

Double-stranded DNA viruses typically package their genomes into a preformed capsid. In contrast, single-stranded RNA viruses assemble their coat proteins around their genomes via extensive nucleotide-protein interactions. Single-stranded DNA (ssDNA) viruses appear to blend both strategies, using nucleotide-protein interactions to organize their genomes into preformed shells, likely by a controlled process. Chaotic genome-capsid associations could inhibit packaging or genome release during the subsequent infection. This process appears to be partially controlled by the unessential A* protein, a shorter version of the essential A protein that mediates rolling-circle DNA replication. Protein A* may elevate fitness by ensuring the product fidelity of packaging reactions. This phenomenon may be widespread in ssDNA viruses that simultaneously synthesize and package DNA with rolling circle and rolling circle-like DNA replication proteins. Many of these viruses encode smaller, unessential, and/or functionally undefined in-frame versions of A/A*-like proteins.
KEYWORDS: DNA packaging, microvirus, ϕX174, ssDNA
ABSTRACT
In microviruses, 60 copies of the positively charged DNA binding protein J guide the single-stranded DNA genome into the icosahedral capsid. Consequently, ∼12% of the genome is icosahedrally ordered within virions. Although the internal volume of the ϕX174, G4, and α3 capsids are nearly identical, their genome lengths vary widely from 5,386 (ϕX174) to 6,067 (α3) nucleotides. As the genome size increases, the J protein’s length and charge decreases. The ϕX174 J protein is 37 amino acids long and has a charge of +12, whereas the 23-residue G4 and α3 proteins have respective +6 and +8 charges. While the large ϕX174 J protein can substitute for the smaller ones, the converse is not true. Thus, the smallest genome, ϕX174, requires the more stringent J protein packaging guide. To investigate this further, a chimeric virus (ϕXG4J) was generated by replacing the indigenous ϕX174 J gene with that of G4. The resulting mutant, ϕXG4J, was not viable on the level of plaque formation without ϕX174 J gene complementation. During uncomplemented infections, capsids dissociated during packaging or quickly thereafter. Those that survived were significantly less stable and infectious than the wild type. Complementation-independent ϕXG4J variants were isolated. They contained duplications that increased genome size by as much as 3.8%. Each duplication started at nucleotide 991, creating an additional DNA substrate for the unessential but highly conserved A* protein. Accordingly, ϕXG4J viability and infectivity was also restored by the exogenous expression of a cloned A* gene.
IMPORTANCE Double-stranded DNA viruses typically package their genomes into a preformed capsid. In contrast, single-stranded RNA viruses assemble their coat proteins around their genomes via extensive nucleotide-protein interactions. Single-stranded DNA (ssDNA) viruses appear to blend both strategies, using nucleotide-protein interactions to organize their genomes into preformed shells, likely by a controlled process. Chaotic genome-capsid associations could inhibit packaging or genome release during the subsequent infection. This process appears to be partially controlled by the unessential A* protein, a shorter version of the essential A protein that mediates rolling-circle DNA replication. Protein A* may elevate fitness by ensuring the product fidelity of packaging reactions. This phenomenon may be widespread in ssDNA viruses that simultaneously synthesize and package DNA with rolling circle and rolling circle-like DNA replication proteins. Many of these viruses encode smaller, unessential, and/or functionally undefined in-frame versions of A/A*-like proteins.
INTRODUCTION
Icosahedral double-stranded DNA (dsDNA) viruses typically package their genomes into preformed, volumetrically constrained capsids (1–3). Filled capsids are extremely dense, with internal DNA concentrations reaching approximately ∼500 mg/ml (4). To achieve this density, the packaged DNA takes the form of an ordered spool (5). In contrast, capsid assembly and packaging are concurrent processes in most single-stranded RNA (ssRNA) virus life cycles (6, 7). Coat proteins recognize specific RNA sequences or secondary structures, which likely creates the high concentrations of coat protein needed to nucleate assembly around the genome. The complex branched secondary structures of viral ssRNA genomes make them unusually compact molecules, suggesting that they have evolved to be encapsidated in this manner (8). The ssDNA circoviruses and geminiviruses may employ a similar packaging mechanism (9, 10). In contrast, the ssDNA parvo- and microviruses combine features of both dsDNA and ssRNA packaging strategies. Like dsDNA viruses, genomes are packaged into preformed shells (11, 12); however, they do not form the iconic, highly ordered dsDNA spool. As seen with ssRNA viruses (7, 9, 10), a portion of the packaged genome interacts with the capsid’s inner surface (13–18).
In micro- and parvoviruses, ssDNA synthesis and packaging are concurrent processes. The ϕX174 ssDNA synthesis/packaging reaction commences when protein A covalently attaches to the replication origin of a J protein-coated, dsDNA template (19, 20). Protein J is a DNA binding protein (see below) and is also a virion structural protein. As the daughter strand is synthesized, the parental strand is displaced and enters the capsid along with 60 J proteins. After one round of ssDNA biosynthesis, the replication fork encounters the origin. Protein A then ligates the ends of the packaged parental strand, completing synthesis of the single-stranded circular genome, and reattaches to the origin of the dsDNA template.
The J proteins from the three canonical microvirus clades, represented by ϕX174, G4, and α3, share a similar structural arrangement (Fig. 1). Their N termini are highly basic, ranging in charge from +6 to +1. However, the arrangement of the R and K residues exhibit a high degree of variation. The C termini, which interact with the viral coat protein, are strongly conserved, along with their interacting coat protein residues (16–18). In virions, the proteins appear to guide the DNA between asymmetric units around the 5-fold axes of symmetry (Fig. 1). Consequently, ∼12% of the genome is icosahedrally ordered (15–18, 21). These interactions also suppress intramolecular base pairing found in naked DNA. Approximately 70% of the naked ϕX174 genome is base paired, which decreases to ∼30% within virions (22). In some instances, J proteins can cross-complement. For example, bacteriophage α3 can package with the ϕX174 J protein. This alters the pattern of the icosahedrally ordered DNA density (17, 23, 24). In addition, the chimeric virus does not appear to utilize the entire ϕX174 J protein. Only the homologous regions found in both J proteins is ordered in the virion structure.
FIG 1.
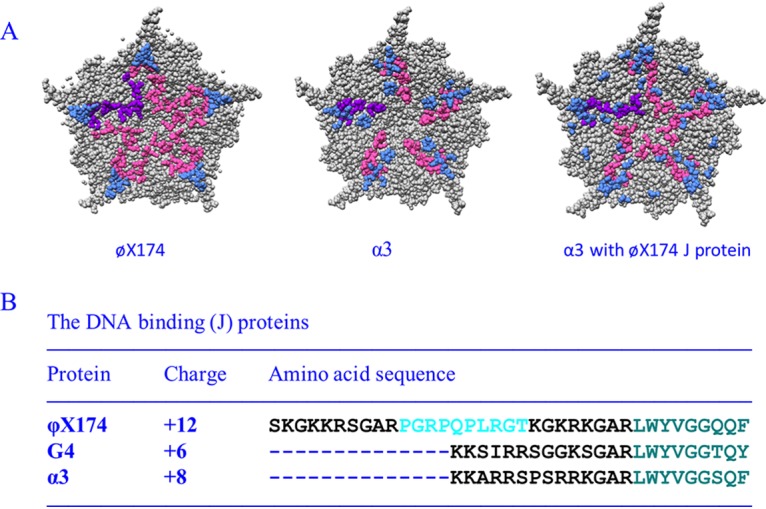
Characteristics of microvirus J proteins and their structural influences on packaged DNA. (A) Fivefold axes of symmetry of ϕX174 filled with its own J protein (PDB 2BPA), α3 filled with its own J protein (PDB 1M06), and α3 filled with the ϕX174 J protein (PDB 1RB8). The coat protein is depicted in gray, and ordered DNA is shown in light blue. The leftmost J protein is depicted in purple, the remaining five are depicted in magenta (B) ϕX174, G4, and α3 J proteins. The conserved, coat protein-interacting, C-terminal amino acids are depicted in teal. The basic DNA-binding regions are depicted in black. The unique proline-rich region in the ϕX174 protein is depicted in cyan.
J protein size and charge neutralization do not directly correlate with genome size (Fig. 1). The ϕX174, G4, and α3 genomes are, respectively, 5,386, 5,577, and 6,089 nucleotides in length. Although ϕX174 has the smallest genome, its J protein is considerably larger and more basic than those found in G4- and α3-like viruses. Cross-complementation studies demonstrate that ϕX174 requires its larger J protein, whereas G4 and α3 can utilize any of the three J proteins (17, 25). Thus, a simple charge neutralization model does not explain packaging capacity. Vis-à-vis volume, a similar packaging discrepancy has been noted between the genome sizes of the adeno-associated and autonomous replicating parvoviruses (26). To more fully elucidate the biophysical and genetic parameters governing this phenomenon, a chimeric virus (ϕXG4J) was generated in which the ϕX174J gene was replaced with that of G4. The results of subsequent biochemical and second-site genetic analyses suggest that the unessential ϕX174 A* protein plays a role during genome packaging.
