

Persistent human papillomavirus infections can cause a variety of significant cancers. The ability of HPV to persist depends on evasion of the host immune system. In this study, we show that the HPV16 E5 protein can suppress an important aspect of the host immune response. In addition, we find that the E5 protein is important for helping the virus avoid integration into the host genome, which is a frequent step along the pathway to cancer development.
KEYWORDS: E5, EGFR, STAT signaling, TGFBR2, innate immunity, interferon kappa, papillomavirus
ABSTRACT
Human papillomaviruses (HPVs) infect keratinocytes of stratified epithelia. Long-term persistence of infection is a critical risk factor for the development of HPV-induced malignancies. Through the actions of its oncogenes, HPV evades host immune responses to facilitate its productive life cycle. In this work, we discovered a previously unknown function of the HPV16 E5 oncoprotein in the suppression of interferon (IFN) responses. This suppression is focused on keratinocyte-specific IFN-κ and is mediated through E5-induced changes in growth factor signaling pathways, as identified through phosphoproteomics analysis. The loss of E5 in keratinocytes maintaining the complete HPV16 genome results in the derepression of IFNK transcription and subsequent JAK/STAT-dependent upregulation of several IFN-stimulated genes (ISGs) at both the mRNA and protein levels. We also established a link between the loss of E5 and the subsequent loss of genome maintenance and stability, resulting in increased genome integration.
IMPORTANCE Persistent human papillomavirus infections can cause a variety of significant cancers. The ability of HPV to persist depends on evasion of the host immune system. In this study, we show that the HPV16 E5 protein can suppress an important aspect of the host immune response. In addition, we find that the E5 protein is important for helping the virus avoid integration into the host genome, which is a frequent step along the pathway to cancer development.
INTRODUCTION
Human papillomavirus (HPV) infections typically cause benign lesions in cutaneous or mucosal squamous epithelia (1), but persistent infection can result in transition from a benign lesion to malignancy (2–4). The mechanisms involved in HPV persistence are largely unknown, but it is known that the virus must remain undetected by the host immune system in order to persist (1, 5, 6). This is accomplished in part by restricted expression of the major immunogenic proteins, the late capsid proteins, to the upper layers of the epithelium with low levels of immune surveillance (1, 7–9). Further, HPV does not cause viremia, cell lysis, or induction of inflammatory responses (10). In addition to life cycle organization, HPV actively suppresses both innate and adaptive immune responses. The HPV E5, E6, and E7 oncogenes mediate a wide array of anti-inflammatory and immune-evasive functions that render HPV infections and HPV-induced lesions profoundly immunosuppressive (11, 12). The importance of the immune response in HPV infection is indicated by the increased risk of HPV infection and invasive cervical cancer in HIV-infected women and other immunosuppressed patients (13–19).
HPV oncogenes contribute to persistent infection in many ways, most prominently by activating, inhibiting, or modifying host gene expression patterns, including innate immune response genes (20). The least understood HPV oncogene, E5, encodes an 83-amino acid transmembrane protein that has been shown to aid the late stages of the viral life cycle through unknown mechanisms (21, 22). E5 has been reported to regulate multiple growth factor signaling pathways. It is best known for enhancing epidermal growth factor receptor (EGFR) activation by increasing levels of EGFR at the cell surface (23–27). E5 increases c-Met levels and activation in an EGFR-dependent manner (28). In addition, overexpressed E5 has been shown to suppress transforming growth factor beta (TGF-β) signaling by reducing levels of TGF-β receptor II (TGFBR2) transcripts (29).
The interferon (IFN) response is a key component of host innate antiviral immunity. Most type I IFNs are induced through the binding of viral products to pattern recognition receptors (PRRs), leading to activation of interferon response factors (IRFs) and NF-κB to drive the synthesis of IFN molecules (30–32). After secretion, type I IFNs bind to the type I IFN receptor (IFNAR), which induces the phosphorylation and dimerization of signal transducer and activator of transcription 1 (STAT1) and STAT2, which then form a complex and translocate to the nucleus to induce transcription of IFN-stimulated genes (ISGs) (33). ISGs are biochemically diverse but generally function to limit viral replication or spread.
Keratinocytes, targets of HPV, are sometimes designated “immune sentinels” because of the wide array of innate immune sensors and cytokines that they can produce (34–37). Even under resting conditions, keratinocytes secrete an array of cytokines to sustain immune surveillance (38–41). One of these is IFN-κ, which is constitutively expressed in unstimulated keratinocytes (unlike IFN-α/β) (42). The constitutive expression of IFN-κ in resting keratinocytes indicates that IFN-κ expression is subject to fundamentally different regulation than other type I IFNs, such as IFN-α and IFN-β. IFN-κ has been shown to signal through the type I IFN receptor and to promote the upregulation of ISGs that can then promote antiviral immunity (42). To evade IFN responses, HPV oncoproteins repress both basal and agonist-induced transcription of IFNs and ISGs (43–51). IFN-κ, in particular, seems to be a target of HPV both in vivo (52) and in cell culture (11, 53, 54).
Cross talk between IFNs and growth factor signaling pathways has been previously reported. EGFR has been reported to inhibit IFN and ISG expression, including that of IFNK, through IRF1 inhibition (55–62). TGF-β has also been shown to cross talk with IFN pathway regulators (63–67). Our laboratory has recently reported that, in HPV16+ keratinocytes, TGF-β signaling can induce IFNK and ISG expression in keratinocytes harboring episomal HPV16 by reversing methylation of the IFNK promoter (65). This finding indicates that growth factor signaling is critical for regulating the levels of IFNK transcription in HPV16+ cells. As E5 is the viral oncogene most associated with the regulation of host growth factor signaling pathways, in this study, we explored the hypothesis that E5 may play a role in the regulation of IFN-κ in cells containing HPV16. We found that, although IFNK is poorly responsive to PRR agonists, levels of IFNK transcripts are suppressed in the presence of E5. We found that suppression of IFN-κ by E5 involves both the EGFR and TGFBR2 signaling pathways. Furthermore, HPV16 genomes lacking E5 tend to integrate in culture over time at a higher rate than the wild type, suggesting that suppression of IFN-κ by E5 may be required for long-term maintenance of viral genomes, revealing a novel function of E5 in the viral life cycle.
RESULTS
IFNK transcripts are upregulated in response to growth factor treatment.
Since human keratinocytes are the targets of HPV, we used primary human foreskin keratinocytes (HFKs) stably maintaining episomally replicating HPV16 genomes (HPV16+ cells) in our studies (65, 68). Unlike IFN-α/β, which require stimulation in order to be expressed at appreciable levels, IFNK is constitutively expressed in resting keratinocytes (50, 53, 65), so it is an important barrier for a pathogen that preferentially targets keratinocytes. Although IFNK transcripts do increase in response to classical Toll-like receptor (TLR) agonists, such as poly(I·C), the induction is weak compared to that of PRR-responsive IFNB (data not shown). We and others have previously reported that IFNK transcription is downregulated upon differentiation (50, 65). We also previously reported, in agreement with others, that IFNK transcript and protein levels are suppressed in HPV-infected cells compared to uninfected HFKs but that this downregulation could be reversed upon treatment with TGF-β (50, 53, 65). These findings show that IFNK is not simply constitutive but can be regulated in response to cellular conditions. We sought to understand the signaling pathways that regulate IFNK in uninfected or HPV16+ cells.
We have previously shown that the growth factor TGF-β could upregulate IFNK in HPV16+ cells (65). We and others have shown that HPV can increase the levels or function of the receptor tyrosine kinases EGFR and c-Met (the hepatocyte growth factor [HGF] receptor) (24, 25, 28). Therefore, we tested whether EGF and HGF could regulate IFNK transcripts in these cells. Values were normalized to untreated controls to determine the effect of each growth factor. Treatment of HPV16+ cells with TGF-β resulted in upregulation of IFNK in cells containing HPV16, as reported previously (Fig. 1) (65). Upregulation was also observed in response to HGF (Fig. 1). EGF could induce IFNK transcripts significantly, but less markedly than TGF-β1 (Fig. 1). These observations are consistent with the premise that IFNK gene expression is not dictated by pathogen recognition, as other type I IFNs are, but rather responds to growth factors present in the epithelium.
FIG 1.
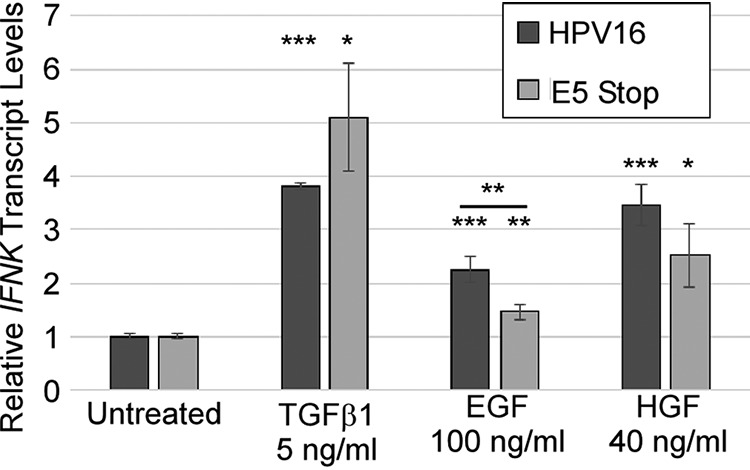
IFNK transcripts are readily induced by growth factors. HPV16+ cells and E5 Stop cells were treated with the indicated growth factors for 24 h. Levels of IFNK mRNAs were measured using RT-qPCR. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars indicate standard error of the mean.
