

Hepatitis C virus (HCV) NS4B protein induces the formation of a membranous web (MW) structure that provides a platform for the assembly of viral replication complexes. The main constituents of the MW are double-membrane vesicles (DMVs). Here, we found that the cellular protein Surf4, which maintains endoplasmic reticulum (ER)-Golgi intermediate compartments and the Golgi compartment, is recruited into HCV RNA replication complexes by NS4B and is involved in the formation of DMVs. Moreover, Surf4 participates in the replication of poliovirus, which uses DMVs as replication sites, but has no effect on the replication of dengue virus, which uses invaginated vesicles as replication sites. These results indicate that the cellular protein Surf4 is involved in the replication of positive-strand RNA viruses that use DMVs as RNA replication sites, providing new insights into DMV formation during virus replication and potential targets for the diagnosis and treatment of positive-strand RNA viruses.
Keywords: hepatitis C virus, Surfeit 4, replication complex, double-membrane vesicles, poliovirus, HCV, NS4B, membranous web, replication
ABSTRACT
A number of positive-strand RNA viruses, such as hepatitis C virus (HCV) and poliovirus, use double-membrane vesicles (DMVs) as replication sites. However, the role of cellular proteins in DMV formation during virus replication is poorly understood. HCV NS4B protein induces the formation of a “membranous web” structure that provides a platform for the assembly of viral replication complexes. Our previous screen of NS4B-associated host membrane proteins by dual-affinity purification, liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS), and small interfering RNA (siRNA) methods revealed that the Surfeit 4 (Surf4) gene, which encodes an integral membrane protein, is involved in the replication of the JFH1 subgenomic replicon. Here, we investigated in detail the effect of Surf4 on HCV replication. Surf4 affects HCV replication in a genotype-independent manner, whereas HCV replication does not alter Surf4 expression. The influence of Surf4 on HCV replication indicates that while Surf4 regulates replication, it has no effect on entry, translation, assembly, or release. Analysis of the underlying mechanism showed that Surf4 is recruited into HCV RNA replication complexes by NS4B and is involved in the formation of DMVs and the structural integrity of RNA replication complexes. Surf4 also participates in the replication of poliovirus, which uses DMVs as replication sites, but it has no effect on the replication of dengue virus, which uses invaginated/sphere-type vesicles as replication sites. These findings clearly show that Surf4 is a novel cofactor that is involved in the replication of positive-strand RNA viruses using DMVs as RNA replication sites, which provides valuable clues for DMV formation during positive-strand RNA virus replication.
IMPORTANCE Hepatitis C virus (HCV) NS4B protein induces the formation of a membranous web (MW) structure that provides a platform for the assembly of viral replication complexes. The main constituents of the MW are double-membrane vesicles (DMVs). Here, we found that the cellular protein Surf4, which maintains endoplasmic reticulum (ER)-Golgi intermediate compartments and the Golgi compartment, is recruited into HCV RNA replication complexes by NS4B and is involved in the formation of DMVs. Moreover, Surf4 participates in the replication of poliovirus, which uses DMVs as replication sites, but has no effect on the replication of dengue virus, which uses invaginated vesicles as replication sites. These results indicate that the cellular protein Surf4 is involved in the replication of positive-strand RNA viruses that use DMVs as RNA replication sites, providing new insights into DMV formation during virus replication and potential targets for the diagnosis and treatment of positive-strand RNA viruses.
INTRODUCTION
Hepatitis C virus (HCV) is an important human pathogen, and its infection can lead to chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (HCC). HCV is a positive-strand RNA virus with seven major natural occurring genotypes. Its genome consists of a 5′ untranslated region (5′ UTR), a single open reading frame (ORF), and a 3′ UTR. The ORF encodes a single polyprotein that is cleaved into at least 10 mature viral proteins in the following order: NH2-C-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5BCOOH (1). HCV cell culture systems containing replicon systems and pseudotyped and virus propagation systems have been established, which provide powerful tools for analyzing viral life cycle and pathogenesis, as well as for developing vaccines and antiviral drugs (2).
Virus-host interactions play important roles in the virus life cycle and pathogenesis. The study of HCV-host interactions is highly valuable, since the HCV life cycle remains largely unclear, anti-HCV drugs exhibit viral genotype-specific differences in efficacy, and drug-resistant mutants rapidly evolve (3). Recent advances in HCV cell culture systems, RNA interference (RNAi) screening, and mass spectrometry interactome approaches have greatly improved HCV-host interaction research, and a number of virus-host interactions have been identified. However, the roles of most of these interactions in the HCV life cycle remain unknown (4). HCV replication causes rearrangements of intracellular membranes and the formation of a replication complex containing viral nonstructural proteins, cellular components, and nascent RNA strands (5, 6). Lipid rafts, detergent-insoluble cholesterol- and sphingolipid-rich microdomains of cellular membranes, have been found to play important roles in the formation of HCV RNA replication complexes (7). HCV NS4B protein triggers a specific membrane rearrangement, designated a “membranous web (MW),” that provides a platform for the assembly of viral replication complexes (8, 9). The main constituents of the MW are single- and double-membrane vesicles (DMVs) (10). DMVs have recently been reported to be the predominant HCV RNA replication sites. However, it remains largely unknown whether and how NS4B recruits cellular proteins for the formation of membrane vesicles.
Surfeit 4 (Surf4) is one of four genes closely spaced in the Surfeit cluster of higher eukaryotic species (11). The gene is conserved between humans and mice and encodes an integral membrane protein that resides in the endoplasmic reticulum (ER), ER-Golgi intermediate compartment (ERGIC), and Golgi compartment (12, 13). ERV29p, the yeast homolog of Surf4, is enriched in COPII vesicles and operates as a transport receptor for specific cargo. Surf4 and Erv29p are multipass proteins with the number of transmembrane domains predicted/reported ranging from four to seven according to basicELM, the UniProt database, and the report (14). They bind soluble cargo proteins with different affinities, enabling prioritization of their trafficking from the ER (15). SFT-4 is the Caenorhabditis elegans homolog of the yeast Erv29p. SFT-4 and Surf4 participate in ER exit site (ERES) organization in animals and regulate ER export of soluble proteins, including lipoproteins (16). Surf4 also modulates STIM1-dependent calcium entry (17). Recent studies have shown that Surf4 has oncogenic potential, is a potential diagnostic biomarker for gastrointestinal stromal tumors, and is involved in the regulation of mammalian lipid homeostasis (13, 18, 19).
We recently showed that prolactin regulatory element binding protein (PREB) is involved in HCV RNA replication by interacting with NS4B (20). In the present study, we identified Surf4 as a novel HCV cofactor involved in HCV RNA replication. We found that Surf4 is recruited into HCV RNA replication complexes by NS4B and is involved in the formation of DMVs and in the structural integrity of replication complexes. Surf4 also promoted the replication of poliovirus that uses DMVs as replication sites like HCV, but it had no effect on the replication of dengue virus that uses invaginated/sphere-type vesicles as replication sites. These results indicate that the cellular protein Surf4 is involved in the replication of positive-strand RNA viruses that use DMVs as RNA replication sites, providing new insights into DMV formation during virus replication and potential novel targets for the diagnosis and treatment of positive-strand RNA viruses.
RESULTS
Surf4 participates in HCV replication.
