

WNV is a mosquito-borne flavivirus that poses a threat to human health across large discontinuous areas throughout the world. Infection with WNV results in febrile illness, which can progress to severe neurological disease. Currently, there are no approved treatment options to control WNV infection. Understanding the cellular immune responses that regulate viral replication is important in diversifying the resources available to control WNV. Here, we show that the elimination of TRIM6 in human cells results in an increase in WNV replication and alters the expression and function of other components of the IFN-I pathway through VAMP8. Dissecting the interactions between WNV and host defenses both informs basic molecular virology and promotes the development of host- and virus-targeted antiviral strategies.
KEYWORDS: flavivirus, TRIM6, type I interferon pathway, ubiquitin, VAMP8, West Nile virus, immunology
ABSTRACT
Several members of the tripartite motif (TRIM) family of E3 ubiquitin ligases regulate immune pathways, including the antiviral type I interferon (IFN-I) system. Previously, we demonstrated that TRIM6 is involved in IFN-I induction and signaling. In the absence of TRIM6, optimal IFN-I signaling is reduced, allowing increased replication of interferon-sensitive viruses. Despite having evolved numerous mechanisms to restrict the vertebrate host’s IFN-I response, West Nile virus (WNV) replication is sensitive to pretreatment with IFN-I. However, the regulators and products of the IFN-I pathway that are important in regulating WNV replication are incompletely defined. Consistent with WNV’s sensitivity to IFN-I, we found that in TRIM6 knockout (TRIM6-KO) A549 cells, WNV replication is significantly increased and IFN-I induction and signaling are impaired compared to wild-type (wt) cells. IFN-β pretreatment was more effective in protecting against subsequent WNV infection in wt cells than TRIM6-KO, indicating that TRIM6 contributes to the establishment of an IFN-induced antiviral response against WNV. Using next-generation sequencing, we identified VAMP8 as a potential factor involved in this TRIM6-mediated antiviral response. VAMP8 knockdown resulted in reduced JAK1 and STAT1 phosphorylation and impaired induction of several interferon-stimulated genes (ISGs) following WNV infection or IFN-β treatment. Furthermore, VAMP8-mediated STAT1 phosphorylation required the presence of TRIM6. Therefore, the VAMP8 protein is a novel regulator of IFN-I signaling, and its expression and function are dependent on TRIM6 activity. Overall, these results provide evidence that TRIM6 contributes to the antiviral response against WNV and identify VAMP8 as a novel regulator of the IFN-I system.
IMPORTANCE WNV is a mosquito-borne flavivirus that poses a threat to human health across large discontinuous areas throughout the world. Infection with WNV results in febrile illness, which can progress to severe neurological disease. Currently, there are no approved treatment options to control WNV infection. Understanding the cellular immune responses that regulate viral replication is important in diversifying the resources available to control WNV. Here, we show that the elimination of TRIM6 in human cells results in an increase in WNV replication and alters the expression and function of other components of the IFN-I pathway through VAMP8. Dissecting the interactions between WNV and host defenses both informs basic molecular virology and promotes the development of host- and virus-targeted antiviral strategies.
INTRODUCTION
West Nile virus (WNV) is an enveloped, positive-sense, single-stranded RNA virus and a member of the family Flaviviridae (1, 2). Mosquitoes competent for WNV (predominantly Culex) transmit the virus through blood feeding (3). Enzootic transmission cycles between birds and mosquitoes maintain the virus in the environment, but mosquitoes also incidentally infect humans and other mammals that act as dead-end hosts. In 1999, WNV was introduced to North America and has since become an endemic pathogen, causing annual outbreaks in human populations, and it is the leading cause of mosquito-borne encephalitis (4). Although primarily asymptomatic, WNV infection causes flu-like symptoms in approximately 20% of infected humans, with fewer than 1% of symptomatic cases progressing to neurological manifestations (5). The case fatality rate for symptomatic cases is approximately 10% (1). Currently, no WNV vaccines or antiviral treatments are approved for human use (6–10).
Understanding the molecular mechanisms of WNV replication at the host cellular level, and, specifically, WNV-host type I interferon (IFN-I) interactions, may identify targets for antiviral development. Many groups have demonstrated that interferon-stimulated gene (ISG) products, such as ISG54 (IFIT2) (11), IFITM3 (12), Oas1b (13), and others (reviewed in reference 14), restrict WNV replication. Furthermore, in mouse models of WNV infection, the lack of IFN-I induction through signaling factors such as Toll-like receptor 3 (TLR3) (15), interferon regulatory factor 7 (IRF7) (16), RIG-I (17), IFN-β (18), the type I interferon receptor (IFNAR) (16), STAT1 (19), and IκB kinase ε (IKKε) (11) increases susceptibility to WNV. Mutations in IFN-I pathway genes and ISGs have also been associated with increased disease during WNV infections in humans (20, 21). Despite WNV’s sensitivity to IFN-I, WNV has evolved several mechanisms to antagonize IFN-I, including NS1 interference with RIG-I and MDA5 function (22), NS3 helicase impairment of Oas1b activity (13), NS5 disruption of the IFNAR surface expression (23), STAT1 phosphorylation (24), and subgenomic flavivirus RNA (25). Since WNV’s resistance to IFN-I contributes to virulence (16, 26), defining the IFN-I signaling pathway components required to respond to WNV infection is important in aiding the development of WNV-specific therapies.
Upon WNV infection, pathogen recognition receptors (PRRs), including RIG-I and MDA5, recognize viral RNA (17) and signal through their adaptor MAVS to activate the downstream IKK-like kinases TRAF family member-associated NF-κB activator (TANK) binding kinase 1 (TBK1) and IKKε. The activation of TBK1 and IKKε promotes IFN-I production through the activation of the transcription factors IRF3 and IRF7 (27). IFN-I is then secreted and engages the IFN-I receptor to induce IFN-I signaling. Early in the IFN-I signaling cascade, the kinases Jak1 and Tyk2 phosphorylate STAT1 (Y701) and STAT2 (Y690), which dimerize and interact with IRF9 to form the interferon-stimulated gene factor 3 (ISGF3) complex (28, 29). ISGF3 then translocates to the nucleus, where it interacts with the interferon-stimulated response element (ISRE) present in the promoter of ISGs. In addition, upon IFN-I stimulation, IKKε plays an essential nonredundant role in the phosphorylation of STAT1 on S708, which is required for the induction of IKKε-dependent ISGs (30). Several IKKε-dependent ISGs, including ISG54 (30), are involved in restricting WNV (11).
TRIM6, an E3 ubiquitin (E3-Ub) ligase in the tripartite motif (TRIM) protein family, plays a crucial role in facilitating the activation of the IKKε-dependent branch of the IFN-I signaling pathway (31). In concert with the ubiquitin-activating (E1) enzyme and the ubiquitin-conjugating (E2) enzyme UbE2K, TRIM6 synthesizes unanchored K48-linked polyubiquitin chains that promote the oligomerization and autophosphorylation of IKKε (T501) (31). Following the phosphorylation of T501, IKKε is activated and phosphorylates STAT1 (S708) to promote the transcription of the IKKε-dependent ISGs (30, 31). Due to TRIM6’s role in activating IKKε-dependent IFN-I signaling and the importance of IKKε-specific ISGs in restricting WNV, we hypothesized that WNV replication would be enhanced in the absence of TRIM6.
Here, we show that the WNV viral load is increased and that IFN-I induction and IKKε-dependent IFN-I signaling are impaired in TRIM6 knockout (TRIM6-KO) cells. Next-generation RNA sequencing (NGS) identified several ISGs expressed at lower levels in TRIM6-KO cells than in wild-type (wt) cells, including several ISGs previously described to restrict WNV replication. In addition to ISGs, the absence of TRIM6 also affects the expression of genes not known to regulate WNV replication or IFN-I signaling, including Vamp8. We investigated the role of a TRIM6-dependent gene not previously described to regulate WNV replication or IFN-I signaling, Vamp8. VAMP8 is a vesicle-associated-membrane protein in the SNARE (soluble-N-ethylmaleimide-sensitive factor attachment protein) family known to modulate endocytosis (32), exocytosis of secretory (33–36) and lytic (37) granules, thymus development (38), receptor exocytosis (39), and cross-presentation by antigen-presenting cells (40). We found that VAMP8 does not directly affect WNV replication but promotes optimal IFN-I signaling. Overall, we conclude that TRIM6 is important in promoting optimal IFN-I induction and signaling during WNV infection and that VAMP8 is a novel TRIM6-dependent factor involved in regulating IFN-I signaling.
(This article was submitted to an online preprint archive [41].)
RESULTS
WNV replication is increased in IFN-I induction- and signaling-impaired TRIM6-KO cells.
