

Without an effective means of eliminating HSV-1 from latently infected neurons, efforts to control the virus have centered on preventing viral reactivation from latency. Virus-specific CD8+ T cells within the infected TG have been shown to play a crucial role in inhibiting viral reactivation, and with a portion of these cells exhibiting functional impairment, checkpoint molecule immunotherapies have presented a potential solution to enhancing the antiviral response of these cells. In pursuing this potential treatment strategy, we found that Tim-3 (often associated with CD8+ T cell functional exhaustion) is not upregulated on impaired cells but instead is upregulated on highly functional cells that have recently received antigenic stimulation. These findings support a role for Tim-3 as a marker of activation rather than exhaustion in this model, and we provide additional evidence for the hypothesis that there is persistent viral gene expression in the HSV-1 latently infected TG.
KEYWORDS: CD8+ T cells, checkpoint molecule, HSV-1, immunology, virology
ABSTRACT
Herpes simplex virus 1 (HSV-1) causes a lifelong infection of neurons that innervate barrier sites like the skin and mucosal surfaces like the eye. After primary infection of the cornea, the virus enters latency within the trigeminal ganglion (TG), from which it can reactivate throughout the life of the host. Viral latency is maintained, in part, by virus-specific CD8+ T cells that nonlethally interact with infected neurons. When CD8+ T cell responses are inhibited, HSV-1 can reactivate, and these recurrent reactivation events can lead to blinding scarring of the cornea. In the C57BL/6 mouse, CD8+ T cells specific for the immunodominant epitope from glycoprotein B maintain functionality throughout latency, while CD8+ T cells specific for subdominant epitopes undergo functional impairment that is associated with the expression of the inhibitory checkpoint molecule programmed death 1 (PD-1). Here, we investigate the checkpoint molecule T cell immunoglobulin and mucin domain-containing 3 (Tim-3), which has traditionally been associated with CD8+ T cell exhaustion. Unexpectedly, we found that Tim-3 was preferentially expressed on highly functional ganglionic CD8+ T cells during acute and latent HSV-1 infection. This, paired with data that show that Tim-3 expression on CD8+ T cells in the latently infected TG is influenced by viral gene expression, suggests that Tim-3 is an indicator of recent T cell stimulation, rather than functional compromise, in this model. We conclude that Tim-3 expression is not sufficient to define functional compromise during latency; however, it may be useful in identifying activated cells within the TG during HSV-1 infection.
IMPORTANCE Without an effective means of eliminating HSV-1 from latently infected neurons, efforts to control the virus have centered on preventing viral reactivation from latency. Virus-specific CD8+ T cells within the infected TG have been shown to play a crucial role in inhibiting viral reactivation, and with a portion of these cells exhibiting functional impairment, checkpoint molecule immunotherapies have presented a potential solution to enhancing the antiviral response of these cells. In pursuing this potential treatment strategy, we found that Tim-3 (often associated with CD8+ T cell functional exhaustion) is not upregulated on impaired cells but instead is upregulated on highly functional cells that have recently received antigenic stimulation. These findings support a role for Tim-3 as a marker of activation rather than exhaustion in this model, and we provide additional evidence for the hypothesis that there is persistent viral gene expression in the HSV-1 latently infected TG.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a prevalent human pathogen, with over 50% of adults in the United States testing positive by late adulthood (1). HSV-1 infection can result in a number of pathologies that depend on both the mucosal site of primary infection as well as the neurons that ultimately harbor the latent virus and are the site of sporadic reactivation events that lead to recurrent peripheral disease. One potential site of viral infection, the cornea, plays a crucial role in vision by transmitting and focusing light so that images can be processed by the retina and brain. Without an optically clear cornea, visual acuity and quality of life are severely reduced.
During acute infection, HSV-1 replicates in the corneal epithelium and gains access to the axons of sensory neurons before being cleared from the cornea by innate immune cells like macrophages and NK cells (2–5). Using retrograde axonal transport, HSV-1 travels to neuronal cell bodies in the trigeminal ganglion (TG), where it establishes latency. Although the virus is able to be maintained in a latent state indefinitely, various stressors, such as UV light exposure, hormone fluctuation, or trauma, can cause viral reactivation, which results in active viral replication within infected neurons and anterograde transport of infectious virus down axons back to the periphery (6–8). These reactivation events can, in some individuals, result in the development of recurrent herpes stromal keratitis (HSK), which is characterized by a progressive development of corneal opacity, neovascularization, edema, and hypoesthesia, which can ultimately lead to loss of vision (9). As there is no effective and approved vaccine for HSV-1, therapeutic efforts have primarily focused on controlling viral replication, restricting inflammation, and preventing reactivation from viral latency.
Viral latency is maintained by several factors, including host microRNAs (miRNAs), viral miRNAs derived from the latency-associated transcript (LAT), and immune cells such as virus-specific CD8+ T cells (10–12). Virus-specific CD8+ T cells have been shown to infiltrate the infected TG (13) and interact with infected neurons to prevent viral reactivation (14). In the C57BL/6 (B6) mouse, 50% of TG-associated CD8+ T cells are specific for an immunodominant epitope found on glycoprotein B (gB) (15). Previous work from our laboratory identified the remaining epitope specificities of the CD8+ T cell repertoire in this model, which consists of 18 subdominant HSV-1 epitopes from viral proteins, including ribonucleotide reductase 1 (RR1), glycoprotein C (gC), infected cell protein 8 (ICP8), and others (16). While the mechanism governing the immunodominance hierarchy remains incompletely understood, 80% of the epitopes recognized by HSV-1-specific CD8+ T cells are from proteins expressed before DNA synthesis. These data, together with our previous studies, support the notion that CD8+ T cell activity is required to prevent viral reactivation from latency (17). When functionality between the gB-specific (gB-CD8+ T cells) and subdominant epitope-specific (Subdom-CD8+ T cells) CD8+ T cell groups was assessed at latency, a higher frequency of gB-CD8+ T cells produced granzyme B (GzmB) directly ex vivo and interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) after in vitro peptide stimulation than Subdom-CD8+ T cells (18). Since CD8+ T cell functionality plays an important role in suppressing viral gene expression and preventing reactivation, improving the function of TG-resident Subdom-CD8+ T cells provides a potentially useful strategy for preventing reactivation in the TG.
Loss of functionality in T cells after prolonged exposure to their cognate antigen is a phenomenon that has received considerable attention in recent years in both chronic viral infection and tumor models. In these models, CD8+ T cells progressively lose their capacity to respond to their antigen after repeated stimulations over an extended period of time, with the affected cells being considered “exhausted” (19–22). This development of exhausted cells allows the perpetuation of viral infection or tumor growth. As such, there has been substantial enthusiasm for the development of immunotherapies to reverse this loss in functionality. The major targets of these therapies have centered on checkpoint molecules such as programmed death 1 (PD-1) and cytotoxic T lymphocyte protein 4 (CTLA-4), although numerous others are in development (23, 24). The specific contributions of individual checkpoint molecules are not yet fully understood; however, it is generally accepted that increased expression of single and/or coexpression of multiple checkpoint molecules results in functional compromise (25). Therapies blocking these molecules have successfully reinvigorated exhausted CD8+ T cells in animals and the clinic, resulting in more efficient viral/tumor clearance and increased patient survival (23, 25–28).
Here, we have defined the expression of several classical checkpoint molecules during HSV-1 latency. We show that while the expression levels of the majority of assessed molecules are low in ganglionic CD8+ T cell populations during HSV-1 latency, T cell immunoglobulin and mucin domain-containing 3 (Tim-3) is preferentially upregulated on functional gB-CD8+ T cells rather than impaired Subdom-CD8+ T cells. Although other laboratories have reported similar expression levels of Tim-3 on these populations (29, 30), our study is the first to correlate the expression pattern of Tim-3 with functionality in this model. We found that Tim-3-positive (Tim-3+) cells can readily respond to peptide stimulation and are in fact highly multifunctional. Furthermore, during latency, we were able to modulate Tim-3 expression on TG-resident CD8+ T cells by using strains of the virus with altered expression patterns of viral CD8+ T cell epitopes, suggesting that Tim-3 may serve as a T cell activation marker in this model. Our data also suggest that functionally compromised cells may acquire this phenotype during acute infection rather than gradually throughout latency. Collectively, our results indicate that despite the traditional classification of Tim-3 as an inhibitory checkpoint molecule, its expression should not automatically imply functional impairment. Instead, Tim-3 expression in the TG may serve as a marker of recent T cell activation and may be helpful in determining the levels of expression of viral genes during latency.
RESULTS
Tim-3 and 2B4 are preferentially expressed on gB-CD8+ T cells during latent HSV-1 infection.