All known microviruses produce an A protein variant known as protein A* (27). Translated from in-frame internal start codons, A* proteins are N-terminally truncated versions of A proteins. Eliminating ϕX174 A* expression appears to have no discernible phenotype (28). Thus, like Greta Garbo, a star who wanted to be left alone, A*’s private life has remained mysterious. Similar A/A*-like arrangements are seen in some parvo- and circoviruses, encoding N-terminally truncated versions of their rep proteins, which also mediate rolling circle and rolling-circle like DNA replication (29, 30).
RESULTS
The ϕXG4J chimera requires the exogenous expression of the cloned ϕX174 J gene.
To create ϕXG4J, the G4 J gene was engineered into the ϕX174 viral genome, completely replacing the endogenous J gene. Only the coding sequence was replaced to ensure that gene expression remained under wild-type (WT) control. The chimera displayed a complementation-dependent phenotype: plaque formation required the exogenous expression of the cloned ϕX174 J gene (Table 1). Rescue was species specific. Cloned G4 J gene expression, which rescued a G4 am(J) mutant, did not support ϕXG4J plaque formation. J proteins gain access to the procapsid by first coating the replicative-form (RF) dsDNA template at a stoichiometry similar to those found in virions (19, 20). There are 100 to 120 J proteins per RF dsDNA versus 60 proteins per ssDNA genome in the virion. Within the G4 system, the G4 J protein is obviously functional. Thus, the differences in the inherent ability of the two J proteins to bind DNA before packaging commences do not adequately explain the lethal phenotype. Since viable complementation-independent ϕXG4J variants, which did not alter the G4 J protein, were ultimately isolated (see below), the lethal phenotype most likely reflects packaging defects and/or the production of uninfectious packaged particles.
TABLE 1.
Rescue of ϕXG4J by the expression of cloned ϕX174 J and A* genes
| Mutant | Rescuea of ϕXG4J by expression of various cloned ϕX174 J and A* genes |
||||
|---|---|---|---|---|---|
| None | ϕXJ | G4J | ϕXA* |
||
| +b | – | ||||
| ϕXG4J | <10−3 | 1.0 | <10−3 | 0.8 | <10−3 |
| ϕX174 am(J) | <10−3 | 1.0 | <10−3 | <10−3 | <10−3 |
| G4 am(J) | <10−3 | 0.9 | 1.0 | <10−3 | <10−3 |
Rescue, assay titer/titer on cells expressing the cloned ϕX174 J gene.
With + and – cloned gene induction.
The ϕXG4J chimera formed unstable, less infectious virions at reduced yields.
To examine the ϕXG4J infection products, lysis-resistant cells were infected at a multiplicity of infection (MOI) of 3.0 and incubated for 3 h at 33°C. Soluble extracts were then prepared from concentrated infected cells. Before sucrose gradient sedimentation, extracts were treated with chloroform to remove the external scaffolding protein from unfilled procapsids (108S). The resulting degraded procapsids sediment at 70S, which can be easily distinguished from virions (114S) after rate zonal sedimentation. Assembled particles were detected by UV spectroscopy after gradient fractionation. As shown in Fig. 2A, the ϕXG4J 114S particle yield (magenta) was greatly reduced compared to the wild-type control (blue). Moreover, the specific infectivity (PFU/A260) of particles was reduced by 90% compared to wild-type particles: 2.0 × 1011 PFU/A260 (ϕXG4J) versus 2.0 × 1012 PFU/A260 (WT). Titers were determined in cells expressing the wild-type ϕX174 J gene: thus, any particle capable of infecting should form a plaque.
FIG 2.
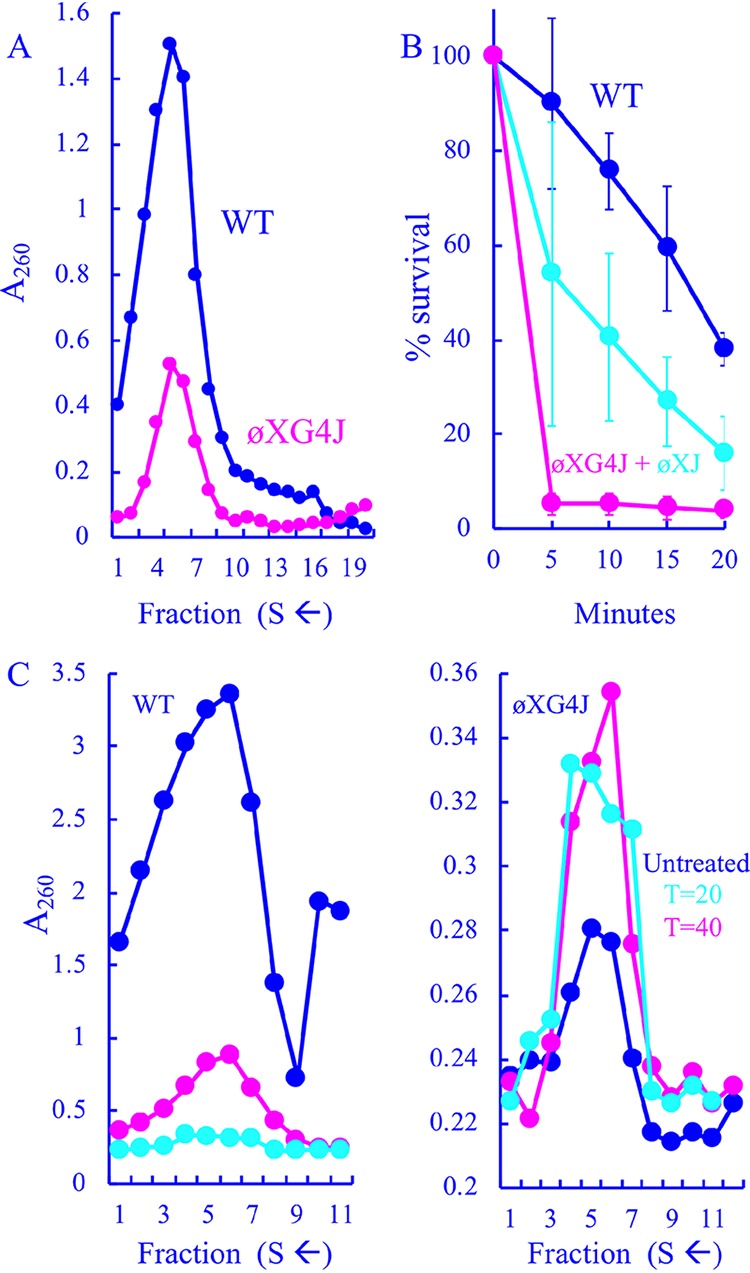
Particles generated in ϕXG4J-infected cells. (A) Sedimentation profiles of extracts generated from WT- and ϕXG4J-infected cells. Gradients were fractionated from the bottom; thus, lower fractions contain particles with higher S values. (B) Heat stability as a function of time of the wild type, ϕXG4J exclusively filled with the G4 J protein (magenta), and ϕXG4J generated in cells expressing a cloned ϕX174 J gene (cyan). (C) Sedimentation profiles of particles produced in wild-type (left)- and ϕXG4J (right)-infected cells in the presence of nalidixic acid, with a gyrase inhibitor added 20 (cyan) or 40 min postinfection (cyan), and untreated infections (blue).
Blocking DNA packaging typically leads to an accumulation of procapsids (108S) or, when chloroform is used during purification, 70S degraded procapsids (31–33). However, no peak was observed within the relevant region of the gradient, which typically spans fractions 12 to 15. In addition, the coat protein level in ϕXG4J whole-cell lysates was comparable to that observed in the wild-type infection (data not shown), indicating that the cells were infected at approximately the same level as the wild type. This suggests that ϕX174 particles filled with G4 J may be unstable and/or dissociate during DNA packaging. Two experiments were performed to address this hypothesis. In the first experiment, WT and ϕXG4J particles isolated from the gradients depicted in Fig. 2A were assayed for heat stability at 42°C. ϕXG4J particles generated in cells expressing the ϕX174 J gene were also tested. ϕXG4J particles were considerably less stable than the WT (Fig. 2B). The ϕXG4J particles generated in cells expressing the cloned ϕX174 J gene exhibited an intermediate level of stability, which suggests that the presence of the ϕX174 J protein in these heterogeneous particles can partially restore stability. Although two J proteins are evident when complemented progeny are analyzed by SDS-PAGE, precise protein ratios could not be established. There was too much variability between samples and too many technical difficulties associated with separating these small basic proteins.