IFNK transcripts are suppressed by E5.
E5 is the main HPV gene product known to regulate growth factor signaling (69). Because E5 can regulate all three of the pathways shown in Fig. 1 (TGF-β, EGF, and HGF) (23–29), we sought to determine whether E5 can affect IFNK transcription in response to these factors. In order to address the role of E5 in the HPV life cycle, we created keratinocyte-derived cell lines maintaining a mutant HPV16 genome with a stop codon in the E5 open reading frame (ORF) (E5 Stop cells) (28). These mutant viral genomes can immortalize HFKs and maintain themselves as episomes with normal levels of E6/E7 expression (28) but are genetically unable to express E5. We found that the response of IFNK mRNA to TGF-β and HGF was similar to that seen in wild-type HPV16+ cells but that the response of IFNK to EGF in these cells was significantly reduced (Fig. 1), consistent with lower levels of EGFR activity in the cells due to the absence of E5 (28).
To better understand the role of E5 in the regulation of IFNK, we compared the total levels of IFN transcripts in HFKs, HPV16+ cells, and E5 Stop cells. Because the cells were unstimulated, basal levels of IFNA and IFNB transcripts were very low (CT values of 30 to 33). IFNA levels were reduced by the presence of HPV16, and IFNB levels were unchanged; neither differed in the absence of E5 (Fig. 2a). Next, we confirmed previous data showing that HPV16 can inhibit IFNK transcription compared to HFKs. As previously reported, mRNA levels of IFNK were reduced by the presence of HPV16 (65), but this suppression was not as efficient in E5 Stop cells, indicating that E5 contributes to suppression of IFNK by HPV16 (Fig. 2b). Failure of E5 Stop cells to suppress IFN-κ was observed at the protein level, as well (Fig. 2c and d).
FIG 2.
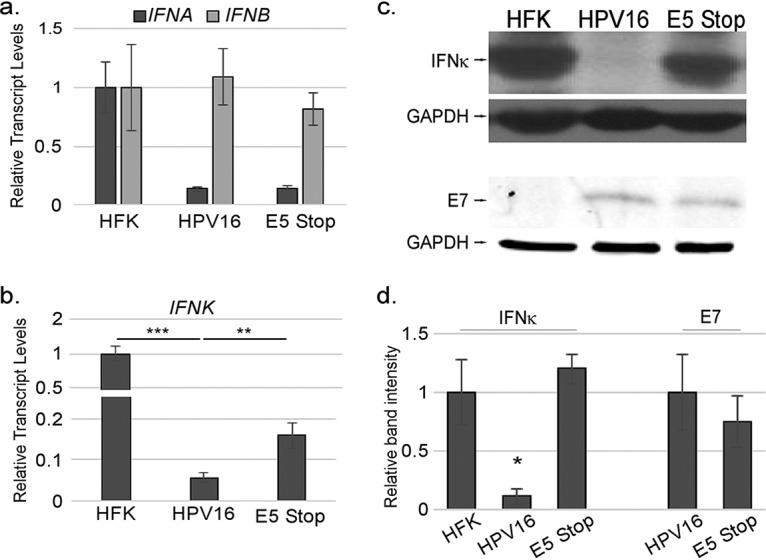
IFN-κ is suppressed in an E5-dependent manner. HPV16-containing and E5 Stop cells were isolated, and their RNA or protein was harvested. (a) Levels of IFNA and IFNB mRNAs were measured by RT-qPCR. (b and c) IFNK mRNA (b) and IFN-κ protein (c) levels were measured by RT-qPCR and immunoblotting, respectively. Levels of E7 were also measured by immunoblotting. (d) Quantification of immunoblots. n > 5 for all PCRs, and n > 3 for Western blots. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars indicate standard error of the mean.
These experiments indicate that E5 contributes to IFNK suppression in HPV16+ cells. To determine whether E5 alone is sufficient for IFNK suppression, we examined IFNK transcript levels in h-Tert-immortalized normal oral keratinocytes (NOKs) (70) that were stably transduced with retroviruses expressing HPV16 E5 alone. As E6 is the HPV oncogene previously shown to inhibit IFNK (50, 53), NOKs expressing E6 alone were included as a positive control. Comparing immortalized NOKs with primary HFKs revealed no significant differences in IFNK levels (reference 65 and data not shown), indicating that immortalization by itself does not affect IFNK expression. We observed a reduction in IFNK transcripts in cells expressing E6, consistent with previous reports that E6 can inhibit IFNK transcription (50, 53) (Fig. 3a). Importantly, IFNK transcript levels were reduced significantly in cells expressing E5, at an efficiency at least equal to that of E6 (Fig. 3a). To confirm these results, we transduced primary HFKs with retroviruses that express either E6/E7 alone or E6/E7 along with E5 expressed from the same transcript with an internal ribosome entry site (IRES). Following immortalization, IFNK mRNA levels in these cells were measured, and we observed that E5 suppresses IFNK suppression, even in the presence of E6 (Fig. 3b). These data provide evidence that E5 can suppress IFNK, a novel function of E5.
FIG 3.
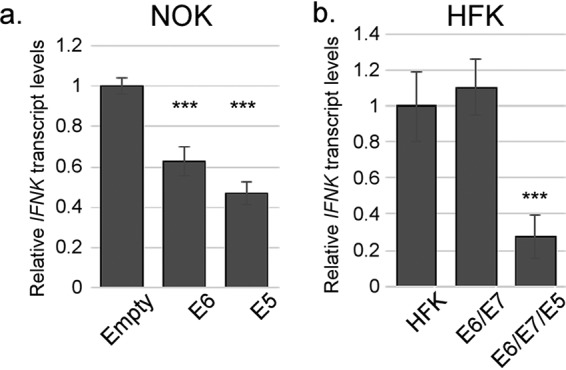
HPV16 E5 is sufficient to suppress IFN-κ expression. (a) NOKs that were previously hTert immortalized were transduced with retroviruses containing an empty vector (LXSN-Empty), a vector expressing the HPV16 E6 protein alone (LXSN-16E6), or a vector expressing the wild-type HPV16 E5 protein (LXSN-16E5). Following selection and outgrowth, IFNK levels were measured using RT-qPCR. (b) Primary HFKs were transduced with retroviruses expressing either E6/E7 alone or E6/E7 IRES E5. Following selection, the immortalized colonies were pooled, and IFNK levels were measured using RT-qPCR. ***, P < 0.001. The error bars indicate standard error of the mean.
ISG transcripts are suppressed by E5.
Because IFN-κ is constitutively expressed in keratinocytes, it can drive basal expression of ISGs (65). Since IFNK transcript and protein levels are higher in E5 Stop cells than inHPV16+ cells, we tested whether ISG levels were also increased. Levels of transcripts for several ISGs, including some that have been shown to inhibit the HPV life cycle (71–73), were suppressed as expected in HPV16+ cells but increased in E5 Stop cells (Fig. 4a). This increase was also observed at the protein level for two of these factors (Fig. 4b). Both isoforms of OAS2 (p69/p71) (74) were usually observed, but differential effects of E5 were not consistently seen. To determine whether E5 alone is sufficient to suppress ISG expression, ISG transcript levels from E5-expressing NOKs were examined by reverse transcription-quantitative PCR (RT-qPCR). Significant reductions were observed in the levels of these ISG mRNAs in E5-expressing NOKs versus NOKs containing the empty vector, similar to what was seen with E6 expression alone (Fig. 4c). Similarly, expression of E5, along with E6/E7, in HFKs resulted in additional inhibition of ISGs, showing that E5 can contribute to ISG suppression even in the presence of E6 and E7 (Fig. 4d). Since IFNK is the only IFN differentially expressed in these cells (Fig. 2), we concluded that increased IFNK transcripts in E5 Stop cells were responsible for the increased levels of ISGs. Further, we concluded that IFN-κ-driven ISG expression was inhibited by E5.
FIG 4.

Suppression of ISGs in HPV16+ cells is E5 dependent. (a) Levels of the indicated ISG mRNAs in HFK, HPV16+, and E5 Stop cells were measured using RT-qPCR. (b) Immunoblots showing the levels of OAS2 protein (top) and IFIT1 (bottom) in HFK, HPV16, and E5 Stop cells. (c) Levels of the indicated ISG mRNAs in NOKs transduced with empty or E6- or E5-expressing retroviruses were measured by RT-qPCR. (d) Levels of the indicated ISG mRNAs in HFKs immortalized with retroviruses expressing E6/E7 alone or E6/E7 IRES E5 were measured using RT-qPCR. The statistics in panels c and d are relative to HFK. **, P < 0.01; ***, P < 0.001. The error bars indicate standard error of the mean.