The expression of a single checkpoint molecule does not necessarily indicate functional impairment; however, the simultaneous expression of multiple checkpoint molecules is a hallmark of CD8+ T cell exhaustion (25). Our laboratory has recently shown that during latent HSV-1 infection within the TG, CD8+ T cells recognizing 18 subdominant epitopes from HSV-1 viral proteins (Subdom-CD8+ T cells) collectively express high levels of the inhibitory checkpoint molecule PD-1. In contrast, CD8+ T cells specific for the immunodominant epitope gB498–505 (gB-CD8+ T cells) found in the same TGs express low levels of PD-1 (31). The expression of other checkpoint molecules on TG-CD8+ T cells remained largely unknown, so we assessed the expression of six additional checkpoint molecules on gB-CD8+ T cells (gB tetramer-positive cells) and Subdom-CD8+ T cells (gB tetramer-negative cells) at latency (33 to 43 days postinfection [dpi]) (Fig. 1A).
FIG 1.

Tim-3 and 2B4 are preferentially expressed on gB-CD8+ T cells during latent HSV-1 infection. TGs from latently infected mice (33 to 43 dpi) were removed; processed into single-cell suspensions; stained for viability, CD45, CD8, CTLA-4, BTLA, LAG3, TIGIT, 2B4, or Tim-3 and the gB tetramer to distinguish gB-CD8+ T cells from Subdom-CD8+ T cells; and analyzed by flow cytometry. Representative dot plots from one mouse showing the expression of the indicated checkpoint molecules on the gB-CD8+ T cells and Subdom-CD8+ T cells are shown along with the FMO gating control, with a graph from one representative experiment shown on the right (B to G). In the graphs, bars represent means ± standard error of the mean (SEM). Differences between groups were assessed by unpaired t tests (*, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001). (A) Gating strategy is as follows: lymphocytes were first gated by size, doublets were excluded, dead cells were excluded, CD45+ cells were gated, CD8+ cells were gated, and CD8+ cells were split into gB-CD8+ T cell or Subdom-CD8+ T cell groups by using the gB tetramer before looking at the various checkpoint molecules. FSC, forward scatter; SSC, side scatter. (B) Expression of CTLA-4 (stained both extracellularly and intracellularly) (n = 5). Data are representative of results from two independent experiments. (C) Expression of BTLA (n = 6). Data are representative of results from two independent experiments. (D) Expression of LAG3 (n = 14). Data are representative of results from of two independent experiments. (E) Expression of TIGIT (n = 13). Data are representative of results from two independent experiments. (F) Expression of 2B4 (n = 6). Data are representative of results from four independent experiments. (G) Expression of Tim-3 (n = 11). Data are representative of results from 10 independent experiments.
Similar to the expression pattern of PD-1, CTLA-4 and B and T lymphocyte attenuator (BTLA) were expressed on a higher percentage of Subdom-CD8+ T cells than gB-CD8+ T cells, although overall levels of both checkpoint molecules were relatively low in both groups (Fig. 1B and C). In addition, gB-CD8+ T cells and Subdom-CD8+ T cells had equivalent expression levels of lymphocyte activation gene 3 (LAG3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT) (Fig. 1D and E). In contrast to PD-1, CTLA-4, and BTLA expression, a higher percentage of gB-CD8+ T cells expressed 2B4 and Tim-3 than Subdom-CD8+ T cells (Fig. 1F and G). Therefore, despite the canonical expression pattern of checkpoint molecules such as PD-1, BTLA, and CTLA-4 on Subdom-CD8+ T cells, which exhibit functional impairment, we show that Tim-3 and 2B4 are enriched on gB-CD8+ T cells, which retain functionality throughout latent infection. These data call into question the association of these molecules with functional exhaustion in this model.
Tim-3 is primarily expressed on PD-1− CD8+ T cells in the latently infected TG.
Tim-3 has been shown to mark functionally exhausted CD8+ T cells during infection with lymphocytic choriomeningitis virus (LCMV), HIV, and hepatitis C virus (HCV) as well as in tumors (26, 32–34). In several of these models, the severity of exhaustion in Tim-3-expressing cells is dependent on the coexpression of PD-1, with cells expressing both checkpoint molecules being notably less functional than cells that express either PD-1 or Tim-3 alone (26, 34). This is consistent with the idea that functional exhaustion is a result of accumulating expression of multiple inhibitory checkpoint molecules. It was previously reported that Tim-3 marks exhausted CD8+ T cells in the latent TG during HSV-1 infection (29, 30); however, we wanted to assess Tim-3 expression on TG-CD8+ T cells that expressed or did not express the canonical exhaustion marker PD-1. When we assessed the expression of these two checkpoint molecules on CD8+ T cells in the latently infected TG (Fig. 2A), it was apparent that there was little overlap between the two markers, with fewer than 50 total cells/TG or fewer than 10% of the CD8+ T cells being Tim-3+ PD-1+ in both the gB-CD8+ T cell and Subdom-CD8+ T cell groups (Fig. 2B and C). This lack of strong coexpression between Tim-3 and PD-1, and the preferential expression of Tim-3 on gB-CD8+ T cells and PD-1 on Subdom-CD8+ T cells, indicated that Tim-3 may not be functioning in the typically inhibitory manner that is seen in most chronic diseases.
FIG 2.

Tim-3 is primarily expressed on PD-1− CD8+ T cells in the latently infected TG. TGs from latently infected mice (33 to 35 dpi) were removed; processed into single-cell suspensions; stained for viability, CD45, CD8, gB tetramer, Tim-3, and PD-1; and analyzed by flow cytometry. To assess in vivo T cell proliferation, mice received i.p. injections of 1 mg of BrdU 48 and 24 h prior to sacrifice, and BrdU uptake was measured by flow cytometry. Representative dot plots from one mouse showing the expression of the indicated checkpoint molecules on gB-CD8+ T cells and Subdom-CD8+ T cells are shown along with the FMO gating controls (A). In the graphs, the bars represent the mean number of cells, percentage of CD8+ T cells, or percentage of BrdU+ cells ± SEM. Differences in panels B to D were assessed by two-way analysis of variance (ANOVA) with Tukey’s posttests (*, P ≤ 0.05; ***, P ≤ 0.001; ****, P ≤ 0.0001). (A) Expression of Tim-3 and PD-1. (B) Total number of CD8+ T cells/TG in each of the Tim-3/PD-1 quadrants from panel A (n = 11). The experiment was repeated an additional eight times, with similar results. (C) Percentage of CD8+ T cells in each of the Tim-3/PD-1 quadrants from panel A (n = 11). The experiment was repeated an additional eight times, with similar results. (D) gB-CD8+ T cell and Subdom-CD8+ T cell populations were each gated into Tim-3+ and Tim-3− populations, and the percentages of cells that were BrdU+ in each subgroup are shown (n = 25). Data are from two pooled independent experiments.
As our Tim-3 and PD-1 coexpression data implied that Tim-3 might not be inhibitory in the latently infected TG, we next wanted to determine if Tim-3 plays a role in CD8+ T cell proliferation, which is often diminished during functional exhaustion (22). After a 2-day in vivo bromodeoxyuridine (BrdU) pulse, we found that levels of proliferation in Tim-3+ cells were not significantly different than those in Tim-3-negative (Tim-3−) cells in both the gB-CD8+ T cells and Subdom-CD8+ T cells, implying that these cells are not exhausted (Fig. 2D). Collectively, these results show that Tim-3 expression is largely independent of PD-1 expression and further suggest that Tim-3 may not mark cells that have lost functionality in the latently infected TG.
Tim-3+ gB-CD8+ T cells are multifunctional after ex vivo stimulation.
This study and previous work (29) showed that Tim-3 was primarily expressed on PD-1-negative cells (in both gB-CD8+ T cells and Subdom-CD8+ T cells). Therefore, we were next interested in determining if Tim-3 was instead preferentially expressed on functional cells. After stimulation with cognate antigen, the most functionally competent cells can produce and release multiple factors, including IFN-γ, TNF-α, and lytic granules containing GzmB, while functionally exhausted CD8+ T cells gradually lose this multifunctionality (35). To determine if the Tim-3+ cells in our model were functional, we stained latently infected TGs with anti-Tim-3 antibody and subsequently stimulated the TGs with gB peptide-pulsed B6WT3 cells for 6 h along with fluorescently labeled anti-CD107a and brefeldin A. We then used flow cytometry to identify cells that produced IFN-γ and/or TNF-α and confirmed multifunctionality by assessing lytic granule release (CD107a+) in responding cells (Fig. 3A). When Tim-3 expression was quantified on the responding CD8+ T cells (cells that produced IFN-γ and/or TNF-α after gB peptide stimulation), we found that ∼35% expressed Tim-3 (Fig. 3B). When we calculated the multifunctionality of Tim-3+ cells, we found that ∼70% of the Tim-3+ cells were multifunctional (IFN-γ+ TNF-α+ CD107a+) (Fig. 3C), which suggested that Tim-3 expression was not associated with impaired cells but instead appeared to be associated with functionally competent cells.