In the second experiment, wild-type and ϕXG4J particle production was characterized in lysis-resistant cells treated with nalidixic acid, a gyrase inhibitor that blocks DNA synthesis. Under normal infection conditions, ϕX174 first delivers its ssDNA genome to the cytoplasm, where it becomes associated the host’s inner membrane, the site of stage I DNA synthesis (ssDNA to dsDNA), which produces the parental RF molecule. This molecule is copied in stage II DNA synthesis. After synthesis of the relevant viral proteins, stage III DNA synthesis (dsDNA to ssDNA) and packaging commence. Nalidixic acid inhibits the final two stages of ϕX174 DNA replication (34): thus, delaying its addition to the infection should allow stage II synthesis but block stage III and packaging. At 20 or 40 min postinfection, a portion of each culture was removed, and nalidixic acid was added to a concentration of 50 μg/ml (34). The remaining third was left untreated. After a 3-h incubation, extracts were prepared without chloroform and analyzed by rate zonal sedimentation. To distinguish between virions and procapsids isolated in the sedimentation profiles, material was analyzed by SDS-PAGE, and the external scaffolding/coat protein ratio was calculated. For virions, this ratio is typically ∼0.1. In contrast, it is ∼0.8 for procapsids.
Compared to the untreated control infection (Fig. 3C, blue), wild-type particle production was greatly reduced in nalidixic acid-treated cells (magenta and cyan). This is likely due to reductions in RF DNA, the transcription template, as described above. The particles in the untreated sample were primarily virions, whereas they were primarily procapsids in the treated samples. Unlike the wild-type infections, inhibiting DNA synthesis led to equal or higher ϕXG4J particle yields (Fig. 3D). Again, the predominating particles were virions and procapsids in untreated and treated samples, respectively. Thus, procapsids can be efficiently isolated from ϕXG4J infections if packaging is blocked, suggesting that they dissociate while being filled with the G4 J protein or the resulting virions are unstable and quickly aggregate. Alternatively, the off-pathway products sediment too heterogeneously to form a peak above background. However, the procapsids that survive being filled display reduced heat stability and a lower specific infectivity, which may also reflect stability defects. Particles within the virion peak may degrade after gradients have been fractionated but before plating.
FIG 3.
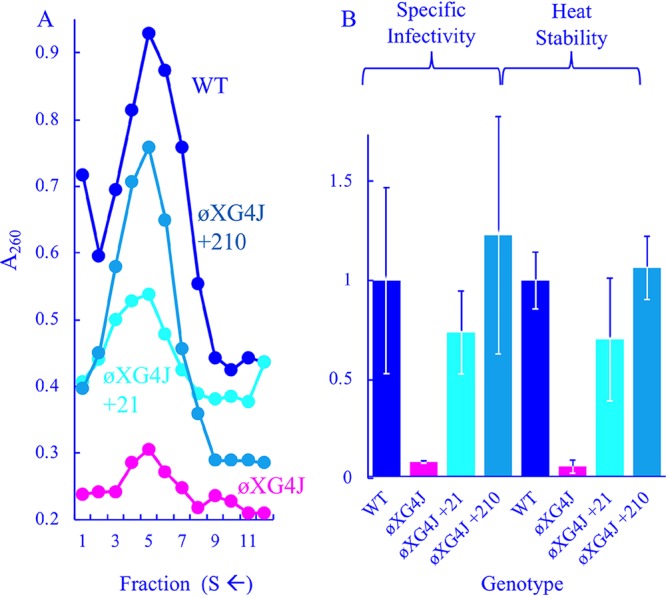
Characteristics of viable ϕXG4J variants with chromosomal duplications. (A) Profiles of extracts generated from WT-, ϕXG4J-, ϕXG4J+210-, and ϕXG4J+21-infected cells. (B) Specific infectivity (PFU/A260) and heat stability (% survival at t = 20) of purified particles from panel A. All values have been normalized to the wild type and incorporate three replicates.
Second-site suppressor analyses suggest two compensatory mechanisms for restoring viability.
The results of second-site suppressor analyses can elucidate mechanistic details that may not be apparent from biochemical results. Thus, viable, complementation-independent ϕXG4J variants were selected on cells without cloned ϕX174J gene expression. Two distinct plaque-forming phenotypes were readily apparent: fuzzy pinprick plaques and medium-sized, clear plaques compared to the wild type. The suppressors with the pinprick plaque phenotype contained missense mutations in gene F, which encodes the viral coat protein. Many were identical or similar to previously isolated suppressors of defective mutations in the internal scaffolding B or J genes (24, 31, 33, 35–37). Table 2 summarizes these mutations, several of which were independently isolated. Without exogenous ϕX174 J expression, the plating efficiencies (calculated as uncomplemented titer/complemented titer) were still low at approximately 10−1. While this indicates that the mutations may stabilize procapsids during the packaging reaction or the resulting virions, the effects were too modest to be detected in the in vivo biochemical characterizations conducted with the stronger suppressors (see below) that conferred changes in the noncoding J-F intercistronic region.
TABLE 2.
Genotypes and characteristics of complementation-independent ϕXG4J isolates
| Genetic changes found in viable strainsa |
No. of independent isolates | Suppressing phenotypeb | Structural/genetic characteristicsc | |
|---|---|---|---|---|
| Nucleotide change | Coat protein amino acidd | |||
| T1044C | L14P | 2 | Pinprick | F-J contact site |
| G1067T | G22C | 1 | Pinprick | J protein suppressor† |
| G1129T | E42D* | 3 | Pinprick | F-J contact site |
| A1134G | D44G | 1 | Pinprick | F-J contact site |
| G1214T | V71F* | 2 | Pinprick | J protein suppressor† |
| Internal scaffolding (B) suppressor† | ||||
| B/J protein binding pocket | ||||
| C1220T | H73Y* | 2 | Pinprick | B/J protein binding pocket |
| A1613G | T204A* | 2 | Pinprick | J protein suppressor† |
| External scaffolding (D) suppressor† | ||||
| A991G | 2 | Pinprick | J-F intercistronic region | |
| G993T | 1 | Pinprick | J-F intercistronic region | |
| G993C | 1 | Pinprick | J-F intercistronic region | |
| 991–1011 duplicated (+21) | None | 5 | Medium | J-F intercistronic region |
| 991–1014 duplicated (+24) | None | 1 | Medium | J-F intercistronic region |
| 991–1202 duplicated (+210) | E42D | 1 | Medium | J-F intercistronic region |
Single base changes in gene F, which encodes the viral coat protein. The nucleotide substitutions are reported with the wild-type nucleotide, the position within the published sequence (69), and the substituted nucleotide. The amino acid substitutions conferred by the mutation are reported with the wild-type amino acid, the position within the protein, and the substituted amino acid. In some strains, a portion of the J-F intercistronic region and 5′ end of the downstream F gene was duplicated. The duplicated nucleotides are numbered. The number within the parentheses refers to the duplication’s length.
The suppressing phenotype reflects plating efficiencies and plaque morphologies. Phenotypes: pinprick, forms weak fuzzy pinprick plaques without exogenous complementation by the cloned ϕX174 J gene at an ∼10−1 frequency (uncomplemented titer/complemented titer); medium, forms clear distinct plaques, but smaller than the wild type. Uncomplemented and complemented titers are comparable.
Information regarding the coat protein amino acids in the virion atomic structure (15, 16) and/or other phenotypes associated with the mutation. †, Mutations previously isolated as suppressors of mutant J proteins (24), mutant internal scaffolding proteins (62), or mutant external scaffolding proteins (31, 33, 35).
*, Missense mutations that were also isolated independently in cis with the 991–1011 (+21) duplication.
Two types of suppressors mapped to the J-F intercistronic region. The weak suppressors conferred single nucleotide substitutions at nucleotides 991 and 993. The suppressors with the medium plaque phenotype all contained duplications within the J-F intercistronic region, some of which included portions of gene F. Every duplication began precisely at nucleotide 991 and were either 21, 24, or 210 nucleotides in length. Unlike the weak suppressors, the duplications exhibited approximately equal plating efficiencies regardless of exogenous cloned ϕX174J gene expression (Table 3).
TABLE 3.
Phenotypes of wild-type ϕX174, ϕXG4J, and ϕXG4J with genome duplications and inserts
| Mutant | Genome (ϕX174) |
Inserted sequence | Plating efficiencya (ϕXJ gene expression) |
||
|---|---|---|---|---|---|
| Size (no. of nucleotides) | Content (%) | With | Without | ||
| ϕX174 | 5,386 | 100 | 1.0 | 1.0 | |
| ϕX174 G4J | 5,344 | 99.2 | 1.0 | 10–5 | |
| ϕXG4J dup21 | 5,364 | 99.6 | AGGATAAATTAATGTCTAAT | 1.0 | 1.0 |
| ϕXG4J dup24 | 5,368 | 99.6 | AGGATAAATTAATGTCTAATATTC | 1.0 | 1.0 |
| ϕXG4J dup210 | 5,554 | 103.8 | AGGATAAATTAATGTCTAAT+190 | 1.0 | 1.0 |
| ϕXG4J scrambled | 5,364 | 99.6 | GAATTATAGTAGTATAACTA | 1.0 | <10–4 |
| ϕXG4J+42H | 5,386 | 100 | Tolerated 42-nucleotide insert in gene H | 1.0 | <10–4 |
Plating efficiency, assay titer/titer on cells expressing the cloned ϕX174 J gene.