A previous screen of NS4B-associated host cofactors by small interfering RNA (siRNA) silencing showed that siRNA targeting Surf4 inhibits the replication of JFH1 subgenomic replicon (SGR2a) (20). To validate the role of endogenous Surf4 in HCV replication, we treated SGR2a cells with different concentrations of siSurf4. siSurf4 specifically decreased Surf4 expression and SGR2a replication in a dose-dependent manner (Fig. 1A). The silencing of dehydrocholesterol reductase (DHCR) has been reported to inhibit HCV replication (21). siRNA targeting DHCR (siDHCR) as a positive control decreased SGR2a replication, whereas nontargeting siRNA (siNT) as a negative control had no effect on SGR2a replication (Fig. 1A). Next, three unique siRNAs targeting different sites of Surf4 (siSurf4), siRNA targeting 24-dehydrocholesterol reductase (siDHCR), or nontargeting siRNA (siNT) were transfected into SGR2a cells. The three siRNAs targeting Surf4 inhibited luciferase activity (an indicator of SGR2a replication), albeit to slightly different degrees (Fig. 1B). None of the siRNAs tested exhibited cytotoxicity against the replicon cells (data not shown). Together, these results suggest that the inhibition of SGR2a replication by siRNA targeting Surf4 is Surf4 specific, and not an off-target effect. To further determine the effects of endogenous Surf4 on HCV replication, Huh7 cells were transfected with a plasmid encoding a short hairpin RNA (shRNA) targeting Surf4 and selected with hygromycin B, resulting in ShSurf4 cells without Surf4 expression (Fig. 1C). ShSurf4 cells and Huh7 cells stably transfected with negative-control pSilencer hygro plasmid (shNC) were electroporated with SGR2a RNA in the presence or absence of an expression plasmid for shRNA-resistant Surf4 (Surf4shr) or wild-type Surf4. Immunoblotting confirmed the expression of Surf4 in cells (Fig. 1D). As expected, the replication of HCV replicons in transfected cells was significantly impaired in ShSurf4 cells with Surf4 knockdown compared with shNC cells (Fig. 1D). The expression of Surf4shr in ShSurf4 cells restored the level of replicon replication. However, transfection of wild-type Surf4 into ShSurf4 cells showed no increase of Surf4 expression and HCV replication (Fig. 1D). We next determined the effects of endogenous Surf4 in HCV genome replication. ShSurf4 cells and shNC cells were transfected with JFH1 genomic RNA in the presence or absence of an expression plasmid for Surf4shr or wild-type Surf4. Immunoblotting demonstrated the expression of Surf4 in transfected cells (Fig. 1E). HCV replication was significantly impaired in ShSurf4 cells compared with shNC cells (Fig. 1E). The expression of Surf4shr in ShSurf4 cells restored the level of HCV replication. However, transfection of wild-type Surf4 in ShSurf4 cells did not increase Surf4 expression and HCV replication (Fig. 1E).
FIG 1.

Surf4 participates in HCV replication. (A) SGR2a cells were transfected with siS-1 (10 nM, 25 nM, and 50 nM), siDHCR (25 nM), or siNT (25 nM). At 3 days posttransfection, inhibition of HCV replication (top blot) and silencing of proteins (middle and bottom blots) were determined by luciferase and immunoblot assays, respectively. The luciferase activity is shown in relative light units (RLU). The positions of molecular mass positions are shown in kilodaltons (kD) to the right of the blots. (B) SGR2a cells were treated with siSurf4, including siS-1, siS-2, and siS-3, or with siDHCR or siNT. Three days later, HCV replication was determined by luciferase assay. (C) Huh7 cells were transfected with pSilencer-shSurf4 or negative-control shNC and selected using hygromycin B. Knockdown of Surf4 in stable transfectants (shSurf4) was determined by immunoblotting. (D and E) shSurf4 and shNC cells were transfected with plasmids expressing Surf4shr, Surf4, or empty vector as indicated. After 24 h, transfected cells were electroporated with SGR2a RNA (D) or JFH1 genomic RNA (E). Three days later, intracellular HCV RNA was determined by real-time RT-PCR (graphs), and Surf4 expression was determined by immunoblotting. (F) Effect of Surf4 silencing on SGR1b. SGR1b cells were transfected with siSurf4 (25 nM), siDHCR (25 nM), or siNT (25 nM). At 3 days posttransfection, inhibition of HCV replication (graph) and silencing of proteins (blots) were determined by luciferase and immunoblot assays, respectively. (G) SGR2a, SGR1b, and Huh7 (control) cells were subjected to immunoblot assays using anti-Surf4, anti-NS5A, and antiactin antibodies. The relative intensity of each Surf4 band after normalization to that of actin is indicated by the number above the lane. Data in the graphs are averages of triplicate values with error bars representing standard deviations (SDs). Values that are significantly different are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01.
To investigate the role of endogenous Surf4 in the replication of HCV genotype 1b, siSurf4, siDHCR, or siNT was transfected into Huh7 cells harboring HCV genotype 1b subgenomic replicon (SGR1b). siSurf4 decreased Surf4 expression and luciferase activity (an indicator of SGR1b replication) (Fig. 1F). siDHCR as a positive control specifically decreased the expression of DHCR and SGR1b replication, whereas siNT as a negative control had no effect on the expression of Surf4 and DHCR or on SGR1b replication (Fig. 1F). None of the siRNAs tested exhibited cytotoxicity against SGR1b cells (data not shown). Since we demonstrated that Surf4 played an important role in HCV replication, we were interested in the effect of HCV replication on the expression of Surf4. As shown in Fig. 1G, the replication of SGR2a and SGR1b did not alter the expression of Surf4.
Surf4 participates in HCV RNA replication but has no effect on entry, translation, assembly, or release.
Having demonstrated that Surf4 participates in HCV replication, we investigated the effect of Surf4 silencing on replication and translation of HCV RNA. Huh7 cells were treated with siSurf4 for 3 days and then electroporated with SGR2a RNA. The luciferase activity 4 h after HCV RNA transfection has been reported to reflect HCV RNA translation levels, but not replication levels (20, 22). Surf4 silencing had no effect on luciferase activity 4 h postelectroporation (Fig. 2A), suggesting that Surf4 does not alter HCV RNA translation. However, Surf4 silencing significantly decreased the SGR2a RNA level 3 days postelectroporation (Fig. 2B), suggesting that Surf4 participates in HCV RNA replication.
FIG 2.

Surf4 participates in HCV RNA replication but has no effect on viral RNA translation, entry, release, or assembly. (A and B) SGR2a RNA was introduced into Huh7 cells pretreated with the indicated siRNAs for 3 days. (A) For analysis of SGR2a translation, transfected cells were harvested to determine luciferase activity (graph) and protein levels (blots) at 4 h posttransfection. (B) For analysis of SGR2a replication, transfected cells were harvested to determine SGR2a RNA levels by real-time RT-PCR at 3 days posttransfection. (C) Huh7 cells were treated with the indicated siRNAs for 3 days and then infected with HCVpp1b, HCVpp2a, or VSVpp, which were pretreated with CD81 antibody (0.5 mg/ml, 2 h) as indicated. At 4 h postinfection, the medium was replaced with DMEM with 10% FBS, and the cells were harvested 3 days later to determine intracellular luciferase activity. (D to F) JFH1-infected cells were transfected with siSurf4 (10 nM, 25 nM, or 50 nM), siDHCR (25 nM), or siNT (25 nM). (D) At 3 days posttransfection, silencing of proteins was determined by immunoblotting. (E) Core proteins of cell lysate (left) and supernatant (middle) were evaluated by ELISA. Viral release was determined by the ratio of extracellular core level relative to that of intracellular core (right). To determine intracellular infectivity, intracellular HCV particles of cells treated with siSurf4 (25 nM) or siNT (25 nM) were collected and used to infect Huh7 cells. (F) At 3 days postinfection, intracellular HCV core antigen was determined by ELISA. (G) For viral assembly, specific infectivity was determined by calculating the ratio of infectivity (panel F) and intracellular core levels (left panel in panel E). siNT (25 nM) treatment was used as a control for siSurf4 (25 nM) treatment. Data are averages of triplicate values with error bars representing SDs. Statistical significance: *, P < 0.05; **, P < 0.01.