Increased ISG levels suggested that the IFNAR/JAK/STAT pathway could be chronically activated in E5 Stop cells. Indeed, levels of both phosphorylated and total STAT1 were higher in E5 Stop cells than in wild-type HPV16+ cells, consistent both with increased JAK signaling and with STAT1 itself being an ISG (Fig. 5a and b). The increase in STAT1 protein levels is also seen at the mRNA level, with higher STAT1 mRNA levels expressed in E5 Stop cells (Fig. 5c). Additionally, levels of STAT1 mRNA were reduced in NOKs expressing E5 (Fig. 5d). These results indicate that E5 can suppress STAT1 activation and expression. Activation of STAT1 suggests that the high levels of ISGs in E5 Stop cells are due to IFN-κ activating the classical JAK/STAT1 pathway (42). If so, treatment with the JAK1/2 inhibitor ruxolitinib should reduce ISG levels in these cells. We treated HFKs, HPV16+ cells, and E5 Stop cells with ruxolitinib for 24 h. Values were normalized to those of dimethyl sulfoxide (DMSO) treatment for each cell line to reveal the effect of drug treatment. Ruxolitinib treatment resulted in a significant reduction of MX1, IFIT1-IFI56, OAS2, and IFI16 transcript levels in all three cell types (Fig. 6a to d), suggesting that ISG levels are governed by classical JAK/STAT regulation in these cells regardless of the presence of E5. The efficacy of ruxolitinib treatment was confirmed by its ability to inhibit STAT1 phosphorylation (Y701) in response to IFN-β treatment (Fig. 6e). The data also show that chronic ISG expression in HFKs is governed by the JAK/STAT1 pathway, as would be expected downstream of IFN-κ signaling.
FIG 5.
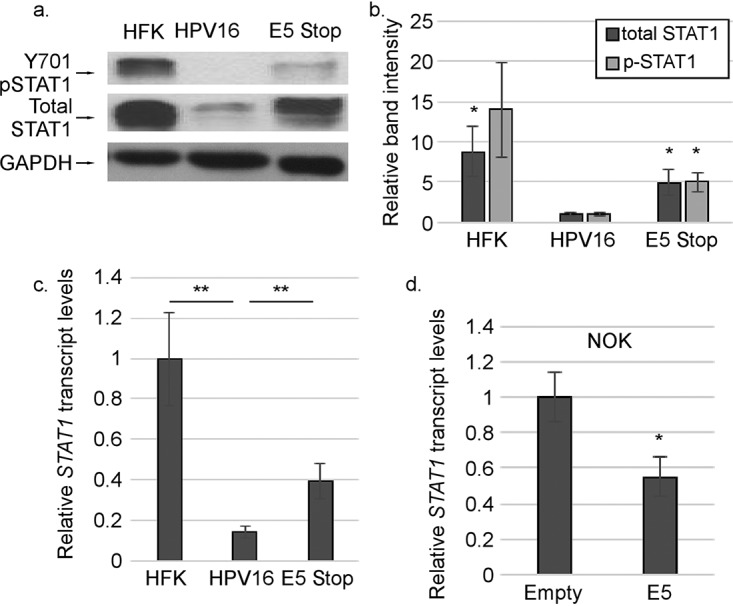
STAT1 levels and signaling are suppressed by E5. (a and b) Levels of IFN-κ in HPV16+ and E5 Stop cells were measured by immunoblotting (a) and quantified (b). (c and d) IFNK mRNA levels in HFK, HPV16+, and E5 Stop cells (c) or NOKs transduced with empty vector or E5 (d) were measured using RT-qPCR. *, P < 0.05; **, P < 0.01. The error bars indicate standard error of the mean.
FIG 6.
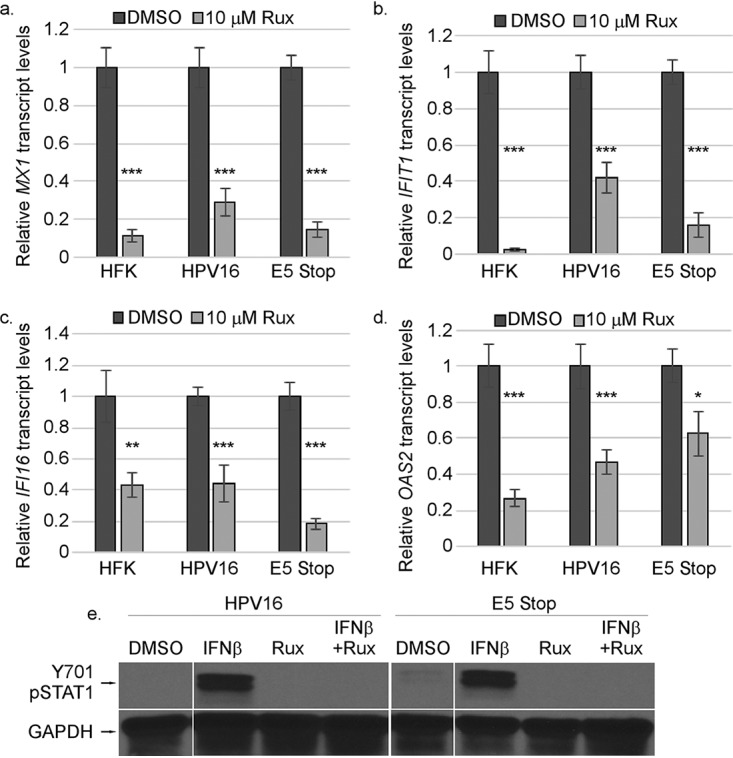
ISG mRNA levels are regulated by JAK/STAT signaling. (a to d) HFK, HPV16+, and E5 Stop cells were treated with DMSO (control) or 10 μM ruxolitinib (Rux) for 24 h, followed by RT-qPCR for the MX1 (a), IFIT1 (b), IFI6 (c), or OAS2 (d) transcripts. (e) The efficacy of Rux treatment was confirmed by its ability to inhibit STAT1 (pY701) in HPV16 and E5 Stop cells treated with IFN-β. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars indicate standard error of the mean.
Phosphoproteomics indicates that E5 regulates EGFR, TGFBR2, and STAT signaling.
Having shown that IFNK gene expression is regulated by growth factors and that E5 can inhibit IFNK gene expression and the resulting IFN response, we sought to understand how alteration in growth factor signaling by E5 can affect IFNK levels. In order to build a more complete picture of the various signaling pathways and interactions occurring in cells containing HPV16 with or without E5 expression, we used an antibody proteomics array (Full Moon Biosystems). The array measures the phosphorylated and total levels of various proteins involved in a variety of pathways, including growth factor signaling, cell cycle regulation, and innate immunity. Protein harvested from uninfected HFKs, episomal HPV16+ cells, and episomal E5 Stop cells was processed according to the Full Moon Biosystems protocol and analyzed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (75, 76). Comparisons between signal intensities allowed us to determine the fold differences of various phosphorylated proteins and total proteins between the samples and the signaling pathways regulated by HPV16 in an E5-dependent manner in the context of the complete viral genome and physiological host cell type.
HPV16+ cells exhibit increased phosphorylation and total protein levels of 142 proteins that are upregulated at least 2-fold versus those in HFKs. There were only 8 proteins that were decreased in HPV16+ cells versus HFKs. Some of the factors of interest are listed in Table 1, (the full, noncurated table is available in File S1 in the supplemental material), and Table 2 lists the pathways impacted in HPV16+ cells compared to HFKs according to DAVID analysis. Factors associated with a variety of growth factor-related signaling pathways were affected by the presence of HPV16, including the HGF-Met receptor pathway and Ras signaling (28, 77–81). Phosphorylated species of MEK1 and other mitogen-activated protein kinases (MAPKs), such as Erk, as well as the increased activation of EGFR signaling, have previously been shown to be targets of the HPV16 E5 protein (23, 28, 77, 81, 82). These results are consistent with and add to studies of the HPV18 E5 protein in the context of the complete viral genome (82). Both inhibitors of κB alpha and beta (IκB-alpha and IκB-beta) were increased by HPV16. We also observed the decrease of the Rel and p100/p52 NF-κB subunits by HPV16, which is also consistent with suppression of NF-κB activity by HPV oncogenes reported in some previous studies (83–92). STAT1 Y701 phosphorylation was also reduced, further confirming the observations shown in Fig. 5.
TABLE 1.