The strain with the largest duplication also contained the G→C nucleotide change at position 1129, which confers an E→D amino acid substitution at position 42 in the viral coat protein. This substitution was also isolated as a single mutation with weak suppressing effects. In the +210 strain, this mutation is present in the duplication and the F gene coding sequence. Thus, it likely arose first. As the +210 insertion only arose once, there is no indication that the E→D mutation is unique in any regard. Indeed, other coat protein suppressors were isolated in cis with the smaller duplications.
The +21 duplication was isolated quite readily, independently arising in four different genetic backgrounds. After the initial selection, the putative weak suppressors were plaque purified without exogenous J gene expression to avoid propagating any residual parental ϕXG4J. Two very distinct plaque morphologies were present on many of the purification plates. Although the most prominent morphology represented the original isolate, large plaque-forming variants typically constituted 1 to 10% of the total population. Genomes producing each representative plaque morphology were sequenced. Although both contained an identical coat gene mutation, the large plaque-forming variants acquired the +21 duplication.
Duplications elevated ϕXG4J infectivity and stability.
The ϕXG4J +21 and +210 variants were purified as described above (Fig. 3A), and their specific infectivities and heat stabilities were determined (Fig. 3B). These variants appeared to produce more assembled particles than the ϕXG4 J parent but less than the wild type. The duplications also appeared to elevate specific infectivity and heat stability to wild-type levels.
Unraveling the mechanism underlying the suppressing duplications.
The J-F intercistronic region contains at least five different regulatory elements: a transcription terminator, which overlaps with an mRNA stability sequence for the upstream J gene, the gene F ribosome binding site (RBS), a weak minor promoter, and the cleavage-covalent attachment site for the unessential A* protein (20, 38, 39). Deletions within this region have been previously characterized. Those that do not affect the gene F RBS are well tolerated and even advantageous under some selective conditions (40–44). Another deletion has been shown to increase intracellular coat protein levels via translational coupling mechanism without altering mRNA levels (40).
As listed above, there are numerous genetic elements that could affect gene expression within the J-F intercistronic region; thus, the duplications could affect either transcription or translation. Altering the relative ratios of interacting structural proteins is a well-documented suppression mechanism (40, 45, 46). In ϕX174, for example, 20 external scaffolding proteins interact with one coat protein pentamer during procapsid assembly. When a mutation lowers the concentration of functional scaffolding proteins, they are too broadly distributed among the pentamers to support assembly. However, reducing the coat protein concentrations restores the optimal scaffolding/pentamer ratio (35). If the duplications operate by altering J/F protein ratios, it may be possible to rescue ϕXG4J plaque formation by recapitulating the optimal ratio via exogenously expressing cloned J or F genes. Similarly, exogenous gene expression may counteract the newly established balance in cells infected with ϕXG4J +21 and +210, which could inhibit plaque formation. As can be seen in Table 4, the exogenous expression of these genes had no effect on plaque formation in any of the tested strains. Although these results argue against this model, they do not eliminate the possibility of subtle expression alterations that cannot be controlled in plating assays nor detected by examining protein levels of infected whole-cell lysates on SDS-PAGE gels (data not shown).
TABLE 4.
ϕXG4J, ϕXG4J+21, and ϕXG4J+210 plating efficienciesa in cells expressing cloned F, ϕX174J, and G4J genes
| Mutant | Plating efficiency in cells expressing various genes |
|||
|---|---|---|---|---|
| None | ϕXJ | G4J | ϕXF | |
| ϕXG4J | <10−3 | 1.0 | <10−3 | <10−3 |
| ϕXG4J+21 | 1.0 | 1.0 | 1.0 | 0.8 |
| ϕXG4J+210 | 1.0 | 1.0 | 1.0 | 1.0 |
Plating efficiency, assay titer/titer on cells expressing the cloned ϕX174 J gene.
Very few places within the ϕX174 genome can tolerate random genetic duplications without affecting gene regulation or encoded protein function. This may explain why the duplications occurred in the noncoding J-F intercistronic region, but it does not address if more DNA, regardless of sequence or location, can assuage ϕXG4J’s lethal phenotype. However, there are coding sequences, primarily in gene H, that tolerate in-frame insertions and duplications that do not affect the protein’s folding and function (47). Although gene H duplications were not recovered as suppressors, a viable H+42 gene was placed directly in the ϕXGJ background. The insertion did not alleviate the complementation-dependent phenotype (Table 3). To determine whether the suppressing duplications were sequence specific, the nucleotides of the +21 insertion were scrambled and placed with the ϕXG4J genome. Again, the resulting mutant retained the complementation-dependent phenotype (Table 3), indicating that suppression required a sequence-specific duplication.
Exogenous A* gene expression rescues ϕXG4J plaque formation.
The J-F intercistronic region also contains the primary cleavage-covalent attachment site of protein A*, an unessential, but highly conserved gene found in all microviruses. A* is an N-terminally truncated version of the essential A protein, which mediates rolling circle DNA replication (27, 28). Each suppressing duplication started at nucleotide 991, to which protein A* is found covalently attached when purified from phage-infected cells (38). If the mechanism of suppression involves duplicating A*’s nucleotide substrate, then elevating intracellular A* levels may also rescue ϕXG4J. Classical genetic approaches are particularly problematic with gene A*. Since A* shares a common reading frame with gene A, modifying the A* start codon or RBS creates substitutions within protein A. However, the A* gene can be cloned if expression of its toxic gene product is tightly controlled (28), as in the pBAD vector system (48). Cloned A* gene expression rescued ϕXG4J on the level of plaque formation (Table 1). The plating efficiency was comparable to that obtained on cells expressing the cloned ϕX174 J gene. Rescue was a function of cloned gene induction, since repressing expression from the pBAD plasmid abolished rescue.
Exogenous expression of gene A* elevated the infectivity of ϕXG4J virions.
To elucidate the mechanism by which elevated A* levels rescue the packaging defects associated with ϕXG4J, infection products were analyzed with or without exogenous A* gene expression. For these experiments, lysis-resistant cells were grown to a concentration of 2.0 × 108 cells/ml in media containing glucose to repress A* gene expression, which is under arabinose induction. Cells were concentrated, washed to remove glucose, and then transferred to prewarmed media containing either glucose or arabinose. Infected-cell extracts were analyzed by rate zonal sedimentation. As shown in Fig. 4A, ϕXG4J particle yield did not vary as a function of cloned A* gene expression. However, wild-type yield was reduced. This may be associated with the toxic effects associated with the A* protein (28).
FIG 4.
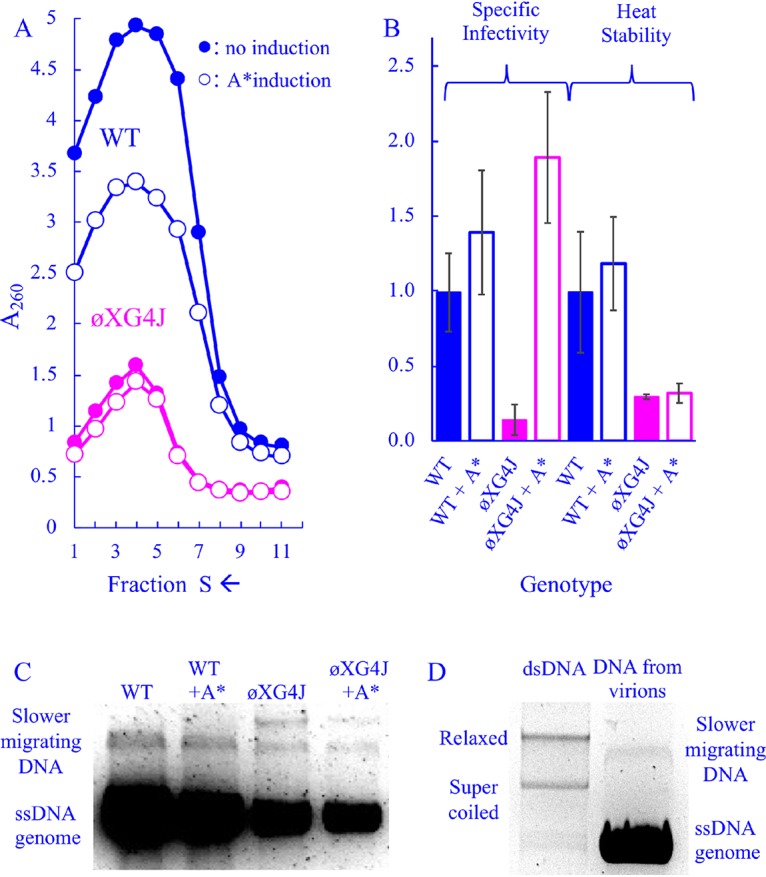
Characteristics of wild-type and ϕXG4J particles in cells expressing a cloned A* gene. (A) Sedimentation profiles of extracts generated from WT- and ϕXG4J-infected cells with or without cloned gene A* induction (open and closed symbols, respectively). (B) Specific infectivity (PFU/A260) and heat stability (% survival at t = 20) of purified particles from panel A. All values have been normalized to the wild type, and error bars represent the standard deviations of three replicates. (C) DNA isolated from purified particles from panel A. (D) DNA from the wild-type particles depicted in panel C run next to purified double-stranded RF ϕX174 DNA.