We next examined the effect of Surf4 on other stages of the HCV life cycle, including viral entry, assembly, and release. To investigate the role of Surf4 in HCV entry, Huh7 cells were treated with siSurf4 and infected with HCVpp1b harboring E1 and E2 glycoproteins of the TH clone (genotype 1b), HCVpp2a harboring E1 and E2 glycoproteins of the JFH-1 clone (genotype 2a), or VSVpp containing vesicular stomatitis virus (VSV) G glycoprotein. Surf4 expression was determined by immunoblot assay (Fig. 2C). Surf4 silencing did not inhibit the entry of HCVpp1b and HCVpp2a (Fig. 2C). As a control, pretreatment with CD81 antibody inhibited the entry of HCVpp1b and HCVpp2a but had no effect on VSVpp entry (Fig. 2C). To investigate the role of endogenous Surf4 in HCV assembly and release, JFH1-infected Huh7 cells were treated with siSurf4, siDHCR, or siNT for 3 days. siSurf4 decreased Surf4 expression (Fig. 2D) and the levels of intra- and extracellular HCV core protein in a dose-dependent manner (Fig. 2E, left and middle) but had no effect on cell viability (data not shown). As a positive control, siDHCR decreased the levels of intra- and extracellular HCV core protein (Fig. 2E, left and middle). To determine the effect of Surf4 silencing on viral release, we calculated core release based on intracellular and extracellular core levels. Surf4 had no significant effect on core release when comparing siSurf4 (25 nM) with siNT (25 nM) transfection (Fig. 2E, right), suggesting that Surf4 does not alter viral release. To determine the effect of Surf4 on viral assembly, virus particles from the above siRNA-treated cells were harvested and used to infect naive Huh7 cells. At 3 days postinfection, the levels of intracellular HCV core protein were detected by an enzyme-linked immunosorbent assay (ELISA) (Fig. 2F). We calculated the specific infectivity of HCV particles (core ratios based on intracellular core levels as shown in Fig. 2E and F). We previously confirmed a good correlation between the levels of core antigen and infectious titers (23). Surf4 had no significant effect on the specific infectivity of HCV particles when comparing siSurf4 (25 nM) treatment with siNT (25 nM) treatment (Fig. 2G), suggesting that Surf4 has no influence on viral assembly.
Surf4 participates in HCV replication by interacting with NS4B.
Our previous screen of NS4B-associated host proteins by dual-affinity purification and liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) showed that Surf4 is a novel NS4B-associated host protein (20). To confirm the interaction between Surf4 and NS4B, we performed coimmunoprecipitation, immunofluorescence, and proximity ligation assays (PLA). Huh7 cells harboring the genotype 2a JFH1 subgenomic replicon were used for coimmunoprecipitation assays of Surf4 and NS4B using antibodies against the two proteins. As expected, Surf4 was coimmunoprecipitated with NS4B (Fig. 3A). NS4B was also coimmunoprecipitated with Surf4 (Fig. 3A). As a control, NS5A and Surf4 did not coimmunoprecipitate (Fig. 3B). To further verify the colocalization between NS4B and Surf4, we performed PLA using antibodies tagged with circular DNA probes. If the antibodies are in close proximity, the probes can ligate together and subsequently be amplified by a polymerase. We were able to detect PLA signals predominantly in the cytoplasm of cells coexpressing NS4B tagged with C-myc (NS4Bmyc) and Surf4 tagged with V5 (Surf4v5). In contrast, PLA signals were not observed with NS4B or Surf4 expression alone (Fig. 3C). Taken together, these results strongly indicated that Surf4 interacts with NS4B.
FIG 3.

Surf4 participates in HCV replication by interacting with NS4B. (A and B) Lysates of SGR2a cells were immunoprecipitated with anti-NS4B antibody (A), anti-NS5A antibody (B), or anti-Surf4 antibody (A and B). The resulting immunoprecipitates (IP) and whole-cell lysates used in immunoprecipitation (Input) were examined by immunoblotting (IB) using anti-NS4B antibody (A), anti-NS5A antibody (B), and anti-Surf4 antibody (A and B). The negative-control sample was generated by using control IgG antibody (A and B). (C) PLA validates the colocalization between NS4B and Surf4. SGR2a cells were transfected with the indicated expression plasmids. Two days posttransfection, cells were fixed, permeabilized with Triton X-100, and then subjected to PLA using anti-myc and anti-V5 antibodies. (D) Diagram of NS4B and Surf4 constructs. Myc and V5 tags are depicted by the black and blue boxes, respectively. Mutant amino acid residues are depicted by the red line. Deleted amino acid residues are depicted by the dotted line. Positions of the amino acid residues are indicated above the black lines. (E and F) Huh7 cells were transfected with the indicated plasmids. Cell lysates were immunoprecipitated with anti-myc or anti-V5 antibody. The resulting immunoprecipitates (IP) and whole-cell lysates (Input) were examined by immunoblotting using anti-myc or anti-V5 antibody. (G) Huh7 cells were cotransfected with NS4B and Surf4 constructs. Three days posttransfection, cells were subjected to immunofluorescence staining using anti-myc (green) and anti-V5 antibodies (red). Nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI). The panels on the left present low-magnification overviews; the boxed areas are enlarged in the corresponding panels on the right. Quantities for the degree of colocalization (overlapping area/NS4B area) are given at the top of the enlarged pictures and are indicated as means plus SDs. (H) SGR2a and SGR1b cells were transfected with the indicated plasmids. At 3 days posttransfection, HCV replication and the expression of proteins were determined by luciferase and immunoblot assays, respectively. For luciferase activity, data are averages of triplicate values with error bars representing SDs. *, P < 0.05.
To determine the interaction region of Surf4 with NS4B, we first prepared V5-tagged full-length Surf4 (Surf4v5) and deletion constructs (Surf4d1, Surf4d2, and Surf4d3) and Myc-tagged NS4B (NS4Bmyc) (Fig. 3D). The expression of NS4Bmyc and Surf4 deletion constructs was confirmed by immunoblotting (Fig. 3E). The interaction of Surf4 constructs and NS4B was analyzed by immunoprecipitation. NS4Bmyc and Surf4d3 coprecipitated strongly, whereas NS4Bmyc and Surf4d2 coprecipitated weakly. On the other hand, the deletion construct Surf4d1 did not coprecipitate (Fig. 3E). The interaction of the proteins was also analyzed by immunofluorescence staining, which showed that colocalization between Surf4d1 and NS4Bmyc was absent. In contrast, Surf4v5, Surf4d2, and Surf4d3 strongly colocalized in the cytoplasm with NS4Bmyc (Fig. 3G). Taken together, these results indicated that the Surf4 region (amino acids 203 to 269 [aa 203–269]) deleted in Surf4d1 could be important for binding to NS4B. A very weak interaction between Surf4d2 and NS4B was detected compared to the interaction between NS4B and Surf4d3. Since Surf4d2 and NS4B are transmembrane proteins, it is possible that nonspecific binding may have been detected. In general, it is known that immunoprecipitation of membrane proteins tends to cause nonspecific binding. Protein conformation plays an important role in protein-protein interactions (24), and Surf4d2 has two transmembrane domains (aa 62–84 and 89–110) based on the prediction of basicELM (http://elm.eu.org/), which are possible NS4B binding sites. Alternatively, Surf4d1 could be misfolded, and an overlapping region (aa 112–157) of Surf4d2 and -d3 could be important for binding to NS4B. To address this possibility, Surf4 constructs Surf4d4 and Surf4d5 were prepared. Surf4d4 is a mutant in which aa 112–157 of Surf4d2 were mutated to alanines or glycines (Fig. 3D). Surf4d5 is a mutant in which aa 112–157 of Surf4 were deleted (Fig. 3D). Like Surf4, Surf4d4 and Surf4d5 coprecipitated with NS4Bmyc (Fig. 3F), strongly suggesting that aa 112–157 of Surf4 are nonessential for binding to NS4B.