Curated list of factors altered by the presence of HPV16 cells versus HFKs
| Protein | Ratio | P value |
|---|---|---|
| P73 (phospho-Tyr99) | 8.29 | 0.0004 |
| Tau (Ab-231) | 8.12 | 0.0442 |
| PAK2 (Ab-192) | 6.72 | 0.0105 |
| MEK1 (Ab-217) | 5.41 | 0.0488 |
| Rel (phospho-Ser503) | 4.95 | 0.0192 |
| Fos (Ab-232) | 4.43 | 0.0132 |
| Chk1 (phospho-Ser301) | 4.14 | 0.0138 |
| MEK1 (phospho-Thr291) | 4.05 | 0.0053 |
| MKK7/MAP2K7 (Ab-271) | 4.03 | 0.0007 |
| AKT1 (phospho-Ser473) | 3.90 | 0.0238 |
| BCL-6 (Ab-333) | 3.84 | 0.0064 |
| IκB-alpha (phospho-Tyr305) | 3.83 | 0.0476 |
| MEK1 (phospho-Ser298) | 3.75 | 0.0033 |
| MDM4 (phospho-Ser367) | 3.71 | 0.0057 |
| Integrin beta-3 (Ab-773) | 3.64 | 0.0021 |
| p44/42 MAPK (Ab-204) | 3.58 | 0.0398 |
| Mnk1 (Ab-385) | 3.47 | 0.0049 |
| MEK2 (phospho-Thr394) | 3.46 | 0.0273 |
| GSK3 beta (phospho-Ser9) | 3.46 | 0.0455 |
| Chk1 (phospho-Ser286) | 3.36 | 0.0373 |
| NF-κB–p65 (Ab-311) | 3.31 | 0.0319 |
| AKT1 (phospho-Thr308) | 3.27 | 0.0114 |
| FGFR1 (phospho-Tyr766) | 3.24 | 0.0151 |
| STAT1 (phospho-Ser727) | 3.23 | 0.0045 |
| IκB-beta (phospho-Ser23) | 3.22 | 0.0450 |
| MAP3K7/TAK1 (Ab-439) | 3.12 | 0.0354 |
| JunD (Ab-255) | 3.11 | 0.0142 |
| HER2 (phospho-Thr686) | 3.10 | 0.0256 |
| Fyn (phospho-Tyr530) | 3.09 | 0.0214 |
| IGF2R (Ab-2409) | 3.05 | 0.0215 |
| CREB (Ab-129) | 3.00 | 0.0367 |
| MKK4/SEK1 (phospho-Thr261) | 2.95 | 0.0176 |
| AKT1 (phospho-Tyr474) | 2.94 | 0.0479 |
| MKK4/SEK1 (phospho-Ser257) | 2.72 | 0.0027 |
| p44/42 MAPK (Ab-202) | 2.72 | 0.0037 |
| IGF1R (phospho-Tyr1161) | 2.60 | 0.0283 |
| MEK1 (phospho-Ser221) | 2.54 | 0.0496 |
| MEF2A (phospho-Ser408) | 2.54 | 0.0190 |
| MKK3/MAP2K3 (Ab-189) | 2.53 | 0.0240 |
| Chk1 (Ab-345) | 2.50 | 0.0468 |
| c-Jun (Ab-63) | 2.47 | 0.0284 |
| Elk1 (Ab-383) | 2.43 | 0.0202 |
| Dok-2 (Ab-299) | 2.42 | 0.0036 |
| EGFR (Ab-1197) | 2.30 | 0.0412 |
| c-Jun (Ab-170) | 2.17 | 0.0416 |
| c-Jun (phospho-Thr91) | 2.15 | 0.0044 |
| 4E-BP1 (phospho-Ser65) | 2.12 | 0.0034 |
| FAK (Ab-861) | 2.12 | 0.0197 |
| EGFR (phospho-Tyr1016) | 2.02 | 0.0280 |
| AKT1 (phospho-Ser124) | 2.02 | 0.0308 |
| Src (phospho-Ser75) | 2.01 | 0.0084 |
| Rel (Ab-503) | −1.99 | 0.0109 |
| c-Jun (phospho-Thr93) | −2.04 | 0.0252 |
| STAT1 (phospho-Tyr701) | −2.22 | 0.0467 |
| PDGFR beta (Ab-751) | −2.25 | 0.0242 |
| VAV1 (Ab-160) | −2.30 | 0.0112 |
| NF-κB–p100/p52 (phospho-Ser872) | −3.49 | 0.0394 |
TABLE 2.
Pathways affected in HPV16+ cells versus HFKs
| Regulation | Pathwaya | No. of genes from array | % of total array genes | P value | FDRb |
|---|---|---|---|---|---|
| Up | ErbB signaling | 24 | 20.87 | 3.82E−23 | 4.68E−20 |
| Up | Pathways in cancer | 36 | 31.30 | 1.41E−18 | 1.73E−15 |
| Up | Focal adhesion | 27 | 23.48 | 1.77E−17 | 2.17E−14 |
| Up | Proteoglycans in cancer | 23 | 20.00 | 1.47E−13 | 1.81E−10 |
| Up | Ras signaling | 24 | 20.87 | 2.04E−13 | 2.50E−10 |
| Up | Rap1 signaling | 23 | 20.00 | 4.06E−13 | 4.98E−10 |
| Up | Viral carcinogenesis | 18 | 15.65 | 9.66E−09 | 1.18E−05 |
| Up | Apoptosis | 11 | 9.57 | 2.47E−08 | 3.03E−05 |
| Up | Regulation of actin cytoskeleton | 17 | 14.78 | 9.12E−08 | 1.12E−04 |
| Up | TLR signaling | 17 | 14.78 | 3.22E−12 | 3.95E−09 |
| Up | TNF signaling | 17 | 14.78 | 3.74E−12 | 4.59E−09 |
| Up | cAMP signaling pathway | 20 | 17.39 | 9.36E−11 | 1.15E−07 |
| Up | Keratinocyte differentiation | 13 | 11.30 | 2.53E−06 | 3.18E−03 |
| Up | Signaling of HGF receptor | 12 | 10.43 | 7.64E−07 | 9.62E−04 |
| Up | HIF-1 signaling pathway | 19 | 16.52 | 2.33E−15 | 2.85E−12 |
| Down | Proto-oncogene | 5 | 62.5 | 5.74E−07 | 0.00056479 |
| Down | Response to cytokine | 4 | 50 | 9.72E−07 | 0.00123059 |
TNF, tumor necrosis factor; cAMP, cyclic AMP.
FDR, false-discovery rate.
We compared E5 Stop cells to HPV16+ cells, and 94 total proteins were differentially regulated, with 19 upregulated and 75 downregulated (see File S1). STAT1 Y701 phosphorylation was upregulated, consistent with our observations shown in Fig. 5 (Table 3). We also observed the increased activation of other STATs, including STAT5A and STAT6, which are associated with growth factor signaling and Th2 polarization, respectively (93, 94). Total levels of the Rel NF-κB subunit were increased (Table 3), indicating the potential for NF-κB activity to be increased in E5 Stop cells. The predominant gene ontology pathways associated with downregulated factors were growth factor signaling pathways, including ErbB, Ras, and phosphatidylinositol 3-kinase [PI3K]), MAPK, and vascular endothelial growth factor receptor (VEGFR) (Table 4). Reduced growth factor signaling in E5 Stop cells would be expected based on experiments with overexpressed E5, but these experiments confirm that E5 regulates these pathways in keratinocytes in the context of the complete viral genome.
TABLE 3.
Curated list of factors altered by the presence of E5 Stop versus HPV16 cells
| Protein | Ratio | P value |
|---|---|---|
| p38 MAPK (Ab-182) | 4.44 | 0.0245 |
| Src (Ab-418) | 3.73 | 0.0140 |
| STAT5A (Ab-780) | 3.47 | 0.0119 |
| p53 (Ab-18) | 3.42 | 0.0254 |
| CREB (phospho-Thr100) | 3.34 | 0.0161 |
| STAT6 (Ab-645) | 3.30 | 0.0478 |
| PECAM-1 (Ab-713) | 3.24 | 0.0454 |
| P90RSK (Ab-359/363) | 2.84 | 0.0258 |
| VEGFR2 (Ab-1214) | 2.81 | 0.0236 |
| STAT1 (phospho-Tyr701) | 2.75 | 0.0001 |
| Pyk2 (Ab-580) | 2.55 | 0.0406 |
| Smad3 (phospho-Ser213) | 2.52 | 0.0184 |
| PTEN (Ab-370) | 2.30 | 0.0255 |
| Rel (Ab-503) | 2.24 | 0.0046 |
| PI3-kinase p85-subunit alpha/gamma (Ab-467/199) | −2.03 | 0.0227 |
| Elk1 (Ab-389) | −2.03 | 0.0467 |
| PAK2 (Ab-192) | −2.05 | 0.0355 |
| Integrin beta-4 (phospho-Tyr1510) | −2.05 | 0.0060 |
| JunB (phospho-Ser79) | −2.06 | 0.0270 |
| AKT1 (phospho-Ser124) | −2.08 | 0.0291 |
| Tau (phospho-Thr212) | −2.09 | 0.0054 |
| FLT3 (phospho-Tyr599) | −2.12 | 0.0277 |
| AKT1 (phospho-Thr308) | −2.14 | 0.0216 |
| TYK2 (Ab-1054) | −2.20 | 0.0118 |
| MEK1 (phospho-Ser298) | −2.21 | 0.0146 |
| FGFR1 (phospho-Tyr766) | −2.32 | 0.0288 |
| IGF1R (phospho-Tyr1161) | −2.37 | 0.0289 |
| EEF2 (phospho-Thr56) | −2.42 | 0.0180 |
| MEF2A (phospho-Ser408) | −2.45 | 0.0153 |
| MKK7/MAP2K7 (Ab-271) | −2.47 | 0.0011 |
| Elk1 (Ab-383) | −2.49 | 0.0168 |
| GSK3 alpha (Ab-21) | −2.53 | 0.0348 |
| p44/42 MAPK (Ab-202) | −2.55 | 0.0046 |
| MDM4 (phospho-Ser367) | −2.60 | 0.0081 |
| c-Jun (Ab-170) | −2.62 | 0.0232 |
| c-Jun (Ab-63) | −2.66 | 0.0243 |
| Cyclin E1 (phospho-Thr395) | −2.68 | 0.0030 |
| AKT1 (phospho-Ser473) | −2.68 | 0.0299 |
| MEK1 (Ab-221) | −2.80 | 0.0203 |
| JunD (Ab-255) | −2.81 | 0.0103 |
| FAK (Ab-397) | −2.83 | 0.0137 |
| eIF4G (phospho-Ser1108) | −2.85 | 0.0020 |
| MEK1 (phospho-Thr291) | −2.87 | 0.0105 |
| MEK1 (phospho-Ser221) | −2.89 | 0.0387 |
| MEK2 (phospho-Thr394) | −2.94 | 0.0289 |
| GSK3 alpha (phospho-Ser21) | −3.30 | 0.0223 |
| VEGFR2 (phospho-Tyr951) | −3.69 | 0.0011 |
| GSK3 beta (phospho-Ser9) | −3.89 | 0.0414 |
| Fos (Ab-232) | −3.91 | 0.0160 |
| P73 (phospho-Tyr99) | −4.94 | 0.0001 |
| Tau (Ab-231) | −6.42 | 0.0481 |
TABLE 4.