FIG 3.

Tim-3+ gB-CD8+ T cells are multifunctional after ex vivo stimulation. TGs from latently infected mice (33 to 35 dpi) were removed, processed into single-cell suspensions, stained with anti-Tim-3 antibody, and stimulated for 6 h with gB peptide-pulsed B6WT3 cells in the presence of anti-CD107a and brefeldin A. Cells were then collected and stained for viability, CD45, CD8, IFN-γ, and TNF-α. In graphs, bars represent means ± SEM (n = 24), from two pooled independent experiments. (A) Representative dot plots of IFN-γ and TNF-α responses from an individual TG that received no peptide stimulation (3rd plot from the left) and an individual TG that received gB peptide stimulation (4th plot from the left). Shaded quadrants make up the “responding CD8+ T cells.” Gating for CD107a is shown at the far right, with FMO represented in black and a representative sample in gray. (B) Percentage of responding CD8+ T cells that are positive for Tim-3. (C) Percentage of Tim-3+ responding CD8+ T cells that are multifunctional (IFN-γ+ TNF-α+ CD107a+).
Tim-3+ cells are functional in vivo.
Although our data from Fig. 3 established that Tim-3-expressing cells were highly functional after peptide stimulation, we were also interested in further investigating their ability to produce GzmB, which marks activated CD8+ T cells and contributes to the maintenance of viral latency in host neurons by cleaving the essential HSV-1 protein ICP4 (17). When CD8+ T cells from latently infected TGs were assessed for GzmB expression directly ex vivo, significantly higher percentages of gB-CD8+ T cells expressed GzmB than Subdom-CD8+ T cells (Fig. 4A), consistent with previously reported data (18). When GzmB expression was measured in the Tim-3+ gB-CD8+ T cells and Subdom-CD8+ T cells, we found that the majority of cells in both groups were positive for GzmB; however, a higher frequency of Tim-3+ gB-CD8+ T cells expressed GzmB+ than Tim-3+ Subdom-CD8+ T cells (Fig. 4B). The percentage of Tim-3+ gB-CD8+ T cells that were GzmB+ was similar to the percentage of cells that were multifunctional after stimulation (Fig. 3C), which supported the hypothesis that Tim-3+ cells are functional.
FIG 4.
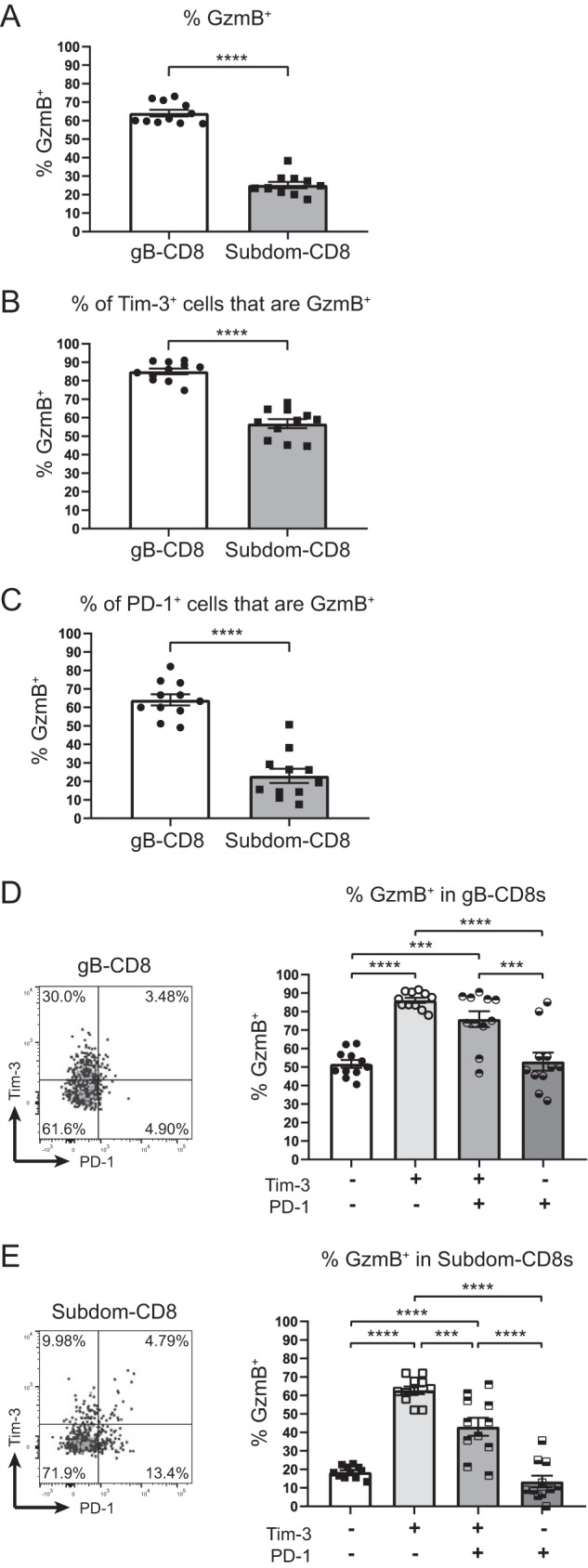
Tim-3+ cells are functional in vivo. TGs from latently infected mice (33 to 35 dpi) were removed; processed into single-cell suspensions; stained for viability, CD45, CD8, gB tetramer, Tim-3, PD-1, and GzmB; and assessed by flow cytometry. In the graphs, the bars represent the mean frequencies of cells ± SEM, with the gB-CD8+ T cell population represented by circles/white bars and the Subdom-CD8+ T cell population represented by squares/gray bars in graphs that contain both groups (n = 11). The experiment was repeated an additional five times, with similar results. Differences were assessed by unpaired t tests (A to C) or one-way ANOVAs with Tukey’s posttests (D and E) (***, P ≤ 0.001; ****, P ≤ 0.0001). (A) Percentages of cells in gB-CD8+ T cell and Subdom-CD8+ T cell groups that are GzmB+. (B) Percentages of Tim-3+ cells in gB-CD8+ T cell and Subdom-CD8+ T cell groups that are GzmB+. (C) Percentages of PD-1+ cells in gB-CD8+ T cell and Subdom-CD8+ T cell groups that are GzmB+. (D) Percentages of gB-CD8+ T cells in Tim-3/PD-1 quadrants that are GzmB+. (E) Percentages of Subdom-CD8+ T cells in Tim-3/PD-1 quadrants that are GzmB+.
As we had unexpectedly found that the Tim-3+ Subdom-CD8+ T cells were significantly less functional in terms of GzmB production than the Tim-3+ gB-CD8+ T cells, we assessed whether PD-1 could be modulating this difference, despite the low levels of coexpression that we observed (Fig. 2). When PD-1+ cells were assessed for GzmB expression, we found that 60% of PD-1+ gB-CD8+ T cells expressed GzmB, while only 20% of PD-1+ Subdom-CD8+ T cells expressed GzmB (Fig. 4C). This corroborated our previous study, which showed a lower overall level of PD-1 expression on gB-CD8+ T cells than on Subdom-CD8+ T cells (31). Therefore, the disparity in functionality between PD-1+ gB-CD8+ T cells and PD-1+ Subdom-CD8+ T cells may be due to the differences in expression levels of PD-1.
With Tim-3+ or PD-1+ Subdom-CD8+ T cells exhibiting a reduced ability to produce GzmB compared to Tim-3+ or PD-1+ gB-CD8+ T cells, we then proceeded to look more specifically at (i) Tim-3− PD-1−, (ii) Tim-3+ PD-1−, (iii) Tim-3+ PD-1+, and (iv) Tim-3− PD-1+ cells with respect to their ability to produce GzmB. In the gB-CD8+ T cells, the subgroup with the highest percentage of cells that produced GzmB was that which expressed Tim-3 alone (Fig. 4D). Cells coexpressing Tim-3 and PD-1 produced slightly less GzmB, although the difference was not significant. The Tim-3− PD-1+ and Tim-3− PD-1− subgroups had significantly lower percentages of GzmB+ cells than the groups expressing Tim-3. Like the gB-CD8+ T cells, Subdom-CD8+ T cells expressing Tim-3 only had the highest percentage of cells that produced GzmB; however, unlike for gB-CD8+ T cells, we observed a significant decrease in the GzmB expression frequency in both the Tim-3+ PD-1+ and Tim-3− PD-1+ subgroups compared to the Tim-3+ PD-1− group (Fig. 4E). We conclude from these data that Tim-3 is associated with HSV-1-specific T cell functionality, while PD-1 is associated with impairment at latency, with cells expressing both checkpoint molecules exhibiting an intermediate functional phenotype.