Having demonstrated that Surf4 participates in HCV RNA replication and interacts with NS4B, we were interested in the role of the Surf4-NS4B interaction in HCV replication. HCV genotype 2a subgenomic replicon (SGR2a) cells were transfected with plasmids expressing Surf4v5, mutant Surf4d1, -d2, or -d3. SGR2a replication was increased by full-length Surf4 (Surf4v5) expression, whereas the expression of Surf4d1, -d2, or -d3 had no effect on HCV RNA replication (Fig. 3H). Likewise, RNA replication of the HCV genotype 1b subgenomic replicon (SGR1b) was increased by elevated expression of Surf4 and Surf4v5, whereas this effect was not observed in the presence of Surf4d1 expression (Fig. 3H). Collectively, these results indicate that the Surf4-NS4B interaction is important for the role of Surf4 in HCV replication.
Surf4 is recruited into the HCV RNA replication complex by NS4B.
Since NS4B triggers the formation of the HCV RNA replication complex and Surf4 interacts with NS4B, it is reasonable to think that Surf4 is involved in the HCV RNA replication complex. The HCV RNA replication complex has been reported to be located in detergent-resistant membranes (DRM) (7). To investigate whether Surf4 is located in DRM, the membrane fractions from Huh7 cells and HCV replicon cells were subjected to immunoblot analysis. Interestingly, Surf4 localized in DRM in Huh7 cells and SGR2a cells. As expected, caveolin, a DRM marker, was detected in the DRM fraction, and calnexin, a detergent-soluble membrane (DSM) marker, was present in the DSM fraction (Fig. 4A). To visualize whether Surf4 localized in the HCV RNA replication complex, we examined the colocalization of Surf4 and double-stranded RNA (dsRNA), a marker of the HCV RNA replication complex. Surf4 colocalized with dsRNA in SGR2a cells (Fig. 4B). As a positive control, NS5A residing in the HCV RNA replication complex colocalized with dsRNA in SGR2a cells (Fig. 4B). As a negative control, Huh7 cells without the HCV replicon exhibited no dsRNA signal (Fig. 4B).
FIG 4.

Surf4 is recruited into the HCV RNA replication complex by NS4B. (A) Surf4 localization in DRM in Huh7 cells. Cell lysates from Huh7 and SGR2a cells were treated with 1% Triton X-100 (TX100) at 4°C or 37°C as indicated. Membrane fractions were isolated by membrane flotation assays and then analyzed by immunoblotting with anti-Surf4, anti-NS4B, anticaveolin, and anticalnexin antibodies. MB, membrane fraction; NM, nonmembrane fraction; RM, DRM fraction; SM, DSM fraction. (B) Colocalization of Surf4 with dsRNA. SGR2a and Huh7 cells were fixed and subjected to immunofluorescence staining using anti-Surf4 (green), anti-NS5A (green), and anti-dsRNA antibodies (red). (C) Surf4 colocalized with dsRNA via NS4B. SGR2a cells were transfected with plasmids expressing Surf4v5 or Surf4d1. At 3 days posttransfection, cells were subjected to immunofluorescence staining using anti-V5 (green) and anti-dsRNA antibodies (red). The four panels on the left present low-magnification overviews; the boxed areas in the Merge panels are enlarged in the corresponding panels on the right. Quantities for the degree of colocalization (overlapping area/dsRNA area) are given above the enlarged pictures and are indicated as means ± SDs.
To investigate whether the interaction of Surf4 with NS4B is necessary for Surf4 colocalization with dsRNA, SGR2a cells were transfected with Surf4v5 or Surf4d1, which lacks the ability to bind to NS4B. Surf4v5 colocalized with dsRNA (Fig. 4C, top), but Surf4d1 did not (Fig. 4C, bottom). These results together provided evidence that Surf4 is recruited into the HCV RNA replication complex via its interaction with NS4B.
Surf4 is involved in the formation of DMVs and in the structural integrity of the HCV RNA replication complex.
Since Surf4 is recruited into the HCV RNA replication complex by NS4B, we investigated the effect of Surf4 silencing on the formation and number of DMVs, which have been reported to be the predominant HCV RNA replication sites (25, 26). SGR2a cells were treated with siSurf4 or siNT and analyzed by transmission electron microscopy (TEM). Compared to siNT treatment, siSurf4 treatment decreased the level of formation of DMVs (Fig. 5A, left) and the number of DMVs (Fig. 5A, right). We next validated the effect of Surf4 silencing on the formation of DMVs by membrane flotation assays. SGR2a cells were treated with siSurf4 or siNT, and then the membrane fraction was isolated. Interesting structures, presumably from formation-hindered or structure-impaired DMVs, appeared in siSurf4-treated cells (Fig. 5B, right side of left panel). Fewer DMVs were detected in siSurf4-treated cells compared to siNT-treated cells (Fig. 5B, right panel), suggesting that Surf4 is involved in the formation of HCV-induced DMVs. To exclude the possibility that Surf4 knockdown decreases the formation of DMVs by inhibiting HCV replication, we used an HCV replication-independent system to examine the effect of Surf4 on membrane web formation. Huh7 cells were infected with AdexCAT7 and AxCAwt (a control virus), transfected with pSGRlucneoGND, which contains an inactivating mutation within NS5B, and analyzed by immunoblotting (Fig. 5C, top side of left panel) and TEM (Fig. 5C, right panel). HCV protein-induced membrane vesicles were observed in siNT-treated cells, but siSurf4 treatment decreased the formation of membrane vesicles. Fewer DMVs were detected in siSurf4-treated cells compared to siNT-treated cells (Fig. 5C, lower side of left panel). These results together indicated that Surf4 is involved in the formation of membrane vesicles.
FIG 5.


Surf4 is involved in the formation of DMVs and structural integrity of HCV RNA replication complexes. (A) SGR2a cells were treated with 25 nM siSurf4 or siNT. At 3 days posttransfection, cells were examined by TEM (left). The right panels are magnified views of the black squares in the left panels. Arrows indicate HCV protein-induced membrane vesicles. The size and number of DMVs in the boxed areas of siRNA-treated cells in panel A were quantified (right). Values were obtained from three samples and are means plus SDs. (B) SGR2a cells were treated with siSurf4 or siNT for 3 days. Membrane fractions from treated cells were examined by TEM. Representative membranous structures are shown (left). The number of membrane structures in 10 randomly chosen squares (3,600 μm2/square) was determined. The percentage of DMV and non-DMV in siSurf4-treated or control cells was determined for more than 200 membrane structures (right). (C) Huh7 cells were infected with AdexCAT7, a recombinant adenovirus containing the bacteriophage T7 RNA polymerase, or AxCAwt, a control virus, and transfected with pSGRlucneoGND. The cells were then transfected with 25 nM siSurf4 or siNT. At 3 days posttransfection, cells were examined by immunoblotting and TEM. Arrows indicate HCV protein-induced membrane vesicles. The micrographs present low-magnification overviews; the boxed areas are enlarged in the corresponding panels on the right. The size and number of DMVs in the areas in left panel were quantified. Values were obtained from three samples and are means ± SDs. (D and E) SGR2a cells were treated with siSurf4 or siNT. Three days later, DRM from treated cells were digested with RNase A (500 ng/ml) at 37°C for 5 min as indicated, and then used to determine the HCV RNA levels by real-time RT-PCR (graph in panel E) and protein levels by immunoblotting using anti-Surf4, anti-NS5A, and anti-caveolin antibodies (D). SGR2a cells were treated with siSurf4 or siNT. Three days later, DRM from transfected cells were treated with trypsin (25 μg/ml) at 37°C for 10 min as indicated and then used to determine protein levels by immunoblotting using anti-Surf4, anti-NS4B, and anti-NS5A antibodies (blots in panel E). For real-time RT-PCR, data are averages of triplicate values with error bars representing SDs.