To test our hypothesis that the absence of TRIM6 facilitates WNV replication, growth kinetics at different multiplicities of infection (MOIs) were determined in wt and TRIM6-KO A549 cells. A significant increase in viral replication was detected in TRIM6-KO cells at 48 h postinfection (p.i.) in comparison to wt cells when infections were performed at both low (0.1 PFU/cell) and high (5.0 PFU/cell) MOIs (Fig. 1A and B). To address the effect of the absence of TRIM6 on the IFN-I pathway, protein expression and phosphorylation of IFN-I pathway components in WNV-infected cells were assessed (Fig. 1C to I). The activation of both TBK1 and IKKε kinases is known to contribute to efficient IRF3 phosphorylation and IFN-I induction. While the levels of phosphorylated TBK1 (pTBK1) (S172) were substantially higher in TRIM6-KO than in wt cells at late time points p.i. (Fig. 1C and D), the TRIM6-dependent phosphorylation on IKKε (T501) was significantly attenuated in TRIM6-KO cells (Fig. 1C and F), as we previously reported in a model of influenza virus infection (31). Although less striking, phosphorylation on IKKε (S172) was also reduced in TRIM6-KO cells (Fig. 1C and E). These data suggest that TRIM6 is important for promoting the efficient activation of IKKε but not TBK1 in the context of WNV infection.
FIG 1.

Increased WNV replication in TRIM6-KO cells is associated with impaired IFN-I induction and signaling. TRIM6-KO or wt A549 cells were infected with WNV 385-99 at an MOI of 0.1 (A, C to I, and K) or 5.0 (B, J, and L). (A and B) The viral load in supernatants of infected cells was measured by a plaque assay on Vero ATCC CCL-81 cells. (C) Whole-cell lysates from WNV (MOI of 0.1)-infected cells were run on Western blots for analysis of protein expression and phosphorylation. (D to I) The densitometry area under the curve (AUC) for each band was calculated using Fiji (59). For the phosphorylated proteins, the AUC for the respective total protein was normalized to the actin AUC (AUC total/AUC actin), and the AUC for the phosphorylated protein was then normalized to the total/actin ratio. The AUC reported is the average from three separate measurements. (J) RNA isolated from mock- and WNV-infected cells was isolated to assess gene expression of Ifnb; the ISGs Isg54, Oas1, Irf7, and Stat1; and a non-IFN-I-regulated gene, Il6. Change in expression is represented as fold induction. (K and L) IFN-β was measured via an ELISA of irradiated supernatants infected with WNV at MOIs of 0.1 (K) and 5.0 (L). Error bars represent standard deviations (n = 3). For statistical analysis, two-way ANOVA with Tukey’s posttest for multiple comparisons was used (****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05). All experiments were performed in triplicate, and immunoblots are for representative samples. All experiments were repeated at least 2 times. LOD, limit of detection.
Although the amount of pTBK1 (S172) is increased in TRIM6-KO cells, the level of phosphorylation of IRF3 (S386) is lower in TRIM6-KO than in wt cells (Fig. 1C and G), suggesting that TBK1 cannot completely compensate for reduced IKKε activity under these conditions of WNV infection. In line with impaired IRF3 activation in TRIM6-KO cells, Ifnb mRNA levels were reduced in TRIM6-KO cells only at early time points postinfection (6 h p.i.) (Fig. 1J). However, the expression levels of Ifnb in TRIM6-KO cells significantly increased over time compared to those in wt cells and correlated with the increased virus titers at 48 h p.i. (Fig. 1J). Despite the increase in Ifnb mRNA levels at 48 h p.i. in TRIM6-KO cells, the amount of secreted IFN-β protein was significantly decreased in TRIM6-KO compared to wt cells (Fig. 1K and L).
Consistent with the reduced IFN-β protein accumulation in supernatants of infected TRIM6-KO cells, the amount of pSTAT1 (Y701) is lower in TRIM6-KO cells than in wt cells at all time points evaluated (Fig. 1C and H). In contrast, the expression of total STAT1 is substantially increased in TRIM6-KO cells compared to wt cells, which is consistent with the reported accumulation of unphosphorylated STAT1 in IKKε-KO cells (42). Phosphorylation on STAT1 (S708), an IKKε- and TRIM6-dependent modification (30, 31), is nearly undetectable in the TRIM6-KO cells upon WNV infection (Fig. 1C and I).
Consistent with this defect in the TRIM6-IKKε branch of the IFN-I signaling pathway in TRIM6-KO cells, the TRIM6/IKKε-dependent ISGs Isg54 and Oas1 (30, 31) have different patterns of induction. In the case of Isg54, the level of induction is significantly lower in TRIM6-KO cells than in wt cells at 24 h p.i., but this pattern is reversed at 72 h p.i., mirroring Ifnb expression (Fig. 1J). In contrast, the induction of Oas1 is attenuated in TRIM6-KO cells at 24 to 72 h p.i. (Fig. 1J). The IKKε-independent ISGs Irf7 and Stat1 and the non-ISG Il6 are expressed at higher levels in TRIM6-KO cells at later time points (Fig. 1J), again in correlation with Ifnb induction. As pIKKε (T501) plays a nonredundant role in facilitating the expression of multiple ISGs (30, 31) that are known to be important in restricting WNV replication (11, 13, 43), the reduced capacity of TRIM6-KO cells to phosphorylate this residue likely contributes to the impaired antiviral activity against WNV. Overall, the absence of TRIM6 augments WNV replication and impairs the IKKε-dependent branch of the IFN-I pathway, in line with our previous findings with other viruses, including influenza A virus (IAV), Sendai virus (SeV), and encephalomyocarditis virus (EMCV) (31).
IFN-I has reduced anti-WNV activity in TRIM6-deficient cells.
Next, we sought to evaluate further the impact of TRIM6 on the antiviral efficiency of IFN-I against WNV. Prior to infection, wt and TRIM6-KO A549 cells were treated with 100 U of recombinant human IFN-β for 4 h prior to infection with WNV (MOI of 5.0) for 24 h (Fig. 2). Pretreatment with IFN-β decreased viral loads in both wt and TRIM6-KO cells; however, IFN-I pretreatment was significantly less effective in inhibiting WNV replication in TRIM6-KO cells (40-fold) than in wt controls (63-fold). As expected, this result indicates that IFN-I signaling is suboptimal in the absence of TRIM6, enabling WNV to replicate to higher titers, and suggests that the expression of TRIM6-dependent ISGs may be involved in establishing an optimal IFN-I-mediated anti-WNV response.
FIG 2.
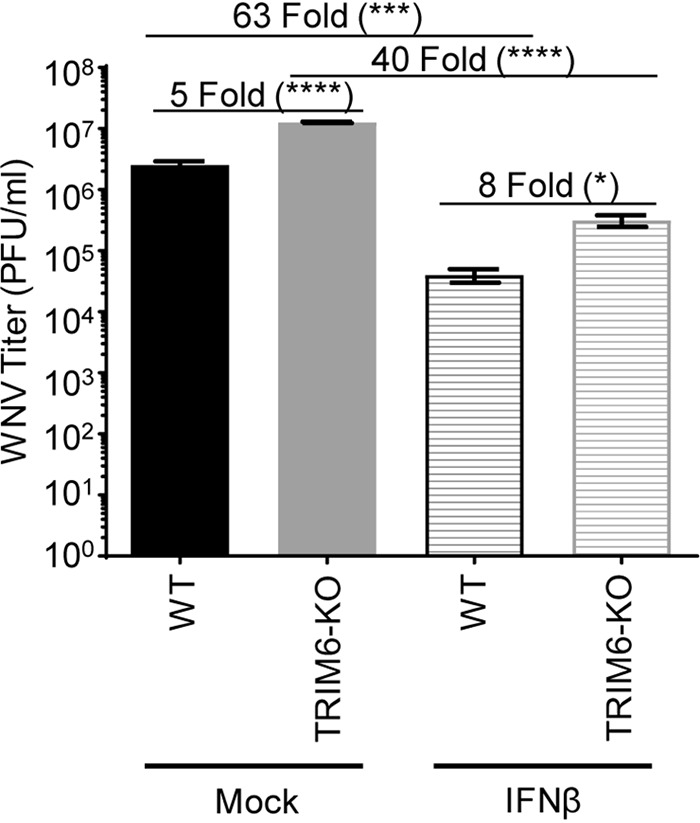
IFN-I pretreatment is less efficient in antagonizing WNV replication in TRIM6-KO cells. TRIM6-KO or wt A549 cells were treated with recombinant human IFN-β-1a (100 U) for 4 h prior to infection with WNV 385-99 (MOI of 5.0) for 24 h. Supernatants from infected cells were titrated, and the viral load was calculated via a plaque assay. Error bars represent standard deviations (n = 3). One-way ANOVA with Tukey’s posttest was performed to assess statistical significance (****, P < 0.0001; *, P < 0.05). Fold changes are reported in parentheses. All experiments were performed in triplicate.