Viral gene expression controls Tim-3 expression in the latently infected TG.
Even though lytic viral proteins have not been reliably detected in the latently infected TG, our laboratory previously showed that only HSV-1-specific CD8+ T cells are retained within the TG during latency and that retention requires antigen expression, leading to the conclusion that there are consistent low levels of lytic viral gene expression during latent HSV-1 infection (36, 37). Since we observed that Tim-3 expression was associated with functionality, we were next interested in determining if changes in antigen availability during latency could alter Tim-3 expression on virus-specific CD8+ T cells. To investigate this, we manipulated the virus-specific CD8+ T cell repertoire and viral gene expression by using genetically modified strains of HSV-1 expressing different levels of the gB498–505 epitope and assessed Tim-3 expression on TG-resident CD8+ T cells during latent infection.
We first used a recently detailed virus (S1L) that contains a single point mutation within the gB epitope that completely abrogates T cell recognition of gB498–505 and therefore prevents the priming and expansion of gB-specific CD8+ T cells but does not affect viral fitness or the expression profile of other viral genes (36). This lack of gB-specific CD8+ T cells allows RR1-specific CD8+ T cells (RR1982–989 and RR1822–829) to expand and infiltrate the TG in greater numbers than in wild-type (WT) infection (36). We observed that the increase in cell numbers extended beyond acute infection and into latency (Fig. 5A). With this, we were able to assess alterations in Tim-3 expression when there are more CD8+ T cells responding to the same level of viral gene expression, which effectively reduces the chances that any one RR1-specific CD8+ T cell would be antigenically stimulated. Tim-3 expression on RR1-CD8+ T cells was reduced in S1L infection compared to wild-type infection, and there was a concurrent reduction in GzmB expression in the RR1-CD8+ T cells in S1L-infected TGs compared to wild-type-infected TGs (Fig. 5B and C). Despite this, the percentages of Tim-3+ RR1-CD8+ T cells that were GzmB+ remained similar between the two infections (Fig. 5D). While it is unknown how the presentation of viral epitopes on major histocompatibility complex (MHC) molecules may change when the gB498–505 epitope is deleted and how this may influence the total number of cells that are stimulated, overall Tim-3 expression, albeit reduced in S1L infection, continued to correlate with the expression pattern of GzmB. Therefore, we can conclude from these data that Tim-3 expression is likely linked with recent antigenic stimulation of TG-resident CD8+ T cells.
FIG 5.

Viral gene expression controls Tim-3 expression in the latently infected TG. TGs from mice latently infected with wild-type KOS or viruses containing mutations (S1L, gC, or ICP0) were removed; processed into single-cell suspensions; stained for viability, CD45, CD8, gB or RR1 tetramer, Tim-3, and GzmB; and assessed by flow cytometry. Bars represent the mean total numbers or frequencies of cells ± SEM. Differences were assessed by unpaired t tests (A to D) or one-way ANOVAs with Tukey’s posttests (E to H) (**, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001) (for panels A to D, n = 16 [KOS] and n = 17 [S1L] [data were pooled from two independent experiments]; for panels E to H, n = 18 [KOS], n = 20 [ICP0], and n = 20 [gC] [data were pooled from two independent experiments]). (A) Total numbers of cells that are positive for RR1 tetramers in KOS- and S1L-infected TGs. (B) Percentages of RR1-CD8+ T cells that are positive for Tim-3 in KOS- and S1L-infected TGs. (C) Percentages of RR1-CD8+ T cells that are positive for GzmB in KOS- and S1L-infected TGs. (D) Percentages of Tim-3+ RR1-CD8+ T cells that are positive for GzmB in KOS- and S1L-infected TGs. (E) Percentages of CD8+ T cells that are positive for gB tetramer in KOS-, gC-, and ICP0-infected TGs. (F) Percentages of gB-CD8+ T cells that are positive for Tim-3 in KOS-, gC-, and ICP0-infected TGs. (G) Percentages of gB-CD8+ T cells that are positive for GzmB in KOS-, gC-, and ICP0-infected TGs. (H) Percentages of Tim-3+ gB-CD8+ T cells that are positive for GzmB in KOS-, gC-, and ICP0-infected TGs.
We next sought to determine if increasing the expression of viral antigen within the TG could result in an upregulation of Tim-3. To study this, we made use of two viruses that express four copies of the gB epitope under the control of the viral promoter ICP0 or gC (B. R. Treat, S. M. Bidula, R. L. Hendricks, and P. R. Kinchington, submitted for publication). Compared to the wild-type virus, TGs infected with these viruses maintained a similar frequency of gB-CD8+ T cells during latency; however, significantly higher percentages of gB-CD8+ T cells expressed Tim-3 in the gC- and ICP0-infected TGs (Fig. 5E and F). The gB-CD8+ T cells in the ICP0- and gC-infected TGs also had significantly higher percentages of GzmB expression while maintaining similar percentages of Tim-3+ cells that were GzmB+ (Fig. 5G and H). Since these viruses express higher levels of the gB antigen but the TGs possess similar numbers of gB-CD8+ T cells to WT infection, these results suggest that increasing antigen availability to the gB-CD8+ T cells increases the expression of Tim-3 on the gB population as a whole. These data, together with the data from the S1L virus, support the notion that Tim-3 expression correlates with the overall level of T cell stimulation during HSV-1 latency.
Tim-3+ cells preferentially upregulate genes that are associated with T cell activation.
Before regulating functional impairment in models of chronic viral infection or persistent tumors, checkpoint molecules are transiently expressed after T cell activation and may function in tempering the response to their cognate antigens during acute infection (38). As Tim-3 was preferentially upregulated in functional cells during latent infection, we next assessed whether Tim-3 expression was associated with functionality during acute infection. To do this, we used NanoString technology to compare the transcriptional profiles of fluorescence-activated cell sorter (FACS)-sorted Tim-3+ PD-1− cells and Tim-3− PD-1− cells within the gB-CD8+ T cells and Subdom-CD8+ T cells from TGs at 8 days postinfection, when latency has just been established and the immune infiltrate has peaked. As we observed in latency, granzymes were upregulated in Tim-3-expressing cells from both groups, indicating effector functionality (Fig. 6A and B). This was further supported by the upregulation of genes such as Notch1, Zap70, and Runx3, all of which are known to contribute to T cell activation (39–41). When we compared Tim-3+ PD-1− gB-CD8+ T cells to Tim-3+ PD-1− Subdom-CD8+ T cells, we found that there was an overall upregulation of genes associated with T cell functionality in the gB-CD8+ T cell population, which was consistent with a higher level of functionality during latency (Fig. 6C). This is indicative of very early differences between the gB-CD8+ T cells and Subdom-CD8+ T cells rather than an acquired loss of functionality during latency, as is typical during chronic viral infection.
FIG 6.

Tim-3+ cells preferentially upregulate genes that are associated with T cell activation. TGs from acutely infected mice (8 dpi) were removed, processed into a single-cell suspension, and stained for viable cells, CD45, CD8, gB tetramer, Tim-3, and PD-1. CD8+ T cells were gated on gB-CD8+ T cells or Subdom-CD8+ T cells using gB tetramer; Tim-3− PD-1−, Tim-3+ PD-1−, Tim-3+ PD-1+, and Tim-3− PD-1+ cells were sorted, and their transcriptional profile was assessed by using NanoString technology. (A) Genes of interest that averaged a fold change of ≥1.5 over 3 independent replicates between Tim-3+ PD-1− gB-CD8+ T cells and Tim-3− PD-1− gB-CD8+ T cells. (B) Fold change of genes of interest between Tim-3+ PD-1− Subdom-CD8+ T cells and Tim-3− PD-1− Subdom-CD8+ T cells. (C) Fold change of genes of interest between Tim-3+ PD-1− gB-CD8+ T cells and Tim-3+ PD-1− Subdom-CD8+ T cells.
Tim-3+ cells are functional at early times postinfection.