Pathways affected in E5 Stop versus HPV16+ cells
| Regulation | E5 Stop vs HPV16 pathway | No. of genes from array | % of total array genes | P value | FDRa |
|---|---|---|---|---|---|
| Down | ErbB signaling | 16 | 25.0 | 2.40E−16 | 2.66E−13 |
| Down | VEGF signaling | 11 | 17.19 | 6.25E−11 | 7.55E−08 |
| Down | PI3K-Akt signaling | 19 | 29.69 | 2.56E−10 | 3.09E−07 |
| Down | MAPK signaling | 16 | 25 | 1.89E−09 | 2.28E−06 |
| Down | Ras signaling | 15 | 23.44 | 4.09E−09 | 4.94E−06 |
FDR, false-discovery rate.
To determine if the differential regulation of any factors was E5 dependent, we compared the analysis of the HPV16+ cells versus HFKs to the analysis of the E5 Stop cells versus HPV16+ cells and noted factors that were regulated in opposite directions. Table 5 lists the factors either up- or downregulated by HPV16 in an E5-dependent manner. The most notable factors regulated in an E5-dependent manner included STAT1 Y701 phosphorylation, Rel, and activation of various MEKs and MAP kinases. Phosphorylation of p73, a protein that can regulate host cell differentiation via unknown mechanisms (95, 96), was strongly upregulated in an E5-dependent manner.
TABLE 5.
Factors regulated in an E5-dependent manner
| Protein | HPV16 cells vs HFKs |
E5 Stop vs HPV16 cells |
||
|---|---|---|---|---|
| Ratio | P value | Ratio | P value | |
| P73 (phospho-Tyr99) | 8.29 | 0.00037 | −4.94 | 0.0001 |
| Tyrosine hydroxylase (phospho-Ser8) | 4.38 | 0.01271 | −3.29 | 0.01558 |
| MEK1 (phospho-Thr291) | 4.05 | 0.00526 | −2.87 | 0.01047 |
| MKK7/MAP2K7 (Ab-271) | 4.03 | 0.00072 | −2.47 | 0.00109 |
| STAM2 (Ab-192) | 3.99 | 0.02290 | −2.41 | 0.04608 |
| VEGFR2 (phospho-Tyr951) | 3.83 | 0.00289 | −3.69 | 0.00113 |
| MEK1 (phospho-Ser298) | 3.75 | 0.00334 | −2.21 | 0.01458 |
| MDM4 (phospho-Ser367) | 3.71 | 0.00571 | −2.60 | 0.00813 |
| MEK2 (phospho-Thr394) | 3.46 | 0.02731 | −2.94 | 0.02893 |
| FGFR1 (phospho-Tyr766) | 3.24 | 0.01506 | −2.32 | 0.02878 |
| Integrin beta-4 (phospho-Tyr1510) | 2.56 | 0.00319 | −2.05 | 0.00597 |
| c-Jun (Ab-63) | 2.47 | 0.02835 | −2.66 | 0.02428 |
| Elk1 (Ab-383) | 2.43 | 0.02015 | −2.49 | 0.01678 |
| c-Jun (Ab-170) | 2.17 | 0.04156 | −2.62 | 0.02321 |
| Rel (Ab-503) | −2.00 | 0.01088 | 2.24 | 0.00465 |
| STAT1 (phospho-Tyr701) | −2.22 | 0.04666 | 2.75 | 0.00014 |
JAK/STAT signaling is not responsible for increased IFNK in E5 Stop cells.
The phosphoproteomics array, coupled with our previous data, showed that the EGFR/MAPK and IFNAR/STAT pathways are deregulated in E5 Stop cells compared to wild-type HPV16+ cells. We next sought to determine the impacts of these pathways on IFNK expression. We showed previously that ISGs are dependent on IFNAR/JAK/STAT signaling (Fig. 6), but to determine whether IFNK is similarly dependent, we treated HFK, HPV16+, and E5 Stop cells with ruxolitinib for 24 h. We also treated them with IFN-β to confirm that the inhibitor was working and to determine whether IFNK could respond to other type I IFNs. The RT-qPCR results were normalized to the DMSO treatment for each cell line to reveal the effect of drug treatment. We observed that all three cell types could increase IFNK transcripts a modest 2- to 2.5-fold in response to IFN-β (Fig. 7a). Thus, the presence of the virus does not affect the essential functions of the JAK pathway with respect to IFNK levels. Ruxolitinib alone did not suppress or only weakly suppressed IFNK mRNA. There was modest but consistent downregulation in HPV16+ cells treated with ruxolitinib and IFN-β together. We do not understand the basis for this effect. Knocking down JAK1 resulted in only a modest reduction of IFNK transcripts in HPV16 cells, but not E5 Stop cells (Fig. 7b and c). Together, these data indicate that signaling through the JAK pathway is not a major player in controlling the levels of IFNK.
FIG 7.

Basal IFNK is modestly affected by JAK/STAT signaling. (a) HFK, HPV16+, and E5 Stop cells were treated with IFN-β with or without 10 μM ruxolitinib (Rux) for 24 h. IFNK transcript levels were measured using RT-qPCR. (b) The efficacy of JAK1-targeting siRNA (siJAK1) versus nontargeting control (NTC) was tested in HPV16-containing cells by Western blotting. (c) The levels of IFN-κ transcripts in cells transfected with siJAK1 were measured by RT-qPCR. NS, not significant; *, P < 0.05; ***, P < 0.001. The error bars indicate standard error of the mean.
Impact of EGFR signaling on IFNK.
We found previously that IFNK was upregulated in response to EGF in HPV16+ and E5 Stop cells (Fig. 1). As MAPK signaling pathways were reduced in E5 Stop cells compared to wild-type HPV16+ cells in our array experiments, we used inhibitors to target EGFR to determine the effect on IFNK transcripts. Cells were treated with AG1478 (a specific EGFR inhibitor) for 24 h. The RT-qPCR results were normalized to the DMSO treatment for each cell line to reveal the effect of drug treatment. In HPV16+ cells treated with AG1478, IFNK transcript levels were increased, consistent with a suppressive effect of EGFR on IFNK that was previously reported by others (Fig. 8a) (55, 97). Upregulation was also seen in HFKs treated with AG1478. Interestingly, in E5 Stop cells, no such increase in IFNK transcripts was observed (Fig. 8a), indicating that EGFR kinase activity does not suppress IFNK in E5 Stop cells. Consistent with previous findings (28), Western blotting revealed that total EGFR levels in the absence of E5 were at or below the levels seen in HFKs, in contrast to the high levels observed in HPV16+ cells (Fig. 8b). Knocking down EGFR with small interfering RNAs (siRNAs) resulted in increased IFNK transcripts in HPV16 cells, but not in E5 Stop cells (Fig. 8c and d). These data indicate that EGFR plays an important role in the ability of E5 to suppress IFNK in HPV-containing cells. It may also play a role in suppressing IFNK in uninfected cells. Treatment of HPV16+ and E5 Stop cells with the Met inhibitor SU11274 resulted in no appreciable change in IFNK transcript levels (data not shown).
FIG 8.

IFNK mRNA levels are suppressed by EGFR kinase activity. (a) HFK, HPV16+, and E5 Stop cells were treated for 24 h with 1 μM AG1478. IFNK transcript levels were then measured using RT-qPCR. (b) Levels of EGFR in the indicated cell types were measured by Western blotting (left) and quantified (right). (c) The efficacy of EGFR-targeting siRNA (siEGFR) versus NTC was tested in HPV16-containing cells by Western blotting. (d) The levels of IFN-κ transcripts in cells transfected with siEGFR were measured by RT-qPCR. NS, not significant; *, P < 0.05; **, P < 0.01. The error bars indicate standard error of the mean.
IFN signaling in E5 Stop cells is increased in a TGF-β-dependent manner.
We have previously observed that TGF-β1 treatment of HPV16 cells promotes the upregulation of IFNK transcripts (65) (Fig. 1a). We also observed alterations in both canonical and noncanonical TGF-β pathway signaling intermediates in the phosphoproteomics array, consistent with prior observations that HPV16 E5 can suppress the TGF-β signaling pathway by downregulating the TGFBR2 receptor (29). These observations suggested that suppression of the TGF-β pathway may be important for the ability of E5 to suppress IFNK. To determine if TGF-β signaling contributes to IFNK transcript levels, we treated cells with the drug SB431542, a TGFBR2-specific inhibitor. We observed significant reductions in IFNK transcript levels in HFKs, E5 Stop cells, and HPV16+ cells (Fig. 9a). A similar reduction in IFNK transcripts was seen when we knocked down TGFBR2 with siRNA (Fig. 9b and c). These findings indicate that TGF-β signaling is critical for IFNK expression in keratinocytes regardless of the presence of the virus. Analysis of E5 Stop cells showed increased phospho-SMAD3 levels (Fig. 10) and TGFBR2 protein levels (Fig. 10b) versus wild-type HPV16+ cells. These increases were consistent and significant (Fig. 10c), showing that the TGF-β signaling pathway is increased in E5 Stop cells versus wild-type HPV16+ cells. Analysis of HFKs and HPV16 and E5 Stop cells for TGFB1, TGFB2, and TGFBR2 transcript levels showed significant suppression of TGFB1 and an increase of TGFB2 in HPV16+ cells versus HFKs, as previously reported (65) (Fig. 10d). Significantly, both the suppression of TGFB1 and activation of TGFB2 by HPV were E5 dependent. Patterns of TGFBR2 mRNA levels reflected those of TGFB1 and were also E5 dependent, as previously reported (29). NOKs expressing HPV16 E5 also exhibited significantly reduced TGFBR2 transcript levels (Fig. 10e), showing that E5 is sufficient to reduce expression of the gene in the absence of the other viral oncogenes. These data show that TGF-β-induced signaling is suppressed in an E5-dependent manner in cells containing the complete HPV16 genome.