VAMP8 is induced in a TRIM6-dependent manner.
To identify other genes affected as a consequence of TRIM6’s absence, NGS of mock-infected (Fig. 3A) and WNV-infected (Fig. 3B) wt and TRIM6-KO cells was performed. During WNV infection, canonical ISGs were identified as being expressed at lower levels in TRIM6-KO than in wt cells, which validates the methodology (Fig. 3B). Several canonical ISGs downregulated in TRIM6-KO cells have previously been described to inhibit WNV replication, including Ifitm2 (43) and Ifitm3 (12), or their loss of function is associated with increased WNV susceptibility, including Mx1 (21, 44) and OasL (44). We elected to investigate other genes not previously described to regulate WNV replication or IFN-I pathways. One of the most strongly downregulated genes in both mock and infected cells, VAMP8, was chosen as a target for further mechanistic validation (Fig. 3A and B). Although VAMP8 has previously been noted to play an antiviral role in response to Japanese encephalitis virus (JEV) (45), the mechanism has not been reported. Furthermore, VAMP8’s described roles are diverse, but a connection to the antiviral IFN-I pathway has not been described.
FIG 3.

Transcription of canonical interferon-stimulated genes (ISGs) and VAMP8 is downregulated in TRIM6 knockout cells. (A and B) Transcriptional profiling of cellular mRNA by next-generation sequencing of mock-infected (A) or WNV 385-99-infected (MOI of 5.0) (B) wt or TRIM6-KO A549 cells at 24 h postinfection. The log2-fold change was calculated as TRIM6-KO/wt, with genes downregulated in TRIM6-KO cells on the left (negative values) and those upregulated in TRIM6-KO cells on the right (positive values). The −log10 P value represents the significance. VAMP8 data points are represented as light gray squares, and ISGs are represented as dark gray triangles. (C and D) Validation of VAMP8 expression at the protein (immunoblotting) (C) or RNA (RT-qPCR) (D) levels in wt or TRIM6-KO cells. Error bars represent standard deviations (n = 3), and VAMP8 expression validation experiments were performed in triplicate and repeated three times.
After confirming the lower expression of VAMP8 at the translational (Fig. 3C) and transcriptional (Fig. 3D) levels in TRIM6-KO cells, the role of VAMP8 in regulating WNV replication was interrogated. Therefore, wt A549 cells were transfected with a VAMP8-targeting small interfering RNA (siRNA) pool (siVAMP8) or nontargeting control (NTC) siRNA (siControl) for 24 h prior to WNV infection (MOI of 0.1). VAMP8 knockdown (VAMP8-kd) had no measurable effect on WNV replication (Fig. 4A), suggesting that VAMP8 does not have a direct anti-WNV function. VAMP8 knockdown was validated by Western blotting, showing undetectable levels of protein, with a clear upregulation of VAMP8 protein by 24 h p.i. in NTC-transfected cells (Fig. 4B). Upon infection, phosphorylation of IRF3 in VAMP8-kd cells was not significantly different from that in NTC cells, suggesting that VAMP8 is not involved in the IFN-I induction arm of the pathway (Fig. 4B and C). In contrast, the amount of pSTAT1 (Y701) was notably smaller in the VAMP8-kd cells, suggesting impairment in the IFN-I signaling pathway (Fig. 4B and D).
FIG 4.

Depletion of VAMP8 impairs STAT1 phosphorylation downstream of IFN-I signaling but does not alter WNV replication. (A and B) wt A549 cells were treated with nontargeting control (control) or VAMP8-targeting (VAMP8) siRNAs for 24 h, followed by infection with WNV 385-99 (MOI of 0.1) for 72 h. Supernatants and lysates of infected cells were collected at 1, 6, 24, 48, and 72 h p.i. to assess viral loads by plaque assays (A) and protein expression and phosphorylation by Western blotting (B). (C and D) The bands in the Western blot were quantified by densitometry as described in the legend of Fig. 1. (E to J) wt A549 (E to I) or ATCC HTB-15 (J) cells were treated with nontargeting control (siControl) or VAMP8-targeting (siVAMP8) siRNAs for 24 h, followed by treatment with recombinant human IFN-β-1a (500 U/ml) (E, F, I, and J) or human IFN-γ (500 U/ml) (G and H). IFN treatments shown in panels E, F, I, and J were done for 16 h. Cells were lysed, and either protein (E to H) or RNA (I and J) was isolated for analysis by Western blotting or qRT-PCR, respectively. Error bars represent standard deviations. Gene expression data were analyzed using one-way ANOVA with Tukey’s posttest to assess statistical significance (I and J) (****, P < 0.0001; **, P < 0.01. No statistical significance was found in panel A. The experiment was performed in triplicate.
To further confirm whether VAMP8 is involved in the regulation of the IFN-I signaling pathway in the absence of virus infection, wt A549 cells were transfected with VAMP8-targeting or NTC siRNAs, followed by treatment with IFN-β for 16 h. As expected, total STAT1 was induced in both VAMP8 and NTC siRNA-treated cells following IFN-β stimulation, but the level of total STAT1 in VAMP8-kd cells was slightly attenuated (Fig. 4E). VAMP8’s effect on STAT1 activation is more evident, however, with a strong reduction in the amount of pSTAT1 (Y701) (Fig. 4E and F). To evaluate whether the observed impairment in pSTAT1 (Y701) is specific for type I IFN signaling, we treated NTC and VAMP8-kd cells with type II interferon (IFN-γ) for 16 h. In contrast to the observed defect in the response to IFN-β, there were no detectable differences in pSTAT (Y701) levels between siControl- and siVAMP8-treated cells following IFN-γ stimulation (Fig. 4G and H). Consistent with a potential role of VAMP8 in regulating STAT1 phosphorylation downstream of IFN-I signaling, mRNA expression levels upon IFN-β treatment of ISGs, including Isg54, Oas1, and Stat1, were significantly reduced in VAMP8-kd cells compared to controls for both A549 cells (Fig. 4I) and a brain-derived cell line (ATCC HTB-15 glioblastoma cells) (Fig. 4J).
In an effort to determine whether VAMP8 functions at the level of STAT1 or upstream in the pathway, we assessed the effects of VAMP8 on JAK1 activation, which is responsible for the phosphorylation of STAT1 on Y701. In the absence of VAMP8, phosphorylation of JAK1 (pY1034/1035) was reduced compared to that in control cells (Fig. 5A and B). Furthermore, ectopic expression of FLAG-tagged VAMP8 in HEK293T cells enhanced pJAK1 following IFN-β stimulation compared to empty-vector-transfected cells (Fig. 5C and D). Overall, the above-described evidence supports that (i) VAMP8 expression is TRIM6 dependent, (ii) VAMP8 does not directly affect WNV replication, and (iii) VAMP8 is involved in positive regulation of IFN-I signaling on or upstream of JAK1 phosphorylation.
FIG 5.

VAMP8 regulates JAK1 phosphorylation downstream of IFN-I signaling. (A and B) wt A549 cells were treated with nontargeting control (control) or VAMP8-targeting (VAMP8) siRNAs for 24 h, followed by treatment with recombinant human IFN-β-1a (500 U/ml) for 0, 15, and 30 min. (C and D) FLAG-tagged VAMP8 or an empty vector was transfected into HEK293T cells for 24 h prior to treatment with human IFNβ-1a (1,000 U/ml) for 1 h, and protein lysates were collected to assess JAK1 activation. The bands in the Western blot were quantified by densitometry (B and D) as described in the legend of Fig. 1.
VAMP8 knockdown enhances WNV replication in cells pretreated with type I IFN.
Since VAMP8 modulates the IFN-I system but does not appear to alter WNV replication, we examined whether exogenous IFN-I pretreatment would reveal a functional defect in IFN-I signaling in VAMP8-kd cells during WNV infection. Prior to infection, wt A549 or brain-derived ATCC HTB-15 cells were treated with siRNA (VAMP8 or NTC), followed by IFN-β treatment. Although IFN-β pretreatment reduced viral production in both groups, it was less efficient in protecting VAMP8-kd cells against WNV replication than NTC cells (508-fold for NTC siRNA and 79-fold for VAMP8 siRNA in A549 cells [Fig. 6A]; 430-fold for NTC siRNA and 20-fold for VAMP8 siRNA in ATCC HTB-15 cells [Fig. 6B]). As opposed to previous experiments showing no impact on WNV replication following VAMP8-kd, the combination of IFN-β pretreatment and VAMP8 siRNA showed an 8-fold (A549) (Fig. 6A) or 22-fold (ATCC HTB-15) (Fig. 6B) increase in the replication of WNV over cells treated with NTC siRNA and IFN-β. This result suggests that VAMP8 plays a functional role in IFN-I signaling during WNV infection.
FIG 6.