The viral proteins and genomic RNAs in HCV RNA replication complexes have been reported to be resistant to cellular proteases and nucleases, respectively (27). To investigate whether Surf4 silencing affects the structure of the HCV RNA replication complex, we investigated the effect of Surf4 silencing on the enzymatic resistance of HCV replicase proteins and genomic RNAs. SGR2a cells were treated with siSurf4 or siNT, and then the cellular DRM fraction was isolated and its resistance to enzymatic degradation was analyzed. Immunoblot assays confirmed Surf4 silencing in siSurf4-treated SGR2a cells (Fig. 5D), which decreased the RNase resistance of HCV RNA in DRM (Fig. 5E, left). Next, we investigated the effect of Surf4 silencing on the protease resistance of viral replicase proteins, including NS4B and NS5A, in DRM, and found that it significantly decreased protease resistance (Fig. 5E, right). Collectively, these data demonstrate that Surf4 silencing may impair the integrity of HCV-induced DMVs and thus increase the degradation of HCV RNA and proteins in the replication complex.
Effect of Surf4 silencing on replication of poliovirus and dengue virus.
A number of positive-strand RNA viruses, such as HCV and poliovirus, use DMVs as replication sites (20). Since we demonstrated that Surf4 participates in HCV replication by promoting DMV formation, we next examined the effect of Surf4 silencing on poliovirus replication. Dengue virus, which uses invaginated/sphere-type vesicles as replication sites, was also used as a negative control. As expected, Surf4 silencing significantly decreased poliovirus replication but had no inhibitory effect on dengue virus replication, which is consistent with a previous finding in which PREB silencing decreases the replication of poliovirus but not dengue virus (20). In addition, as positive controls, alpha interferon (IFN-α) dramatically decreased the replication of poliovirus and dengue virus (Fig. 6). These results together demonstrate that Surf4 has a pan-specific effect on DMV formation during the replication of positive strand viruses.
FIG 6.
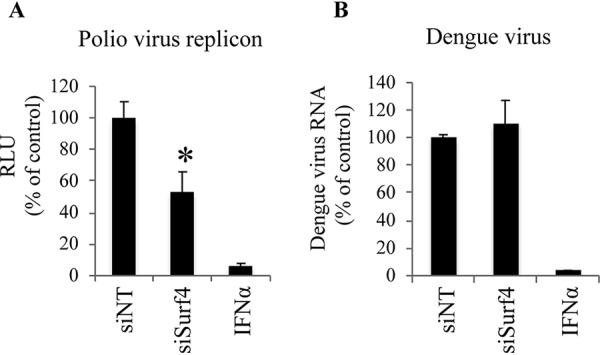
Effect of Surf4 silencing on replication of poliovirus and dengue virus. (A) Naive Huh7 cells were transfected with poliovirus replicon RNA and then treated with siRNAs as indicated. After 8 h, the replication of poliovirus replicon RNA in cells was analyzed by luciferase activity. (B) Naive Huh7 cells were infected with dengue virus and then treated with siRNAs as indicated. After 3 days, the amount of dengue virus RNA in cells was determined by TaqMan RT-PCR. IFN-α was used as a positive control. siNT was used as a negative-control siRNA. Values were obtained from quadruplicate wells in two independent experiments and are means plus SDs. *, P < 0.05.
DISCUSSION
HCV NS4B rearranges cellular membranes to form a MW where HCV replication complexes reside (28). However, NS4B-associated host proteins and their roles in the HCV life cycle, particularly in replication, remain largely unclear. We previously identified Surf4 as a novel NS4B-associated host protein by tandem affinity purification and LC-MS/MS analysis. Furthermore, preliminary siRNA screening showed that siRNA targeted to Surf4 inhibits the replication of the HCV genotype 2a replicon (20). Here, we further investigated the effect of Surf4 on various stages of the HCV life cycle in detail. A strong relationship was observed between Surf4 silencing by siSurf4 and the inhibition of replication of HCV genotype 1b and 2a replicons (Fig. 1), suggesting that Surf4 is a common cofactor in HCV genotype 2a and 1b replication. Similarly, PI4KIIIα and PREB have been reported to act as common cofactors of HCV genotypes 1b and 2a (4, 20). Since replication of HCV replicons includes RNA translation and HCV RNA synthesis stages, we investigated the effect of Surf4 on RNA translation and replication, respectively. Surf4 participated in HCV RNA replication but had no effect on RNA translation (Fig. 2). NS4B was recently reported to be involved in virus production (29), which led us to examine whether Surf4, an NS4B-associated host membrane protein, affected various stages of virus production, including viral entry, release, and assembly. HCVpp and propagation assays showed that Surf4 had no effect on viral entry, release, or assembly (Fig. 2). However, consistent with the data from HCV replicons, Surf4 silencing decreased HCV propagation, which was likely caused by the inhibition of virus replication (Fig. 2), suggesting that Surf4 is a potential anti-HCV drug target.
Since we demonstrated that Surf4, an NS4B-associated membrane protein, participates in HCV RNA replication (Fig. 2) (20) and NS4B is necessary for HCV RNA replication, we next examined whether Surf4 played a role in HCV RNA replication by interacting with NS4B. Heterogenous expression of Surf4, but not Surf4d1 lacking NS4B binding ability, increased HCV replication (Fig. 3H), suggesting that the interaction of Surf4 with NS4B is necessary for the role played by Surf4 in HCV replication. Surf4d2 and Surf4d3 were weakly and strongly bound to NS4B, respectively (Fig. 3E). On the other hand, Surf4d2 and Surf4d3 were not involved in HCV replication (Fig. 3H). Luminal loop(s) in positions amino acids aa 130–137 (L1) and/or aa 221–247 (L2) of ERV29p, the yeast homolog of Surf4, are available for interactions with secretory cargo of COPII vesicles (30). ERV29p has four transmembrane domains (aa 108–130 [TM1], aa137-156 [TM2], aa 137–156 [TM3], aa 202–221 [TM3], aa 247–269 [TM4]). Some of them are necessary to transport cargo protein from ER lumen to cis-Golgi lumen. Therefore, Surf4d2 (deleting L1, L2, TM3, and TM4) and Surf4d3 (deleting TM1) may have lost the function of full-length Surf4. Further research is needed to understand the functional domains of Surf4 as a COPII protein and their role for HCV DMV formation.
The question then arose as to what the underlying mechanism is. Since NS4B has been proposed to be a scaffold protein of HCV replication complexes (8), it is probable that Surf4 is recruited into HCV RNA replication complexes by NS4B. Indeed, indirect immunofluorescence assays showed that full-length Surf4 colocalized with dsRNA, a marker of HCV RNA complexes, whereas no colocalization of Surf4d1 without NS4B binding was detected (Fig. 4). Collectively, these results indicate that Surf4 is recruited into HCV replication complexes by NS4B, which provides an explanation for why the interaction of Surf4 with NS4B is necessary for Surf4 to promote HCV RNA replication.
Host cofactors involved in the HCV replication complex have been reported to affect HCV replication by regulating the formation, structure, and activity of the complex (20). It is possible that Surf4 is involved in the formation of membranous replication compartments, since NS4B triggers the formation of MW. As expected, ultrastructural analysis showed that Surf4 silencing decreased the formation of DMVs, the predominant HCV replication complex sites (Fig. 5). Furthermore, we showed that Surf4 knockdown decreased DMVs independent from HCV replication by an HCV replication-independent system (Fig. 5C). We observed the accumulation of small membrane vesicles in HCV protein-inducing cells (Fig. 5C). These clusters of membrane vesicles were similar to the MW initially reported for Huh7 cells harboring subgenomic replicons (9) and to double- and multimembrane vesicles reported for HCV-infected Huh7 cells (10). HCV replicase proteins and genomic RNA in HCV replication complexes are protected from degradation by cellular proteinases and nucleases, respectively (20). Since NS4B, a scaffold protein involved in forming MW, recruits Surf4 to the HCV RNA replication complex, we hypothesized that Surf4 silencing could impair the structural integrity of the NS4B-induced membranous replication compartment, thus leading to easy access by degradative enzymes. Indeed, our data showed that Surf4 silencing decreased the nuclease and protease resistance of HCV genomic RNA and replicase proteins (NS4B and NS5A) (Fig. 5), respectively, in DRM where HCV RNA replication complexes reside.