FIG 9.
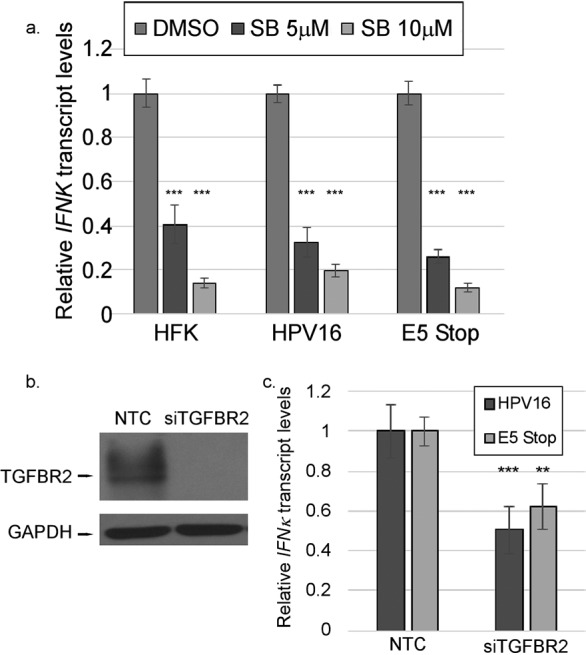
TGF-β signaling is crucial for IFN-κ in keratinocytes. (a) HFK, HPV16+, and E5 Stop cells were treated with 5 or 10 μM SB31542 (SB) for 24 h. IFNK mRNA levels were measured using RT-qPCR. (b) The efficacy of TGFBR2-targeting siRNA (siTGFBR2) versus NTC was tested in HPV16-containing cells by Western blotting. (c) The levels of IFN-κ transcripts in cells transfected with siTGFBR2 were measured by RT-qPCR. **, P < 0.01; ***, P < 0.001. The error bars indicate standard error of the mean.
FIG 10.
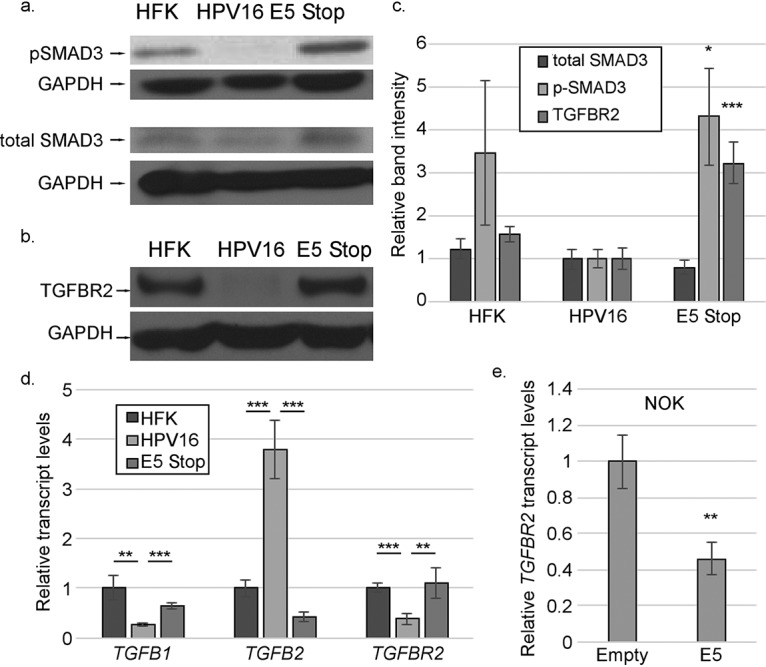
TGF-β signaling is increased in E5 Stop cells. (a and b) Western blots showing the levels of SMAD3/phosphorylated SMAD3 (a) and TGFBR2 (b). (c) Quantification of results of immunoblotting experiments. (d) mRNAs for TGFB1, TGFB2, and TGFBR2 in HFK, HPV16+, and E5 Stop cells were measured by RT-qPCR. (e) Levels of TGFBR2 mRNA in hTert-immortalized NOKs stably transduced with an empty vector or a vector expressing 16E5 were measured using RT-qPCR. The blots shown are representative. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars indicate standard error of the mean.
E5 does not regulate IFNK promoter methylation.
Since HPV16 E6 represses IFNK transcription by inducing promoter methylation and TGF-β increases IFNK transcripts, at least in part, by reversing that methylation (53, 65) (Fig. 11a), we examined whether higher levels of IFNK transcripts in E5 Stop cells were due to reduced levels of promoter methylation. Using bisulfite-sequencing analysis on DNA derived from wild-type HPV16+ cells or E5 Stop cells, we confirmed that CpGs in the IFNK promoter are mostly methylated in wild-type HPV16+ cells (53, 65). We did not observe a significant difference in IFNK promoter methylation in E5 Stop cells compared to wild-type HPV16+ cells, as measured by bisulfite sequencing (Fig. 11b). This finding suggests that higher levels of IFNK expression in E5 Stop cells are not due to demethylation of the promoter. It also suggests that processes other than methylation can regulate IFNK levels in HPV16+ cells.
FIG 11.
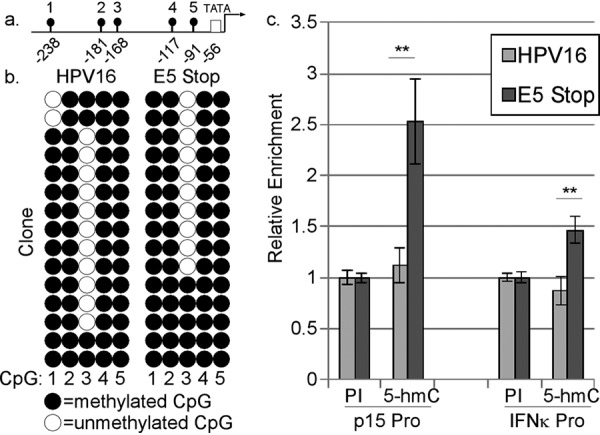
E5 does not substantially affect IFNK promoter methylation. (a) Diagram of the IFNK promoter showing the locations of the TATA box and five CpGs. (b) Bisulfite-sequencing analysis of the IFNK promoter in wild-type HPV16+ cells and E5 Stop cells. Following bisulfite modification of total DNA, the IFNK promoter was isolated by PCR and inserted into a cloning vector, and individual clones were sequenced. Each row corresponds to a separate clone, and each column corresponds to one of the five CpG sites in the promoter. Five clones each from 3 donor backgrounds were analyzed in the experiment. (c) MeDIP analysis of the CDKN2B (p15) promoter (Pro) and IFNK promoter DNA from HPV16+ cells and E5 Stop cells. DNA was subjected to immunoprecipitation with 0.25 μg/μl 5-methylcytosine-specific (5-hm-C) or preimmune (PI) serum. The immunoprecipitants were subjected to qPCR using primers specific for the p15 or IFNK promoter. n = 9. **, P < 0.01. The error bars indicate standard error of the mean.
Our previous work showed that the IFNK promoter is actively demethylated through a TDG-dependent mechanism (65). Hydroxymethylcytosine (5-hmC) is an intermediate in the TET-TDG-mediated active demethylation pathway (98, 99). Bisulfite sequencing is unable to distinguish between methylated and hydroxymethylated cytosines (100), but unlike methylated C, hydroxymethylation can still permit nucleosomes to be in an open and transcriptionally active state due to the weakening of interactions between DNA and histones (101). Therefore, it is possible that the IFNK promoter is hydroxymethylated in E5 Stop cells. We used methylated DNA immunoprecipitation (MeDIP) to test this idea. Total DNA from wild-type HPV16+ cells or E5 Stop cells was immunoprecipitated using an antibody specific for 5-hmC. Following purification, the DNA was subjected to qPCR analysis to determine enrichment compared to preimmune serum. Thillainadesan et al. showed, using MeDIP, that TGF-β signaling induces 5-hmC conversion at the CDKN2A (p15) promoter during active demethylation in keratinocytes (102), so the p15 promoter was used as a positive control for the presence of hydroxymethylation. As shown in Fig. 11c, the p15 promoter showed enrichment for 5-hmC in E5 Stop cells compared to wild-type HPV16+ cells, as expected. A modest but statistically significant increase in hydroxymethylation at the IFNK promoter was also observed in E5 Stop cells compared to wild-type HPV16+ cells (Fig. 11c). Whether this modest increase is sufficient to explain the increased IFNK transcripts found in E5 Stop cells remains to be determined.
E5 Stop cells have higher levels of viral genome integration over time.