Since Tim-3 and PD-1 appeared differentially regulated in the TG during latency, and our transcriptional analysis indicated that Tim-3 expression at early times postinfection was associated with the transcription of genes that are generally involved in T cell activation, we next evaluated the relationship between Tim-3, PD-1, and GzmB at early times postinfection. Despite PD-1 being primarily expressed on Subdom-CD8+ T cells during latency, a higher frequency of gB-CD8+ T cells expressed PD-1 than Subdom-CD8+ T cells during the acute response at 8 days postinfection (Fig. 7A and B). Of note, the expression patterns of PD-1 and GzmB were different between gB-CD8+ T cells and Subdom-CD8+ T cells, with most PD-1+ cells in the gB-CD8+ T cell group also expressing GzmB, consistent with transient upregulation after activation. In contrast, there was a distinct population of PD-1+ cells that were GzmB− in the Subdom-CD8+ T cell group (Fig. 7A). Accordingly, a higher percentage of PD-1+ gB-CD8+ T cells expressed GzmB than PD-1+ Subdom-CD8+ T cells (Fig. 7C). Furthermore, the levels of PD-1 expression were significantly higher in GzmB− Subdom-CD8+ T cells than in both GzmB+ Subdom-CD8+ T cells and GzmB− gB-CD8+ T cells (Fig. 7D).
FIG 7.

Tim-3+ cells are functional at early times postinfection. TGs from acutely infected mice (8 dpi) were removed; processed into single-cell suspensions; stained for viability, CD45, CD8, gB tetramer, Tim-3, PD-1, and GzmB; and assessed by flow cytometry. In graphs, bars represent means ± SEM (n = 20). Data were pooled from two independent experiments. Data in panels D, H, and J were analyzed by two-way ANOVAs with Tukey’s posttests; data in panels K and L were analyzed by one-way ANOVAs with Tukey’s posttests; and data in panels B, C, F, and G were analyzed by unpaired t tests (*, P ≤ 0.05; ***, P ≤ 0.001; ****, P ≤ 0.0001). (A) Representative dot plots from one mouse showing the expression of PD-1 and GzmB on gB-CD8+ T cells and Subdom-CD8+ T cells along with the FMO gating controls. (B) Percentages of cells in gB-CD8+ T cell and Subdom-CD8+ T cell groups that are PD-1+. (C) Percentages of PD-1+ cells in gB-CD8+ T cell and Subdom-CD8+ T cell groups that are GzmB+. (D) Mean fluorescence intensities (MFI) of PD-1 in PD-1+ GzmB+ and PD-1+ GzmB− gB-CD8+ T cell and Subdom-CD8+ T cell groups. (E) Representative dot plots from one mouse showing the expression of Tim-3 and GzmB on gB-CD8+ T cells and Subdom-CD8+ T cells along with the FMO gating controls. (F) Percentages of cells in gB-CD8+ T cell and Subdom-CD8+ T cell groups that are Tim-3+. (G) Percentages of Tim-3+ cells in gB-CD8+ T cell and Subdom-CD8+ T cell groups that are GzmB+. (H) MFI of Tim-3 in Tim-3+ GzmB+ and Tim-3+ GzmB− gB-CD8+ T cell and Subdom-CD8+ T cell groups. (I) Representative dot plots from one mouse showing the expression of Tim-3 and PD-1 on gB-CD8+ T cells and Subdom-CD8+ T cells along with the FMO gating controls. (J) Total numbers of CD8+ T cells/TG in each of the Tim-3/PD-1 quadrants from panel I. (K) Percentages of gB-CD8+ T cells in Tim-3/PD-1 quadrants that are GzmB+. (L) Percentages of Subdom-CD8+ T cells in Tim-3/PD-1 quadrants that are GzmB+.
Like PD-1, the overall percentage of cells expressing Tim-3 was elevated at 8 days postinfection in both groups, with a significantly higher percentage of cells in the gB-CD8+ T cell group being Tim-3+ than in the Subdom-CD8+ T cell group, similar to what was observed during latency (Fig. 7E and F). Tim-3 and GzmB expression appeared to be strongly correlated, as over 75% of gB-CD8+ T cells expressed both Tim-3 and GzmB (Fig. 7E). Similarly, when we considered only Tim-3+ gB-CD8+ T cells, we found that over 90% of the cells also expressed GzmB (Fig. 7G). Even though a majority of Tim-3+ Subdom-CD8+ T cells also coexpressed GzmB, a lower overall frequency of Tim-3+ Subdom-CD8+ T cells expressed GzmB than their Tim-3+ gB-CD8+ T cell counterparts (Fig. 7G). Despite the differences in GzmB expression between gB-CD8+ T cells and Subdom-CD8+ T cells, a consistent observation in latent and acute infection was that GzmB was preferentially expressed in Tim-3+ cells, suggesting that Tim-3 did not inhibit functionality during HSV-1 infection (Fig. 7H).
During latency, we observed in both the gB-CD8+ T cells and Subdom-CD8+ T cells that cells expressing only Tim-3 were the most functional, cells expressing only PD-1 were the least functional, and cells expressing both Tim-3 and PD-1 exhibited an intermediate functionality, suggesting that Tim-3 was associated with functionality and that PD-1 was associated with impairment (Fig. 4D and E). At 8 days postinfection, the Tim-3+ PD-1+ population made up a much more substantial proportion of the total CD8+ T cells in both groups; however, unlike in latency, the relationship between Tim-3, PD-1, and GzmB differed between the gB-CD8+ T cells and Subdom-CD8+ T cells (Fig. 7I and J). Despite a drop in GzmB expression in Tim-3− PD-1+ cells compared to Tim-3+ cells in the gB-CD8+ T cells, 80% of Tim-3− PD-1+ cells still expressed GzmB (Fig. 7K). This is consistent with the idea that Tim-3 is associated with functionality and that PD-1 is transiently upregulated in gB-CD8+ T cells upon T cell activation during acute infection. The Subdom-CD8+ T cell group had a somewhat different response in which both the Tim-3+ PD-1+ and Tim-3− PD-1+ groups had significantly fewer GzmB+ cells than the Tim-3+ PD-1− group (Fig. 7L). Together, these data support the idea that functional impairment in the Subdom-CD8+ T cell group may not be a result of continuous stimulation during latent HSV-1 infection. Instead, our data suggest that functional impairment may be imprinted on Subdom-CD8+ T cells during acute infection, possibly due to suboptimal priming, resulting in a state of functional impairment that is perpetuated into latency.
DISCUSSION
Checkpoint molecules play an important role in balancing the ability of the adaptive immune system to rapidly respond to insult while also preventing damaging immunopathology. Not surprisingly, this control is not limited to the action of a single checkpoint molecule but instead relies on the collective contributions of many such molecules. Checkpoint molecules can be transiently upregulated after T cell activation and periodically thereafter when the cell reencounters its cognate antigen. Expression of these checkpoint molecules can have different effects, ranging from stimulation to inhibition depending on the expression of ligand(s) (38). In most viral infections, this control works efficiently, and the infection is quickly cleared. However, in cases of persistent antigenic exposure, like those found in LCMV or various cancers, the host fails to clear the infection or tumor, resulting in extended stimulation of T cells and aberrant upregulation of checkpoint molecules (25, 26, 34, 42, 43). This process then begins to limit the T cell response, effectively creating a stalemate between the immune response and the antigenic source.
With both acute and latent life cycles, HSV-1 infection exhibits characteristics of both a short-term, normally cleared viral infection and a chronic infection. Although actively replicating virus is cleared quickly from the periphery after primary infection, the virus infiltrates and establishes latency in host neurons, where it persists for the life of the host. In its latent state, HSV-1 does not produce infectious virus; however, our laboratory and others have shown that low levels of antigenic expression during latency stimulate ganglion-resident CD8+ T cells throughout the life of the host (14, 44–47). Specifically, we know that CD8+ T cells cluster their T cell receptors (TCRs) toward latently infected TG neurons and that TG-CD8+ T cells require periodic antigenic stimulation to remain within the TG (14, 37, 44). Based on previous studies, one would expect that these repeated stimulations of virus-specific CD8+ T cells would lead to a loss of function over time (35). Indeed, studies from our laboratory and others have shown that functional impairment of HSV-1-specific CD8+ T cells does occur in the TG and the brain; however, in the TG, this functional impairment is largely limited to the 50% of CD8+ T cells that recognize subdominant epitopes (18, 31, 48). The other 50% of CD8+ T cells that recognize the immunodominant epitope gB498–505 remain highly functional and actually become more functional throughout latency (18, 49). Similar to chronic viral infection models, PD-1 expression demarks impaired CD8+ T cells in the latently infected TG (Subdom-CD8+ T cells), and functional CD8+ T cells (gB-CD8+ T cells) express little to no PD-1 (31). This disparate phenotype between Subdom-CD8+ T cells and gB-CD8+ T cells led us to investigate how functional impairment may develop in HSV-1 infection. Thus, we sought to identify the expression patterns of other checkpoint molecules to better understand how they may dictate the level of functionality in HSV-1-specific CD8+ T cells.
