virus sequence. Barcode counts were enumerated with the “grep” command line function, with each barcode sequence serving as a search string. Pilot studies indicated a sequencing depth of 1,000 reads/sample or more was sufficient to recapitulate barcode ratios; we set this as the threshold. Because of the variations in sequencing depth, we set 0.0005 viral barcode reads/total reads/sample as the cutoff to count a barcode as present. Barcode frequencies are presented as fractions of reads for a specific barcode over total reads containing a viral barcode. To test if barcodes became truncated during generation of our viral stocks, we used Illumina sequencing to analyze all of the sequences in the barcode locus rather than only the known barcode sequences. We found a single truncation at very low abundance; that was the viral sequence used for cloning and was not the wild-type sequence. Thus, truncation of barcodes was not prevalent during growth in HeLa cells.
Graphing and statistics.
All graphs and statistics were produced with GraphPad Prism. Shannon diversity index data were calculated using the R package “vegan” (22). Figures were prepared using Inkscape.
ACKNOWLEDGMENTS
We thank members of the Pfeiffer laboratory for helpful discussions.
This work was supported by a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases award (J.K.P.) and by R01 AI74668 (J.K.P.). J.K.P. is a Howard Hughes Medical Institute Faculty Scholar. M.R.L. was supported in part by NIH grant T32 GM109776. B.T.M. was supported by NIH grants T32 AI007520 and F32 AI138392.
REFERENCES
- 1.Pfeiffer JK, Kirkegaard K. 2005. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog 1:e11. doi: 10.1371/journal.ppat.0010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. 2006. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCune BT, Lanahan MR, tenOever BR, Pfeiffer JK. 2019. Rapid dissemination and monopolization of viral populations in mice revealed using a panel of barcoded viruses. bioRxiv https://www.biorxiv.org/content/10.1101/769976v1.article-info. [DOI] [PMC free article] [PubMed]
- 4.Varble A, Albrecht RA, Backes S, Crumiller M, Bouvier NM, Sachs D, García-Sastre A, TenOever BR. 2014. Influenza A virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe 16:691–700. doi: 10.1016/j.chom.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfeiffer JK, Kirkegaard K. 2006. Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain. Proc Natl Acad Sci U S A 103:5520–5525. doi: 10.1073/pnas.0600834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erickson AK, Pfeiffer JK. 2013. Dynamic viral dissemination in mice infected with yellow fever virus strain 17D. J Virol 87:12392–12397. doi: 10.1128/JVI.02149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuss SK, Etheredge CA, Pfeiffer JK. 2008. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog 4:e1000082. doi: 10.1371/journal.ppat.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luethy LN, Erickson AK, Jesudhasan PR, Ikizler M, Dermody TS, Pfeiffer JK. 2016. Comparison of three neurotropic viruses reveals differences in viral dissemination to the central nervous system. Virology 487:1–10. doi: 10.1016/j.virol.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zwart MP, Elena SF. 2015. Matters of size: genetic bottlenecks in virus infection and their potential impact on evolution. Annu Rev Virol 2:161–179. doi: 10.1146/annurev-virology-100114-055135. [DOI] [PubMed] [Google Scholar]
- 10.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping L-H, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A 105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bull RA, Luciani F, McElroy K, Gaudieri S, Pham ST, Chopra A, Cameron B, Maher L, Dore GJ, White PA, Lloyd AR. 2011. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog 7:e1002243. doi: 10.1371/journal.ppat.1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weger-Lucarelli J, Garcia SM, Rückert C, Byas A, O'Connor SL, Aliota MT, Friedrich TC, O'Connor DH, Ebel GD. 2018. Using barcoded Zika virus to assess virus population structure in vitro and in Aedes aegypti mosquitoes. Virology 521:138–148. doi: 10.1016/j.virol.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aliota MT, Dudley DM, Newman CM, Weger-Lucarelli J, Stewart LM, Koenig MR, Breitbach ME, Weiler AM, Semler MR, Barry GL, Zarbock KR, Haj AK, Moriarty RV, Mohns MS, Mohr EL, Venturi V, Schultz-Darken N, Peterson E, Newton W, Schotzko ML, Simmons HA, Mejia A, Hayes JM, Capuano S, Davenport MP, Friedrich TC, Ebel GD, O'Connor SL, O'Connor DH. 2018. Molecularly barcoded Zika virus libraries to probe in vivo evolutionary dynamics. PLoS Pathog 14:e1006964. doi: 10.1371/journal.ppat.1006964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCrone JT, Woods RJ, Martin ET, Malosh RE, Monto AS, Lauring AS. 3 May 2018, posting date Stochastic processes constrain the within and between host evolution of influenza virus. Elife 7:1–19. doi: 10.7554/eLife.35962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson CM, Wang Y, Pfeiffer JK. 2017. Sex-dependent intestinal replication of an enteric virus. J Virol 91:e02101-16. doi: 10.1128/JVI.02101-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohka S, Igarashi H, Nagata N, Sakai M, Koike S, Nochi T, Kiyono H, Nomoto A. 2007. Establishment of a poliovirus oral infection system in human poliovirus receptor-expressing transgenic mice that are deficient in alpha/beta interferon receptor. J Virol 81:7902–7912. doi: 10.1128/JVI.02675-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, Yoneyama M, Fujita T, Taya C, Yonekawa H, Koike S. 2005. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol 79:4460–4469. doi: 10.1128/JVI.79.7.4460-4469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Pfeiffer JK. 2016. Emergence of a large-plaque variant in mice infected with coxsackievirus B3. mBio 7:e00119. doi: 10.1128/mBio.00119-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lancaster KZ, Pfeiffer JK. 2010. Limited trafficking of a neurotropic virus through inefficient retrograde axonal transport and the type I interferon response. PLoS Pathog 6:e1000791. doi: 10.1371/journal.ppat.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao Y, Dolan PT, Goldstein EF, Li M, Farkov M, Brodsky L, Andino R. 2017. Poliovirus intrahost evolution is required to overcome tissue-specific innate immune responses. Nat Commun 8:375. doi: 10.1038/s41467-017-00354-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beaurepaire C, Smyth D, McKay DM. 2009. Interferon-gamma regulation of intestinal epithelial permeability. J Interferon Cytokine Res 29:133–144. doi: 10.1089/jir.2008.0057. [DOI] [PubMed] [Google Scholar]
- 22.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H. 2018. vegan: Community Ecology Package. R Package Version 2.5-3. https://CRAN.R-project.org/package=vegan.