

HSV-1 ocular infections are the leading cause of corneal blindness. Eye disease is the result of a prolonged immune response to the replicating virus. HSV-1, on the other hand, has evolved several mechanisms to evade clearance by the host immune system. We describe a novel mechanism of HSV-1 immune evasion via ICP22-dependent downregulation of the host T cell costimulatory molecule CD80. However, the exact role of CD80 in HSV-1 immune pathology is not clear. In this study, we show that eye disease is independent of the level of HSV-1 replication and that viral expression of CD80 has a detrimental role in corneal scarring, likely by increasing CD8+ T cell recruitment and activation.
KEYWORDS: cornea, virus replication, viral transcripts, transcriptome, latency reactivation, ocular
ABSTRACT
We recently reported that herpes simplex virus 1 (HSV-1) infection suppresses CD80 but not CD86 expression in vitro and in vivo. This suppression required the HSV-1 ICP22 gene. We also reported that overexpression of CD80 by HSV-1 exacerbated corneal scarring in BALB/c mice. We now show that this recombinant virus (HSV-CD80) expressed high levels of CD80 both in vitro in cultured rabbit skin cells and in vivo in infected mouse corneas. CD80 protein was detected on the surface of infected cells. The virulence of the recombinant HSV-CD80 virus was similar to that of the parental strain, and the replication of HSV-CD80 was similar to that of control virus in vitro and in vivo. Transcriptome analysis detected 75 known HSV-1 genes in the corneas of mice infected with HSV-CD80 or parental virus on day 4 postinfection. Except for significantly higher CD80 expression in HSV-CD80-infected mice, levels of HSV-1 gene expression were similar in corneas from HSV-CD80-infected and parental virus-infected mice. The number of CD8+ T cells was higher, and the number of CD4+ T cells was lower, in the corneas of HSV-CD80-infected mice than in mice infected with parental virus. HSV-CD80-infected mice displayed a transient increase in dendritic cells. Transcriptome analysis revealed mild differences in dendritic cell maturation and interleukin-1 signaling pathways and increased expression of interferon-induced protein with tetratricopeptide repeats 2 (Ifit2). Together, these results suggest that increased CD80 levels promote increased CD8+ T cells, leading to exacerbated eye disease in HSV-1-infected mice.
IMPORTANCE HSV-1 ocular infections are the leading cause of corneal blindness. Eye disease is the result of a prolonged immune response to the replicating virus. HSV-1, on the other hand, has evolved several mechanisms to evade clearance by the host immune system. We describe a novel mechanism of HSV-1 immune evasion via ICP22-dependent downregulation of the host T cell costimulatory molecule CD80. However, the exact role of CD80 in HSV-1 immune pathology is not clear. In this study, we show that eye disease is independent of the level of HSV-1 replication and that viral expression of CD80 has a detrimental role in corneal scarring, likely by increasing CD8+ T cell recruitment and activation.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) ocular infections are the most common cause of corneal blindness in developed countries (1, 2). An estimated 70 to 90% of the population is HSV-1 seropositive. HSV-1 ocular infection causes a robust immune response, resulting in clearance from the site of initial infection. However, HSV-1 establishes lifelong latency in the trigeminal ganglia (TG), from which it can periodically reactivate (3–6). Recurrent infections and the prolonged inflammatory response after viral clearance both contribute to corneal scarring (7). Although antiviral medication and topical corticosteroid therapies can effectively shorten the duration of infection and dampen the inflammatory response, corticosteroids have side effects (8, 9). Further, because no current medications can prevent HSV-1 ocular recurrence, it is essential to understand the mechanisms that lead to HSV-1 ocular pathology so that effective therapies can be developed.
T cell activation is crucial for clearance of the initial infection (10–12). HSV-1 is recognized by antigen-presenting cells, such as dendritic cells (DCs), natural killer cells, and macrophages (13–15), which induce the secretion of interferons (IFNs) and cytokines and the activation of CD4+ and CD8+ T cells (15). This T cell activation is tightly controlled and requires at least two signals (16). The first signal, antigen bound to the major histocompatibility complex class II receptor on the surface of an antigen-presenting cell (APC), is recognized by T cell receptors (17–19). The second signal involves the binding of CD28, CTLA-4, or PD-1 on the surfaces of T cells to either of the APC costimulatory molecules, CD80 (B7-1) or CD86 (B7-2) (20, 21). T cell activation can also be modulated by the interaction of CD80 with programmed death ligand 1 (PD-L1), which inhibits T cell proliferation and cytokine production (22). T cell activation results in rapid cell proliferation, differentiation into different T cell subtypes, and cytokine production (15).
HSV-1 has evolved several mechanisms to evade clearance by the immune system, which allows it to establish a lifelong latency. For example, the HSV-1 ICP0 gene product inhibits the type I IFN pathway at multiple levels during the lytic cycle by activating NF-κB and increasing the levels of the adaptor protein MyD88 (23–27). Further, the HSV-1 latency-associated transcript (LAT) inhibits the type I IFN pathway during latency, which likely contributes to LAT antiapoptotic activity and efficient establishment of latency and reactivation (28–31).
We recently reported an additional mechanism of HSV-1 immune evasion by downregulating the host costimulatory molecule CD80 in an ICP22-dependent manner (32). We have also demonstrated that overexpression of CD80 leads to productive infection in normally nonpermissive DCs in vitro (33) and to more severe eye disease in mice (32). Consistent with these findings, ocular infection of mice with HSV-1 lacking the ICP22 gene produced similar levels of eye disease and angiogenesis, as did infection with wild-type virus, despite the fact that ICP22-null virus replicates less efficiently and has lower latency and reactivation than does wild-type virus (34). These results support the hypothesis that the host immune response, not viral replication per se, is responsible for eye disease.
In this study, we ocularly infected mice with a recombinant virus expressing CD80 under the LAT promoter and found that while overexpression of CD80 had no effect on CD80 cell surface expression or viral replication, HSV-CD80 virus establishes latency more efficiently and reactivates with faster kinetics than its parental virus in vivo. Using fluorescence-activated cell sorting (FACS) analysis and host transcriptome analysis by RNA sequencing, we demonstrate that overexpression of CD80 by HSV-CD80 leads to increased recruitment and activation of CD8+ T cells. These results support our previous findings and the conclusion that corneal scarring is a consequence of host immune response, not viral replication, because infection with HSV-CD80 resulted in more severe scarring (32), despite its replication being similar to that of the parental virus. Thus, we have extended our previous work and shown that overexpression of CD80 has a pathogenic effect during HSV-1 ocular infection.
RESULTS
CD80 is expressed on the surfaces of RS cells infected with HSV-CD80.
To determine whether CD80 expression driven by the HSV-1 LAT promoter in the LAT–/– mutant is expressed on the surfaces of infected cells, we infected rabbit skin (RS) cells with 0.1, 1.0, or 10 PFU of HSV-CD80 or 10 PFU of the parental dLAT2903 virus as described in Materials and Methods. CD80 expression in infected cells was visualized using immunofluorescence confocal microscopy. Cell surface expression of HSV-1 gC was used as a control. CD80 expression was found on the surfaces of cells infected with HSV-CD80, but not on mock-infected cells or on cells infected with parental virus (Fig. 1A). As expected, CD80 expression increased in a viral dose-dependent manner. Parallel infected cells stained with anti-HSV-1 gC antibody showed cell surface expression of gC in both HSV-CD80- and parental virus-infected cells (Fig. 1B). The expression of gC increased in a dose-dependent manner, as expected. Further, increased gC expression correlated with increased CD80 expression, as expected.
FIG 1.

Expression of CD80 on the cell surface of RS cells infected with HSV-CD80. RS cells were either mock infected or infected with 0.1, 1, or 10 PFU/cell of HSV-CD80 or parental virus. At 16 h p.i., the cells were stained with antibodies against CD80 (A) or gC (B) and examined for fluorescence. (C) RS cell monolayers were infected with 1 PFU/cell of recombinant HSV-CD80 or parental virus or were mock infected for 24 h. Infected cells were harvested and stained with anti-CD8 and anti-gC antibodies and then analyzed by flow cytometry.
We also analyzed cells infected with either HSV-CD80 or parental virus or mock infected for the expression of CD80 and gC by FACS (Fig. 1C). Six percent of cells infected with HSV-CD80 stained positive for CD80 but not for gC, similar to what occurred with parental virus- and mock-infected cells (1 and 4%, respectively). A higher percentage of HSV-CD80-infected cells coexpressed gC and CD80 than did parental virus-infected or mock-infected cells (27, 1, and 0%, respectively). This difference is likely due to the two extra copies of CD80 expressed from HSV-1 genome. Together, these results suggest that infection of RS cells with HSV-CD80 results in cell surface CD80 expression. Further, this CD80 is largely expressed from the viral gene.
CD80 expression by HSV-CD80 virus does not alter virus replication in mouse eyes.
We have previously shown that the kinetics of HSV-CD80 replication in RS cells is similar to that of parental virus (33). To determine whether HSV-CD80 virus replication is similar to that of parental virus in vivo, we infected mouse eyes with 105 PFU/eye of HSV-CD80 or parental virus and collected corneas on days 3 and 5 postinfection (p.i.). The total RNA was isolated as described in Materials and Methods. We then measured viral glycoprotein B (gB) expression as an indicator of viral replication using quantitative reverse transcription-PCR (qRT-PCR) and found that levels of gB expression did not differ significantly in HSV-CD80- and parental virus-infected corneas on day 3 or 5 p.i. (Fig. 2A, P = 0.4 or P = 0.7).
FIG 2.