Using a system in which we can delineate between the largely functional (gB-CD8+ T cell) and functionally impaired (Subdom-CD8+ T cell) populations, we found relatively few differences in the expression of classical checkpoint molecules, with low overall expression levels of most of these markers in both groups. One exception to this was Tim-3, which we observed to be expressed in both populations of CD8+ T cells. Conventionally, Tim-3 has been considered to be an inhibitory checkpoint molecule, with a number of studies showing that Tim-3 expression correlates with functionally exhausted cells in chronic viral infections, cancer models, and HSV-1 infections (26, 29, 30, 32–34). While the evidence for functional compromise in Tim-3+ cells in chronic viral infections and cancer is compelling, the HSV-1 studies assigning functional compromise to Tim-3+ HSV-1-specific CD8+ T cells lacked substantial functional data to support the inhibitory nature of the checkpoint molecule (29, 30). In light of recent evidence that Tim-3 can be stimulatory in some infection models and our previous observation that half of the CD8+ T cells in the latently infected TG remain highly functional, we were interested in identifying whether Tim-3 expression truly implied functional compromise during HSV-1 latency (50–53). Our study revealed that cells expressing Tim-3 or cells negative for both Tim-3 and PD-1 remained highly functional during latency, while cells expressing only PD-1 appeared functionally compromised. Together, these data suggest that PD-1 expression denotes functional compromise in the latently infected TG, while Tim-3 expression may be an effect of recent antigenic stimulation.
When it became apparent that Tim-3 was not mitigating functionality, we asked if viral factors were governing Tim-3 expression in the latently infected TG. Using genetically modified viruses, we were able to moderate the levels of antigenic stimulation during latency. We first showed that the percentage of cells expressing Tim-3 is reduced when CD8+ T cell numbers are increased relative to antigen expression. Next, by overexpressing antigen, we were able to show that Tim-3 expression increased on TG-resident CD8+ T cells. Collectively, these data led to the conclusion that Tim-3 expression in the TG is governed, in part, by viral gene expression. In this model, Tim-3 does not appear to regulate T cell functionality; however, our data suggest that Tim-3 expression during latency may be a marker of recent antigenic stimulation. Therefore, our data support the notion that leaky viral gene expression occurs throughout latency and that expression of immune activation markers like Tim-3 on CD8+ T cells may be useful for determining which viral genes are expressed in the TG during latency, as the virus exits latency, and during viral reactivation. It will be intriguing in future studies to use microscopy to assess the expression levels of Tim-3 and PD-1 on either HSV-1-specific CD8+ T cells that have formed immune synapses with neurons, indicating active stimulation, or cells that are not associated with neurons and are patrolling the TG.
While our initial investigations into Tim-3 were focused on latent time points, we became interested in how Tim-3 may contribute to the T cell response during acute infection. Transcriptional analysis, as well as functional studies directly ex vivo, showed that, like in latent infection, Tim-3 expression at early times after activation is associated with functionality. This was not unexpected, as many checkpoint molecules are transiently upregulated after T cell activation and may serve to hone the functionality of these responding cells (38). During acute infection, Tim-3+ HSV-1-specific CD8+ T cells were highly functional, with about half of these cells concurrently expressing PD-1. In the Subdom-CD8+ T cell group, there was an unexpected, but distinct, population of PD-1+ cells that did not coexpress Tim-3 and had diminished functionality compared to Tim-3+ PD-1+ or Tim-3+ PD-1− cells. The progression to T cell exhaustion/impairment is usually described as a stepwise reduction in functionality with concurrent increased expression of inhibitory checkpoint molecules. However, our results suggest that the T cell impairment observed selectively in Subdom-CD8+ T cells at late times postinfection may be initiated during very early infection and persists into latency. This implies that T cell impairment can result not only from repeated stimulations but also from early defects in development, potentially in priming, that are somehow able to persist despite mechanisms to cull cells with suboptimal performance during activation and expansion. Previous results from our laboratory have shown that regulatory cytokines like interleukin-10 (IL-10) may play a role in the disparate functionality between populations of CD8+ T cells, and it will be of interest to determine how cytokine signaling, in addition to TCR signaling, may affect the expression of checkpoint molecules and subsequent losses of functionality (18).
Checkpoint molecules are generally ascribed inhibitory or stimulatory roles, and for molecules such as PD-1 that have clearly defined negative effects on T cell signaling, their classification as inhibitory is fitting. However, in the case of Tim-3, where downstream signaling and even its ligands have yet to be fully elucidated, classification remains complicated, and claims about inhibition/activation should be supported by T cell functionality data. Although there has been clear evidence showing that Tim-3 can indeed function in an inhibitory manner in many viral infections, functional assessment of Tim-3+ CD8+ T cells from the latently infected TG, until now, has never been performed. Our data support an alternative scenario in which Tim-3 is not a dominant inhibitor or an enhancer of CD8+ T cell function but instead marks cells that are responding to antigenic stimulation and are highly functional.
MATERIALS AND METHODS
Mice.
Six-week-old C57BL/6 female mice were purchased from Jackson Laboratories and infected at 7 to 8 weeks of age via administration of 1 × 105 PFU of purified HSV-1 in 3 μl of phosphate-buffered saline (PBS) onto a cornea that was scarified in a crosshatch pattern using a 30-gauge needle, with the viral inoculant massaged into the cornea with the eyelids. Prior to infection, mice were anesthetized with 2.5 mg of ketamine and 0.25 mg of xylazine intraperitoneally (i.p.). After infection, 6.25 μg of atipamezole (Antisedan) was administered i.p. Mice were infected bilaterally, with one eye receiving HSV-1 strain RE and one eye receiving HSV-1 strain KOS for experiments shown in all figures except for Fig. 5. In Fig. 5, mice were bilaterally infected with the viruses indicated in the figure legend. For clarity, data in all figures except Fig. 5 are from RE-infected TGs, as no notable differences were found between the RE- and KOS-infected TGs. All experimental animal procedures were reviewed and approved by the University of Pittsburgh Institutional Animal Care and Use Committee, and the animals were handled in accordance with guidelines established by the Institutional Animal Care and Use Committee.
Virus.
Viruses used were wild-type HSV-1 KOS and RE strains and genetically modified HSV-1 strains S1L, gC, and ICP0, which were made on the KOS background. The S1L virus lacks the immunodominant gB498–505 epitope through the mutation of residue 498 (SSIEFARL to LSIEFARL). Characterization of the T cell infiltrates to S1L and its construction were recently described (36). The S1L virus was then used to generate the recombinant gC and ICP0 HSV-1 strains. These viruses contain four copies of the gB epitope and flanking sequences (residues 494 to 509) linked to enhanced green fluorescent protein (EGFP) that is expressed from the indicated viral promoters in the gC locus. The construction and characterization of these viruses have been described in detail elsewhere (Treat et al., submitted). Briefly, the following pUC19-based recombinant HSV-1 KOS gC locus-targeting plasmids were constructed: (i) p.gCp-pep4-EGFP, which contains a 4×gB494–509-EGFP expression cassette immediately downstream of the native gC promoter, and (ii) p.gC-ICP0p-pep4-EGFP, which has an interrupted gC promoter and an ectopically placed ICP0 promoter driving 4×gB494–509-EGFP expression. Clean virus was then generated by classical homologous DNA recombination with the above-described linearized gC-targeting plasmids and S1L infectious viral DNA, followed by three rounds of plaque purification based on gain of fluorescence and confirmation of recombinant promoter viruses by Southern blotting. Virus strains were prepared as follows. Flasks with Vero cell monolayers were infected at a multiplicity of infection (MOI) of 0.01 and monitored until cytopathic effect was observed in more than 90% of cells. NaCl (5 M) was added to the existing culture medium in each flask to a final concentration of 0.45 M, flasks were rocked for 1 h at room temperature to release cells into the medium, and cells were collected into 50-ml conical tubes and centrifuged at 6,000 × g for 10 min at 4°C. Supernatants were then filtered through a 0.8-μm filter, overlaid onto a 50% sucrose cushion (0.22-μm filtered) in 38.5-ml polypropylene tubes (catalog number 326823; Beckman Coulter), and pelleted at 142,000 × g for 1 h. After careful removal of most of the supernatant, the virus pellet was resuspended in the remaining sucrose cushion and medium, aliquoted, and stored at −80°C. Before use, the titers of viral stocks were determined on Vero cells. Promoter kinetics and epitope expression in the gC and ICP0 HSV-1 strains were confirmed by in vitro gB-CD8+ T cell stimulations with infected B6 fibroblasts at 4, 8, and 24 h postinfection. The strong ICP0 promoter-driven epitope is detected at 4 h, while no significant gC promoter-driven epitope is detectable until 24 h, as measured by gB-CD8+ T cell activation.
Reagents.