VAMP8 is important for efficient establishment of an anti-WNV response mediated by IFN-β. Wild-type A549 (A) or ATCC HTB-15 (B) cells were treated with nontargeting control (siControl) or VAMP8-targeting (siVAMP8) siRNAs for 24 h and then treated with recombinant human IFN-β-1a (500 U/ml) for 16 h prior to infection with WNV 385-99 (MOI of 5.0) for 24 h. Supernatants from infected cells were titrated, and the viral load was calculated via a plaque assay. Error bars represent standard deviations. One-way ANOVA with Tukey’s posttest was performed to assess statistical significance (****, P < 0.0001; *, P < 0.05). Fold changes are reported in parentheses. The experiment was completed in triplicate.
VAMP8 overexpression attenuates WNV replication.
To further investigate whether VAMP8 overexpression can induce an anti-WNV response and whether this requires the presence of TRIM6, we transfected wt or TRIM6-KO A549 cells with either an empty vector (pCAGGS) or FLAG-tagged VAMP8 (FLAG-VAMP8) 30 h prior to infection with WNV (MOI of 5.0). As predicted, in wt A549 cells, the viral load was significantly decreased (8.5-fold) in cells overexpressing FLAG-VAMP8 compared to the empty vector control (Fig. 7A). In contrast, when FLAG-VAMP8 was overexpressed in TRIM6-KO cells, the viral load was not significantly different from that in cells transfected with the empty vector (Fig. 7A). This result suggests that VAMP8’s attenuation of WNV replication requires TRIM6 activity and can be explained by VAMP8’s inability to compensate for the defect in pSTAT1 (S708) in TRIM6-KO cells (Fig. 7B and C). This phosphorylation on S708 of STAT1 is required for the stabilization of the ISGF3 complex (42) and induction of the complete set of ISGs (30), including ISGs known to be involved in the anti-WNV response (11, 13, 43). Therefore, while VAMP8 is responsible for promoting STAT1 (Y701) phosphorylation, which can account for only limited antiviral effects, induction of the complete set of anti-WNV ISGs does not occur unless the TRIM6-IKKε-STAT1-S708 arm of the pathway is active. To rule out a potential role of TRIM6 in VAMP8-mediated promotion of STAT1 (Y701) phosphorylation, we overexpressed FLAG-VAMP8 in wt or TRIM6-KO A549 cells prior to treatment with IFN-β for 16 h. Unexpectedly, overexpression of VAMP8 induced pSTAT1 (Y701) following IFN-β stimulation in wt but not TRIM6-KO A549 cells (Fig. 7D and E), suggesting that TRIM6 is required for not only VAMP8 expression (as described above) (Fig. 3) but also VAMP8 activity. To further test this possibility, we investigated whether TRIM6 interacts with VAMP8. Coimmunoprecipitation (coIP) assays showed that FLAG-VAMP8 coprecipitated hemagglutinin (HA)-TRIM6 (Fig. 7F) and that HA-TRIM6 coprecipitated FLAG-VAMP8 in the reverse coIP (Fig. 7G). Taken together, these data suggest that TRIM6 is important for VAMP8 expression levels as well as VAMP8 activity downstream of the IFN-I receptor and is also important for an optimal anti-WNV response.
FIG 7.

VAMP8 overexpression attenuates WNV replication. (A to E) Wild-type or TRIM6-KO A549 cells were transfected with 250 ng of an empty vector or FLAG-VAMP8 for 30 h and then infected with WNV 385-99 at an MOI of 5.0 for 24 h (A to C) or treated with IFN-β (500 U/ml) for 16 h (D and E). (A) Supernatants from infected cells were titrated, and the viral load was calculated via a plaque assay. (B and D) Protein lysates were collected to measure STAT1 activation [pSTAT1 (S708) (B) or pSTAT1 (Y701) (D)] and to confirm equal levels of VAMP8 overexpression in wt and TRIM6-KO cells. (C and E) The bands in the Western blots were quantified by densitometry as described in the legend of Fig. 1. (F and G) FLAG-tagged VAMP8 was cotransfected with an empty vector or HA-tagged TRIM6 into HEK293T cells for 24 h. Protein lysates from the cotransfected cells were immunoprecipitated (IP) with either anti-FLAG (F) or anti-HA (G) beads overnight. Whole-cell lysates (WCE) and IP samples were immunoblotted to assess expression and the VAMP8-TRIM6 interaction. Error bars represent standard deviations. Student’s t test was used to assess statistical significance (***, P < 0.001; **, P < 0.01; ns, no significance). The experiment was performed in triplicate.
DISCUSSION
Our study demonstrates the relevance of TRIM6 in regulating the IFN-I pathway during WNV infection and identifies VAMP8 as a factor functionally involved in IFN-I signaling. Extensive research has implicated the TRIM family of proteins in both the regulation of the innate immune response and viral replication (46–51). Specifically, TRIM6 has been shown to facilitate the formation of unanchored K48-linked polyubiquitin chains that provide a scaffold for IKKε binding, ultimately resulting in IKKε activation and STAT1 phosphorylation at S708 (31). Phosphorylation of STAT1 at S708 is important to sustain IFN-I signaling and to express a unique subset of ISGs (30, 42). The relevance of IKKε-dependent gene expression has previously been described for WNV, and in the absence of ISG54 or IKKε, mice have an increased susceptibility (11). These experiments served as a rationale for exploring the functional role of TRIM6 during WNV infection.
As expected, we observed an increase in WNV replication in TRIM6-KO cells in parallel with attenuated TRIM6-dependent activation of IKKε (T501 phosphorylation), IKKε-dependent STAT1 (S708) phosphorylation, and IKKε-dependent gene expression. There was impaired Ifnb and Isg54 mRNA induction in TRIM6-KO cells at 6 and 24 h p.i., respectively, but higher levels of induction at 72 h p.i. The transient effect of TRIM6 on the IFN-I pathway may be related to the increase in pTBK1 (S172) in TRIM6-KO cells compensating for the impaired IKKε-dependent response to WNV infection. Another possible explanation is that the ISGs that are present at higher levels in TRIM6-KO cells either are pSTAT1 (S708) independent (STAT1 and IRF7 [30]) or can be induced directly via IRF3 or IRF7 by binding their promoters (e.g., ISG54 [52]), while Oas1 is a TRIM6-IKKε-pSTAT1 (S708)-dependent ISG (30, 31) that is absent in TRIM6-KO cells. Furthermore, redundant or alternative pathways not investigated here may be enacted in the TRIM6-KO cells to control WNV replication. Although no other factor has been shown to synthesize the unanchored K48-linked polyubiquitin chains required for IKKε activation, we cannot exclude that other TRIM members or other E3-Ub ligases may compensate for the loss of TRIM6. Alternatively, TRIM6 may play important roles in other pathways (i.e., NF-κB), resulting in cytokine dysregulation and/or the induction of IFN-β by TRIM6-independent pathways. Furthermore, emergent WNV strains encode a functional 2-O-methylase in their nonstructural protein 5 that prohibits IFIT proteins, specifically murine ISG54 and human ISG58, from suppressing viral mRNA expression (53). Since WNV antagonizes components of this pathway, we cannot rule out the possibility that WNV proteins target TRIM6 to impede the IKKε-dependent expression of WNV-restricting ISGs. For example, the matrix protein of Nipah virus (family Paramyxoviridae) works to promote the degradation of TRIM6 during viral infection to promote viral replication through impaired IKKε signaling and, thus, causes a blunted IFN-I response (47). WNV protein antagonism of TRIM6 could also preclude observing more severe differences in WNV replication between wt and TRIM6-KO cells. Alternatively, TRIM6 could play an essential role in IKKε-dependent signaling but could also be hijacked by a virus to facilitate viral replication. In a previous study, we showed that TRIM6 directly promotes the replication of ebolavirus (family Filoviridae) through interactions with VP35 and that VP35 antagonizes TRIM6’s capacity to promote IFN-I signaling (46).
Importantly, although the experiments presented here were performed with cell lines, our previous studies suggest that TRIM6 plays an important antiviral role via the IFN-I system in primary human monocyte-derived dendritic cells (hMDDC) and the antiviral response to two other viruses, SeV and EMCV, and to IAV in lungs of mice (31). Therefore, the effects of TRIM6 are not restricted to a particular cell line or to WNV.