The specific infectivity and heat stability of all particle types were analyzed (Fig. 4B). Unlike the genome duplications, exogenous A* expression did not affect product heat stability, but it appeared to elevate specific infectivity. ϕXG4J particles synthesized in A*-expressing cells exhibited a specific infectivity 10-fold higher than those in which cloned A* gene expression was repressed. Although these particles also appear to be slightly more infectious than the wild type, this is likely an experimental artifact. When calculating specific infectivity (PFU/A260), lower particle yields reduce the denominator (A260). Thus, the contribution of background absorbance may artificially elevate the specific infectivity of low-yield, short peaks, which makes comparing their specific infectivity values to high-yield, tall peaks difficult.
Aberrant DNAs associated with wild-type and ϕXG4J assembled particles.
To further characterize the DNA associated with particles, DNA was extracted from particles that sedimented within the virion peaks depicted in Fig. 4A. DNA was collected from particles found in the fraction at the center of the absorbance peak and the two adjacent fractions, which likely spans the 90–140S region of the gradient. The extracted DNA was analyzed by agarose electrophoresis (Fig. 4C). In all samples, single-stranded genomic DNA predominated. However, one slower-migrating species was observed in the DNA extracted from wild-type particles, and two slower species were observed in the ϕXG4J populations. The slower-migrating band did not migrate like supercoiled or relaxed RF DNA, suggesting that it is not RF DNA (Fig. 4D). These slower-migrating DNAs are very minor species, which can only be detected on vastly overloaded gels. Despite numerous attempts, it was not possible to purify the requisite quantity for a PCR-independent analyses, such as endonuclease and S1 nuclease digestion. Thus, insights are limited, but include the following: (i) the off-pathway mechanism that produces these species occurs more often in ϕXG4J-infected cells than in wild-type infected cells, and (ii) while particles associated with these abnormal DNAs may contribute to ϕXG4J’s lower specific infectivity, which is approximately 10% of the wild type, they do not fully account for the reduction.
The +210 insertion is genetically unstable in an otherwise wild-type ϕX174 background.
The ϕXG4J+210 genome is ∼103.8% in length compared to the wild-type genome, a value beyond the previously described 101% packaging capacity of ϕX174 determined in vitro (49). If the insertion was placed into a wild-type background, genome size would increase slightly due to the longer ϕX174 J gene. To determine whether the +21 and +210 duplications were stable in a wild-type background, the G4 J gene was replaced with the ϕX174 J gene in an otherwise wild-type genetic background (see Materials and Methods). The ϕX174 +21 phage was easily recovered. However, recovering the ϕX174 + 210 was much more problematic. PCR amplification of ϕX174 + 210 plaques using primers flanking the +210 insertion produced multiple bands, suggesting the duplication was in the process of being lost during plaque formation. As this had never been observed when ϕXG4J+210 was used as the template, the instability of +210 duplication in a wild-type background was investigated in a more rigorous manner.
A ϕX174 + 210 plaque that yielded a homogenous PCR product was first identified. The product was sequenced and the +210 insertion was maintained. Eight progeny plaques were selected from this “genetically clean” parent and the PCRs were repeated. Wild-type ϕX174 and ϕXG4J+210, which had never displayed genetic instability, were used as controls. Ten ϕXG4J+210 plaques and 10 wild-type ϕX174 plaques were pooled into separate PCRs. Thus, 20 progeny of each were analyzed. Three additional purified DNA templates were included: ϕX174, ϕXG4J, and ϕXG4J+210. As can be seen in Fig. 5, the PCR products from the eight ϕX174 + 210 plaques varied widely, indicating that the +210 insertion was lost in full or in part within the phage populations. In contrast, ϕXG4J+210 and wild-type ϕX174 plaques yielded homogenous products, as did the purified DNA controls. These results demonstrate that ϕXG4J+210 is genetically much more stable than ϕX174 + 210. However, it is packaged with the G4 J protein and duplication loss would likely eliminate viability.
FIG 5.

PCR products generated from purified DNA and plaques. Primers annealed to genes D and F, flanking the region containing the +210 duplication. The template DNAs used in each reaction are given in the figure and are described in the text.
DISCUSSION
Factors governing Microviridae DNA packaging capacity.
Although genome size varies between ϕX174-, G4-, and α3-like viruses, i.e., ∼5,400, ∼5,600, and ∼6,100 bases, respectively (27), their capsid volumes remain relatively constant (15–18, 50). Larger genomes are packaged with less basic and smaller J proteins. Thus, direct correlations between genome size, capsid volume, and/or charge neutralization do not account for packaging capacity. There may exist an interplay between genome and J protein size: smaller J proteins volumetrically compensate for their larger genomes. However, the larger ϕX174 J protein effectively operates in the α3 and G4 systems (17, 25). Moreover, the lethal ϕXG4J phenotype can be reversed by exogenous expression of the A* gene, which does not increase genome size.
Microvirus packaging is a three-component system: the capsid’s inner surface, the J protein guide, and the displaced ssDNA.
In virions, protein J appears to guide the genome around the 5-fold axes of symmetry. This path leads the genome between successive nucleic acid binding sites, one in each coat protein (15–18). Packaging is probably not a chaotic process. If DNA randomly associated with coat proteins, the entry site could become obstructed. Moreover, packaged DNA must assume a conformation that can easily exit through the virus’s genome delivery tube, a 22-Å wide proteinaceous tube that spans the host’s cell wall (51, 52). ϕXG4J’s lethal phenotype and molecular characteristics are consistent with suboptimal or chaotic packaging. Particles are unstable, dissociating during either packaging or purification; those that do survive are considerably less stable and infectious than the wild type. However, stability and infectivity may be related phenomena: particles within the virion peak may degrade before plating assays but after gradients have been fractionated.
From X-ray structures, the location of nucleic acid binding sites in relation to J proteins strongly suggests that J proteins guide the incoming nucleic acid strand into position. The contribution of the ssDNA substrate to packaging efficiency is more difficult to define. When placed into plasmids containing no other ϕX174 DNA sequences, the 30-base replication/packaging origin is both necessary and sufficient to fill a procapsid with a ssDNA version of the plasmid (11, 49, 53). However, there are differences between the products of in vitro packaging reactions with genome-length packaging plasmids versus the bona fide ϕX174 genome. The genomic substrate yields particles that are indistinguishable from in vivo-generated virions. In contrast, when using a unit-length packaging plasmid, >50% of the final products do not fully encapsidate the DNA and lack infectivity. Moreover, the fully filled particles exhibit a lower specific infectivity: only 10% of the products are infectious compared to those of the genome-substrate control. Thus, changing the packaging substrate appears to lower fitness without totally eliminating viability (49). A similar phenomenon has been documented with the ssDNA adeno-associated parvoviruses (54).
The aforementioned in vitro experiments utilized a packaging substrate composed primarily of foreign DNA. Results suggest that the maximal packaging capacity of ϕX174 is 100.2%, albeit the efficiency was poor. Similar in vivo studies have been conducted to define ϕX174’s upper packaging limit. However, in these studies the packaging substrates were phage genomes containing insertions, which ranged in size from a 100.7 to a 103% unit length (47, 55, 56). The ϕXG4J+210 genome is ∼104% unit length. Combined, these data suggest that along with the capsid’s inner surface and the J protein guide, the ssDNA substrate can also influence packaging capacity. As demonstrated with ssRNA viruses that assemble around their genomes, (8), single-stranded viral DNA with enhanced packaging efficiency may be adaptive. Indeed, bioinformatic results suggest that the genome contains 60 degenerate consensus sequences (57, 58). The role of these sequences have yet to be experimentally investigated, but they probably do not govern protein J’s initial interactions with the double-stranded RF DNA by which it gains access to the procapsid. J protein appears to bind all nucleic acids in a sequence-independent manner (19). However, these sequences could affect properties of the ssDNA, such as its local persistence length, to optimize its organization within the capsid’s icosahedral symmetry.