Ketamine, xylazine, and atipamezole were purchased from Henry Schein. BrdU (catalog number B5002), DNase I (catalog number D5025), and Liberase (catalog number 5401119001) were purchased from Millipore-Sigma. Brefeldin A (catalog number B7450), anti-CD45-peridinin chlorophyll protein (PerCP) (clone 30-F11), anti-CD8-allophycocyanin-H7 or -allophycocyanin-Cy7 (clone 53-6.7), anti-IFN-γ–allophycocyanin (clone XMG1.2), anti-TNF-α–phycoerythrin (PE)–Cy7 (clone MP6-XT22), anti-CD107a-fluorescein isothiocyanate (FITC) (clone 1DB4), anti-granzyme B-Brilliant Violet (BV) 421 (clone GB11), and FITC anti-BrdU sets containing FITC anti-BrdU (clone 3D4) and the FITC mouse IgG1 κ isotype (clone MOPC-21), were purchased from BD Biosciences. Anti-Tim-3–PE (clone 215008) was purchased from R&D Systems. Anti-PD-1–PE–Cy7 (clone RMP1-30) was purchased from BioLegend. Anti-PD-1–FITC (clone RMP1-30), anti-TIGIT-FITC (clone GIGD7), and anti-LAG3-FITC (clone eBioC9B7W) were purchased from eBioscience. Anti-CLTA-4–PE–Cy7 (clone UC10-4F10-11) was purchased from Tonbo Biosciences. Anti-BTLA-PE-Vio770 (clone REA224) and anti-2B4-VioBright FITC or -allophycocyanin-Vio770 (clone REA388) were purchased from Miltenyi Biotec. Aqua Live Dead (catalog number L34957) and anti-granzyme B-allophycocyanin (clone GB11) were purchased from Invitrogen. iScript (catalog number 170-8898) was purchased from Bio-Rad. The NIH Tetramer Core Facility provided the H2-Kb tetramers containing the immunodominant gB498–505 peptide conjugated to BV421 and the subdominant RR1982–989 and RR1822–829 peptides conjugated to allophycocyanin. All peptides were purchased from Invitrogen. The BD Biosciences Cytofix/Cytoperm kit (catalog number 554714) was used for all intracellular staining, and the Cytofix/Cytoperm Plus reagent (catalog number 561651) was additionally used for BrdU staining experiments. B6WT3 fibroblasts were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 5% fetal bovine serum (FBS), 1% penicillin and streptomycin, and 1% GlutaMAX (54). Vero cells were maintained in DMEM containing 10% FBS, 1% penicillin and streptomycin, and 1% GlutaMAX.
Flow staining.
TGs were excised and digested in 100 μl of DMEM containing 0.2 U/ml Liberase for 50 min at 37°C before being triturated into a single-cell suspension. TG suspensions were filtered through 35-μm-filter-top flow tubes and stained for surface markers, gB tetramer, and viable cells with Aqua Live Dead for 1 h at room temperature in the dark in PBS or PBS containing 10% FBS. For experiments that included intracellular staining, cells were fixed in Cytofix/Cytoperm for 20 min at 4°C, washed with perm wash, stained with intracellular antibodies in perm wash for 30 min at 4°C, washed with perm wash, and resuspended in FACS buffer (1% FBS and 0.1% sodium azide in PBS). For BrdU staining, cells were fixed in Cytofix/Cytoperm as described above, washed in perm wash, incubated in Cytofix/Cytoperm Plus reagent for 10 min at 4°C, washed in perm wash, and refixed in Cytofix/Cytoperm for 5 min at 4°C before being incubated with 0.3 mg/ml DNase I in PBS for 1 h at 37°C. After DNase I treatment, cells were washed in perm wash, incubated with anti-BrdU antibody for 20 min at room temperature, washed with perm wash, and washed and resuspended in FACS buffer. For experiments that did not involve intracellular staining, cells were fixed in 1% paraformaldehyde for 20 min at 4°C, washed in FACS buffer, and resuspended in FACS buffer before being run.
Stimulations.
For stimulation experiments, 1 × 106 B6WT3 cells/ml were pulsed with 0.9 μg/ml gB peptide in stim medium (RPMI containing 10% FBS, 1% penicillin and streptomycin, and 1% GlutaMAX) for 60 min at 37°C with 5% CO2, with shaking every 10 min. B6WT3 cells were then washed with stim medium and resuspended in stim medium containing 5 μg/ml brefeldin A, 50 μM 2-mercaptoethanol, and CD107a-FITC antibody, and 5 × 105 B6WT3 cells were added to each dissociated TG. Stimulation mixtures were incubated for 6 h at 37°C with 5% CO2 and then stained for surface markers and intracellular cytokines as described above.
NanoString technology.
TGs excised from 10 to 12 mice at 8 dpi were dissociated and stained as described above for viable cells, CD45, CD8, gB tetramer, Tim-3, and PD-1. Since essentially all CD8+ T cells in the HSV-1-infected TG are HSV-1 specific (16), gB tetramer staining was used to gate the CD8+ T cell population into gB-CD8+ T cells or Subdom-CD8+ T cells, and Tim-3− PD-1−, Tim-3+ PD-1−, Tim-3+ PD-1+, and Tim-3− PD-1+ cells from each population were sorted into PBS. Cells were pelleted and resuspended in iScript at a concentration of 250 cells/μl, followed by vortexing for 30 s to lyse cells. The lysate was then pelleted to clear cell debris and stored at −80°C until use. RNA from 1 μl of this cell lysate was amplified by using the nCounter Low RNA input kit (NanoString) with 10 cycles of amplification. This amplified sample was then run with the nCounter PanCancer Immune Profiling Panel. After normalization to housekeeping genes, the indicated populations were assessed for fold change differences, and a threshold of a 1.5-fold change averaged over the three experimental replicates (each replicate consisted of cells from the pooled TGs of 10 to 12 mice for a total of 20 to 24 TGs/replicate) was used to identify genes of interest in the gB-CD8+ T cell population. These genes were further used to assess the Subdom-CD8+ T cell population as well as to compare the gB-CD8+ T cells and Subdom-CD8+ T cells.
Data analysis.
All flow samples were run on a BD FACSAria instrument. Total cell numbers were determined using counting beads. Gates were set on fluorescence-minus-one (FMO) controls. All flow analysis was done in FlowJo, and graphs and statistical analysis were done in Prism (GraphPad). Statistical tests used and numbers for each experiment are indicated in the figure legends.
Data availability.
The NanoString data have been deposited in the NCBI Gene Expression Omnibus (GEO) under GEO series accession number GSE137973.
ACKNOWLEDGMENTS
We thank Moira Geary for assistance with tissue processing and Nancy Zurowski for acquisition and sorting of cells in flow cytometry experiments. We also thank the NIH Tetramer Core Facility (Emory University Vaccine Center, Atlanta, GA) for supplying the gB and RR1 tetramers.
The work was supported by NIH grants R01 EY005945 (R.L.H.), R01 AI138504 (L.P.K.), R01 CA206517 (L.P.K.), R01 EY015291 (P.R.K.), R00 EY025761 (A.J.S.), T32 EY017271 (K.L.C. and B.R.T.), T32 AI049820 (B.R.T.), and P30 EY08098. In addition, it was supported by The Eye and Ear Foundation of Pittsburgh and by an unrestricted grant from Research To Prevent Blindness, New York, NY.