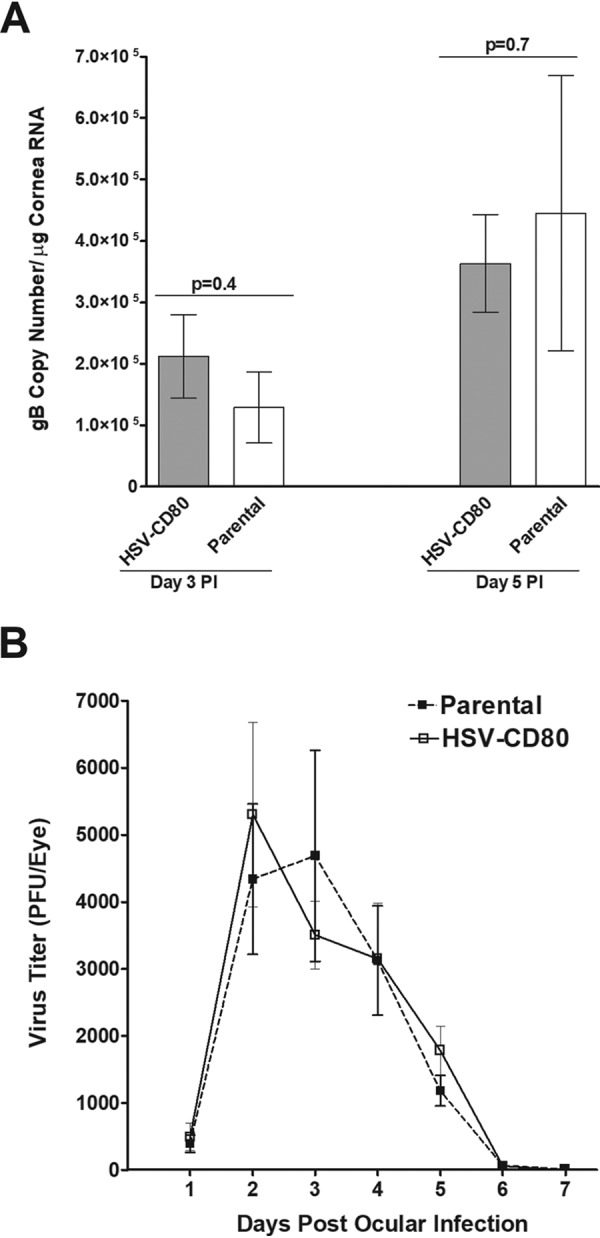
Levels of replication of HSV-CD80 virus and parental virus in mouse corneas are indistinguishable. (A) Corneas of female BALB/c mice were ocularly infected with 105 PFU/eye HSV-CD80 or parental virus and harvested on days 3 and 5 p.i. The gB copy number was determined by qPCR. No differences in gB copy number were seen between the two groups (P = 0.4 and P = 0.7 [Fisher exact test] on days 3 and 5 p.i.). (B) Virus titers were determined from tears of mice infected with either HSV-CD80 or parental virus on days 1 to 7 p.i. Viral titers peaked around days 2 to 3 p.i., and virus was cleared from tears by day 7 p.i. No significant differences were seen in titers from mice infected with HSV-CD80 or parental virus (P > 0.05 [Fisher exact test]). Error bars represent the SEM.
To determine whether overexpression of CD80 affects the amount of viral shedding, we measured virus titers from the tears of mice infected with HSV-CD80 or parental virus from days 1 to 7 after ocular infection (Fig. 2B). Viral titers in both HSV-CD80- and parental virus-infected mice peaked at days 2 and 3 p.i., declined thereafter, and were cleared by day 7 p.i. (Fig. 2B). No significant differences in viral titers were seen between mice infected with either HSV-CD80 or parental virus throughout the time course. These results show that, similarly to gB expression (Fig. 2A), viral replication and shedding in tears are not affected by CD80 expression.
We next determined infectious viral load in the corneas, TG, and brains of infected mice by infecting mice as described above and collecting these tissues on days 3, 5, and 7 p.i. Viral titers from the supernatants of homogenized cornea, TG, and brains were determined using standard plaque assay as described in Materials and Methods. Viral titers in the corneas of mice infected with HSV-CD80 or parental virus did not differ on days 3 and 5 p.i. (Fig. 3A, P > 0.05). However, on day 7 p.i., the viral titers were significantly higher in the corneas of mice infected with parental virus (Fig. 3A, P < 0.01). Mice infected with HSV-CD80 had higher viral titers in TG on day 3 p.i. than did those infected with parental virus (Fig. 3B, P < 0.001), but no statistically significant differences between these groups were seen on day 5 or 7 p.i. (Fig. 3B, P > 0.05). Viral titers in the brains of mice infected with either virus (Fig. 3C, P > 0.05) were also not statistically different. These results suggest that HSV-CD80 may be cleared from the cornea faster or enter the TG via faster kinetics than the parental virus.
FIG 3.
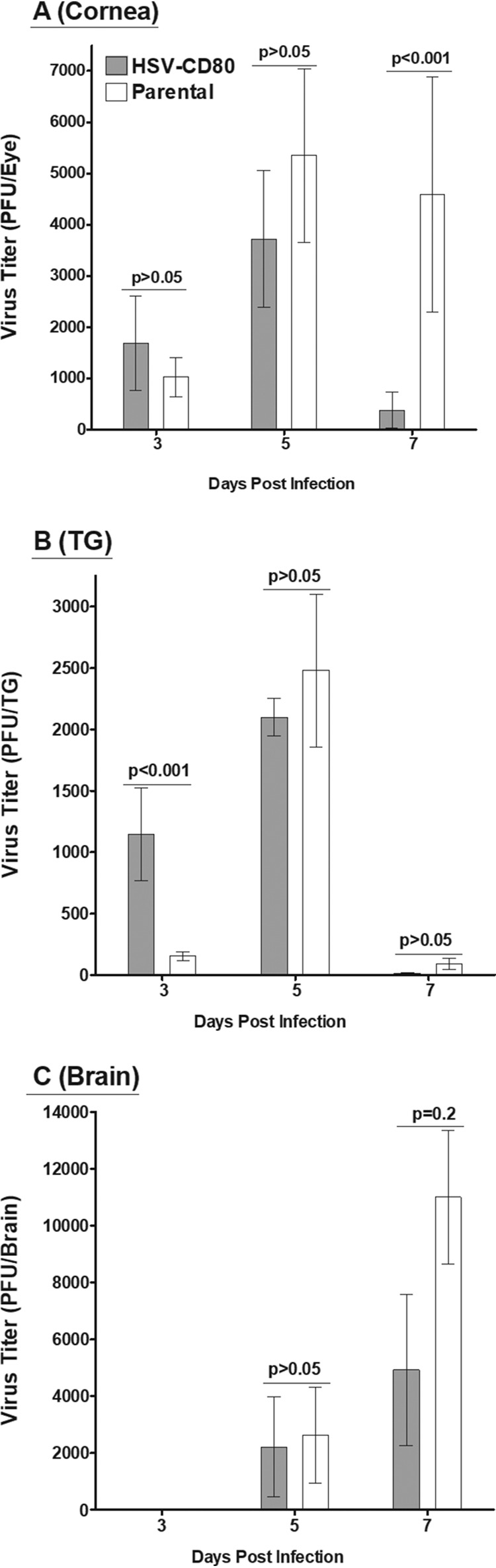
Viral expression of CD80 results in faster viral clearance from the cornea. Mice ocularly infected with either HSV-CD80 or parental virus as described in Fig. 2 were euthanized on days 3, 5, and 7 p.i., and viral titers were determined in the corneas (A), TG (B), and brains (C) using standard plaque assays. Error bars represent the SEM, and results were considered significant for P < 0.05 (Fisher exact test).
CD80 is expressed on the surfaces of corneas and TG of mice infected with HSV-CD80.
Our in vitro studies described above (Fig. 1) show CD80 expression in infected RS cells. To determine whether HSV-CD80 infection also results in CD80 expression in vivo, we used FACS analysis to measure CD80 expression in corneas and TG of mice infected as described above with HSV-CD80 or parental virus. The expression of the HSV-1 gC was used as an indicator of infection. The percentage of gC-positive cells increased from days 3 to 5 p.i. in mouse corneas (from 1.8 to 33.4% in HSV-CD80-infected corneas and from 1.3 to 33.4% in parental virus-infected corneas) and mouse TG (from 2.4 to 25.1% in HSV-CD80-infected TG and from 0.9 to 15.4% in parental virus-infected TG (Fig. 4). The percentage of gC-positive cells was slightly higher on day 3 p.i. in the corneas and TG of mice infected with HSV-CD80 than in parental virus-infected mice (Fig. 4, 1.8% versus 1.3% and 2.4% versus 0.9%, respectively). The proportion of gC-positive cells remained slightly higher in the TG of mice infected with HSV-CD80 than in the parental virus-infected group (Fig. 4, 25.1% versus 15.4%, respectively). However, the percentages of infected cells on day 5 did not differ between mice infected with HSV-CD80 and parental virus-infected corneas (Fig. 4, 33.4 and 33.4%). These results correlate with the viral shedding data in Fig. 2B and support that viral expression of CD80 does not impair infectivity or viral replication in vivo. Further, CD80 expression was higher in corneas of mice infected with HSV-CD80 than in parental virus-infected mice (Fig. 4, 2 and 1.6% on day 3 p.i. and 0.4 and 0.1% on day 5 p.i.). CD80 expression was also higher in HSV-CD80-infected mouse TG than in those of parental virus-infected mice on day 3 (Fig. 4, 0.7% versus 0.2%) but not on day 5 p.i. (Fig. 4, 0.2% versus 0.2%), suggesting that CD80 is expressed in mouse corneas and TG infected with HSV-CD80 in vivo.
FIG 4.

CD80 and HSV-1 gC are expressed on cells of the cornea and TG in mice infected with HSV-CD80. Mice were ocularly infected as described in Fig. 2. Corneas and TG from infected mice were harvested on days 3 and 5 p.i. Corneas or TG from each mouse were combined, and single cell suspensions were stained with anti-CD80 and anti-gC antibodies and analyzed by flow cytometry. The percentages of CD80+, gC+, and CD80+gC+ cells on days 3 and 5 p.i. in the corneas and TG of HSV-CD80- and parental virus-infected mice are shown. Experiments were repeated three times.
HSV-CD80 infection produces increased CD8+ T cells and decreased CD4+ T cells in corneas of infected mice.
Previous results from our lab suggested that viral expression of CD80 resulted in increased severity of eye disease (32). However, it is not clear how HSV-CD80 causes increased pathology. CD80 expression is associated with increased CD8+ T cell production (35), which is beneficial in clearing viral infections, and we previously reported that exacerbation of corneal scarring in HSV-1 gK-immunized mice was associated with increased CD8+ T cells in infected mouse corneas (36–39). To determine the effect of viral CD80 expression on different T cell subtypes and investigate a possible mechanism for increased eye disease, we measured CD4+ and CD8+ T cell numbers by FACS analysis. The percentage of CD4+ T cells in HSV-CD80-infected corneas was similar to that in parental virus-infected corneas on days 3 and 5 p.i. (Fig. 5A, day 3, 1.5% versus 1.4%; day 5, 0.1% versus 0.3%). The percentages of CD8+ T cells were similar in HSV-CD80- and parental virus-infected corneas on day 3 p.i. (Fig. 5A, 48% versus 54%) but higher on day 5 p.i. (73% versus 51%). The experiment was repeated twice more as described above, with the results depicted in Fig. 5B. On days 3 and 5 p.i., HSV-CD80 infection resulted in a moderate but statistically significant increase in CD8+ T cells compared to the level in mice infected with parental virus (Fig. 5B, P = 0.04 and P < 0.05). The numbers of CD4+ T cells did not differ between HSV-CD80- and parental virus-infected corneas on day 3 p.i. (Fig. 5B, P = 0.8), but on day 5 p.i., the HSV-CD80-infected corneas had significantly fewer CD4+ T cells than did the parental virus-infected corneas (Fig. 5B, P = 0.03). Thus, increased CD8+ T cell numbers correlated with increased CD80 expression by HSV-CD80 virus and with increased corneal scarring, as we reported previously (32).
FIG 5.