Although we identified several ISGs differentially expressed in TRIM6-KO compared to wt cells following WNV infection, we also identified Vamp8 to be significantly downregulated both under basal conditions and during WNV infection. VAMP8 has not been previously described to affect WNV replication or the IFN-I pathway. Although its role was not described, VAMP8 had been identified as an antiviral factor in an siRNA screen for JEV, another mosquito-borne flavivirus (45). Here, we show that, in contrast to what was seen with JEV, VAMP8 knockdown does not directly affect WNV replication. The impairment of STAT1 (Y701) phosphorylation during WNV infection in VAMP8-kd cells lent evidence that VAMP8 could be involved in IFN-I signaling. Since WNV efficiently impairs IFN-I induction until nearly 24 h p.i., VAMP8 depletion may not substantially impede IFN-I signaling during WNV infection in a tissue culture system. Evaluation of VAMP8’s role in IFN-I signaling in the absence of WNV infection revealed a striking impairment in STAT1 (Y701) phosphorylation and modest inhibition of ISG expression in VAMP8-depleted cells. The impairment of STAT1 activation (Y701) in the absence of VAMP8 occurs specifically downstream of IFN-I but not IFN-II signaling, and a delay and reduction of JAK1 activation may contribute to this phenotype. Furthermore, following IFN-β treatment, VAMP8 knockdown less efficiently inhibited WNV replication, which provides support that VAMP8 mediates a functional step in the IFN-I signaling pathway. We also show that VAMP8 overexpression modestly inhibits WNV replication in wt but not TRIM6-KO cells, which suggests that both VAMP8 and TRIM6 are necessary for the induction of the full set of antiviral ISGs. There are two possible explanations for these results. First, perhaps VAMP8 is important for efficient phosphorylation on STAT1 (Y701) [independent of STAT1 (S708) phosphorylation by the TRIM6-IKKε arm], and in parallel, the TRIM6-IKKε arm is required for phosphorylation on STAT1-S708. Both of these events would be necessary for the efficient induction of the full set of ISGs and antiviral activity. Alternatively, TRIM6 could be important for directly regulating VAMP8-mediated JAK1-STAT1 (Y701) phosphorylation and activation, in addition to promoting IKKε-dependent STAT1 (S708) phosphorylation. We favor the latter explanation because VAMP8 requires the presence of TRIM6 to attenuate WNV replication (Fig. 7A) and to induce IFN-dependent STAT1 (Y701) phosphorylation (Fig. 7D and E). In addition, TRIM6 interacts with VAMP8 (Fig. 7F and G), and TRIM6 deficiency impedes the induction of STAT1 (S708) phosphorylation independent of VAMP8 expression (Fig. 7B and C).
Although our results suggest that TRIM6 and VAMP8 interact and that VAMP8’s regulation of the IFN-I pathway is TRIM6 dependent, the detailed mechanism of how VAMP8 promotes JAK1 activation downstream of the IFN-I receptor is unknown. VAMP8 is involved in endocytosis (32), vesicle-vesicle fusion (35), and exocytosis (33, 34, 36) in various cell types, including leukocytes (37, 40) and various secretory cells (33, 34, 36), including human lung goblet cells. As a vesicular SNARE (v-SNARE), VAMP8 on the surface of a vesicle interacts with SNAREs on the target membrane surface to facilitate membrane fusion (33, 35, 37). Potential mechanisms of the IFN-I pathway, which VAMP8 may regulate, include surface expression of the IFNAR or recycling of receptor components to the plasma membrane. Although we did not find an interaction of VAMP8 with the IFNAR or JAK1 (data not shown), it is still possible that VAMP8 could regulate JAK1 activity or IFNAR function. VAMP8 has been described to regulate the surface expression of a water transport channel, aquaporin 2, in the kidney (39). Despite the reduced surface expression, the total amount of aquaporin 2 is larger in the cell, but it is retained in vesicles below the plasma membrane (39). Alternatively, VAMP8 might influence the secretion of factors or the oxidative condition of the microenvironment important to maintain IFN-I signaling. In phagocytic cells infected with Leishmania, VAMP8 regulates the transport of NADPH oxidase to the phagosome to facilitate optimal conditions for peptide loading into major histocompatibility complex (MHC) class I molecules (40). Although VAMP8 would not be regulating phagocytosis in this model of WNV infection, it is possible that VAMP8 regulates NADPH oxidase localization, thereby affecting the oxidative environment of the infected cell and, consequently, IFN-I signaling (54–56). TRIM6 may either directly affect VAMP8 expression or act indirectly through an as-yet-unidentified secondary factor. Knowing that TRIM6 is important in regulating IKKε activation and function (31), we assessed whether IKKε alters VAMP8 expression. There was no difference in VAMP8 expression between IKKε-wt and -KO murine embryonic fibroblasts under basal conditions (Fig. 8A). We did identify two transcription factors, HNF4α and HNF4γ, with binding sites upstream of VAMP8’s transcription initiation site and within a VAMP8 intron, that have significantly lower gene expression levels in TRIM6-KO than in wt A549 cells (Fig. 8B to D). The expression of wt TRIM6, but not the TRIM6 catalytic mutant (C15A), partially rescues the expression of VAMP8 in TRIM6-KO A549 cells, suggesting that the presence of TRIM6 and its ubiquitin ligase activity are required for VAMP8 expression (Fig. 8E). Our study indicates a new role for VAMP8 in the IFN-I pathway, which is regulated by TRIM6 during viral infection (Fig. 9).
FIG 8.

VAMP8 expression is associated with the presence of TRIM6 but is IKKε independent. (A) Protein lysates from wt (+/+) or IKBKE knockout (−/−) murine embryonic fibroblasts were collected under basal conditions to evaluate the expression of IKKε, VAMP8, and actin via immunoblotting. (B and C) Next-generation sequencing of mRNA collected from mock-infected wt or TRIM6 KO A549 cells. Analysis of the experiment is shown in Fig. 3. The gene expression levels of two transcription factors, HNF4α (B) and HNF4γ (C), with binding sites within the VAMP8 gene region (GenBank accession number NG_022887.1) are presented. FPKM, fragments per kilobase per million. (D) Schematic showing the HNF4α and -γ binding sites within the VAMP8 gene (GenBank accession number NG_022887.1) related to the transcript (GenBank accession number NM_003761.5). The binding sites identified in the VAMP8 genetic region were compared to the consensus sequences of HNF4α (MA0114.1; http://jaspar.genereg.net/matrix/MA0114.1/) and HNF4γ (MA0484.1; http://jaspar.genereg.net/matrix/MA0484.1/) available through JASPAR (2018). nt, nucleotides. (E) The overexpression of wt TRIM6, but not the catalytic mutant (C15A), in TRIM6-KO A549 cells partially rescues the protein expression of VAMP8 to the levels observed in wt A549 cells.
FIG 9.

Graphical summary. Following virus infection, viral RNA is recognized by pathogen recognition receptors (PRRs). PRRs then signal through their adaptors, triggering the activation of the kinases TBK1 and IKKε, which phosphorylate and activate the transcription factor IRF3. Once activated, IRF3 translocates to the nucleus and, in concert with other factors not indicated, promotes the transcription of IFN-β. IFN-β is then secreted and signals in an autocrine or paracrine manner through the type I IFN receptor (IFNAR). The kinases (JAK1 and Tyk2) associated with the IFNAR then facilitate the phosphorylation of STAT1 at tyrosine 701 (Y701) and STAT2 in an IKKε-independent manner. Phosphorylated STAT1 and STAT2 interact with IRF9 to form the ISGF3 complex, which translocates to the nucleus to promote the transcription of genes with interferon-stimulated response elements (ISREs), including Stat1, Oas1, and Isg54. In addition to IKKε-independent IFN-I signaling, the E3 ubiquitin ligase TRIM6 facilitates IKKε-dependent IFN-I signaling. TRIM6, in coordination with the ubiquitin-activating (UbE1) and -ligating (UbE2K) enzymes, facilitates the formation of K48-linked unanchored polyubiquitin chains, which act as a scaffold for the oligomerization and cross-phosphorylation of IKKε at threonine 501 (T501) (30). TRIM6 also facilitates the activation of IKKε during IFN-I induction. During IFN-I signaling, activated IKKε phosphorylates STAT1 at serine 708 (S708). STAT1 phosphorylation at S708, an IKKε-dependent modification, facilitates the formation of an ISGF3 complex with different biophysiological properties, which allows the ISGF3 complex to have enhanced binding to certain ISRE-containing promoters, ultimately inducing the complete ISG profile. When STAT1 is phosphorylated only at Y701 (in the absence of IKKε and/or TRIM6), IFN-I signaling results in the induction of a different and incomplete ISG profile (30, 31, 42). Although the mechanism is currently unknown (question mark), TRIM6 induces VAMP8 expression and VAMP8 activity. VAMP8, in turn, is important for the optimal activation of JAK1 and, subsequently, STAT1 (Y701) required for an efficient antiviral response.
Elucidating the interactions of the human immune system with viral infection is essential to understanding viral pathology as well as identifying cellular targets for antiviral drug development. Our work has identified a novel IFN-I-related host factor that is important in the regulation of WNV replication and in the life cycles of other viruses. This may provide a conserved target for the development of antiviral strategies and for the elucidation of further conserved pathways in host-pathogen interactions.