REFERENCES
- 1.Bradley H, Markowitz LE, Gibson T, McQuillan GM. 2014. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J Infect Dis 209:325–333. doi: 10.1093/infdis/jit458. [DOI] [PubMed] [Google Scholar]
- 2.Carr DJ, Wuest T, Ash J. 2008. An increase in herpes simplex virus type 1 in the anterior segment of the eye is linked to a deficiency in NK cell infiltration in mice deficient in CXCR3. J Interferon Cytokine Res 28:245–251. doi: 10.1089/jir.2007.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank GM, Buela K-AG, Maker DM, Harvey SAK, Hendricks RL. 2012. Early responding dendritic cells direct the local NK response to control herpes simplex virus 1 infection within the cornea. J Immunol 188:1350–1359. doi: 10.4049/jimmunol.1101968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kassim SH, Rajasagi NK, Ritz BW, Pruett SB, Gardner EM, Chervenak R, Jennings SR. 2009. Dendritic cells are required for optimal activation of natural killer functions following primary infection with herpes simplex virus type 1. J Virol 83:3175–3186. doi: 10.1128/JVI.01907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeon S, Rowe AM, Carroll KL, Harvey SAK, Hendricks RL. 2018. PD-L1/B7-H1 inhibits viral clearance by macrophages in HSV-1-infected corneas. J Immunol 200:3711–3719. doi: 10.4049/jimmunol.1700417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blyth WA, Hill TJ, Field HJ, Harbour DA. 1976. Reactivation of herpes simplex virus infection by ultraviolet light and possible involvement of prostaglandins. J Gen Virol 33:547–550. doi: 10.1099/0022-1317-33-3-547. [DOI] [PubMed] [Google Scholar]
- 7.Hill TJ, Blyth WA, Harbour DA. 1978. Trauma to the skin causes recurrence of herpes simplex in the mouse. J Gen Virol 39:21–28. doi: 10.1099/0022-1317-39-1-21. [DOI] [PubMed] [Google Scholar]
- 8.Cherpes TL, Busch JL, Sheridan BS, Harvey SA, Hendricks RL. 2008. Medroxyprogesterone acetate inhibits CD8+ T cell viral-specific effector function and induces herpes simplex virus type 1 reactivation. J Immunol 181:969–975. doi: 10.4049/jimmunol.181.2.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rowe AM, St Leger AJ, Jeon S, Dhaliwal DK, Knickelbein JE, Hendricks RL. 2013. Herpes keratitis. Prog Retin Eye Res 32:88–101. doi: 10.1016/j.preteyeres.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. 2000. CD8+ T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med 191:1459–1466. doi: 10.1084/jem.191.9.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan D, Flores O, Umbach JL, Pesola JM, Bentley P, Rosato PC, Leib DA, Cullen BR, Coen DM. 2014. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe 15:446–456. doi: 10.1016/j.chom.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verjans GMGM, Hintzen RQ, van Dun JM, Poot A, Milikan JC, Laman JD, Langerak AW, Kinchington PR, Osterhaus ADME. 2007. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci U S A 104:3496–3501. doi: 10.1073/pnas.0610847104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. 2003. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18:593–603. doi: 10.1016/s1074-7613(03)00112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace ME, Keating R, Heath WR, Carbone FR. 1999. The cytotoxic T-cell response to herpes simplex virus type 1 infection of C57BL/6 mice is almost entirely directed against a single immunodominant determinant. J Virol 73:7619–7626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.St Leger AJ, Peters B, Sidney J, Sette A, Hendricks RL. 2011. Defining the herpes simplex virus-specific CD8+ T cell repertoire in C57BL/6 mice. J Immunol 186:3927–3933. doi: 10.4049/jimmunol.1003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. 2008. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322:268–271. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.St Leger AJ, Jeon S, Hendricks RL. 2013. Broadening the repertoire of functional herpes simplex virus type 1-specific CD8+ T cells reduces viral reactivation from latency in sensory ganglia. J Immunol 191:2258–2265. doi: 10.4049/jimmunol.1300585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. 1998. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM. 1999. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med 5:677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 21.Gallimore A, Glithero A, Godkin A, Tissot AC, Plückthun A, Elliott T, Hengartner H, Zinkernagel R. 1998. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med 187:1383–1393. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wherry EJ. 2011. T cell exhaustion. Nat Immunol 12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 23.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJM, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curran MA, Montalvo W, Yagita H, Allison JP. 2010. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DAA, Wherry EJ. 2009. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin H-T, Anderson AC, Tan WG, West EE, Ha S-J, Araki K, Freeman GJ, Kuchroo VK, Ahmed R. 2010. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 107:14733–14738. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, Morton KE, Mavroukakis SA, Duray PH, Steinberg SM, Allison JP, Davis TA, Rosenberg SA. 2003. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A 100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian SL. 2010. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen SJ, Hamrah P, Gate D, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, BenMohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus 1. J Virol 85:4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy PBJ, Sehrawat S, Suryawanshi A, Rajasagi NK, Mulik S, Hirashima M, Rouse BT. 2011. Influence of galectin-9/Tim-3 interaction on herpes simplex virus-1 latency. J Immunol 187:5745–5755. doi: 10.4049/jimmunol.1102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeon S, St Leger AJ, Cherpes TL, Sheridan BS, Hendricks RL. 2013. PD-L1/B7-H1 regulates the survival but not the function of CD8+ T cells in herpes simplex virus type 1 latently infected trigeminal ganglia. J Immunol 190:6277–6286. doi: 10.4049/jimmunol.1300582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, Wong JC, Satkunarajah M, Schweneker M, Chapman JM, Gyenes G, Vali B, Hyrcza MD, Yue FY, Kovacs C, Sassi A, Loutfy M, Halpenny R, Persad D, Spotts G, Hecht FM, Chun T-W, McCune JM, Kaul R, Rini JM, Nixon DF, Ostrowski MA. 2008. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med 205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, Castelblanco N, Kuchroo V, Gretch DR, Rosen HR. 2009. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol 83:9122–9130. doi: 10.1128/JVI.00639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. 2010. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wherry EJ, Kurachi M. 2015. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Treat BR, Bidula SM, Ramachandran S, St Leger AJ, Hendricks RL, Kinchington PR. 2017. Influence of an immunodominant herpes simplex virus type 1 CD8+ T cell epitope on the target hierarchy and function of subdominant CD8+ T cells. PLoS Pathog 13:e1006732. doi: 10.1371/journal.ppat.1006732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheridan BS, Cherpes TL, Urban J, Kalinski P, Hendricks RL. 2009. Reevaluating the CD8 T-cell response to herpes simplex virus type 1: involvement of CD8 T cells reactive to subdominant epitopes. J Virol 83:2237–2245. doi: 10.1128/JVI.01699-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, Flies DB. 2013. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HES, Hsu L-Y, Freedman TS, Weiss A. 2010. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol 2:a002279. doi: 10.1101/cshperspect.a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, Groner Y, Rao A. 2009. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med 206:51–59. doi: 10.1084/jem.20081242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsukumo S, Yasutomo K. 2018. Regulation of CD8+ T cells and antitumor immunity by Notch signaling. Front Immunol 9:101. doi: 10.3389/fimmu.2018.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. 2009. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 44.Cantin EM, Hinton DR, Chen J, Openshaw H. 1995. Gamma interferon expression during acute and latent nervous system infection by herpes simplex virus type 1. J Virol 69:4898–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu T, Tang Q, Hendricks RL. 1996. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J Virol 70:264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen S-H, Garber DA, Schaffer PA, Knipe DM, Coen DM. 2000. Persistent elevated expression of cytokine transcripts in ganglia latently infected with herpes simplex virus in the absence of ganglionic replication or reactivation. Virology 278:207–216. doi: 10.1006/viro.2000.0643. [DOI] [PubMed] [Google Scholar]
- 47.Halford WP, Gebhardt BM, Carr DJ. 1996. Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J Immunol 157:3542–3549. [PubMed] [Google Scholar]
- 48.Menendez CM, Jinkins JK, Carr DJ. 2016. Resident T cells are unable to control herpes simplex virus-1 activity in the brain ependymal region during latency. J Immunol 197:1262–1275. doi: 10.4049/jimmunol.1600207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frank GM, Lepisto AJ, Freeman ML, Sheridan BS, Cherpes TL, Hendricks RL. 2010. Early CD4+ T cell help prevents partial CD8+ T cell exhaustion and promotes maintenance of herpes simplex virus 1 latency. J Immunol 184:277–286. doi: 10.4049/jimmunol.0902373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Avery L, Filderman J, Szymczak-Workman AL, Kane LP. 2018. Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc Natl Acad Sci U S A 115:2455–2460. doi: 10.1073/pnas.1712107115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorman JV, Starbeck-Miller G, Pham N-LL, Traver GL, Rothman PB, Harty JT, Colgan JD. 2014. Tim-3 directly enhances CD8 T cell responses to acute Listeria monocytogenes infection. J Immunol 192:3133–3142. doi: 10.4049/jimmunol.1302290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu Y, Chen J, Liao H, Zhang Y, Wang H, Li S, Luo Y, Fang D, Li G, Zhou B, Shen L, Chen CY, Huang D, Cai J, Cao K, Jiang L, Zeng G, Chen ZW. 2012. Tim-3-expressing CD4+ and CD8+ T cells in human tuberculosis (TB) exhibit polarized effector memory phenotypes and stronger anti-TB effector functions. PLoS Pathog 8:e1002984. doi: 10.1371/journal.ppat.1002984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gorman JV, Colgan JD. 2018. Acute stimulation generates Tim-3-expressing T helper type 1 CD4 T cells that persist in vivo and show enhanced effector function. Immunology 154:418–433. doi: 10.1111/imm.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pretell J, Greenfield RS, Tevethia SS. 1979. Biology of simian virus 40 (SV40) transplantation antigen (TrAg). V. In vitro demonstration of SV40 TrAg in SV40 infected nonpermissive mouse cells by the lymphocyte mediated cytotoxicity assay. Virology 97:32–41. doi: 10.1016/0042-6822(79)90370-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The NanoString data have been deposited in the NCBI Gene Expression Omnibus (GEO) under GEO series accession number GSE137973.