The corneas of mice infected with HSV-CD80 virus have an increased number of CD8+ T cells. (A) Mice were ocularly infected as described for Fig. 2, and corneas or TG were harvested on days 3 and 5 p.i. The corneas or TG from each mouse were combined, and single cell suspensions were stained with anti-CD3 and anti-CD4 or anti-CD3 and analyzed by flow cytometry. (B) The experiment in panel A was repeated two more times. The graph represents the average results from three independent experiments. Error bars represent the SEM, and P values were calculated using a Fisher exact test and were considered significant for P < 0.05.
Viral expression of CD80 is associated with a transient increase in DCs.
To determine whether CD80 expression affects the recruitment of DCs, mice were infected with HSV-CD80 or parental virus. Corneas from infected mice were isolated on days 3 and 5 p.i., and single cell suspensions were prepared and stained with antibodies against CD11b and CD11c. FACS analysis showed a higher percentage of CD11b+ CD11c+ cells in HSV-CD80 virus-infected corneas than in parental virus-infected corneas (Fig. 6A, 61% versus 43%). By day 5 p.i., the number of CD11b+ CD11c+ cells had decreased in both HSV-CD80-infected and parental virus-infected corneas. The numbers of CD11b+ CD11c+ cells did not differ between the two groups on day 5 p.i., suggesting that virally expressed CD80 induces a transient increase in DCs.
FIG 6.

Overexpression of CD80 is not associated with increased DCs or cell death. The corneas from mice infected with HSV-CD80 or parental virus were harvested on days 3 or 5 p.i. and processed for flow cytometry. (A) Single cell suspensions were stained with CD11b and CD11c. The percentages of CD11b+, CD11c+, and CD11b+ CD11c+ cells are shown. (B) Mice were infected as in panel A, and single cell corneal suspensions were stained with annexin-V and 7-ADD dye. The percentages of nonviable and apoptotic cells are shown.
CD80 expression does not affect cell survival.
Enhanced eye disease and faster viral clearance from the corneas of mice infected with HSV-CD80 could be due to increased cell death. To determine whether the viral expression of CD80 affects cell survival, corneas were isolated from infected mice on days 3 and 5 p.i. Single cell suspensions of infected corneas were prepared, and the numbers of nonviable, apoptotic, and viable cells in the corneas of infected mice were determined by flow cytometry. Although the percentage of apoptotic and nonviable cells increased from day 3 to day 5 p.i. (from 8 and 6% to 12 and 8% in HSV-CD80-infected corneas and from 9 and 6% to 17 and 7% in parental virus-infected corneas) (Fig. 6B), there were no differences in the proportions of nonviable and apoptotic cells between the two groups on either day, indicating that CD80 expression does not have deleterious effects on cell survival. These results also suggest that viral clearance and ocular pathology are not due to increased cell death.
HSV-CD80 ocular infection does not affect the survival of mice.
To determine whether CD80 overexpression plays a role in BALB/c mouse survival, groups of 30 and 20 mice in two separate experiments were ocularly infected with of HSV-CD80 or parental virus at 105 PFU/eye, and infected-mouse survival was monitored for 28 days. The mortality of HSV-CD80-infected mice (17/30, 57% survival) did not differ from the mortality of mice infected with parental virus (13/20, 65% survival) (Fig. 7, P = 0.8 [Fisher exact test]), suggesting that overexpression of CD80 does not affect the survival of infected mice.
FIG 7.
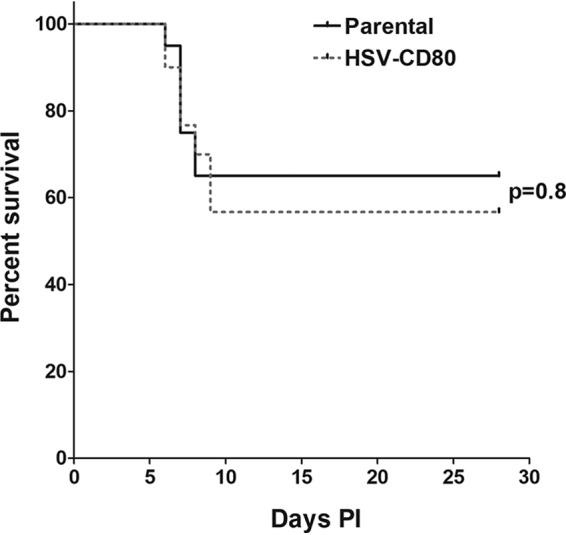
Viral expression of CD80 does not affect mortality. Mice were ocularly infected with HSV-CD80 or parental virus as described in legend for Fig. 2. Mouse survival was monitored during a 28-day period after infection. The graph represents the average results from two independent experiments (P = 0.8 [Fisher exact test]).
CD80 overexpression enhances latency and reactivation in mice.
To determine whether CD80 overexpression affects latency establishment, we measured the HSV-1 gB copy number on day 28 p.i. in the TG of mice latently infected with HSV-CD80 or parental virus. The gB copy number was significantly higher in mice infected with HSV-CD80, than in mice infected with parental virus (Fig. 8A, P = 0.004 [Fisher exact test]).
FIG 8.
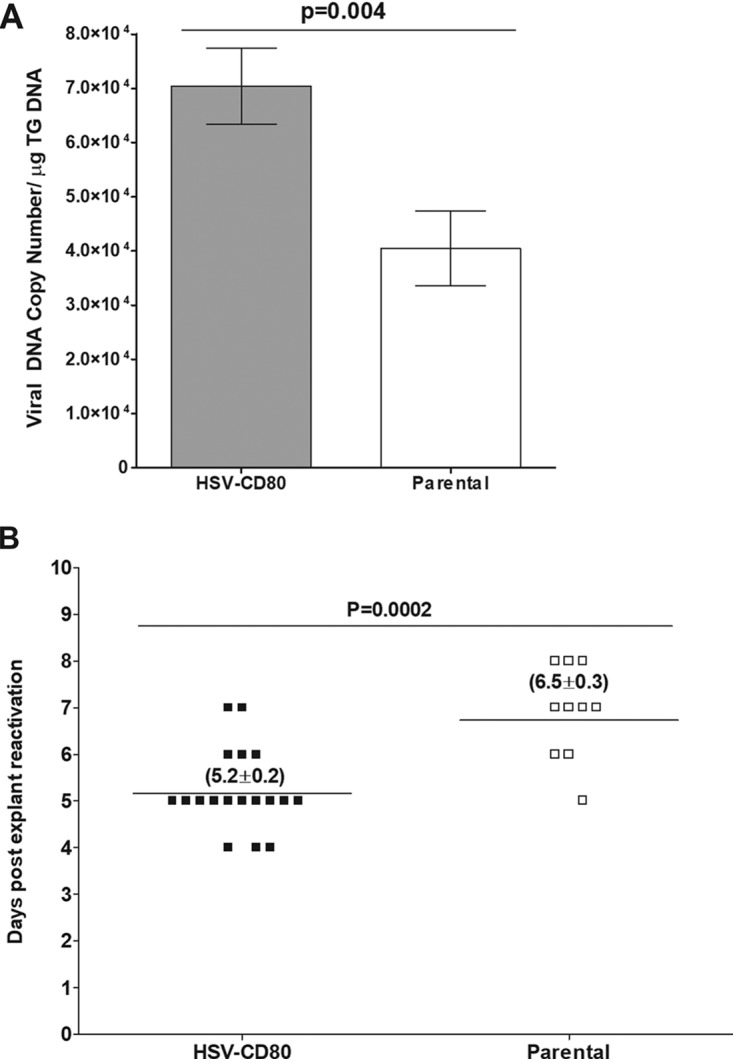
Overexpression of CD80 enhances latency and reactivation. (A) The viral gB DNA copy number in the TG of mice latently infected with HSV-CD80 or parental virus was determined by qPCR. The graph represents an average of 16 TG. Error bars represent the SEM (P = 0.004 [Fisher exact test]). (B) TG from surviving mice latently infected with HSV-CD80 or parental virus were collected at 28 days p.i. and cultured. Aliquots of TG culture supernatant were plated daily on RS cells and monitored for cell death. Each point represents an activation event for a TG. Of HSV-CD80-infected TG, 18/18 reactivated, and 10/10 TG infected with parental virus reactivated. The horizontal line represents the mean time to reactivation. Values in parentheses represent the mean time to reactivation ± the SEM (P = 0.0002 [Fisher exact test]).
Because increased latency correlates with faster time to reactivation (40), we looked at the time to reactivation in HSV-CD80- and parental virus-infected mice. The TG from mice that survived ocular infection were isolated on day 28 p.i. and monitored for the presence of infectious virus by explant reactivation, as described in Materials and Methods. The average time to reactivation for mice infected with parental virus was 6.5 ± 0.3 days, whereas for mice infected with HSV-CD80, the time to reactivation was significantly shorter at 5.2 ± 0.2 days (Fig. 8B, P = 0.0002 [Fisher exact test]). These results suggest that CD80 overexpression enhances latency and reactivation.
CD80 overexpression results in minimal changes in the host transcriptome.
To investigate the effects of CD80 overexpression on the regulation of gene expression in mouse corneas, 15 mice per group were infected with HSV-CD80 or parental virus or were mock infected. On day 5 p.i., corneas from five mice per treatment group were combined, and the total RNA was isolated. cDNA libraries were prepared from each treatment group and sequenced, and a comparison of parental virus-infected to uninfected mice identified 2,069 differentially regulated genes (1,253 upregulated, 816 downregulated, P < 0.05) (Fig. 9A and D; see also Table S1 in the supplemental material). Comparison of HSV-CD80-infected mice to uninfected mice identified 1,262 differentially expressed genes, with 596 upregulated and 666 downregulated genes (Fig. 9B and D; see also Table S2 in the supplemental material, P < 0.05). CD80 expression in mice infected with HSV-CD80 was 2.8-fold higher than in mice infected with parental virus, as predicted based on the two copies of CD80 present in the HSV-CD80 virus. Between HSV-CD80- and parental virus-infected mice, nine host genes (including CD80 expressed by HSV-CD80) showed significant differences in expression (Fig. 9C and Table S3). Of these genes, six were downregulated in HSV-CD80-infected mice (those encoding major intrinsic protein of lens fiber [Mip], proline-rich transmembrane protein 2 [Prrt2], and lactase-like protein [Lctl] and the predicted genes Gm45234, Gm11175, and Gm37988), and three were upregulated (GTPase, very large IFN inducible 1 [Gvin1], and the predicted genes Gm26690 and Gm20479) (Fig. 9C and D). Principal-component analysis results for various samples (parental versus uninfected, HSV-CD80 versus uninfected, and parental versus HSV-CD80) are shown in Fig. 9E and reveal some variability between the samples despite combining corneas from five different animals for each sample.
FIG 9.