Type I IFN signaling can promote episomal loss and integration of the HPV16 genome (103, 104). Because IFN signaling is chronically activated in E5 Stop cells, we predicted that long-term maintenance of viral episomes would be disrupted. Southern blots are routinely and periodically performed on HPV-containing cell lines in the laboratory to ensure that cells used in experiments contain episomal rather than integrated HPV16 genomes (Fig. 12a). Following a review and tabulation of these Southern blots, we found that the frequency of integration in E5 Stop samples was significantly higher than in wild-type HPV16 cells (Table 6). Furthermore, when we considered only the samples that contained episomal viral DNA, quantification of band intensities showed that E5 Stop cells had a significantly lower copy number than wild-type HPV16 cells (Fig. 12b). A low copy number accompanied by a high rate of integration is consistent with the known response of HPV to type I IFN (103–108).
FIG 12.

(a) Representative Southern blot indicating the supercoiled (episomal) and nicked circular genome bands. (b) Quantification of episomal viral DNA in wild-type HPV16-containing cells or E5 Stop cells, with HPV16 set to 1. (c) Levels of IFNK mRNA were measured in HFKs and both integrated (Int) and episomal (Epi) HPV16+ and E5 Stop cells (Fig. 1). (d) Model: IFN-κ drives a host response that leads to integration of the viral episome. IFN-κ is positively regulated by the TGF-β pathway and negatively regulated by the EGFR pathway. E5 helps promote long-term episomal maintenance by inhibiting TGF-β signaling and enhancing EGFR signaling, thus reducing levels of IFN-κ. NS, not significant; **, P < 0.01; ***, P < 0.001. The error bars indicate standard error of the mean.
TABLE 6.
HPV genome status in HPV16+ and E5 Stop cells
| Genotype | No. of samples |
% integrated (P = 3.4 × 10−6) | ||
|---|---|---|---|---|
| Total | Episomal | Integrated | ||
| HPV16+ | 81 | 70 | 11 | 13.5 |
| E5 Stop | 78 | 38 | 40 | 51.3 |
To determine how genome integration correlates with IFNK expression, IFNK transcript levels were measured in E5 Stop cell RNA harvested from both episomal and integrated samples. Interestingly, while both integrated and episomal E5 Stop samples showed higher IFNK transcripts than wild-type HPV16+ cells (Fig. 12c), integrated E5 Stop samples showed significantly higher IFNK than E5 Stop cells with episomal genomes (Fig. 12c). This finding is consistent with a model in which the increased IFNK transcripts observed in E5 Stop cells could be driving the increased integration rates of the cells (Fig. 12d). Further, the significant upregulation of IFNK in episomal E5 Stop cells suggests that the loss of E5-induced suppression of IFNK occurs prior to, and may be driving, genome integration. To address the question of whether integration itself may drive IFNK upregulation, we analyzed integrated wild-type HPV16-containing cells and observed no significant change in IFNK transcript levels versus episomal HPV16+ samples (Fig. 12c). This finding confirms that integration per se is not associated with the upregulation of IFNK but that the lack of E5 causes upregulated IFNK and increased IFNK-stimulated signaling.
DISCUSSION
Our findings support a model (Fig. 12d) in which the E5 oncoprotein suppresses TGF-β signaling through inhibition of the TGFBR2 complex and stimulates the MAPK pathway through enhancement of EGFR signaling. Together, these activities prevent induction of IFNK transcription. In the absence of E5, EGFR signaling is reduced and TGFBR2 signaling is increased, which results in increased production of IFN-κ. IFN-κ activates the JAK/STAT signaling pathway, which results in higher levels of ISG expression. Chronic ISG expression results in HPV genome instability and integration (104–108). Together, these findings demonstrate a previously unrecognized role for E5 in the viral life cycle—as an innate immune evasion factor. This role of E5 may explain why the acute phenotypes of E5 mutants in culture systems have generally been mild, despite E5 being conserved among high-risk HPVs (21, 22): the virus can immortalize, maintain itself episomally, and replicate to a reasonable degree in the absence of E5, but it cannot evade the accumulating effects of the innate immune response over time.
This study adds to a growing body of literature showing a multitude of mechanisms encoded by HPVs to evade the IFN response (11, 20, 109). Which particular ISGs are most important for HPV to evade is a question whose answer remains only partially understood. In particular, which are responsible for driving HPV integration and which particular downstream mechanisms drive growth factor-dependent IFNK regulation remain to be determined, although ISGs that are capable of inhibiting HPV have been described (72, 73, 110). The transcription factors involved in controlling IFNK promoter activity have yet to be identified. Our findings suggest that the IFNK promoter utilizes transcription factor complexes other than canonical IRFs to control its expression, in contrast to other type I IFNs. We have found potential IRF1 and AP-1 binding sites in the IFNK promoter through computational studies, but they have not yet been confirmed (data not shown). Further work will be needed to investigate what transcription factors are involved in IFNK expression and activity.
Since we observed increased IFNK transcripts and protein in E5 Stop cells (Fig. 4), we also examined the levels of IFN-α/β, which were found to be equivalent to those in wild-type HPV16+ cells (Fig. 2a). We previously found that increased IFN-κ resulted in increased IFN-α/β transcription (65). E5 Stop cells have levels of E6 and E7 transcripts and protein equivalent to those of wild-type HPV-containing cells (28). Therefore, the activities of E6 and E7, which have been previously reported to inhibit the activities of IRFs and IFN-α/β production (43, 111–115), are sufficient to prevent increased IFN-α/β production in E5 Stop cells. Regardless, we were able to detect increased ISG transcripts and STAT1 phosphorylation in E5 Stop cells (Fig. 4), indicating that IFN-κ upregulation can drive a downstream type I IFN response in HPV-containing keratinocytes, even in the absence of IFN-α/β and in the presence of the other viral oncogenes. Many attempts to knock down IFN-κ have failed (data not shown), so we have not formally proven that high ISG levels are due to IFN-κ, but since IFN-κ is the only type I IFN that is upregulated in E5 Stop cells, we feel that it is reasonable to conclude that IFN-κ is responsible.
Most of what is known about the effects of E5 on cellular pathways has been determined using overexpression systems. We have now confirmed that many of those pathways are in fact regulated by E5 in keratinocytes containing the complete HPV16 genome. We used phosphoproteomics to create a more detailed picture of the cellular effects of E5 than was previously available. Our data reveal that growth factor signaling plays an important role in the regulation of IFNK by HPV16. Some of the findings seem paradoxical. First, the treatment of HPV16+ cells with EGF promoted the upregulation of IFNK transcripts (Fig. 1), but treatment with an EGFR inhibitor also increased IFNK transcripts (Fig. 8). This paradoxical finding may be due to the complexity of the EGFR family and its ligands. EGF-induced EGFR activation drives the EGFR to form heterodimers with the HER2 (ErbB2) receptor (116–118), another member of the EGFR/ErbB family of receptors; however, treatment with AG1478 stimulates the formation of EGFR-EGFR homodimers that are functionally inactive (119). Furthermore, other agonists for EGFR in addition to EGF may result in functionally different consequences (116). Regulation of other ErbB family members by E5 has been reported but is not fully understood (23, 120). Our results also show that E5 Stop cells do not have the high levels of EGFR protein seen in wild-type HPV16+ cells (Fig. 8b), consistent with our previous report but in contrast to cells containing HPV18 (28, 82). In many (but not all) E5 Stop cell lines, EGFR levels are even below that seen in HFKs (Fig. 8b), which may explain why inhibition of EGFR has no effect on IFNK levels in E5 Stop cells, as it does in HFKs (Fig. 8a). These findings suggest that the effects of E5 may differ between HPV types and that E5 may have both qualitative and quantitative effects on EGFR signaling. Much remains to be done to understand how E5 impacts the full EGFR family.
Induction of IFNK by TGF-β1 is associated with promoter demethylation (65). Since E5 Stop cells have increased levels of TGF-β signaling (Fig. 10), we expected that high IFNK transcript levels in E5 Stop cells would be due to low levels of promoter methylation. In contrast to our expectations, the IFNK promoter was methylated in E5 Stop cells (Fig. 11). This finding is reminiscent of our previous finding that TGF-β2 (in contrast to TGF-β1) could increase IFNK levels and IFN signaling without inducing IFNK promoter demethylation (65). We believe there are two possible explanations for these data. Loss of E5 expression could enable recruitment of an unknown transcription factor complex to the promoter that can stimulate IFNK expression despite the presence of promoter methylation. One possibility is the SMAD2/3 complex, which is activated by TGFBR2. Alternatively, the demethylation pathway may actually be active at the IFNK promoter but arrested at the hydroxymethylation step. Our data are consistent with at least some increased level of hydroxymethylation at the IFNK promoter in E5 Stop cells (Fig. 11c). Hydroxymethylation is increasingly recognized as a stable modification of DNA and not just as a transient intermediate of demethylation (121). Whether either or both of these mechanisms is responsible for increased IFNK transcription in the absence of E5 remains to be determined.