The fourth packaging component is temporal.
Packaging and ssDNA biosynthesis are concurrent processes. The parental plus DNA strand is displaced into the procapsid as the daughter strand is synthesized. If synthesis outpaces packaging, the displaced ssDNA could form local hairpin loops that may inhibit the reaction or lead to two plus strands competing for interactions with the single minus strand. The plot thickens, and the stage is set for the heroine’s entrance, which can only be performed by A*.
ϕXG4J was rescued by duplicating protein A*’s DNA substrate or by elevating intracellular A* levels. Protein A* is an unessential gene product in ϕX174, as determined by A*-null mutants having a wild-type phenotype in plaque assays (28). However, the gene is highly conserved in all known (n > 60) microviruses (27). Thus, it likely confers a fitness advantage, which may be too small detect in standard lab assays but which has had a large impact over evolutionary time. The α3-clade viruses, with their considerably larger genomes, have two conserved A* genes, with two separate start codons each optimally placed downstream from two separate very strong Shine-Dalgarno sequences (27).
The biochemical properties of proteins A and A* have been investigated in vitro. Enzymatically, both proteins bind DNA, covalently attach to it, and ligate the ends when they dissociate. However, they differ in substrate recognition. Protein A’s substrate is very specific: the origin of replication (59, 60); whereas protein A*’s substrate specificity is broad: at least 30 origin-like sequences. However, it displays a pronounced preference for the one within the J-F intercistronic region (39). Once attached, protein A mediates rolling circle DNA replication, whereas protein A* inhibits it by preventing DNA unwinding (61).
The data presented here imply that protein A* plays a regulatory role during packaging that affects product fidelity. Although the molecular details remain to be elucidated, some speculation is warranted. As the daughter strand is synthesized, the parental strand is displaced and guided into the capsid via its interactions with the J and viral coat proteins. Random haphazard associations could impede this process. Based on its ability to inhibit DNA synthesis, protein A* may ensure synthesis does not outpace displacement. If this does not occur, external, unpackaged, ssDNA would accumulate. This could lead to (i) less than optimal DNA arrangements affecting particle instability or infectivity and (ii) the daughter and parental plus strands competing to base pair with the single minus strand. The latter may account for the higher degree of missed termination events observed with ϕXG4J. By halting ssDNA biosynthesis, protein A* may mitigate these damages either by allowing displacement to catch up to synthesis or by removing the particle entirely from the pathway, conserving time and resources. The results presented here do not distinguish between these two mechanisms.
MATERIALS AND METHODS
Phage plating, media, buffers, stock preparation, bacterial strains.
The plating protocol, media, buffers, stock preparation, and the wild-type Escherichia coli C strain C122 have been previously described (62). The RY7211 cell line contains a mutation in the mraY gene, conferring resistance to viral E protein-mediated lysis (63).
ssDNA, RF dsDNA, and assembled particle-associated DNA purification.
The ssDNA was generated and purified with the OLT (Our Little Trick) protocol, which has been previously described in detail (64), as has the protocol used for RF dsDNA purification (65).
To isolate DNA associated with assembled particles, gradient fractions containing 90–150S particles were diluted 5-fold in 10 mM Tris-HCl (pH 7.5)/10 mM NaCl/1.0 mM EDTA. SDS, pronase, and additional EDTA were added to respective final concentrations of 0.5% (wt/vol), 0.5 mg/ml, and 10 mM. Samples were incubated at 37°C for 2.5 h before successive equal volume extractions with equilibrated phenol, a phenol-chloroform-isoamyl alcohol cocktail (24:24:1), and chloroform-isoamyl alcohol (24:1). The aqueous phase was retained after each extraction. DNA was precipitated by first adding sodium acetate to a concentration of 250 mM and 2 volumes of ice-cold ethanol. Samples were stored over night at –20°C and then spun for 10 min at 16,000 × g. The resulting pellets were resuspended in 0.4 ml of Tris-HCl (pH 7.5)/10 mM NaCl/1.0 mM EDTA for a second precipitation with 0.8-ml volumes of ice-cold ethanol, stored over night at –20°C, and spun again. The resulting pellets were resuspended in 100 μl of Tris-HCl (pH 7.5)/10 mM NaCl/1.0 mM EDTA.
Cloning of the ϕX174 J, G4 J, and ϕX174 A* and F genes.
The ϕX174 and G4 J genes were PCR amplified using primers that introduced upstream NcoI and downstream HindIII sites. The resulting fragments were digested with NcoI and HindIII and ligated into pSE420 (Invitrogen) treated with the same enzymes. Gene expression was induced by the addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to a concentration of 0.1 mM. The complementing clone of the F gene has been previously described (66).
The A* gene, along with its RBS, was PCR amplified with primers that introduced upstream XbaI and downstream HindIII sites. The template DNA contained an ochre mutation in the third codon of the overlapping B gene. The resulting fragment was digested with XbaI and HindIII and ligated into pBAD33 (Thermo Fisher) treated with the same enzymes. Gene expression was induced by supplementing media with 0.2% arabinose and then repressed by supplementing media with 0.2% glucose.
Construction of ϕXG4J, ϕXG4J+42H, ϕXG4J+21 scrambled, ϕX174 + 21, and ϕX174 + 210/F-E42D strains.
For all three strains, the entire genome was PCR amplified with Q5 DNA polymerase (NEB) using abutting primers (described below). The PCR products’ 5′-hydroxyl termini were phosphorylated, and the ends were ligated by using T4 polynucleotide kinase and ligase (NEB), respectively. To generate ϕXG4J, the G4 J sequence was divided between the 5′ ends of the abutting primers. The 3′ ends annealed to the upstream and downstream regions of the ϕX174 genome. The mutant was recovered in cells expressing the ϕX174 J gene. The ϕXG4J+42H contains a viable insertion in gene H (47). It was constructed as described above using ϕXG4J DNA as a template and abutting primers introducing the gene H insertion as previously described (47). The mutant was recovered in cells expressing the ϕX174 J gene. To construct ϕXG4J+21 scrambled, the scrambled insert was divided between the 5′ ends of the abutting primers. The 3′ ends annealed to the upstream and downstream regions of the ϕXG4J genome. The ϕX174 + 21 strain contains the ϕX174 J gene, and the 21-nucleotide duplication that restores viability to ϕXG4J. ϕXG4J+21 DNA served as the template in the Q5 polymerase reactions. The ϕX174 J sequence was divided between the 5′ ends of two abutting primers. The 3′ ends annealed to the upstream and downstream regions of the ϕX174 genome. Most likely due to genetic instability (see Results), the ϕX174 + 210/F-E42D strain, which contains the ϕX174 J gene and the +210/F-E42D duplication that restores viability to ϕXG4J, could not be generated via a one-step mutagenesis. Thus, a more complicated genetic scheme had to be used. Primers were designed to simultaneously remove the G4 J gene and reintroduce ϕX174 nucleotides 848 to 876 and nucleotides 936 to 964 using PCR with Q5 polymerase. The resulting PCR product was circularized as described above and transfected into cells expressing the ϕX174 J gene. The transfection was plated at 37°C, and a plaque of the resulting mutant, ϕX1741/2J, was isolated. The mutant was propagated in cells expressing pϕX174DJ to facilitate genetic recombination (67). J gene recombinants were selected by plating the progeny on C122. Recombinant plaques were isolated and sequenced to confirm that the full-length ϕX174 J gene and the +210/F-E42D duplication were present.
Infected cell extracts, rate zonal sedimentation, and protein electrophoresis.
The protocols for rate zonal sedimentation and protein electrophoresis have been previously described (31, 65, 68). To generate infected cell extracts for rate zonal sedimentation, 100 ml of lysis-resistant cells were infected at an MOI of 3.0, followed by incubation for 3 h at 33°C. Infected cells were concentrated, resuspended in 3.0 ml of sucrose gradient buffer (100 mM NaCl, 5.0 mM EDTA, 6.4 mM Na2HPO4, 3.3 mM KH2PO4 [pH 7.0]), and then lysed by overnight incubation with lysozyme (2.0 mg/ml). After cellular debris was removed by centrifugation (10 min at 16,000 × g), the resulting supernatant was concentrated to 200 μl in a Nanosep centrifugal filter column (100-kDa cutoff). The extract was then loaded atop a 5.0-ml, 5 to 30% sucrose gradient (wt/vol) and spun at 192,000 × g for 1 h. Gradients were divided into approximately 40 125-μl fractions, and assembled particles were detected by UV spectroscopy (A260 and A280).