Differential gene expression in mice infected with HSV-CD80 and parental virus. Mice were ocularly infected as described above. Corneas were harvested on day 4 p.i., and the total mRNA was isolated and sequenced. Transcriptomes of infected mice were compared to those of uninfected naive BALB/c mice. (A to C) The x axis shows the fold change in gene expression between parental virus-infected and uninfected cells (A), HSV-CD80-infected and uninfected cells (B), and HSV-CD80-infected and parental virus-infected cells (C), and the y axis shows the statistical significance of the differences. Dots represent different genes, with red dots representing genes that are statistically different (P < 0.05 [Wald test]) between the different groups and black dots representing genes that are not statistically significant (P > 0.05 [Wald test]). (D) A heatmap was generated from three replicates of differentially expressed genes (P < 0.05 [analysis of variance]). (E) A principal-component analysis plot was generated based on the top 500 genes by variance across all samples. Red and blue dots represent parental virus-infected and uninfected samples (left panel), HSV-CD80-infected and uninfected samples (middle panel), and parental virus-infected and HSV-CD80-infected samples (right panel), respectively.
Pathway analysis reveals minor differences in DC maturation and IL-1 signaling pathways.
To determine whether CD80 overexpression alters the activation of specific pathways, we used the comparison analysis feature of Ingenuity Pathway Analysis software. Canonical pathways having the largest z scores in mice infected with HSV-CD80 or parental virus included DC maturation, TREM1 signaling, TH1 pathway, PKCΘ, and iNOS signaling (Fig. 10). The calcium-dependent signaling pathway was the top downregulated pathway in both groups. Pathways differentially regulated in mice infected with HSV-CD80 include tumor necrosis factor receptor 1 (TNFR1), TNFR2, and interleukin-1 (IL-1) signaling, which were downregulated compared to their expression in the parental virus-infected group (Fig. 10). In addition, signaling by Rho family GTPases and Rho A were downregulated, whereas Rho GDI signaling was upregulated in HSV-CD80-infected mice compared to parental virus-infected mice (Fig. 10).
FIG 10.

Differential pathway activation in mice infected with HSV-CD80 or parental virus. Differentially regulated genes identified by RNA sequencing results were analyzed using the comparison analysis feature of Ingenuity Pathway Analysis software (Qiagen). Bars represent the z-score, and pathways are ranked according to significance. The top 42 pathways with a P < 0.05 (Wald test) are shown.
To verify the RNA sequencing results, we measured the expression of selected transcripts in corneas of mice infected with HSV-CD80 or parental virus by using qRT-PCR. As expected, CD80 expression was significantly higher in corneas of mice infected with HSV-CD80 than in those infected with parental virus (Fig. 11A, P < 0.001). Although RNA sequencing results suggested significant differences in the levels of expression of Gvin1 and Mip between HSV-CD80- and parental virus-infected mice, these differences did not reach statistical significance using qRT-PCR (Fig. 11, P > 0.05). We also measured the expression of Ifit1 and Ifit2 (Fig. 11A). Although the expression of Ifit2 was significantly higher (P = 0.001) in the corneas of mice infected with HSV-CD80 than in those infected with parental virus, the upregulation of Ifit1 did not reach statistical significance (P = 0.05). Overall, these results suggest that CD80 expression by HSV-CD80 has a mild effect on host gene expression.
FIG 11.
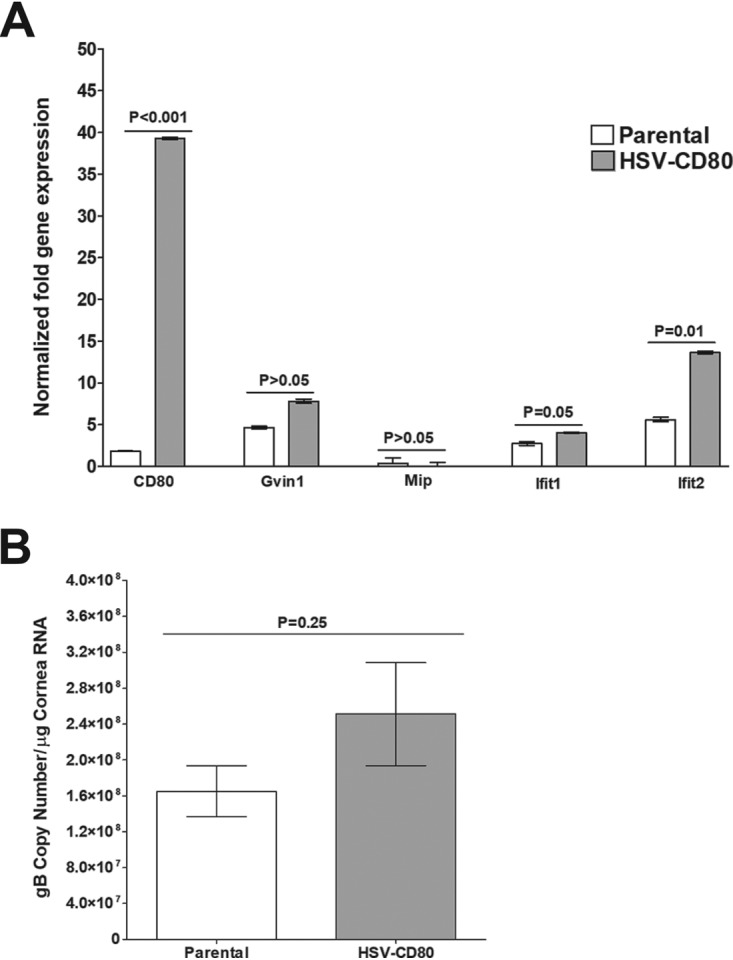
Ifit2 is upregulated in mouse corneas infected with HSV-CD80. RNA was isolated from corneas of uninfected, HSV-CD80-infected, or parental virus-infected corneas. qRT-PCR was performed using total RNA. (A) Expression of CD80, Gvin1, Mip, Ifit1, and Ifit2 in naive mice was used to estimate the relative expression of each transcript in cornea, and GAPDH was used to normalize the expression of each transcript. Each bar represents the mean from 30 TG ± the SEM. Results were considered significant if P < 0.05 (Student t test). (B) The estimated relative copy number of the HSV-1 gB transcripts was calculated using standard curves generated from pAc-gB1. Each bar represents the mean from 30 TG, and error bars represent the SEM (P = 0.25 [Student t test]).
HSV-CD80 infection represses viral gene expression.
To determine whether overexpression of CD80 affects viral gene expression, we annotated the RNA sequencing results against the published HSV-1 strain 17 genome described in Materials and Methods. The expression of 75 viral genes in mouse corneas was ∼2-fold lower in mice infected with HSV-CD80 than in mice infected with parental virus, although these results did not reach statistical significance (Table 1). To verify these results, we determined the number of viral gB transcripts using qPCR. Consistent with the RNA sequencing results, levels of gB transcript expression did not significantly differ in the corneas of HSV-CD80- or parental virus-infected mice (Fig. 11B, P = 0.25).
TABLE 1.
Relative HSV-1 gene expressions in the corneas of mice infected with parental virus or HSV-CD80 virusa
| Gene | Mean expression count ± SEM |
P | |
|---|---|---|---|
| Parental virus | HSV-CD80 | ||
| UL1 (gL) | 1,058.2 ± 281.9 | 804.5 ± 278.6 | 0.82 |
| UL2 | 759.9 ± 204.0 | 454.26 ± 192.2 | 0.70 |
| UL3 | 122.4±35.2 | 69.2 ± 29.1 | 0.70 |
| UL4 | 126.4 ± 39.4 | 66.9 ± 37.9 | 0.74 |
| UL5 | 437.6 ± 134.0 | 260.3 ± 131.9 | 0.75 |
| UL6 | 206.2 ± 63.8 | 102.6 ± 48.1 | 0.69 |
| UL7 | 131.8 ± 42.5 | 62.7 ± 28.3 | 0.69 |
| UL8 | 91.7 ± 22.4 | 46.5 ± 19.5 | 0.69 |
| UL9 | 422.9 ± 117.6 | 233.7 ± 110.8 | 0.71 |
| UL10 (gM) | 338.9 ± 102.8 | 185.2 ±9 3.0 | 0.75 |
| UL11 | 109.5 ± 37.5 | 54.2 ± 28.0 | 0.72 |
| UL12 | 646.6 ± 232.0 | 285.4 ± 144.7 | 0.69 |
| UL13 | 875.9 ± 301.0 | 409.0 ± 207.0 | 0.70 |
| UL14 | 898.6 ± 311.8 | 419.9 ± 212.9 | 0.70 |
| UL15 | 438.3 ± 138.3 | 255.0 ± 125.4 | 0.75 |
| UL16 | 158.1 ± 50.9 | 75.1 ± 38.5 | 0.71 |
| UL17 | 267.9 ± 76.6 | 131.7 ± 66.6 | 0.71 |
| UL18 | 750.2 ± 236.3 | 349.0 ±170.3 | 0.69 |
| UL19 | 1,662.0 ± 574.5 | 714.1 ± 352.4 | 0.69 |
| UL20 | 1,797.2 ± 609.7 | 805.3 ± 396.5 | 0.69 |
| UL21 | 325.5 ± 108.7 | 155.3 ± 73.1 | 0.69 |
| UL22 (gH) | 684.4 ± 231.3 | 325.3 ± 172.0 | 0.70 |
| UL23 | 1,196.6 ± 257.5 | 656.3 ± 379.4 | 0.73 |
| UL24 | 806.8 ± 256.7 | 385.2 ± 180.8 | 0.69 |
| UL25 | 568.9 ± 191.6 | 262.9 ± 117.8 | 0.69 |
| UL26 | 402.2 ± 141.8 | 159.6 ± 66.4 | 0.69 |
| UL26.5 | 316.9 ± 113.5 | 122.6 ± 46.9 | 0.69 |
| UL27 (gB) | 1,850.0 ± 629.5 | 819.4 ± 401.4 | 0.69 |
| UL28 | 2031.4 ± 705.9 | 885.7 ± 434.7 | 0.69 |
| UL29 | 836.5 ± 263.1 | 438.3 ± 232.7 | 0.70 |
| UL30 | 492.4 ± 120.7 | 330.9 ± 167.1 | 0.81 |
| UL31 | 358.3 ± 88.1 | 183.0 ± 87.6 | 0.70 |
| UL32 | 433.6 ± 109.6 | 215.92 ± 106.8 | 0.70 |
| UL33 | 802.3 ± 194.8 | 366.4 ± 183.8 | 0.69 |
| UL34 | 773.2 ± 180.1 | 355.4 ± 177.5 | 0.69 |
| UL35 | 377.4 ± 78.5 | 174.7 ± 84.1 | 0.69 |
| UL36 | 213.2 ± 79.5 | 111.8 ± 60.0 | 0.70 |
| UL37 | 224.2 ± 84.4 | 81.5 ± 45.8 | 0.69 |
| UL38 | 83.4 ± 30.1 | 38.8 ± 20.2 | 0.72 |
| UL39 | 4,078.2 ± 1,272.5 | 2,297.2 ± 1,229.4 | 0.74 |
| UL40 | 1,955.2 ± 256.2 | 1,205.2 ± 629.2 | 0.77 |
| UL41 | 309.0 ± 77.8 | 219.1 ± 87.5 | 0.79 |
| UL42 | 830.0 ± 232.5 | 430.2 ± 218.5 | 0.71 |
| UL43 | 30.7 ± 14.6 | 13.8 ± 7.2 | 0.70 |
| UL44 (gC) | 1,771.2 ± 608.7 | 959.9 ± 454.1 | 0.72 |
| UL45 | 710.2 ± 249.2 | 376.8 ± 188.4 | 0.73 |
| UL46 | 542.5 ± 170.7 | 227.0 ± 107.7 | 0.69 |
| UL47 | 656.3 ± 196.8 | 276.3 ± 129.4 | 0.69 |
| UL48 | 1,323.0 ± 364.8 | 851.8 ± 425.7 | 0.81 |
| UL49 | 755.9 ± 222.9 | 354.8 ± 188.9 | 0.70 |
| UL49A (gN) | 1,151.1 ± 3 17.6 | 646.3 ± 324.4 | 0.73 |
| UL50 | 618.4 ± 180.3 | 266.5 ± 137.8 | 0.69 |
| UL51 | 80.0 ± 26.2 | 30.0 ± 14.2 | 0.69 |
| UL52 | 643.6 ± 154.5 | 398.0 ± 192.7 | 0.77 |
| UL53 (gK) | 393.3 ± 84.9 | 258.7 ± 126.1 | 0.81 |
| UL54 (ICP27) | 315.8 ± 123.6 | 129.8 ± 62.4 | 0.69 |
| UL55 | 150.9 ± 42.4 | 79.3 ± 36.3 | 0.70 |
| UL56 | 211.1 ± 67.5 | 102.2 ± 43.7 | 0.69 |
| US1 (ICP22) | 1,415.5 ± 401.2 | 688.5 ± 334.5 | 0.70 |
| US2 | 297.2 ± 90.8 | 183.2 ± 85.0 | 0.76 |