MATERIALS AND METHODS
Viruses and cells.
West Nile virus (WNV) isolate 385-99 was obtained from the World Reference Center for Emerging Viruses and Arboviruses (Robert Tesh, University of Texas Medical Branch [UTMB]). A549, HEK293T, ATCC HTB-15 (U-118MG), and ATCC CCL-81 lines were obtained from the American Type Culture Collection. Wild-type and IKBKE−/− murine embryonic fibroblasts were a kind gift from Benjamin tenOever at the Icahn School of Medicine at Mount Sinai and were described previously (30, 31). TRIM6 knockout cells were prepared as previously described (46). All lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals). Infections were performed in DMEM supplemented with 2% FBS and 1% penicillin-streptomycin (Gibco). For growth kinetics experiments, 150,000 wt or TRIM6-KO A549 or ATCC HTB-15 cells/well were infected with 100 μl of WNV at a multiplicity of infection (MOI) of 0.1 or 5.0 for 1 h at 37°C with 5% CO2, the inoculum was then removed, and cells were washed 3 times with 1 ml of 1× phosphate-buffered saline (PBS). After the cells were washed, 1 ml of DMEM supplemented with 2% FBS was added to each well. The supernatant (150 μl) was collected at the designated time points for plaque assays. Plaque assays were performed in 12-well plates containing 200,000 ATCC CCL-81 cells/well. Viral samples were diluted log10-fold and applied to the monolayer. Following 1 h in a humidified incubator at 37°C with 5% CO2, a semisolid overlay containing minimal essential medium (MEM), 2% FBS, 1% penicillin-streptomycin, and 0.8% tragacanth (Sigma-Aldrich) was applied. The overlay was removed after 72 h, and monolayers were fixed and stained with 10% neutral buffered formalin (Thermo Fisher Scientific) and crystal violet (Sigma-Aldrich). Plaques were enumerated by counting and graphed. All manipulations of infectious WNV were performed in biological safety level 3 (BSL-3) facilities at UTMB.
IFN treatment.
Cells were treated with either 100 U (wt versus TRIM6-KO) or 500 U (VAMP8) of recombinant human IFN-β-1a (PBL Assay Science) or 500 U/ml of IFN-γ (PBL Interferon Source) for 15 and 30 min to assess JAK1 activation, 16 h to investigate STAT1 activation, and 4 h or 16 h prior to WNV infection.
RNA isolation and quantitative real-time PCR (qRT-PCR).
At the indicated time points per experiment, medium was removed from the cell monolayer, and 1 ml TRIzol reagent (Thermo Fisher Scientific) was added. RNA was isolated using Zymo Direct-zol RNA miniprep kits, according to the manufacturer’s instructions, with in-column DNase treatment. Isolated RNA was then reverse transcribed using the high-capacity cDNA reverse transcription kit (Applied Biosystems). The cDNA was then diluted 1:3 in nuclease-free water (Corning). Relative gene expression levels (primers 18S F [GTAACCCGTTGAACCCCATT], 18S R [CCATCCAATCGGTAGTAGCG], Ifnb F [TCTGGCACAACAGGTAGTAGGC] Ifnb R [GAGAAGCACAACAGGAGAGCAA], Il6 F [AGAGGCACTGGCAGAAAACAAC], Il6 R [AGGCAAGTCTCCTCATTGAATCC], Irf7 F [CGCGGCACTAACGACAGGCGAG], Irf7 R [GCTGCCGTGCCCGGAATTCCAC], Isg54 F [ATGTGCAACCCTACTGGCCTAT], Isg54 R [TGAGAGTCGGCCCAGTGATA], Oas1 F [GATCTCAGAAATACCCCAGCCA], Oas1 R [AGCTACCTCGGAAGCAGGTT], Stat1 F [ACAGCAGAGCGCCTGTATTG], Stat1 R [CAGCTGATCCAAGCAAGCAT], Vamp8 F [CTTGGAACATCTCCGCAACA], and Vamp8 R [CGCTGAACACAGAACTTGAG]) were determined using iTaq universal SYBR green (Bio-Rad) with the CFX384 instrument (Bio-Rad). The relative mRNA expression levels were analyzed using CFX Manager software (Bio-Rad). The change in the threshold cycle (ΔCT) was calculated, with the 18S gene serving as the reference mRNA for normalization. When indicated, the fold change was calculated by dividing the value for the treated sample by that of its respective mock.
RNA sequencing and analysis.
A549 (wt and TRIM6KO) cells were infected at a high MOI (5.0), and RNA was isolated at 24 h postinfection. RNA quality was assessed by visualization of 18S and 28S RNA bands using an Agilent 2100 bioanalyzer (Agilent Technologies, CA); the electropherograms were used to calculate the 28S/18S ratio and the RNA integrity number. Poly(A)+ RNA was enriched from total RNA (1 μg) using oligo(dT)-attached magnetic beads. First- and second-strand synthesis, adaptor ligation, and amplification of the library were performed using the Illumina TruSeq RNA sample preparation kit as recommended by the manufacturer (Illumina, Inc.). Library quality was evaluated using an Agilent DNA-1000 chip on an Agilent 2100 bioanalyzer. Quantification of library DNA templates was performed using quantitative PCR (qPCR) and a known-size reference standard. Cluster formation of the library DNA templates was performed using TruSeq PE cluster kit v3 (Illumina) and the Illumina cBot workstation under conditions recommended by the manufacturer. Paired-end 50-base sequencing by synthesis was performed using TruSeq SBS kit v3 (Illumina) on an Illumina HiSeq 1500 instrument using protocols defined by the manufacturer. The alignment of NGS sequence reads was performed using the Spliced Transcript Alignment to a Reference (STAR) program, version 2.5.1b, using default parameters (57). We used the human hg38 assembly as a reference with the UCSC gene annotation file, both downloaded from the Illumina iGenomes website. The –quantMode GeneCounts option of STAR provided read counts per gene, which were input into the DESeq2 (version 1.12.1) (58) differential expression analysis program to determine expression levels and differentially expressed genes.
Transfections and immunoprecipitations.
Transient knockdown of endogenous VAMP8 in wt A549 cells was done in 12-well plates. Briefly, 20 pmol of Smartpool ON-TARGETplus nontargeting (catalog number D-001810-10-05) or ON-TARGETplus VAMP8 (catalog number L-013503-00-0005) siRNA (Dharmacon) was transfected with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Cells were transfected with siRNA 24 h prior to infection or IFN-β treatment. The efficiency of VAMP8 knockdown was monitored by qRT-PCR or Western blotting. Wild-type or TRIM6-KO A549 or HEK293T cells were transfected with 250 ng of pCAGGS (empty vector) or FLAG-tagged VAMP8 (Origene) using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. Cells were transfected with plasmid DNA 30 h prior to WNV infection or 24 h prior to human IFN-β-1a treatment. For immunoprecipitations, cells were lysed in radioimmunoprecipitation assay (RIPA) complete buffer and centrifuged at 15,000 rpm at 4°C for 20 min to clarify the lysates. The clarified lysates were incubated with anti-HA- or anti-FLAG-coated beads (Sigma) overnight at 4°C. The beads were washed with RIPA buffer (N-ethylaemide [NEM] and iodoacetamide [IA]) seven times, resuspended in 2× Laemmli buffer with 5.0% beta-mercaptoethanol (β-ME), and boiled at 100°C for 10 min.
Western blotting.
Infected or IFN-β-treated cells were lysed in 2× Laemmli buffer with β-ME and boiled at 100°C for 10 min prior to removal from BSL-3 facilities. For immunoblotting, proteins were resolved using SDS-polyacrylamide gel electrophoresis (PAGE) (4 to 15% SDS-PAGE) and transferred onto a methanol-activated polyvinylidene difluoride (PVDF) membrane (Bio-Rad). The following primary antibodies were used: anti-pIRF3 (S386) (1:1,000) (Abcam), anti-total IRF3 (1:1,000) (Immuno-Biological), anti-TRIM6 N terminus (1:1,000) (Sigma), antiactin (1:2,000) (Abcam), anti-pSTAT1 (Y701) (1:1,000) (Cell Signaling), anti-pSTAT1 (S708) (1:2,000), anti-total STAT1 (1:1,000) (BD Biosciences), anti-VAMP8 (Cell Signaling) (1:500), anti-IKKε (T501) (1:1,000) (Novus Biologicals), anti-IKKε (S172) (1:1,000), anti-total IKKε (1:1,000) (Abcam), anti-pTBK1 (S172) (1:1,000) (Epitomics), anti-total TBK1 (1:1,000) (Novus Biologicals), anti-total JAK1 (BD Biosciences), and anti-pJAK1 (Y1034/1035) (Cell Signaling). Immunoblots were developed with the following secondary antibodies: enhanced chemiluminescence (ECL) anti-rabbit IgG horseradish peroxidase-conjugated whole antibody from donkey (1:10,000) and ECL anti-mouse IgG horseradish peroxidase-conjugated whole antibody from sheep (1:10,000) (GE Healthcare, Buckinghamshire, England). The proteins were visualized with either Pierce or SuperSignal West Femto luminol chemiluminescence substrates (Thermo Scientific). The amount of protein expressed was determined using Fiji (59) to calculate the area under the curve (AUC).