DMVs have recently been reported to be the replication sites of a number of positive-strand RNA viruses, including HCV (5, 10, 31). NS5A is the only HCV protein with the ability to induce DMVs and predominantly multimembrane vesicles on its own, but with very low efficiency compared to that obtained by the expression of the NS3-5B polyprotein, implying that NS3, NS4A, NS4B, and NS5B help in the formation of DMVs (10). The cleavage kinetics of HCV polyproteins such as NS4B-5A play important roles in the formation of DMVs (32), highlighting the role of NS4B in DMV formation. The interaction between NS4B and NS5A is essential for proper NS5A localization and hepatitis C virus RNA replication, indicating the importance of the NS4B-NS5A interaction in HCV replication complex organization and function (30). In addition, it is well-known that NS4B induces the formation of MW to provide a platform for the formation of HCV replication complexes. These findings together prompted us to focus on the role of NS4B-associated cellular proteins in the formation of DMVs during the replication of positive-strand RNA viruses. We demonstrated that Surf4 interacted with HCV NS4B and that Surf4 silencing decreased the formation of DMVs in the context of HCV replication. Moreover, Surf4 silencing also decreased the replication of poliovirus, which uses DMVs as replication sites, but had no effect on the replication of dengue virus, which uses invaginated/sphere-type vesicles as replication sites, highlighting the role of Surf4 in DMV formation during virus replication. Interestingly, Surf4 has recently been reported to interact with NS4B of Zika virus (ZIKV), a flavivirus that is related to HCV, although the role for this interaction remains unknown (33). Based on the roles of Surf4 in HCV and poliovirus replication, it is reasonable to speculate that Surf4 is involved in ZIKV replication by regulating DMV formation. However, how Surf4 participates in the formation of DMVs by interacting with NS4B remains to be determined. Some studies suggest that DMVs of coronaviruses (34, 35) and arteriviruses (36) originate from ER membranes. Accumulating evidence has revealed that sphingolipids from Golgi membranes move into HCV RNA replication membrane vesicles originating from the ER (7, 37, 38). Anterograde transport from the ER to the ERGIC is mediated by COPII vesicles at ERES. Interestingly, the cis-Golgi has been found to contact the ERES for cargo capture and delivery from the ER (39). Surf4 is a conserved integral membrane protein that is essential for maintaining the architecture of the ERGIC and Golgi membranes (11, 12). We previously found that PREB, an ER protein, participates in DMV formation by interacting with NS4B (20). Taken together, it is conceivable that NS4B participates in the formation of DMVs by interacting with PREB located at ERES and Surf4 located at the cis-Golgi simultaneously to promote DMV formation, which provides mechanistic insight into the formation of DMVs induced by HCV. It should be noted that NS4B, Surf4, and PREB coordinate to efficiently induce DMV formation with the help of NS5A, since NS5A is the only HCV protein with the ability to induce DMV. Further research is needed to elucidate the mechanism by which HCV induces the formation of DMVs by use of viral and cellular proteins.
In summary, for the first time we identified Surf4 as a novel NS4B-associated host cofactor involved in HCV RNA replication and revealed a novel mechanism by which Surf4 and NS4B coordinate to promote the formation of DMVs for HCV replication. More importantly, we found that Surf4 also promotes the replication of poliovirus, a positive-strand virus that uses DMVs as replication sites, suggesting that Surf4 widely participates in the replication of viruses that use DMVs as replication sites. The formation of HCV replication compartments that include DMVs are induced by the concerted action of viral and cellular proteins (40). Therefore, it is reasonable to think that NS4B, Surf4, PREB, NS5A, and possibly other unknown viral and cellular proteins coordinate closely to induce the efficient formation of DMVs during HCV replication. Our current findings provide new insights into HCV host cofactors, Surf4 function, anti-HCV drug targets, and the formation of DMVs during the replication of positive-strand RNA viruses.
MATERIALS AND METHODS
Cell culture.
Human hepatoma cells Huh7 (a generous gift from Francis V. Chisari) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). Huh7 cells harboring the genotype 1b subgenomic replicon (SGR1b) or genotype 2a JFH1 subgenomic replicon (SGR2a) were maintained in complete DMEM supplemented with 0.5 mg/ml G418 (20).
Plasmids.
Plasmids pJFH1, containing the full-length HCV genotype2a JFH1 strain cDNA, and pSGRlucneo, containing the genotype 2a JFH1 subgenomic replicon, were previously described (20). The poliovirus replicon encoding firefly luciferase (Fluci) in place of the capsid genes was previously described (20). Myc-tagged full-length NS4B was previously constructed (20). Surf4 cDNA was amplified using total RNAs from Huh7 cells as a template and inserted into pCAGGS. Surf4 was predicted to have six transmembrane domains (amino acids 62 to 84 [aa 62–84], aa 89–111, aa 158–175, aa 179–198, aa 203–225, and aa 240–262) using basicELM software, which are possible sites to interact with the transmembrane protein NS4B. Therefore, we constructed V5-tagged Surf4 (Surf4v5) and Surf4 deletion constructs, including Surf4d1 (lacking aa 203–269), Surf4d2 (lacking aa 158–269), and Surf4d3 (lacking aa 1–111). cDNAs encoding full-length Surf4 or Surf4 deletion mutant sequences with a V5 tag at the N terminus were amplified using pCAG-Surf4 as a template. The resultant fragments were cloned into pCAGGS. Surf4d2 and Surf4d3 have an overlapping region containing aa 112–157 of Surf4. To generate the mutants of the overlapping region, Surf4d4 and Surf4d5 were constructed by overlapping PCR. Surf4d4 is a mutant in which amino acids 112 to 157 of Surf4d2 (WDLKFLMRNLALGGGLLLLLAESRSEGKSMFAGVPTMRESSPKQ YM) were mutated to alanines or glycines (AGGAGGAGGGGGAAAAAAAGGGAGAGAGAGAGAAGGAAAGAGAGAA). Surf4d5 is a mutant in which amino acids 112–157 of Surf4 were deleted. pSilencer-shSurf4 expressing an shRNA targeted to Surf4 under the control of the U6 promoter was constructed by cloning the oligonucleotide pair 5′-GATCCGACGATTGCCTACAGCATTTTCAAGAGAAATGCTGTAGGCAATCGTCTTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAAAAAGACGATTGCCTACAGCATTTCTCTTGAAAATGCTGTAGGCAATCGTCG-3′ between the BamHI and HindIII sites of pSilencer 2.1-U6 hygro (Ambion, Austin, TX). To generate the pCAG-shrSurf4 construct expressing shRNA-resistant Surf4, the cDNA fragment encoding Surf4 in which the shRNA targeting region (5′-GACGATTGCCTACAGCATT-3′) was replaced with 5′-AACTATCGCGTATAGTATC-3′ that causes no amino acid change, was amplified by PCR using pCAG-Surf4 as a template. The resulting fragment was confirmed by sequencing and then cloned into pCAGGS.
Antibodies.