| US3 | 1,381.5 ± 419.4 | 707.6 ± 364.9 | 0.70 |
| US4 (gG) | 1,036.1 ± 314.8 | 525.5 ± 282.6 | 0.70 |
| US5 (gJ) | 3,620.4 ± 1,100.6 | 2,178.6 ± 1,081.0 | 0.75 |
| US6 (gD) | 3,574.1 ± 1,091.2 | 2,142.6 ± 1,065.8 | 0.74 |
| US7 (gI) | 2,072.5 ± 664.1 | 1,223.0 ± 621.3 | 0.74 |
| US8 (gE) | 1,764.4 ± 556.4 | 993.7 ± 503.5 | 0.73 |
| US8A | 1,073.2 ± 334.4 | 607.9 ± 304.1 | 0.73 |
| US9 | 704.0 ± 237.0 | 347.1 ± 173.7 | 0.70 |
| US10 | 1,475.9 ± 414.7 | 777.8 ± 387.4 | 0.72 |
| US11 | 1,783.2 ± 508.9 | 913.7 ± 454.6 | 0.71 |
| US12 (ICP47) | 1,823.7 ± 521.1 | 930.7 ± 462.4 | 0.71 |
| LAT | 1,213.3 ± 320.1 | 654.6 ± 273.9 | 0.69 |
| RL (ICP34.5) | 8.9 ± 0.6 | 9.4 ± 5.0 | 0.98 |
| RL2 (ICP0) | 1,077.6 ± 284.9 | 576.2 ± 239.3 | 0.69 |
| RS1 (ICP4) | 106.4 ± 46.4 | 61.0 ± 38.2 | 0.75 |
Female BALB/c mice were ocularly infected with the parental virus or HSV-CD80 at 105 PFU/eye without corneal scarification. Mice were euthanized on day 28 postinfection, and corneas were excised. The corneas of five mice were combined into one sample, with three samples for each condition. RNA was isolated from each sample and sequenced as described in Materials and Methods. Viral genomes were annotated against HSV-1 strain 17 JN555585.1 (PMID 22417106) (80). Values represent averages of three replicate samples’ normalized expression counts ± the SEM. A Student t test was used to obtain the P values.
DISCUSSION
A striking feature of HSV-1 is its ability to establish lifelong latency with periodic reactivation (41, 42). To accomplish this, HSV-1 has evolved several mechanisms to evade host immune responses (27). Results from our laboratory suggested an additional mechanism by which HSV-1 evades the host immune system by downregulating the costimulatory molecule CD80 but not CD86 (32). This was dependent on ICP22, because infection of 293 cells with viruses lacking ICP22 showed increased CD80 promoter activity compared to wild-type virus-infected cells. Despite reduced viral replication, reduced latency, and delayed reactivation, infection of mice with HSV-1 lacking ICP22 resulted in eye disease similar to that caused by the parental virus (32, 34). Overexpression of CD80, on the other hand, resulted in increased ocular pathology (32).
Here, we set out to determine the effects of CD80 overexpression on the course of HSV-1 infection and to identify the mechanism by which CD80 overexpression increases corneal pathology in mice. To study the role of CD80 more directly, we used a recombinant virus with intact ICP22 and two copies of CD80 under the control of the LAT promoter. Since LAT is the only viral gene expressed at high levels during both acute and latent infections (43), this approach allowed us to determine the effect of CD80 during both primary and latent infection. In addition, because ICP22 is intact in this virus, we can rule out an initial growth defect as a cause for any latency defects.
Our results show that corneal scarring is not a result of increased viral replication or cell death. Instead, exacerbated pathology is likely caused by increased CD8+ T cell recruitment in mice infected with HSV-CD80. This could be a direct consequence of CD80 overexpression because CD80 is a potent costimulatory molecule of CD8+ T cells (35). The increase in CD8+ T cells is consistent with a study from the Morrison lab that reported an increase in HSV-specific CD8+ T cells after immunization with a replication-deficient HSV-1 expressing CD80 (ICP8− vhs− B7+ HSV-1) (44). Schrimpf et al. reported less severe eye disease in mice immunized with the CD80-expressing recombinant virus than found by us. This discrepancy could be due to the fact that our study used ocular infection of naive mice to study eye disease, while the Schrimpf study looked at the effect of ocular infection of mice that had been immunized with recombinant virus expressing CD80 and challenged with wild-type virus.
The correlation between increased eye disease and an increase in CD8+ T cells agrees with our previous findings (36–39, 45, 46). For example, overexpression of gK, a viral glycoprotein that functions in viral egress (reviewed in Jaggi et al. [47]), produced increased eye disease that was ameliorated by depleting CD8+ T cells (36).
Despite HSV-CD80 virus replicating with similar kinetics in vitro and in vivo, mice infected with HSV-CD80 had significantly higher latency and a faster time to reactivation. This is likely a consequence of the slightly higher gC expression in TG of mice infected with HSV-CD80 (Fig. 4), because viral load is known to correlate with latency and reactivation (3). The positive correlation between latency and reactivation, on the other hand, is consistent with the literature (48). Higher latency and reactivation have also been correlated with increased pathology (49). In addition, it is tempting to speculate that increased latency could be caused by the transient increase in DCs seen in this study, since we have previously reported that DCs drive latency and reactivation (50–52).
HSV-CD80 may function similarly to LAT. HSV-1 LAT functions in latency establishment and reactivation because viruses lacking LAT do not establish latency as efficiently as WT viruses and have delayed reactivation (53, 54). Here, we show that HSV-CD80, in which LAT is replaced with CD80, establishes higher latency and reactivates faster than its parental virus lacking LAT (dLAT2903). Further studies are under way to establish the link between LAT and CD80.
We found that mice infected with HSV-CD80 had a transient increase in DC infiltrates. Increased DCs could account for the increased T cell recruitment, as DCs are considered the most potent activators of CD8+ T cells (55, 56). We have shown previously that DCs, which do not support HSV-1 productive infection, are permissive to the recombinant HSV-CD80 in vitro (33). CD8+ T cells cocultured with HSV-CD80-infected DCs expressed more IFN-γ than did T cells cocultured with parental virus-infected DCs, suggesting that viral expression of CD80 can enhance T cell activation (33). Since our results showed an increase in the total number of CD8+ T cells, it is possible that these were activated by infected DCs. Infection of bone marrow-derived DCs with HSV-CD80 lysed infected DCs in vitro (33); however, in the present study we did not observe a difference in cell death within corneas of mice infected with HSV-CD80 or parental virus, suggesting that some effects may be context dependent. We have shown that HSV-CD80 infection leads to exacerbated eye disease (32). The increased ocular pathology seen in HSV-CD80-infected mice could be due to reduced availability of PD-L1 for interaction with PD-1. We previously established that virally expressed CD80 binds to its ligand PD-L1 in DCs (32). PD-L1 binding to its receptor, PD-1, dampens T cell activity (57–59). Therefore, CD80 overexpression may sequester PD-L1, thereby reducing inhibition of T cell activation.
This idea is consistent with a recent report showing that higher CD80 expression reduces binding of PD-1 to PD-L1 (60) and with a report from the Hendricks group showing that blocking PD-1–PD-L1 interaction resulted in increased survival of HSV-1-specific CD8+ T cells recognizing a dominant HSV epitope (gB-Tet-CD8+ T cells) but did not protect from HSV-1 reactivation (61). We also observed lower viral titers on day 7 p.i. in the corneas of mice infected with HSV-CD80 compared to parental virus-infected samples (Fig. 3A). This could also be a result of sequestering PD-L1, because Jeon et al. reported a negative effect of PD-L1 expression on viral clearance (62).
Based on these results, we propose a model where virally expressed CD80 could contribute to CD8+ T cell activation and exacerbation of eye disease via three mechanisms (illustrated in Fig. 12). First, CD80 is a potent costimulatory molecule of CD8+ T cells (35). Second, CD80 binds to and sequesters PD-L1, resulting in reduced inhibition of T cell activation (57–59). Third, infection with HSV-CD80 resulted in a transient increase in DCs, which are considered the most potent activators of CD8+ T cells (55, 56). We have previously demonstrated a strong correlation between CD8+ T cells and eye disease (36–39, 45), and depletion of CD8+ T cells ameliorated eye disease caused by overexpression of viral glycoprotein gK (36).
FIG 12.