ACKNOWLEDGMENTS
This research was supported by National Science Foundation grant MCB-1408217 (B.A.F.) and the BIO5 Institute at the University. We also acknowledge the support of the Maximizing Access to Research Careers program at the University of Arizona.
REFERENCES
- 1.Gope R, Serwer P. 1983. Bacteriophage P22 in vitro DNA packaging monitored by agarose gel electrophoresis: rate of DNA entry into capsids. J Virol 47:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiang Y, Morais MC, Battisti AJ, Grimes S, Jardine PJ, Anderson DL, Rossmann MG. 2006. Structural changes of bacteriophage ϕ29 upon DNA packaging and release. EMBO J 25:5229–5239. doi: 10.1038/sj.emboj.7601386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newcomb WW, Homa FL, Thomsen DR, Brown JC. 2001. In vitro assembly of the herpes simplex virus procapsid: formation of small procapsids at reduced scaffolding protein concentration. J Struct Biol 133:23–31. doi: 10.1006/jsbi.2001.4329. [DOI] [PubMed] [Google Scholar]
- 4.Speir JA, Johnson JE. 2012. Nucleic acid packaging in viruses. Curr Opin Struct Biol 22:65–71. doi: 10.1016/j.sbi.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerritelli ME, Studier FW. 1996. Assembly of T7 capsids from independently expressed and purified head protein and scaffolding protein. J Mol Biol 258:286–298. doi: 10.1006/jmbi.1996.0250. [DOI] [PubMed] [Google Scholar]
- 6.Garmann RF, Comas-Garcia M, Knobler CM, Gelbart WM. 2016. Physical principles in the self-assembly of a simple spherical virus. Acc Chem Res 49:48–55. doi: 10.1021/acs.accounts.5b00350. [DOI] [PubMed] [Google Scholar]
- 7.Koning RI, Gomez-Blanco J, Akopjana I, Vargas J, Kazaks A, Tars K, Carazo JM, Koster AJ. 2016. Asymmetric cryo-EM reconstruction of phage MS2 reveals genome structure in situ. Nat Commun 7:12524. doi: 10.1038/ncomms12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gopal A, Egecioglu DE, Yoffe AM, Ben-Shaul A, Rao AL, Knobler CM, Gelbart WM. 2014. Viral RNAs are unusually compact. PLoS One 9:e105875. doi: 10.1371/journal.pone.0105875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horn WT, Tars K, Grahn E, Helgstrand C, Baron AJ, Lago H, Adams CJ, Peabody DS, Phillips SE, Stonehouse NJ, Liljas L, Stockley PG. 2006. Structural basis of RNA binding discrimination between bacteriophages Qβ and MS2. Structure 14:487–495. doi: 10.1016/j.str.2005.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hesketh EL, Saunders K, Fisher C, Potze J, Stanley J, Lomonossoff GP, Ranson NA. 2018. The 3.3 Å structure of a plant geminivirus using cryo-EM. Nat Commun 9:2369. doi: 10.1038/s41467-018-04793-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aoyama A, Hamatake RK, Hayashi M. 1981. Morphogenesis of ϕX174: in vitro synthesis of infectious phage from purified viral components. Proc Natl Acad Sci U S A 78:7285–7289. doi: 10.1073/pnas.78.12.7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King JA, Dubielzig R, Grimm D, Kleinschmidt JA. 2001. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J 20:3282–3291. doi: 10.1093/emboj/20.12.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman MS, Rossmann MG. 1995. Single-stranded DNA-protein interactions in canine parvovirus. Structure 3:151–162. doi: 10.1016/s0969-2126(01)00146-0. [DOI] [PubMed] [Google Scholar]
- 14.Mietzsch M, Penzes JJ, Agbandje-McKenna M. 2019. Twenty-five years of structural parvovirology. Viruses 11(4):362. doi: 10.3390/v11040362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna R, Ilag LL, Rossmann MG. 1994. Analysis of the single-stranded DNA bacteriophage ϕX174, refined at a resolution of 3.0 Å. J Mol Biol 237:517–543. doi: 10.1006/jmbi.1994.1253. [DOI] [PubMed] [Google Scholar]
- 16.McKenna R, Xia D, Willingmann P, Ilag LL, Krishnaswamy S, Rossmann MG, Olson NH, Baker TS, Incardona NL. 1992. Atomic structure of single-stranded DNA bacteriophage ϕX174 and its functional implications. Nature 355:137–143. doi: 10.1038/355137a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernal RA, Hafenstein S, Esmeralda R, Fane BA, Rossmann MG. 2004. The ϕX174 protein J mediates DNA packaging and viral attachment to host cells. J Mol Biol 337:1109–1122. doi: 10.1016/j.jmb.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 18.Bernal RA, Hafenstein S, Olson NH, Bowman VD, Chipman PR, Baker TS, Fane BA, Rossmann MG. 2003. Structural studies of bacteriophage α3 assembly. J Mol Biol 325:11–24. doi: 10.1016/s0022-2836(02)01201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamatake RK, Aoyama A, Hayashi M. 1985. J gene of bacteriophage ϕX174: in vitro analysis of J protein function. J Virol 54:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayashi M, Aoyama A, Richardson DL, Hayashi NM. 1988. Biology of the bacteriophage ϕ X174, p 1–71. In Calendar R. (ed), The bacteriophages, vol 2 Plenum Press, New York, NY. [Google Scholar]
- 21.Jennings B, Fane BA. 1997. Genetic analysis of the ϕX174 DNA binding protein. Virology 227:370–377. doi: 10.1006/viro.1996.8351. [DOI] [PubMed] [Google Scholar]
- 22.Benevides JM, Stow PL, Ilag LL, Incardona NL, Thomas GJ Jr. 1991. Differences in secondary structure between packaged and unpackaged single-stranded DNA of bacteriophage ϕX174 determined by Raman spectroscopy: a model for phi X174 DNA packaging. Biochemistry 30:4855–4863. doi: 10.1021/bi00234a004. [DOI] [PubMed] [Google Scholar]
- 23.Hafenstein S, Fane BA. 2002. ϕX174 genome-capsid interactions influence the biophysical properties of the virion: evidence for a scaffolding-like function for the genome during the final stages of morphogenesis. J Virol 76:5350–5356. doi: 10.1128/jvi.76.11.5350-5356.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hafenstein SL, Chen M, Fane BA. 2004. Genetic and functional analyses of the ϕX174 DNA binding protein: the effects of substitutions for amino acid residues that spatially organize the two DNA binding domains. Virology 318:204–213. doi: 10.1016/j.virol.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 25.Fane BA, Head S, Hayashi M. 1992. Functional relationship between the J proteins of bacteriophages ϕX174 and G4 during phage morphogenesis. J Bacteriol 174:2717–2719. doi: 10.1128/jb.174.8.2717-2719.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grieger JC, Samulski RJ. 2005. Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J Virol 79:9933–9944. doi: 10.1128/JVI.79.15.9933-9944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rokyta DR, Burch CL, Caudle SB, Wichman HA. 2006. Horizontal gene transfer and the evolution of microvirid coliphage genomes. J Bacteriol 188:1134–1142. doi: 10.1128/JB.188.3.1134-1142.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colasanti J, Denhardt DT. 1985. Expression of the cloned bacteriophage ϕX174 A* gene in Escherichia coli inhibits DNA replication and cell division. J Virol 53:807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chejanovsky N, Carter BJ. 1989. Mutagenesis of an AUG codon in the adeno-associated virus rep gene: effects on viral DNA replication. Virology 173:120–128. doi: 10.1016/0042-6822(89)90227-4. [DOI] [PubMed] [Google Scholar]
- 30.Mankertz A, Hillenbrand B. 2001. Replication of porcine circovirus type 1 requires two proteins encoded by the viral rep gene. Virology 279:429–438. doi: 10.1006/viro.2000.0730. [DOI] [PubMed] [Google Scholar]
- 31.Uchiyama A, Fane BA. 2005. Identification of an interacting coat-external scaffolding protein domain required for both the initiation of ϕX174 procapsid morphogenesis and the completion of DNA packaging. J Virol 79:6751–6756. doi: 10.1128/JVI.79.11.6751-6756.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ekechukwu MC, Oberste DJ, Fane BA. 1995. Host and ϕX174 mutations affecting the morphogenesis or stabilization of the 50S complex, a single-stranded DNA synthesizing intermediate. Genetics 140:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uchiyama A, Chen M, Fane BA. 2007. Characterization and function of putative substrate specificity domain in microvirus external scaffolding proteins. J Virol 81:8587–8592. doi: 10.1128/JVI.00301-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taketo A, Kodaira KI. 1978. Synthesis of α3 phage DNA in replication mutants of Escherichia coli. Biochim Biophys Acta 517:55–64. doi: 10.1016/0005-2787(78)90033-3. [DOI] [PubMed] [Google Scholar]