Mouse monoclonal antibodies against actin, flag, and CD81 were obtained from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal double-stranded RNA (dsRNA) antibody was obtained from Biocenter Ltd. (Szirak, Hungary). Mouse monoclonal antibody against caveolin2 was obtained from BD Transduction Laboratories (San Jose, CA). Mouse monoclonal antibody against HCV core and rabbit polyclonal antibody against NS5A are described elsewhere (41). Rabbit polyclonal anti-calnexin was obtained from Stressgen Bioreagents (Victoria, BC, Canada). Polyclonal antibodies against V5 and myc were obtained from Sigma-Aldrich. Rabbit polyclonal antibody against Surf4 and mouse monoclonal antibody against Surf4 were obtained from LifeSpan Biosciences and Abnova (Taipei, Taiwan), respectively. Goat polyclonal antibody against DHCR was obtained from Santa Cruz Biotechnology (Dallas, TX). Anti-NS4B antibody was a gift from M. Kohara (42).
RNA interference, DNA transfection, and cell viability.
siSurf4 (siRNAs targeting Surf4) included siS-1 (5′-GACGAUUGCCUACAGCAUU-3′), siS-2 (5′-CGUCCUUCGUCUUCCUCAA-3′), and siS-3 (5′-GCUCUUUGCCAUCAACGUA-3′). siDHCR (siRNA targeting 24-dehydrocholesterol reductase) was used as a positive control. Nontargeting siRNA (siNT) was used as a negative control. DNA transfection was performed using the Trans LT1 transfection reagent (Mirus, Madison, WI). siRNAs were introduced into cells using Lipofectamine RNAiMAX (Invitrogen, Tokyo, Japan). Cell viability was analyzed using a Cell Titer-Glo Luminescent Cell Viability assay kit (Promega).
Establishment of stable cells expressing shRNA.
Huh7 cells were transfected with pSilencer-shSurf4 or negative-control pSilencer hygrovector (shNC) that expresses a hairpin siRNA with limited homology to any known sequences in the human, mouse, and rat genomes. Drug-resistant clones were selected by treatment with hygromycin B (Wako) at a final concentration of 300 μg/ml for 4 weeks.
Immunoprecipitation, immunoblotting, indirect immunofluorescence, and proximity ligation assays.
Immunoprecipitation, immunoblotting, indirect immunofluorescence, and proximity ligation assays were performed as previously reported (20).
HCV replication assay.
HCV-replicating cells were harvested, and luciferase activity was measured using a Luciferase Reporter Assay system kit (Promega). The HCV RNA level was measured by real-time reverse transcription-PCR (RT-PCR) as described previously (43).
HCV propagation assay.
HCV propagation assays were performed as previously reported (20).
Translation and replication of HCV RNA.
Translation and replication of HCV RNA were analyzed as previously reported (20). For analysis of RNA translation, HCV RNA-transfected cells were harvested to determine luciferase activity at 4 h posttransfection. For analysis of RNA replication, transfected cells were harvested to determine the SGR2a RNA level at 3 days posttransfection.
HCV entry.
Huh7 cells were infected with untreated HCVpp1b harboring E1 and E2 glycoproteins of the TH clone (genotype 1b), HCVpp2a harboring E1 and E2 glycoproteins of the JFH-1 clone (genotype 2a), or VSVpp containing VSV G glycoprotein pretreated with CD81 antibody (0.5 mg/ml) for 2 h as previously reported (20). At 4 h postinfection, the medium was replaced with DMEM with 10% FBS, and the cells were harvested 3 days later to determine intracellular luciferase activity.
Membrane flotation assay.
Membrane flotation assays were performed as previously reported (20).
HCV replication-independent system.
The HCV replication-independent system was described previously (20).
Poliovirus and dengue virus replication assays.
The poliovirus replicon system was described previously (44). Naive Huh7 cells were infected with dengue virus and then treated with siRNAs. After 3 days, the amount of dengue virus RNA in the infected cells was determined by the TaqMan RT-PCR method as described previously (20).
Resistance assays of RNase and protease.
Resistance assays of RNase and protease were performed as previously reported (20).
Ultrastructural analysis.
Cells were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 2 h and then postfixed in 1% osmium tetroxide in 0.1 M phosphate buffer for 1 to 2 h. Samples were dehydrated in ethanol, embedded in Epon 812, and left to polymerize at 60°C for 2 days. Ultrathin sections (50 to 70 nm thick) were picked up on copper grids and were double stained with uranyl acetate and lead citrate by standard procedures. Cellular membrane fractions isolated by membrane flotation assays were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer for 5 min, washed with distilled water, and negatively stained with 1% (wt/vol) uranyl acetate solution on copper grids (300 mesh). These specimens were observed using a transmission electron microscope (H-7100; Hitachi Ltd., Tokyo, Japan).
Statistical analysis.
Statistical analysis was performed using the Statistical Package Social Sciences (SPSS) program version 11.5 by one-way analysis of variance, and significant differences among groups were determined by least significant difference (LSD). The accepted level of statistical significance was P < 0.05.
ACKNOWLEDGMENTS
We thank M. Sasaki and Y. Hirama for their technical assistance. We also thank T. Miyamura for helpful discussions.
The Ministry of Health, Labor and Welfare of Japan provided funding to Hideki Aizaki (10851601, 10850101, and 10850201). The Ministry of Education, Culture, Sports, Science and Technology of Japan provided funding to Hideki Aizaki (15K09034). The Advanced Research & Development Programs for Medical Innovation (AMED, AMED-CREST) provided funding to Hideki Aizaki (19fk0210022j0003, 19fk0210047j0101, 19fk0310112j0703, and 19gm0910005j0005). The China Scholarship Council (CSC) provided funding to Lingbao Kong under grant 201208360008. The Nature Science Foundation of China provided funding to Lingbao Kong under grant 31860038.
We have no conflicts of interest to report.