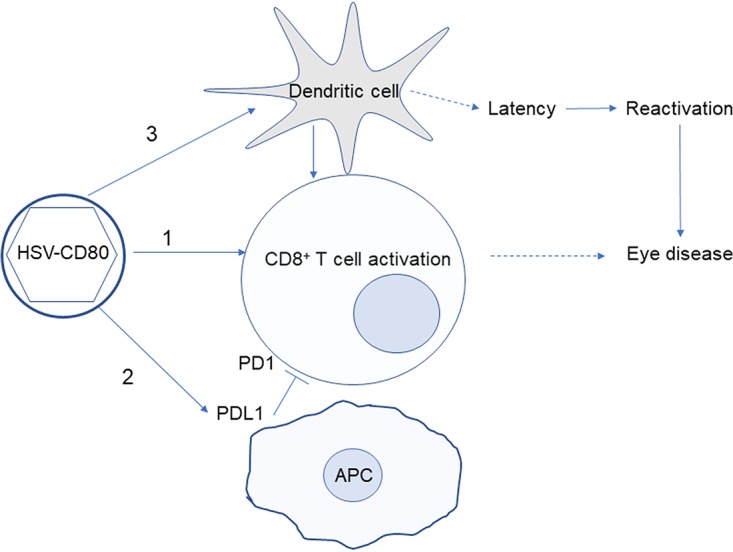
Proposed model for increased eye disease caused by overexpression of CD80. Diagram depicting our model for HSV-CD80's effect on latency, reactivation, and exacerbated eye disease. Infection with HSV-CD80 results in activation of CD8+ T cells (indicated by arrow 1). CD80 may bind to PD-L1 (indicated by arrow 2) as we reported previously (33), reducing interaction between PD-1 and PD-L1, which is known to inhibit T cell activation (blunt arrow). Activated CD8+ T cells are the likely cause of exacerbated eye disease, since CD8+ T cells have been linked with eye disease in other reports (36–39, 45). Infection with HSV-CD80 also results in a transient increase in DCs (arrow 3), which can further activate CD8+ T cells. DCs have been reported to drive latency establishment (52) (dashed arrow), and the level of latency is correlated with reactivation (48).
To identify genes that are differentially expressed during infection with HSV-CD80 and parental virus, we analyzed the host transcriptome at 4 days p.i. To our knowledge, this is the first in vivo analysis of the host transcriptome in HSV-1-infected mouse corneas. Our findings agree with previous reports from high-throughput in vitro studies (63–65). For example, expression of several immunomodulatory genes, including IFN-γ, IL-6, IL-1β, and TLR9, was significantly higher in parental virus-infected corneas than in uninfected samples (Table S1, log2-fold change > 3, P < 0.006). Our results also suggest activation of TLR, JAK-STAT, and TNFR signaling pathways and upregulation of NF-κB upon infection with both HSV-CD80 and parental virus. These pathways are known to be activated upon HSV-1 acute infection, increasing our confidence in the RNA sequencing results (Fig. 10).
The top canonical pathways upregulated in HSV-CD80- and parental virus-infected corneas were DC maturation and TREM1 (triggering receptor expressed on myeloid cells 1) signaling. The upregulation of components in DC maturation agree with a study from our lab demonstrating that CD8α+ DCs drive HSV-1 latency (66). TREM1 is expressed on neutrophils and monocytes, and its activation leads to initiation and amplification of inflammation (67). Linderman et al. demonstrated that the TREM1 pathway is upregulated in HSV-1 latently infected neuronal cultures in response to a reactivating signal (phosphatidylinositol 3-kinase inhibitor LY294002) and IFN-γ but not IFN-β (68). However, because these pathways were similarly upregulated in both HSV-CD80- and parental virus-infected mice, it is unlikely that activation of this pathway is responsible for exacerbated eye disease in HSV-CD80-infected mice.
The fact that we observed a large number of differentially expressed genes in infected and uninfected mice is impressive, considering that the number of infected cells in corneas is low at any given time. This suggests that those few infected cells must express these genes at substantial levels. On the other hand, the relatively small number of genes that are differentially expressed in HSV-CD80 and parental virus-infected mice could be because these viruses differ by only one gene, which is also present in the host.
Comparison of transcriptomes of mice infected with HSV-CD80 or parental virus revealed nine differentially expressed genes. Because four of the nine genes were uncharacterized proteins, we did not attempt to verify these findings using alternative methods. The other five genes were those encoding CD80, as predicted, lactase-like protein (Lctl), major intrinsic protein of lens fiber (Mip), proline-rich transmembrane protein 2 (Prrt2), and GTPase, very large interferon inducible 1 (Gvin1). Unfortunately, we were not able to confirm the differential expression of Gvin1 or Mip using qRT-PCR. Interestingly, interferon-induced protein with tetratricopeptide repeats 2 (Ifit2) was slightly upregulated in the corneas of mice infected with HSV-CD80, although this did not reach statistical significance. However, qRT-PCR verification of RNA sequencing results showed a 2.6-fold increase in expression of Ifit2, but not Ifit1 (Fig. 11A). Increased expression of Ifit2 is consistent with our previous study showing increased IFN-γ expression in DCs upon HSV-CD80 infection (33) and increased expression of several immune infiltrates upon infection with ICP22-null virus (34).
Analysis of viral gene expression revealed no statistically significant differences in viral transcripts between HSV-CD80- and parental virus-infected mouse corneas. These results are striking considering that infection with HSV-CD80 exacerbated eye disease (32). The lack of correlation between eye disease severity and viral copy number also supports our overall conclusion that corneal scarring is a consequence of host immune response, not viral replication, and that HSV-1 downregulation of CD80 is a novel immune evasion mechanism.
In summary, this work is the first to offer insight into the effects of HSV-1 infection on the mouse cornea transcriptome in vivo. We also provide strong evidence supporting the conclusions that (i) corneal pathology is caused by the immune response, not viral replication or viral gene expression; (ii) HSV-1 downregulation of CD80 is a novel mechanism of viral immune evasion; (iii) transient reduction of DCs could explain enhanced latency and reactivation; and (iv) exacerbated eye disease is likely caused by enhanced recruitment and activation of CD3+ CD8+ T cells.
MATERIALS AND METHODS
Ethics statement.
All animal procedures were performed in strict accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (ISBN 0-309-05377-3). Our animal research protocol was approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center (protocol 5030).
Viruses, cells, and mice.
Triply plaque-purified dLAT2903 (parental virus), and HSV-CD80 viruses were used in this study (33). RS cells (used to prepare virus stocks, culture mouse tear films, and determine growth kinetics) were grown in Eagle minimal essential medium supplemented with 5% fetal calf serum. Six-week-old female inbred BALB/c mice were used in this study (The Jackson Laboratory, Bar Harbor, ME).
Immunofluorescence.
RS cell monolayers grown on Lab-Tex chamber slides were infected with 0.1, 1, or 10 PFU/cell of recombinant HSV-CD80 or parental virus for 16 h. To examine cell surface immunofluorescence, unfixed, unpermeabilized cells were incubated with FITC-gC (Genway, 20-902-170310) or FITC-CD80 (BioLegend, San Diego, CA) antibody for 1 h at 25°C. Slides were washed three times with phosphate-buffered saline (PBS), air dried, fixed with acetone, mounted with Prolong Gold DAPI mounting medium (Invitrogen), and examined for fluorescence. Images were acquired with a Zeiss ApoTome-equipped Axio Imager Z1 (Carl Zeiss).
Flow cytometric analysis of infected cells.
RS cell monolayers were infected with 1 PFU/cell of recombinant HSV-CD80 or parental virus or were mock infected for 24 h. Infected cells were harvested and stained with anti-CD80 and anti-gC antibodies. Stained cells were washed 2× with FACS buffer (1× PBS with 0.1% sodium azide), resuspended in 4% paraformaldehyde, and analyzed in a BD LSR II flow cytometer using BD FACSDiva software (BD Biosciences). Postexperiment data analysis was performed using FlowJo software (TreeStar). Experiments were repeated three times.
Ocular infection and virus titration in the eye.
Mice were infected ocularly with 2 μl of tissue culture media containing 105 PFU/eye of HSV-CD80 recombinant or parental virus without corneal scarification (69). Tear films were collected from 40 eyes on days 1 to 7 for each group, as described previously (70). Each swab was placed in 1 ml of tissue culture medium, and the amount of virus in the medium was determined using a standard plaque assay on RS cells.
Detection of infectious virus in the eye, TG, and brain.
BALB/c mice were infected ocularly with 105 PFU/eye of HSV-CD80 recombinant or parental virus. On days 3, 5, and 7 p.i., infected mice were euthanized, and the eyes, TG, and brain from each mouse were isolated individually. Eyes, TG, and brains were homogenized, and the debris was removed by centrifugation at 3,000 rpm for 10 min in a Beckman TA10 rotor. Viral titers in the supernatants of the eyes, TG, and brains from day 3, 5, or 7 p.i. were determined using a standard plaque assay on RS cells as described previously (71).
Detection of CD4+ and CD8+ T cells and CD11b+ CD11c+ cells in the corneas and TG of infected mice by flow cytometric analysis.
Mice were ocularly infected and infected cells were harvested on days 3 and 5 p.i. Corneas or TG from each mouse were combined, and single cell suspensions of corneas and TG were prepared as we described previously (48, 66, 72). Single cell suspensions of corneas and TG were stained with anti-CD3, anti-CD4, anti-CD8α or anti-CD11c, and anti-CD11b antibodies (BioLegend, San Diego, CA) and then analyzed by flow cytometry as we described previously (73). Experiments were repeated three times.
Analysis of viable cells by flow cytometric analysis.
Mice were ocularly infected, and on days 3 and 5 p.i., infected corneas were harvested and processed for single cell suspension, as we described previously (48, 66, 72). Single cell corneal suspensions were stained with annexin-V and 7-aminoactinomycin D (7-ADD) dye to determine the proportions of viable, apoptotic, and nonviable cells as we described previously (74).
RNA extraction, cDNA synthesis, and TaqMan RT-PCR.
Infected and mock-infected mice were euthanized on days 3 and 5 p.i., and TG were harvested and suspended in 500 μl of RNAlater (Qiagen). Suspensions were stored at –80°C until processing. Tissue processing and RNA extraction were performed using QIAzol RNA reagent (Qiagen) and 1-bromo-2-chloropropane (BCP) as described previously (36, 75, 76). Then, 1 μg of RNA was reverse transcribed using random hexamer primers and murine leukemia virus reverse transcriptase from a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s recommendations. The expression of five transcripts—Ifit1 (Mm00515153_m1; amplicon length, 80 bp), Ifit2 (Mm00492606_m1; amplicon length, 66 bp), Gvin1 (Mm02527660_s1; amplicon length, 81 bp), Mip (Mm00434949_m1; amplicon length, 136 bp), and CD80 (Mm00711660_m1; amplicon length, 117 bp)—was determined using TaqMan gene expression assays (Applied Biosystems, Foster City, CA). Primer-probe sets consisted of two unlabeled PCR primers and FAM dye-labeled TaqMan MGB probe in a single mixture. The relative gene expression was normalized to the expression of the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) housekeeping gene (Mm999999.15_g1; amplicon length, 107 bp).