- 35.Fane BA, Shien S, Hayashi M. 1993. Second-site suppressors of a cold-sensitive external scaffolding protein of bacteriophage ϕX174. Genetics 134:1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gordon EB, Knuff CJ, Fane BA. 2012. Conformational switch-defective ϕX174 internal scaffolding proteins kinetically trap assembly intermediates before procapsid formation. J Virol 86:9911–9918. doi: 10.1128/JVI.01120-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uchiyama A, Heiman P, Fane BA. 2009. N-terminal deletions of the ϕX174 external scaffolding protein affect the timing and fidelity of assembly. Virology 386:303–309. doi: 10.1016/j.virol.2009.01.030. [DOI] [PubMed] [Google Scholar]
- 38.van Mansfeld AD, van Teeffelen HA, Fluit AC, Baas PD, Jansz HS. 1986. Effect of SSB protein on cleavage of single-stranded DNA by ϕX gene A protein and A* protein. Nucleic Acids Res 14:1845–1861. doi: 10.1093/nar/14.4.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langeveld SA, van Mansfeld AD, van der Ende A, van de Pol JH, van Arkel GA, Weisbeek PJ. 1981. The nuclease specificity of the bacteriophage ϕX174 A* protein. Nucleic Acids Res 9:545–562. doi: 10.1093/nar/9.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doore SM, Fane BA. 2015. The kinetic and thermodynamic aftermath of horizontal gene transfer governs evolutionary recovery. Mol Biol Evol 32:2571–2584. doi: 10.1093/molbev/msv130. [DOI] [PubMed] [Google Scholar]
- 41.Hayashi MN, Hayashi M, Muller UR. 1983. Role for the J-F intercistronic region of bacteriophages phi X174 and G4 in stability of mRNA. J Virol 48:186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bull JJ, Badgett MR, Wichman HA, Huelsenbeck JP, Hillis DM, Gulati A, Ho C, Molineux IJ. 1997. Exceptional convergent evolution in a virus. Genetics 147:1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wichman HA, Badgett MR, Scott LA, Boulianne CM, Bull JJ. 1999. Different trajectories of parallel evolution during viral adaptation. Science 285:422–424. doi: 10.1126/science.285.5426.422. [DOI] [PubMed] [Google Scholar]
- 44.Wichman HA, Millstein J, Bull JJ. 2005. Adaptive molecular evolution for 13,000 phage generations: a possible arms race. Genetics 170:19–31. doi: 10.1534/genetics.104.034488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Floor E. 1970. Interaction of morphogenetic genes of bacteriophage T4. J Mol Biol 47:293–306. doi: 10.1016/0022-2836(70)90303-7. [DOI] [PubMed] [Google Scholar]
- 46.Sternberg N. 1976. A genetic analysis of bacteriophage lambda head assembly. Virology 71:568–582. doi: 10.1016/0042-6822(76)90382-2. [DOI] [PubMed] [Google Scholar]
- 47.Roznowski AP, Fane BA. 2016. Structure-function analysis of the ϕX174 DNA-piloting protein using length-altering mutations. J Virol 90:7956–7966. doi: 10.1128/JVI.00914-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aoyama A, Hayashi M. 1985. Effects of genome size on bacteriophage ϕX174 DNA packaging in vitro. J Biol Chem 260:11033–11038. [PubMed] [Google Scholar]
- 50.McKenna R, Bowman BR, Ilag LL, Rossmann MG, Fane BA. 1996. Atomic structure of the degraded procapsid particle of the bacteriophage G4: induced structural changes in the presence of calcium ions and functional implications. J Mol Biol 256:736–750. doi: 10.1006/jmbi.1996.0121. [DOI] [PubMed] [Google Scholar]
- 51.Sun L, Rossmann MG, Fane BA. 2014. High-resolution structure of a virally encoded DNA-translocating conduit and the mechanism of DNA penetration. J Virol 88:10276–10279. doi: 10.1128/JVI.00291-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun L, Young LN, Zhang X, Boudko SP, Fokine A, Zbornik E, Roznowski AP, Molineux IJ, Rossmann MG, Fane BA. 2014. Icosahedral bacteriophage ϕX174 forms a tail for DNA transport during infection. Nature 505:432–435. doi: 10.1038/nature12816. [DOI] [PubMed] [Google Scholar]
- 53.Aoyama A, Hamatake RK, Hayashi M. 1983. In vitro synthesis of bacteriophage ϕX174 by purified components. Proc Natl Acad Sci U S A 80:4195–4199. doi: 10.1073/pnas.80.14.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeltner N, Kohlbrenner E, Clement N, Weber T, Linden RM. 2010. Near-perfect infectivity of wild-type AAV as benchmark for infectivity of recombinant AAV vectors. Gene Ther 17:872–879. doi: 10.1038/gt.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Russell PW, Muller UR. 1984. Construction of bacteriophage luminal diameter X174 mutants with maximum genome sizes. J Virol 52:822–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Christakos KJ, Chapman JA, Fane BA, Campos SK. 2016. PhiXing-it, displaying foreign peptides on bacteriophage PhiX174. Virology 488:242–248. doi: 10.1016/j.virol.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chechetkin VR, Lobzin VV. 2017. Large-scale chromosome folding versus genomic DNA sequences: a discrete double Fourier transform technique. J Theor Biol 426:162–179. doi: 10.1016/j.jtbi.2017.05.033. [DOI] [PubMed] [Google Scholar]
- 58.Chechetkin VR, Lobzin VV. 2018. Genome packaging within icosahedral capsids and large-scale segmentation in viral genomic sequences. J Biomol Struct Dyn 37:2322–2338. [DOI] [PubMed] [Google Scholar]
- 59.Van Mansfeld AD, Baas PD, Jansz HS. 1984. Gene A protein of bacteriophage ϕX174 is a highly specific single-strand nuclease and binds via a tyrosyl residue to DNA after cleavage. Adv Exp Med Biol 179:221–230. doi: 10.1007/978-1-4684-8730-5_23. [DOI] [PubMed] [Google Scholar]
- 60.van Mansfeld AD, van Teeffelen HA, Baas PD, Veeneman GH, van Boom JH, Jansz HS. 1984. The bond in the bacteriophage ϕX174 gene A protein-DNA complex is a tyrosyl-5′-phosphate ester. FEBS Lett 173:351–356. doi: 10.1016/0014-5793(84)80804-2. [DOI] [PubMed] [Google Scholar]
- 61.Eisenberg S, Ascarelli R. 1981. The A* protein of ϕX174 is an inhibitor of DNA replication. Nucleic Acids Res 9:1991–2002. doi: 10.1093/nar/9.8.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fane BA, Hayashi M. 1991. Second-site suppressors of a cold-sensitive prohead accessory protein of bacteriophage ϕX174. Genetics 128:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bernhardt TG, Struck DK, Young R. 2001. The lysis protein E of ϕX174 is a specific inhibitor of the MraY-catalyzed step in peptidoglycan synthesis. J Biol Chem 276:6093–6097. doi: 10.1074/jbc.M007638200. [DOI] [PubMed] [Google Scholar]
- 64.Blackburn BJ, Li S, Roznowski AP, Perez AR, Villarreal RH, Johnson CJ, Hardy M, Tuckerman EC, Burch AD, Fane BA. 2017. Coat protein mutations that alter the flux of morphogenetic intermediates through the ϕX174 early assembly pathway. J Virol 91:e01384-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burch AD, Ta J, Fane BA. 1999. Cross-functional analysis of the Microviridae internal scaffolding protein. J Mol Biol 286:95–104. doi: 10.1006/jmbi.1998.2450. [DOI] [PubMed] [Google Scholar]
- 66.Jaschke PR, Lieberman EK, Rodriguez J, Sierra A, Endy D. 2012. A fully decompressed synthetic bacteriophage ϕX174 genome assembled and archived in yeast. Virology 434:278–284. doi: 10.1016/j.virol.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 67.Burch AD, Fane BA. 2000. Foreign and chimeric external scaffolding proteins as inhibitors of Microviridae morphogenesis. J Virol 74:9347–9352. doi: 10.1128/jvi.74.20.9347-9352.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cherwa JE Jr, Organtini LJ, Ashley RE, Hafenstein SL, Fane BA. 2011. In vitro assembly of the ϕX174 procapsid from external scaffolding protein oligomers and early pentameric assembly intermediates. J Mol Biol 412:387–396. doi: 10.1016/j.jmb.2011.07.070. [DOI] [PubMed] [Google Scholar]
- 69.Sanger F, Coulson AR, Friedmann T, Air GM, Barrell BG, Brown NL, Fiddes JC, Hutchison CA, III, Slocombe PM, Smith M. 1978. The nucleotide sequence of bacteriophage ϕX174. J Mol Biol 125:225–246. doi: 10.1016/0022-2836(78)90346-7. [DOI] [PubMed] [Google Scholar]