IFN-β ELISA.
Irradiated supernatants from WNV-infected wt or TRIM6-KO A549 cells were collected at 8, 24, and 48 h p.i. to measure IFN-β using the VeriKine human IFN-β enzyme-linked immunosorbent assay (ELISA) kit (PBL Assay Science) according to the manufacturer’s instructions. Standards were used to generate a standard curve to extrapolate the amount of IFN-β (picograms per milliliter) in the supernatants. The limit of detection for the assay is 50 pg/ml.
Transcription factor binding site analysis.
To identify transcription factors with binding sites within the VAMP8 genetic region, the VAMP8 RefSeqGene (NCBI accession number NG_022887.1) was analyzed with PROMO (version 3.0.2). The transcription factors predicted within a dissimilarity margin of ≤15% were identified and aligned to the VAMP8 RefSeqGene (NG_022887.1). Transcription factors with confirmed binding sites within the VAMP8 RefSeqGene were searched with JASPAR (2018) to confirm that the transcription factor consensus sequence matched the sequence within Vamp8.
Statistical analysis.
All analyses were performed in GraphPad Prism. All experiments were performed in triplicate. Statistical tests and measures of statistical significance are specified in the relevant figure legends. Repeated-measures two-way analysis of variance (ANOVA) with Bonferroni’s posttest was applied for kinetics two-factor comparisons (kinetics experiments). One-way ANOVA with Tukey’s posttest was used for comparing three more groups, and Student’s t test was used for comparing two groups.
Data availability.
The sequencing data from this study have been deposited in the GEO database under accession number GSE138841.
ACKNOWLEDGMENTS
This work was partially supported by grant R33 AI102267 awarded to A.N.F. and grant T32-AI060549 awarded to K.N.J. from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (NIH/NIAID). The Rajsbaum lab is supported by grants R01 AI134907, R21 AI126012, and R21 AI132479 awarded to R.R.; T32-AI060549 awarded to S.V.T.; and T32 AI060549 awarded to A.H. from the NIH/NIAID.
We thank Leopoldo Aguilera for helping with submission of the NGS data to the GEO repository. We thank Linsey Yeager for reading and editing of the manuscript.
REFERENCES
- 1.Gubler DJ, Kuno G, Markoff L. 2007. Flaviviruses, p 1153–1252. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Lindenbach BD, Thiel HJ, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Kramer LD, Styer LM, Ebel GD. 2008. A global perspective on the epidemiology of West Nile virus. Annu Rev Entomol 53:61–81. doi: 10.1146/annurev.ento.53.103106.093258. [DOI] [PubMed] [Google Scholar]
- 4.Anonymous. 2019. West Nile virus. CDC, Atlanta, GA: https://www.cdc.gov/westnile/index.html. [Google Scholar]
- 5.Sejvar JJ. 2014. Clinical manifestations and outcomes of West Nile virus infection. Viruses 6:606–623. doi: 10.3390/v6020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dayan GH, Pugachev K, Bevilacqua J, Lang J, Monath TP. 2013. Preclinical and clinical development of a YFV 17 D-based chimeric vaccine against West Nile virus. Viruses 5:3048–3070. doi: 10.3390/v5123048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayan GH, Bevilacqua J, Coleman D, Buldo A, Risi G. 2012. Phase II, dose ranging study of the safety and immunogenicity of single dose West Nile vaccine in healthy adults ≥ 50 years of age. Vaccine 30:6656–6664. doi: 10.1016/j.vaccine.2012.08.063. [DOI] [PubMed] [Google Scholar]
- 8.Tesh RB, Arroyo J, Travassos Da Rosa AP, Guzman H, Xiao SY, Monath TP. 2002. Efficacy of killed virus vaccine, live attenuated chimeric virus vaccine, and passive immunization for prevention of West Nile virus encephalitis in hamster model. Emerg Infect Dis 8:1392–1397. doi: 10.3201/eid0812.020229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blazquez AB, Vazquez-Calvo A, Martin-Acebes MA, Saiz JC. 2018. Pharmacological inhibition of protein kinase C reduces West Nile virus replication. Viruses 10:E91. doi: 10.3390/v10020091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrey JD, Taro BS, Siddharthan V, Wang H, Smee DF, Christensen AJ, Furuta Y. 2008. Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res 80:377–379. doi: 10.1016/j.antiviral.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perwitasari O, Cho H, Diamond MS, Gale M Jr. 2011. Inhibitor of kappaB kinase epsilon (IKK(epsilon)), STAT1, and IFIT2 proteins define novel innate immune effector pathway against West Nile virus infection. J Biol Chem 286:44412–44423. doi: 10.1074/jbc.M111.285205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorman MJ, Poddar S, Farzan M, Diamond MS. 2016. The interferon-stimulated gene Ifitm3 restricts West Nile virus infection and pathogenesis. J Virol 90:8212–8225. doi: 10.1128/JVI.00581-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mertens E, Kajaste-Rudnitski A, Torres S, Funk A, Frenkiel M-P, Iteman I, Khromykh AA, Desprès P. 2010. Viral determinants in the NS3 helicase and 2K peptide that promote West Nile virus resistance to antiviral action of 2′,5′-oligoadenylate synthetase 1b. Virology 399:176–185. doi: 10.1016/j.virol.2009.12.036. [DOI] [PubMed] [Google Scholar]
- 14.Lazear HM, Diamond MS. 2015. New insights into innate immune restriction of West Nile virus infection. Curr Opin Virol 11:1–6. doi: 10.1016/j.coviro.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daffis S, Samuel MA, Suthar MS, Gale M Jr, Diamond MS. 2008. Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol 82:10349–10358. doi: 10.1128/JVI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daffis S, Lazear HM, Liu WJ, Audsley M, Engle M, Khromykh AA, Diamond MS. 2011. The naturally attenuated Kunjin strain of West Nile virus shows enhanced sensitivity to the host type I interferon response. J Virol 85:5664–5668. doi: 10.1128/JVI.00232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M Jr. 2013. The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol 87:11416–11425. doi: 10.1128/JVI.01488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazear HM, Pinto AK, Vogt MR, Gale M Jr, Diamond MS. 2011. Beta interferon controls West Nile virus infection and pathogenesis in mice. J Virol 85:7186–7194. doi: 10.1128/JVI.00396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larena M, Lobigs M. 2017. Partial dysfunction of STAT1 profoundly reduces host resistance to flaviviral infection. Virology 506:1–6. doi: 10.1016/j.virol.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Lim JK, Lisco A, McDermott DH, Huynh L, Ward JM, Johnson B, Johnson H, Pape J, Foster GA, Krysztof D, Follmann D, Stramer SL, Margolis LB, Murphy PM. 2009. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog 5:e1000321. doi: 10.1371/journal.ppat.1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bigham AW, Buckingham KJ, Husain S, Emond MJ, Bofferding KM, Gildersleeve H, Rutherford A, Astakhova NM, Perelygin AA, Busch MP, Murray KO, Sejvar JJ, Green S, Kriesel J, Brinton MA, Bamshad M. 2011. Host genetic risk factors for West Nile virus infection and disease progression. PLoS One 6:e24745. doi: 10.1371/journal.pone.0024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang HL, Ye HQ, Liu SQ, Deng CL, Li XD, Shi PY, Zhang B. 2017. West Nile virus NS1 antagonizes interferon beta production by targeting RIG-I and MDA5. J Virol 91:e02396-16. doi: 10.1128/JVI.02396-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lubick KJ, Robertson SJ, McNally KL, Freedman BA, Rasmussen AL, Taylor RT, Walts AD, Tsuruda S, Sakai M, Ishizuka M, Boer EF, Foster EC, Chiramel AI, Addison CB, Green R, Kastner DL, Katze MG, Holland SM, Forlino A, Freeman AF, Boehm M, Yoshii K, Best SM. 2015. Flavivirus antagonism of type I interferon signaling reveals prolidase as a regulator of IFNAR1 surface expression. Cell Host Microbe 18:61–74. doi: 10.1016/j.chom.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laurent-Rolle M, Boer EF, Lubick KJ, Wolfinbarger JB, Carmody AB, Rockx B, Liu W, Ashour J, Shupert WL, Holbrook MR, Barrett AD, Mason PW, Bloom ME, Garcia-Sastre A, Khromykh AA, Best SM. 2010. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J Virol 84:3503–3515. doi: 10.1128/JVI.01161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuessler A, Funk A, Lazear HM, Cooper DA, Torres S, Daffis S, Jha BK, Kumagai Y, Takeuchi O, Hertzog P, Silverman R, Akira S, Barton DJ, Diamond MS, Khromykh AA. 2012. West Nile virus noncoding subgenomic RNA contributes to viral evasion of the type I interferon-mediated antiviral response. J Virol 86:5708–5718. doi: 10.1128/JVI.00207-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, Gale M Jr. 2006. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J Virol 80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 28.Paul A, Tang TH, Ng SK. 2018. Interferon regulatory factor 9 structure and regulation. Front Immunol 9:1831. doi: 10.3389/fimmu.2018.01831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu XY, Kessler DS, Veals SA, Levy DE, Darnell JE Jr. 1990. ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc Natl Acad Sci U S A 87:8555–8559. doi: 10.1073/pnas.87.21.8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.tenOever BR, Ng S-L, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. 2007. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 315:1274–1278. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 31.Rajsbaum R, Versteeg GA, Schmid S, Maestre AM, Belicha-Villanueva A, Martínez-Romero C, Patel JR, Morrison J, Pisanelli G, Miorin L, Laurent-Rolle M, Moulton HM, Stein DA, Fernandez-Sesma A, tenOever BR, García-Sastre A. 2014. Unanchored K48-linked polyubiquitin synthesized by the E3-ubiquitin ligase TRIM6 stimulates the interferon-IKKepsilon kinase-mediated antiviral response. Immunity 40:880–895. doi: 10.1016/j.immuni.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antonin W, Holroyd C, Tikkanen R, Honing S, Jahn R. 2000. The R-SNARE endobrevin/VAMP-8 mediates homotypic fusion of early endosomes and late endosomes. Mol Biol Cell 11:3289–3298. doi: 10.1091/mbc.11.10.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang CC, Ng CP, Lu L, Atlashkin V, Zhang W, Seet LF, Hong W. 2004. A role of VAMP8/endobrevin in regulated exocytosis of pancreatic acinar cells. Dev Cell 7:359–371. doi: 10.1016/j.devcel.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Wang CC, Shi H, Guo K, Ng CP, Li J, Gan BQ, Chien Liew H, Leinonen J, Rajaniemi H, Zhou ZH, Zeng Q, Hong W. 2007. VAMP8/endobrevin as a general vesicular SNARE for regulated exocytosis of the exocrine system. Mol Biol Cell 18:1056–1063. doi: 10.1091/mbc.e06-10-0974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behrendorff N, Dolai S, Hong W, Gaisano HY, Thorn P. 2011. Vesicle-associated membrane protein 8 (VAMP8) is a SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) selectively required for sequential granule-to-granule fusion. J Biol Chem 286:29627–29634. doi: 10.1074/jbc.M111.265199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones LC, Moussa L, Fulcher ML, Zhu Y, Hudson EJ, O’Neal WK, Randell SH, Lazarowski ER, Boucher RC, Kreda SM. 2012. VAMP8 is a vesicle SNARE that regulates mucin secretion in airway goblet cells. J Physiol 590:545–562. doi: 10.1113/jphysiol.2011.222091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loo LS, Hwang LA, Ong YM, Tay HS, Wang CC, Hong W. 2009. A role for endobrevin/VAMP8 in CTL lytic granule exocytosis. Eur J Immunol 39:3520–3528. doi: 10.1002/eji.200939378. [DOI] [PubMed] [Google Scholar]
- 38.Kanwar N, Fayyazi A, Backofen B, Nitsche M, Dressel R, von Mollard GF. 2008. Thymic alterations in mice deficient for the SNARE protein VAMP8/endobrevin. Cell Tissue Res 334:227–242. doi: 10.1007/s00441-008-0692-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang CC, Ng CP, Shi H, Liew HC, Guo K, Zeng Q, Hong W. 2010. A role for VAMP8/endobrevin in surface deployment of the water channel aquaporin 2. Mol Cell Biol 30:333–343. doi: 10.1128/MCB.00814-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matheoud D, Moradin N, Bellemare-Pelletier A, Shio MT, Hong WJ, Olivier M, Gagnon E, Desjardins M, Descoteaux A. 2013. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe 14:15–25. doi: 10.1016/j.chom.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 41.van Tol S, Atkins C, Bharaj P, Johnson KN, Hage A, Freiberg AN, Rajsbaum R. 2020. VAMP8 contributes to TRIM6-mediated type-I interferon antiviral response during West Nile virus infection. bioRxiv doi: 10.1101/749853. [DOI] [PMC free article] [PubMed]
- 42.Ng S-L, Friedman BA, Schmid S, Gertz J, Myers RM, tenOever BR, Maniatis T. 2011. IkappaB kinase epsilon (IKK(epsilon)) regulates the balance between type I and type II interferon responses. Proc Natl Acad Sci U S A 108:21170–21175. doi: 10.1073/pnas.1119137109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang D, Weidner JM, Qing M, Pan XB, Guo H, Xu C, Zhang X, Birk A, Chang J, Shi PY, Block TM, Guo JT. 2010. Identification of five interferon-induced cellular proteins that inhibit West Nile virus and dengue virus infections. J Virol 84:8332–8341. doi: 10.1128/JVI.02199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Danial-Farran N, Eghbaria S, Schwartz N, Kra-Oz Z, Bisharat N. 2015. Genetic variants associated with susceptibility of Ashkenazi Jews to West Nile virus infection. Epidemiol Infect 143:857–863. doi: 10.1017/S0950268814001290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang LK, Chai F, Li HY, Xiao G, Guo L. 2013. Identification of host proteins involved in Japanese encephalitis virus infection by quantitative proteomics analysis. J Proteome Res 12:2666–2678. doi: 10.1021/pr400011k. [DOI] [PubMed] [Google Scholar]
- 46.Bharaj P, Atkins C, Luthra P, Giraldo MI, Dawes BE, Miorin L, Johnson JR, Krogan NJ, Basler CF, Freiberg AN, Rajsbaum R. 2017. The host E3-ubiquitin ligase TRIM6 ubiquitinates the Ebola virus VP35 protein and promotes virus replication. J Virol 91:e00833-17. doi: 10.1128/JVI.00833-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bharaj P, Wang YE, Dawes BE, Yun TE, Park A, Yen B, Basler CF, Freiberg AN, Lee B, Rajsbaum R. 2016. The matrix protein of Nipah virus targets the E3-ubiquitin ligase TRIM6 to inhibit the IKKepsilon kinase-mediated type-I IFN antiviral response. PLoS Pathog 12:e1005880. doi: 10.1371/journal.ppat.1005880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uchil PD, Hinz A, Siegel S, Coenen-Stass A, Pertel T, Luban J, Mothes W. 2013. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J Virol 87:257–272. doi: 10.1128/JVI.01804-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Tol S, Hage A, Giraldo MI, Bharaj P, Rajsbaum R. 2017. The TRIMendous role of TRIMs in virus-host interactions. Vaccines (Basel) 5:E23. doi: 10.3390/vaccines5030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Versteeg GA, Rajsbaum R, Sánchez-Aparicio MT, Maestre AM, Valdiviezo J, Shi M, Inn K-S, Fernandez-Sesma A, Jung J, García-Sastre A. 2013. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38:384–398. doi: 10.1016/j.immuni.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Versteeg GA, Benke S, Garcia-Sastre A, Rajsbaum R. 2014. InTRIMsic immunity: positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev 25:563–576. doi: 10.1016/j.cytogfr.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmid S, Mordstein M, Kochs G, Garcia-Sastre A, tenOever BR. 2010. Transcription factor redundancy ensures induction of the antiviral state. J Biol Chem 285:42013–42022. doi: 10.1074/jbc.M110.165936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M Jr, Shi PY, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olofsson P, Nerstedt A, Hultqvist M, Nilsson EC, Andersson S, Bergelin A, Holmdahl R. 2007. Arthritis suppression by NADPH activation operates through an interferon-beta pathway. BMC Biol 5:19. doi: 10.1186/1741-7007-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quiroga AD, Alvarez MDL, Parody JP, Ronco MT, Francés DE, Pisani GB, Carnovale CE, Carrillo MC. 2007. Involvement of reactive oxygen species on the apoptotic mechanism induced by IFN-alpha2b in rat preneoplastic liver. Biochem Pharmacol 73:1776–1785. doi: 10.1016/j.bcp.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 56.Fink K, Martin L, Mukawera E, Chartier S, De Deken X, Brochiero E, Miot F, Grandvaux N. 2013. IFNbeta/TNFalpha synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH oxidase-mediated airway antiviral response. Cell Res 23:673–690. doi: 10.1038/cr.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequencing data from this study have been deposited in the GEO database under accession number GSE138841.