The expression of HSV-1 gB was evaluated using custom-made TaqMan gene expression assays (Applied Biosystems) with forward primer 5′-AACGCGACGCACATCAAG-3′, reverse primer 5′-CTGGTACGCGATCAGAAAGC-3′, and probe 5′-FAM-CAGCCGCAGTACTACC-3′, as we described previously (36). The gB amplicon length was 72 bp. The relative copy numbers of the gB DNA were calculated using standard curves generated from the plasmid pAc-gB1 DNA (77).
qRT-PCR was performed using QuantStudio 5 (Applied Biosystems) in 384-well plates, as we described previously (48, 66). Real-time PCR was performed in triplicate for each tissue sample. The threshold cycle (CT) values, which represent the PCR cycles at which there is a noticeable increase in reporter fluorescence above baseline, were determined using SDS v2.2 software.
Viral DNA isolation and copy number determination.
TGs from ocularly infected BALB/c mice were collected on day 30 p.i. Individual TG from each animal were processed for DNA extraction, and the viral gB copy number was determined as we described previously (36).
In vitro explant reactivation assay.
Mice were sacrificed 28 days after infection, and individual TG were removed and cultured in tissue culture medium, as we described previously (51). Aliquots of culture medium were removed from the explant daily for 15 days and plated on indicator RS cells to monitor the appearance of reactivated virus. Because the medium from explanted TG cultures was plated daily, the time at which reactivated virus first appeared in the explanted TG cultures could be determined.
Library preparation, RNA sequencing, and analysis.
Mice were ocularly infected as described above and euthanized on day 4 p.i. Corneas from five mice for each group were combined into one sample, with three replicates for each experimental group. Corneas were stored in TRIzol at –80°C until they were processed for RNA. RNA was extracted as described previously (36, 75, 76).
Libraries were constructed using the Illumina TruSeq Stranded mRNA library preparation kit (Illumina, San Diego, CA). Briefly, total RNA samples were assessed for concentration using a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA) and for quality using a 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA). Up to 1 μg of total RNA per sample was used for poly(A) mRNA selection. cDNA was synthesized from enriched and fragmented RNA by reverse transcriptase (Invitrogen, Carlsbad, CA) with random primers. The cDNA was further converted into double-stranded DNA (dsDNA), and the resulting dsDNA was enriched by PCR for library preparation. The PCR-amplified library was purified using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA). The concentration of the amplified library was measured with a Qubit fluorometer, and an aliquot of the library was resolved on a bioanalyzer. Sample libraries were multiplexed and sequenced on a NextSeq 500 platform (Illumina) using 75-bp single-end sequencing. On average, about 20 million reads were generated from each sample.
Raw reads obtained from RNA-Seq were aligned to the transcriptome using STAR (v2.5.0) (78) and RSEM (v1.2.25) (79) with default parameters, with a custom human GRCh38 transcriptome reference downloaded from http://www.gencodegenes.org containing all protein coding and long noncoding RNA genes based on a human GENCODE version 23 annotation. The viral genome was annotated against HSV-1 strain 17 JN555585.1 (80).
Expression counts for each gene in all samples were normalized by a modified trimmed mean of the M-value normalization method, and unsupervised principal-component analysis was performed using the DESeq2 Bioconductor package v1.10.1 in R version 3.2.2. Each gene was fitted into a negative binomial generalized linear model, and a Wald test was applied to assess the differential expression between two sample groups by DESeq2. The Benjamini-Hochberg procedure was applied to adjust for multiple-hypothesis testing, and differential expression gene candidates were selected with a false-discovery rate of <0.05. To visualize coordinated gene expression in samples, a two-way hierarchical clustering with Pearson correlation distance matrix was performed with samples and DEG candidates using the Bioconductor g-plots package (v2.14.2) in R. Pathway analysis was performed using Ingenuity Pathway Analysis (Qiagen).
Statistical analysis.
Protective parameters were analyzed with the Student t test and the Fisher exact test using Instat (GraphPad, San Diego, CA). Error bars represent the standard errors of the mean (SEM), and results were considered statistically significant if the P value was <0.05.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIH grants 1RO1 EY026944 and 1RO1 EY013615.
We thank Kevin Mott and Mandana Zandian for data collection and analysis, as well as the Genomics Core at Cedars-Sinai Medical Center for RNA sequencing and analysis.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Liesegang TJ. 1999. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 18:127–143. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Nicoll MP, Proenca JT, Connor V, Efstathiou S. 2012. Influence of herpes simplex virus 1 latency-associated transcripts on the establishment and maintenance of latency in the ROSA26R reporter mouse model. J Virol 86:8848–8858. doi: 10.1128/JVI.00652-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol 61:3820–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wechsler SL, Nesburn AB, Watson R, Slanina S, Ghiasi H. 1988. Fine mapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J Gen Virol 69:3101–3106. doi: 10.1099/0022-1317-69-12-3101. [DOI] [PubMed] [Google Scholar]
- 6.Wechsler SL, Nesburn AB, Watson R, Slanina SM, Ghiasi H. 1988. Fine mapping of the latency-related gene of herpes simplex virus type 1: alternative splicing produces distinct latency-related RNAs containing open reading frames. J Virol 62:4051–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farooq AV, Shukla D. 2012. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol 57:448–462. doi: 10.1016/j.survophthal.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renfro L, Snow JS. 1992. Ocular effects of topical and systemic steroids. Dermatol Clin 10:505–512. doi: 10.1016/S0733-8635(18)30318-8. [DOI] [PubMed] [Google Scholar]
- 9.Daniel BS, Orchard D. 2015. Ocular side-effects of topical corticosteroids: what a dermatologist needs to know. Australas J Dermatol 56:164–169. doi: 10.1111/ajd.12292. [DOI] [PubMed] [Google Scholar]
- 10.Bonneau RH, Jennings SR. 1989. Modulation of acute and latent herpes simplex virus infection in C57BL/6 mice by adoptive transfer of immune lymphocytes with cytolytic activity. J Virol 63:1480–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen HS, Feng MF, Horohov DW, Moore RN, Rouse BT. 1984. Role of T-lymphocyte subsets in recovery from herpes simplex virus infection. J Virol 50:56–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banerjee K, Biswas PS, Kumaraguru U, Schoenberger SP, Rouse BT. 2004. Protective and pathological roles of virus-specific and bystander CD8+ T cells in herpetic stromal keratitis. J Immunol 173:7575–7583. doi: 10.4049/jimmunol.173.12.7575. [DOI] [PubMed] [Google Scholar]
- 13.Pozzi LA, Maciaszek JW, Rock KL. 2005. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J Immunol 175:2071–2081. doi: 10.4049/jimmunol.175.4.2071. [DOI] [PubMed] [Google Scholar]
- 14.Colonna M, Pulendran B, Iwasaki A. 2006. Dendritic cells at the host-pathogen interface. Nat Immunol 7:117–120. doi: 10.1038/ni0206-117. [DOI] [PubMed] [Google Scholar]
- 15.Chew T, Taylor KE, Mossman KL. 2009. Innate and adaptive immune responses to herpes simplex virus. Viruses 1:979–1002. doi: 10.3390/v1030979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenschow DJ, Walunas TL, Bluestone JA. 1996. CD28/B7 system of T cell costimulation. Annu Rev Immunol 14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 17.Greenfield EA, Nguyen KA, Kuchroo VK. 1998. CD28/B7 costimulation: a review. Crit Rev Immunol 18:389–418. doi: 10.1615/CritRevImmunol.v18.i5.10. [DOI] [PubMed] [Google Scholar]
- 18.Sharpe AH, Freeman GJ. 2002. The B7-CD28 superfamily. Nat Rev Immunol 2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 19.Bretscher PA. 1999. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A 96:185–190. doi: 10.1073/pnas.96.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lumsden JM, Roberts JM, Harris NL, Peach RJ, Ronchese F. 2000. Differential requirement for CD80- and CD80/CD86-dependent costimulation in the lung immune response to an influenza virus infection. J Immunol 164:79–85. doi: 10.4049/jimmunol.164.1.79. [DOI] [PubMed] [Google Scholar]
- 21.Wingren AG, Parra E, Varga M, Kalland T, Sjogren HO, Hedlund G, Dohlsten M. 2017. T cell activation pathways: B7, LFA-3, and ICAM-1 shape unique T cell profiles. Crit Rev Immunol 37:463–481. doi: 10.1615/CritRevImmunol.v37.i2-6.130. [DOI] [PubMed] [Google Scholar]
- 22.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. 2007. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mossman KL, Ashkar AA. 2005. Herpesviruses and the innate immune response. Viral Immunol 18:267–281. doi: 10.1089/vim.2005.18.267. [DOI] [PubMed] [Google Scholar]
- 24.Eidson KM, Hobbs WE, Manning BJ, Carlson P, DeLuca NA. 2002. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J Virol 76:2180–2191. doi: 10.1128/jvi.76.5.2180-2191.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paladino P, Collins SE, Mossman KL. 2010. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS One 5:e10428. doi: 10.1371/journal.pone.0010428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vandevenne P, Sadzot-Delvaux C, Piette J. 2010. Innate immune response and viral interference strategies developed by human herpesviruses. Biochem Pharmacol 80:1955–1972. doi: 10.1016/j.bcp.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Kurt-Jones EA, Orzalli MH, Knipe DM. 2017. Innate immune mechanisms and herpes simplex virus infection and disease. Adv Anat Embryol Cell Biol 223:49–75. doi: 10.1007/978-3-319-53168-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tormanen K, Allen S, Mott KR, Ghiasi H. 2019. The latency-associated transcript inhibits apoptosis via downregulation of components of the type I interferon pathway during latent herpes simplex virus 1 ocular infection. J Virol 93:e00103-19. doi: 10.1128/JVI.00103-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 30.Henderson G, Perng GC, Nesburn AB, Wechsler SL, Jones C. 2004. The latency-related gene encoded by bovine herpesvirus 1 can suppress caspase 3 and caspase 9 cleavage during productive infection. J Neurovirol 10:64–70. doi: 10.1080/13550280490261716. [DOI] [PubMed] [Google Scholar]