REFERENCES
- 1.Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liang TJ. 2013. Current progress in development of hepatitis C virus vaccines. Nat Med 19:869–878. doi: 10.1038/nm.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Q, Zhang YY, Chiu S, Hu Z, Lan KH, Cha H, Sodroski C, Zhang F, Hsu CS, Thomas E, Liang TJ. 2014. Integrative functional genomics of hepatitis C virus infection identifies host dependencies in complete viral replication cycle. PLoS Pathog 10:e1004163. doi: 10.1371/journal.ppat.1004163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romero-Brey I, Bartenschlager R. 2014. Membranous replication factories induced by plus-strand RNA viruses. Viruses 6:2826–2857. doi: 10.3390/v6072826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L, Ou JJ. 2018. Regulation of autophagy by hepatitis C virus for its replication. DNA Cell Biol 37:287–290. doi: 10.1089/dna.2017.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aizaki H, Lee KJ, Sung VM, Ishiko H, Lai MM. 2004. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324:450–461. doi: 10.1016/j.virol.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 8.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984. doi: 10.1128/jvi.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 77:5487–5492. doi: 10.1128/jvi.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reeves JE, Fried M. 1995. The surf-4 gene encodes a novel 30 kDa integral membrane protein. Mol Membr Biol 12:201–208. doi: 10.3109/09687689509027508. [DOI] [PubMed] [Google Scholar]
- 12.Mitrovic S, Ben-Tekaya H, Koegler E, Gruenberg J, Hauri HP. 2008. The cargo receptors Surf4, endoplasmic reticulum-Golgi intermediate compartment (ERGIC)-53, and p25 are required to maintain the architecture of ERGIC and Golgi. Mol Biol Cell 19:1976–1990. doi: 10.1091/mbc.e07-10-0989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emmer BT, Hesketh GG, Kotnik E, Tang VT, Lascuna PJ, Xiang J, Gingras AC, Chen XW, Ginsburg D. 2018. The cargo receptor SURF4 promotes the efficient cellular secretion of PCSK9. Elife 7:e38839. doi: 10.7554/eLife.38839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foley DA, Sharpe HJ, Otte S. 2007. Membrane topology of the endoplasmic reticulum to Golgi transport factor Erv29p. Mol Membr Biol 24:259–268. doi: 10.1080/09687860601178518. [DOI] [PubMed] [Google Scholar]
- 15.Yin Y, Garcia MR, Novak AJ, Saunders AM, Ank RS, Nam AS, Fisher LW. 2018. Surf4 (Erv29p) binds amino-terminal tripeptide motifs of soluble cargo proteins with different affinities, enabling prioritization of their exit from the endoplasmic reticulum. PLoS Biol 16:e2005140. doi: 10.1371/journal.pbio.2005140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saegusa K, Sato M, Morooka N, Hara T, Sato K. 2018. SFT-4/Surf4 control ER export of soluble cargo proteins and participate in ER exit site organization. J Cell Biol 217:2073–2085. doi: 10.1083/jcb.201708115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujii Y, Shiota M, Ohkawa Y, Baba A, Wanibuchi H, Kinashi T, Kurosaki T, Baba Y. 2012. Surf4 modulates STIM1-dependent calcium entry. Biochem Biophys Res Commun 422:615–620. doi: 10.1016/j.bbrc.2012.05.037. [DOI] [PubMed] [Google Scholar]
- 18.Kim J, Hong CM, Park SM, Shin DH, Kim JY, Kwon SM, Kim JH, Kim CD, Lim DS, Lee D. 2018. SURF4 has oncogenic potential in NIH3T3 cells. Biochem Biophys Res Commun 502:43–47. doi: 10.1016/j.bbrc.2018.05.116. [DOI] [PubMed] [Google Scholar]
- 19.Atay S, Wilkey DW, Milhem M, Merchant M, Godwin AK. 2018. Insights into the proteome of gastrointestinal stromal tumors-derived exosomes reveals new potential diagnostic biomarkers. Mol Cell Proteomics 17:495–515. doi: 10.1074/mcp.RA117.000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong L, Fujimoto A, Nakamura M, Aoyagi H, Matsuda M, Watashi K, Suzuki R, Arita M, Yamagoe S, Dohmae N, Suzuki T, Sakamaki Y, Ichinose S, Suzuki T, Wakita T, Aizaki H. 2016. Prolactin regulatory element binding protein is involved in hepatitis C virus replication by interaction with NS4B. J Virol 90:3093–3111. doi: 10.1128/JVI.01540-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takano T, Tsukiyama-Kohara K, Hayashi M, Hirata Y, Satoh M, Tokunaga Y, Tateno C, Hayashi Y, Hishima T, Funata N, Sudoh M, Kohara M. 2011. Augmentation of DHCR24 expression by hepatitis C virus infection facilitates viral replication in hepatocytes. J Hepatol 55:512–521. doi: 10.1016/j.jhep.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 22.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J Virol 77:3007–3019. doi: 10.1128/jvi.77.5.3007-3019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumoto Y, Matsuura T, Aoyagi H, Matsuda M, Hmwe SS, Date T, Watanabe N, Watashi K, Suzuki R, Ichinose S, Wake K, Suzuki T, Miyamura T, Wakita T, Aizaki H. 2013. Antiviral activity of glycyrrhizin against hepatitis C virus in vitro. PLoS One 8:e68992. doi: 10.1371/journal.pone.0068992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, Katinger H, Parren PW, Robinson J, Van Ryk D, Wang L, Burton DR, Freire E, Wyatt R, Sodroski J, Hendrickson WA, Arthos J. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678–682. doi: 10.1038/nature01188. [DOI] [PubMed] [Google Scholar]
- 25.Paul D, Bartenschlager R. 2013. Architecture and biogenesis of plus-strand RNA virus replication factories. World J Virol 2:32–48. doi: 10.5501/wjv.v2.i2.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. 2013. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J Virol 87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyanari Y, Hijikata M, Yamaji M, Hosaka M, Takahashi H, Shimotohno K. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J Biol Chem 278:50301–50308. doi: 10.1074/jbc.M305684200. [DOI] [PubMed] [Google Scholar]
- 28.Li S, Yu X, Guo Y, Kong L. 2012. Interaction networks of hepatitis C virus NS4B: implications for antiviral therapy. Cell Microbiol 14:994–1002. doi: 10.1111/j.1462-5822.2012.01773.x. [DOI] [PubMed] [Google Scholar]
- 29.Gouttenoire J, Montserret R, Paul D, Castillo R, Meister S, Bartenschlager R, Penin F, Moradpour D. 2014. Aminoterminal amphipathic alpha-helix AH1 of hepatitis C virus nonstructural protein 4B possesses a dual role in RNA replication and virus production. PLoS Pathog 10:e1004501. doi: 10.1371/journal.ppat.1004501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biswas A, Treadaway J, Tellinghuisen TL. 2016. Interaction between nonstructural proteins NS4B and NS5A is essential for proper NS5A localization and hepatitis C virus RNA replication. J Virol 90:7205–7218. doi: 10.1128/JVI.00037-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferraris P, Beaumont E, Uzbekov R, Brand D, Gaillard J, Blanchard E, Roingeard P. 2013. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell Mol Life Sci 70:1297–1306. doi: 10.1007/s00018-012-1213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romero-Brey I, Berger C, Kallis S, Kolovou A, Paul D, Lohmann V, Bartenschlager R. 2015. NS5A domain 1 and polyprotein cleavage kinetics are critical for induction of double-membrane vesicles associated with hepatitis C virus replication. mBio 6:e00759-15. doi: 10.1128/mBio.00759-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scaturro P, Stukalov A, Haas DA, Cortese M, Draganova K, Płaszczyca A, Bartenschlager R, Götz M, Pichlmair A. 2018. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature 561:253–257. doi: 10.1038/s41586-018-0484-5. [DOI] [PubMed] [Google Scholar]
- 34.Knoops K, Kikkert M, Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC. 2002. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J Virol 76:3697–3708. doi: 10.1128/jvi.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knoops K, Barcena M, Limpens RW, Koster AJ, Mommaas AM, Snijder EJ. 2012. Ultrastructural characterization of arterivirus replication structures: reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J Virol 86:2474–2487. doi: 10.1128/JVI.06677-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shanmugam S, Saravanabalaji D, Yi M. 2015. Detergent-resistant membrane association of NS2 and E2 during hepatitis C virus replication. J Virol 89:4562–4574. doi: 10.1128/JVI.00123-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi ST, Lee KJ, Aizaki H, Hwang SB, Lai MM. 2003. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J Virol 77:4160–4168. doi: 10.1128/jvi.77.7.4160-4168.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurokawa K, Okamoto M, Nakano A. 2014. Contact of cis-Golgi with ER exit sites executes cargo capture and delivery from the ER. Nat Commun 5:3653. doi: 10.1038/ncomms4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paul D, Madan V, Bartenschlager R. 2014. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe 16:569–579. doi: 10.1016/j.chom.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki R, Saito K, Kato T, Shirakura M, Akazawa D, Ishii K, Aizaki H, Kanegae Y, Matsuura Y, Saito I, Wakita T, Suzuki T. 2012. Trans-complemented hepatitis C virus particles as a versatile tool for study of virus assembly and infection. Virology 432:29–38. doi: 10.1016/j.virol.2012.05.033. [DOI] [PubMed] [Google Scholar]
- 42.Hugle T, Fehrmann F, Bieck E, Kohara M, Krausslich HG, Rice CM, Blum HE, Moradpour D. 2001. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology 284:70–81. doi: 10.1006/viro.2001.0873. [DOI] [PubMed] [Google Scholar]
- 43.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arita M. 2016. Mechanism of poliovirus resistance to host phosphatidylinositol-4 kinase III beta inhibitor. ACS Infect Dis 2:140–148. doi: 10.1021/acsinfecdis.5b00122. [DOI] [PubMed] [Google Scholar]