- 31.Branco FJ, Fraser NW. 2005. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol 79:9019–9025. doi: 10.1128/JVI.79.14.9019-9025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matundan H, Ghiasi H. 2018. Herpes simplex virus 1 ICP22 suppresses CD80 expression by murine dendritic cells. J Virol 93:e01803-18. doi: 10.1128/JVI.01803-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mott KR, Allen SJ, Zandian M, Akbari O, Hamrah P, Maazi H, Wechsler SL, Sharpe AH, Freeman GJ, Ghiasi H. 2014. Inclusion of CD80 in HSV targets the recombinant virus to PD-L1 on DCs and allows productive infection and robust immune responses. PLoS One 9:e87617. doi: 10.1371/journal.pone.0087617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matundan H, Jaggi U, Wang S, Ghiasi H. 2019. Loss of ICP22 in HSV-1 elicits immune infiltration and maintains stromal keratitis despite reduced primary and latent virus infectivity. Invest Ophthalmol Vis Sci 60:3398–3406. doi: 10.1167/iovs.19-27701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanier LL, O’Fallon S, Somoza C, Phillips JH, Linsley PS, Okumura K, Ito D, Azuma M. 1995. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J Immunol 154:97–105. [PubMed] [Google Scholar]
- 36.Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J Virol 81:12962–12972. doi: 10.1128/JVI.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osorio Y, Mott KR, Jabbar AM, Moreno A, Foster TP, Kousoulas KG, Ghiasi H. 2007. Epitope mapping of HSV-1 glycoprotein K (gK) reveals a T cell epitope located within the signal domain of gK. Virus Res 128:71–80. doi: 10.1016/j.virusres.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mott KR, Chentoufi AA, Carpenter D, Benmohamed L, Wechsler SL, Ghiasi H. 2009. The role of a glycoprotein K (gK) CD8+ T-cell epitope of herpes simplex virus on virus replication and pathogenicity. Invest Ophthalmol Vis Sci 50:2903–2912. doi: 10.1167/iovs.08-2957. [DOI] [PubMed] [Google Scholar]
- 39.Allen SJ, Mott KR, Ljubimov AV, Ghiasi H. 2010. Exacerbation of corneal scarring in HSV-1 gK-immunized mice correlates with elevation of CD8+ CD25+ T cells in corneas of ocularly infected mice. Virology 399:11–22. doi: 10.1016/j.virol.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matundan H, Mott KR, Ghiasi H. 2014. Role of CD8+ T cells and myeloid DCs in protection from ocular HSV-1 challenge in immunized mice. J Virol 88:8016–8027. doi: 10.1128/JVI.00913-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koujah L, Suryawanshi RK, Shukla D. 2019. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell Mol Life Sci 76:405–419. doi: 10.1007/s00018-018-2938-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phelan D, Barrozo ER, Bloom DC. 2017. HSV1 latent transcription and noncoding RNA: a critical retrospective. J Neuroimmunol 308:65–101. doi: 10.1016/j.jneuroim.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Zwaagstra JC, Ghiasi H, Nesburn AB, Wechsler SL. 1991. Identification of a major regulatory sequence in the latency-associated transcript (LAT) promoter of herpes simplex virus 1 (HSV-1). Virology 182:287–297. doi: 10.1016/0042-6822(91)90672-X. [DOI] [PubMed] [Google Scholar]
- 44.Schrimpf JE, Tu EM, Wang H, Wong YM, Morrison LA. 2011. B7 costimulation molecules encoded by replication-defective, vhs-deficient HSV-1 improve vaccine-induced protection against corneal disease. PLoS One 6:e22772. doi: 10.1371/journal.pone.0022772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osorio Y, Cai S, Hofman FM, Brown DJ, Ghiasi H. 2004. Involvement of CD8+ T cells in exacerbation of corneal scarring in mice. Curr Eye Res 29:145–151. doi: 10.1080/02713680490504632. [DOI] [PubMed] [Google Scholar]
- 46.Mott KR, Wechsler SL, Ghiasi H. 2010. Ocular infection of mice with an avirulent recombinant HSV-1 expressing IL-4 and an attenuated HSV-1 strain generates virulent recombinants in vivo. Mol Vis 16:2153–2162. [PMC free article] [PubMed] [Google Scholar]
- 47.Jaggi U, Wang S, Tormanen K, Matundan H, Ljubimov AV, Ghiasi H. 2018. Role of herpes simplex virus type 1 (HSV-1) glycoprotein K (gK) pathogenic CD8+ T cells in exacerbation of eye disease. Front Immunol 9:2895. doi: 10.3389/fimmu.2018.02895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matundan HH, Mott KR, Allen SJ, Wang S, Bresee CJ, Ghiasi YN, Town T, Wechsler SL, Ghiasi H. 2016. Interrelationship of primary virus replication, level of latency, and time to reactivation in the trigeminal ganglia of latently infected mice. J Virol 90:9533–9542. doi: 10.1128/JVI.01373-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. 2009. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J Virol 83:2246–2254. doi: 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mott KR, Ghiasi H. 2008. Role of dendritic cells in enhancement of herpes simplex virus type 1 latency and reactivation in vaccinated mice. Clin Vaccine Immunol 15:1859–1867. doi: 10.1128/CVI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mott KR, UnderHill D, Wechsler SL, Ghiasi H. 2008. Lymphoid-related CD11c+ CD8a+ dendritic cells are involved in enhancing HSV-1 latency. J Virol 82:9870–9879. doi: 10.1128/JVI.00566-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mott KR, Allen SJ, Zandian M, Konda B, Sharifi BG, Jones C, Wechsler SL, Town T, Ghiasi H. 2014. CD8a dendritic cells drive establishment of HSV-1 latency. PLoS One 9:e93444. doi: 10.1371/journal.pone.0093444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kassim SH, Rajasagi NK, Zhao X, Chervenak R, Jennings SR. 2006. In vivo ablation of CD11c-positive dendritic cells increases susceptibility to herpes simplex virus type 1 infection and diminishes NK and T-cell responses. J Virol 80:3985–3993. doi: 10.1128/JVI.80.8.3985-3993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allan RS, Smith CM, Belz GT, van Lint AL, Wakim LM, Heath WR, Carbone FR. 2003. Epidermal viral immunity induced by CD8α+ dendritic cells but not by Langerhans cells. Science 301:1925–1928. doi: 10.1126/science.1087576. [DOI] [PubMed] [Google Scholar]
- 57.Brown KE, Freeman GJ, Wherry EJ, Sharpe AH. 2010. Role of PD-1 in regulating acute infections. Curr Opin Immunol 22:397–401. doi: 10.1016/j.coi.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen L, Flies DB. 2013. Molecular mechanisms of T cell costimulation and co-inhibition. Nat Rev Immunol 13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharpe AH, Pauken KE. 2018. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol 18:153–167. doi: 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 60.Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, Okazaki T. 2019. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364:558–566. doi: 10.1126/science.aav7062. [DOI] [PubMed] [Google Scholar]
- 61.Jeon S, St Leger AJ, Cherpes TL, Sheridan BS, Hendricks RL. 2013. PD-L1/B7-H1 regulates the survival but not the function of CD8+ T cells in herpes simplex virus type 1 latently infected trigeminal ganglia. J Immunol 190:6277–6286. doi: 10.4049/jimmunol.1300582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jeon S, Rowe AM, Carroll KL, Harvey SAK, Hendricks RL. 2018. PD-L1/B7-H1 inhibits viral clearance by macrophages in HSV-1-infected corneas. J Immunol 200:3711–3719. doi: 10.4049/jimmunol.1700417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Terasaka Y, Miyazaki D, Yakura K, Haruki T, Inoue Y. 2010. Induction of IL-6 in transcriptional networks in corneal epithelial cells after herpes simplex virus type 1 infection. Invest Ophthalmol Vis Sci 51:2441–2449. doi: 10.1167/iovs.09-4624. [DOI] [PubMed] [Google Scholar]
- 64.Takeda S, Miyazaki D, Sasaki S, Yamamoto Y, Terasaka Y, Yakura K, Yamagami S, Ebihara N, Inoue Y. 2011. Roles played by Toll-like receptor-9 in corneal endothelial cells after herpes simplex virus type 1 infection. Invest Ophthalmol Vis Sci 52:6729–6736. doi: 10.1167/iovs.11-7805. [DOI] [PubMed] [Google Scholar]
- 65.Shi J, Hu N, Mo L, Zeng Z, Sun J, Hu Y. 2018. Deep RNA sequencing reveals a repertoire of human fibroblast circular RNAs associated with cellular responses to herpes simplex virus 1 infection. Cell Physiol Biochem 47:2031–2045. doi: 10.1159/000491471. [DOI] [PubMed] [Google Scholar]
- 66.Mott KR, Gate D, Matundan HH, Ghiasi YN, Town T, Ghiasi H. 2016. CD8+ T cells play a bystander role in mice latently infected with herpes simplex virus 1. J Virol 90:5059–5067. doi: 10.1128/JVI.00255-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roe K, Gibot S, Verma S. 2014. Triggering receptor expressed on myeloid cells-1 (TREM-1): a new player in antiviral immunity? Front Microbiol 5:627. doi: 10.3389/fmicb.2014.00627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Linderman JA, Kobayashi M, Rayannavar V, Fak JJ, Darnell RB, Chao MV, Wilson AC, Mohr I. 2017. Immune escape via a transient gene expression program enables productive replication of a latent pathogen. Cell Rep 18:1312–1323. doi: 10.1016/j.celrep.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghiasi H, Kaiwar R, Nesburn AB, Slanina S, Wechsler SL. 1994. Expression of seven herpes simplex virus type 1 glycoproteins (gB, gC, gD, gE, gG, gH, and gI): comparative protection against lethal challenge in mice. J Virol 68:2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghiasi H, Bahri S, Nesburn AB, Wechsler SL. 1995. Protection against herpes simplex virus-induced eye disease after vaccination with seven individually expressed herpes simplex virus 1 glycoproteins. Invest Ophthalmol Vis Sci 36:1352–1360. [PubMed] [Google Scholar]
- 71.Ghiasi H, Perng GC, Hofman FM, Cai S, Nesburn AB, Wechsler SL, Pemg GC. 1999. Specific and nonspecific immune stimulation of MHC-II-deficient mice results in chronic HSV-1 infection of the trigeminal ganglia following ocular challenge. Virology 258:208–216. doi: 10.1006/viro.1999.9710. [DOI] [PubMed] [Google Scholar]
- 72.Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Osorio Y, La Point SF, Nusinowitz S, Hofman FM, Ghiasi H. 2005. CD8+-dependent CNS demyelination following ocular infection of mice with a recombinant HSV-1 expressing murine IL-2. Exp Neurol 193:1–18. doi: 10.1016/j.expneurol.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 74.Mott KR, Underhill D, Wechsler SL, Town T, Ghiasi H. 2009. A role for the JAK-STAT1 pathway in blocking replication of HSV-1 in dendritic cells and macrophages. Virol J 6:56. doi: 10.1186/1743-422X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mott KR, Osorio Y, Brown DJ, Morishige N, Wahlert A, Jester JV, Ghiasi H. 2007. The corneas of naive mice contain both CD4+ and CD8+ T cells. Mol Vis 13:1802–1812. [PubMed] [Google Scholar]
- 76.Mott KR, Osorio Y, Maguen E, Nesburn AB, Wittek AE, Cai S, Chattopadhyay S, Ghiasi H. 2007. Role of anti-glycoproteins D (anti-gD) and K (anti-gK) IgGs in pathology of herpes stromal keratitis in humans. Invest Ophthalmol Vis Sci 48:2185–2193. doi: 10.1167/iovs.06-1276. [DOI] [PubMed] [Google Scholar]
- 77.Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1992. Expression of herpes simplex virus type 1 glycoprotein B in insect cells. Initial analysis of its biochemical and immunological properties. Virus Res 22:25–39. doi: 10.1016/0168-1702(92)90087-p. [DOI] [PubMed] [Google Scholar]
- 78.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Davison AJ. 2011. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann N Y Acad Sci 1230:E37–E49. doi: 10.1111/j.1749-6632.2011.06358.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.