For over 30 years, researchers have observed that the portion of the HPV genome containing the E5 gene is frequently lost during the integration of the HPV genome in cervical cancer (122–124). Previous studies investigating the integration of high-risk HPV types in vitro have found that episomal loss of HPV16 and the emergence of clones with integrated HPV16 DNA are associated with transient activation of antiviral response genes inducible by type I IFN (104, 107). Whether this transient response is a cause or a consequence of integration is not clear, but long-term IFN treatment is also associated with accumulation of integrated HPV in culture (103, 108). Integration may be due to the ability of IFN to reduce episome numbers, which allows integrant-bearing cells to dominate the population (103), although another study showed that IFN could induce de novo integration (108). Our data indicate that the loss of E5 results in a TGF-β-mediated increase in IFN signaling. This idea is supported by the fact that the emergence of integrated clones from a previously episomal population correlates not only with an increase in IFN signaling, but also with increased TGF-β signaling (104). Although the authors attributed the consequences of increased TGF-β signaling to cell cycle progression (104), our work supports a model in which TGF-β signaling increases IFN signaling through IFN-κ (65), thereby promoting integration.
Studies investigating integration have generally considered the loss of E2 to be the main driver of cancer progression following integration (125–129). We suggest that, in addition, the loss of E5 expression may have functional consequences as either a cause or a consequence of viral genome integration. One possible scenario notes that the region of the viral genome containing E5 is 180° from the viral replication origin, so it would be the point in theta replication where the replication forks would meet and may be more prone to damage or deletion than other regions of the genome. Loss of E5 would result in failure to suppress TGF-β-mediated IFN signaling and promote integration in a single cell. Since IFNs are secreted factors, an IFN response in a single cell would promote an IFN response in the surrounding population of infected cells, resulting in further episomal loss, driving the selection of integrant-bearing cells until only integrant-bearing cells remained. This idea is supported by in vitro data. Pett et al. reported that increased IFN signaling is observed only when there is a mixed population of episomal and integrated cells; in purely episomal or integrated populations, the IFN response is suppressed (104). The IFN-mediated selection of HPV+ integrants is also likely a very rapid process, since Herdman et al. reported that IFN-β treatment could induce the presence of a previously undetectable integrant in a single passage (103). Therefore, IFN-mediated selection of integrants is likely a rapid process that would be undetectable in the clinic. Our findings suggest that the loss of E5 during integration may have previously uncharacterized functional consequences.
MATERIALS AND METHODS
Cell culture and cell lines used.
HFKs were isolated from discarded and deidentified neonatal foreskins; HFKs containing HPV16 genomes (strain W12) were created by transfection and selection, as previously described (68). HPV16 genomes containing a translational stop codon at amino acid 11 in the E5 open reading frame (E5 Stop) were created as previously described (28). The presence of the mutation in E5 Stop cells was confirmed by PCR followed by sequencing (28). HFKs and keratinocyte-derived cell lines were cultivated in E medium with 5% fetal bovine serum (FBS) in the presence of mitomycin C-treated NIH-3T3 J2 fibroblast feeders (68, 130). Cell lines derived from at least three donors were used in separate experiments, and data for all the figures were compiled from at least three individual experiments. Low-passage (less than 10) HFKs and low-passage HFKs containing HPV16 or E5 Stop genomes (less than 12 passages postselection) were used in each experiment. The episomal or integrated status of the viral DNA was confirmed by Southern blotting on total cellular DNA, as previously described (65, 130). Southern blots in which the supercoiled band was visible were scored as episomal; the others were scored as integrated. The intensity of the supercoiled band was measured using ImageJ.
To generate retrovirus stocks, PT67 cells, grown in Dulbecco’s modified Eagle medium (DMEM) (Gibco) supplemented with 5% bovine growth serum (BGS) (HyClone), were seeded at a concentration of 3 million per 10-cm dish. The following day, the cells were transfected overnight with a mixture containing DMEM plus BGS, polyethylenimine (PEI), Opti-MEM medium (Gibco), 1 μg each of the pGag/Pol and pEnv packaging plasmids, and 3 μg of the retroviral plasmid to be packaged. The following day, the medium was aspirated, and fresh DMEM plus BGS was added. After a further 48 h, the supernatant was collected, and centrifuged at 1,000 rpm for 5 min. The debris-free supernatant was then filtered through a 33-mm sterile 0.22-μm syringe filter (Fisher; 09-720-004) and then flash frozen in a dry ice and ethanol bath before being stored at −80°C until it was used for infections.
To create the LXSN-wtE5 retrovirus vector, wild-type HPV16 E5 was cloned from the pUC-HPV16 (strain W12) plasmid using primers (wt16E5 5′ and wt16E5 3′ [Table 7]) that added 5′ EcoRI and 3′ BamHI restriction enzyme sites via PCR. The PCR product was then purified using a Qiagen gel extraction kit and ligated into an EcoRI- and BamHI-double-digested empty LXSN plasmid. After ligation and plasmid propagation, the sequence was verified by DNA sequencing.
TABLE 7.
Oligonucleotides used in this study
| Primer | Sequence (5′-3′) |
|---|---|
| Cyclophilin 5′ | GCAGGAACCCTTATAACCAAATCC |
| Cyclophilin 3′ | CTTGGGCCGCGTCTCC |
| E5 5′ | GTCTGTGTCTACATACACATCATTAATACTATTG |
| E5 3′ | GTAATTAAAAAGCGTGCATGTGTATGT |
| E6/7 5′ | GAACACGTAGAGAAACCCAGCTGTA |
| E6/7 3′ | GCTCATAACAGTAGAGATCAGTTGTCTCT |
| E6 5′ | GCAATGTTTCAGGACCCACA |
| E6 3′ | TCACGTCGCAGTAACTGTTGC |
| IFI16 5′ | GTTCTCACTATATTGTCCAGGCTAGAGT |
| IFI16 3′ | AGTTTATTCTGTTGTCACATCTAGGTTGTT |
| IFIT1 (p56) 5′ | TGCTCCAGACTATCCTTGACCT |
| IFIT1 (p56) 3′ | TCTCAGAGGAGCCTGGCTAA |
| IFNA 5′ | CAAATGAGCAGAATCTCTCCTTCC |
| IFNA 3′ | AGGTTGAAGATCTGCTGGATCAG |
| IFNB 5′ | CAGCAATTTTCAGTGTCAGAAGC |
| IFNB 3′ | TCATCCTGTCCTTGAGGCAGT |
| IFNK 5′ | GATTGTCCGAGTGGAAATCAGAAG |
| IFNK 3′ | GTGCATTTGCGTAGCCACAAT |
| IFNK Promoter 5′ | TCCAAAGTCGGCATAGTCACC |
| IFNK Promoter 3′ | CCTGAATATCTTCAGCAGGAGGAG |
| Mx1 5′ | GCCAGCTGTAGGTGTCCTTG |
| Mx1 3′ | ACCTGATGGCCTATCACCAG |
| OAS2 5′ | ATGGGAAATGGGGAGTCCCA |
| OAS2 3′ | CCAGGGGGAACTGTTCGGGT |
| p15 promoter 5′ | CATGATTCTCGGGATTTTTCTC |
| p15 promoter 3′ | GACAGCTCTGCACCTGTCAT |
| Sp100 5′ | GGGTGGGAAGATGGCAGG |
| Sp100 3′ | GTGTGCAGGAAGATGGTTCATC |
| STAT1 5′ | AAGGTGGCAGGATGTCTCAG |
| STAT1 3′ | ACTGTGCCAGGTACTGTCTG |
| TGFB1 5′ | CACGGGTTCAGGTACCGC |
| TGFB1 3′ | CACGGGTTCAGGTACCGC |
| TGFB2 5′ | GTCCCCCCGGAGGTGA |
| TGFB2 3′ | ATTGCTGAGACGTCAAATCGAAC |
| TGFBR2 5′ | GGAAACTTGACTGCACCGTT |
| TGFBR2 3′ | CTGCACATCGTCCTGTGG |
| wt16E5 5′ | GAATTCATGTCTATATGACAAATCTTGATACTG |
| wt16E5 3′ | GATCCTTATGTAATTAAAAAGCGTGCATG |
hTert-immortalized NOKs were gifts from the laboratory of Rona Scott, who received them from Karl Munger (70). The NOKs were grown in keratinocyte serum-free medium (ThermoFisher) with EGF and bovine pituitary extract added. The NOK lines were seeded overnight at a density of 3 million cells per 10-cm dish and then infected with one of several LXSN retroviral stocks containing either an empty control or the HPV16-E6, HPV16-E6/7, or HPV16-wtE5 ORF. Viral stocks were mixed with Polybrene (8 μg/ml) and medium and then added to the appropriate plate for overnight infection. Following infection, the cultures underwent selection with 250 μg/ml gentamycin (G418; Corning) for 2 weeks, followed by the outgrowth of infected cells. The cell lines were then used for various experiments, and the levels of the viral transcripts were measured by RT-qPCR to confirm expression of the desired ORF (data not shown).
A pLXSN-based retrovirus vector expressing E6/E7 and E5 was created by synthesis of a Gene Block (IDT). The sequence included the HPV16 E6/E7 sequence and E5 sequence (both strain W12) separated by the encephalomyocarditis virus (ECMV) IRES and a Kozak sequence and flanked by EcoRI and BamHI sites. DNA was rehydrated, digested with EcoRI and BamHI, and ligated into the pLXSN vector. Clones were verified by sequencing. Retrovirus particles were generated and used to infect HFK using the protocol described above. Following selection, the immortalized clones were expanded, pooled, and used for analysis. The levels of the viral transcripts were determined by RT-qPCR to confirm expression of the desired ORF (data not shown). The cell lines were maintained in E medium with 5% FBS with feeders.
The drugs used in this study were as follows: poly(I·C) (Sigma; P9582), recombinant IFN-β (PBL Assay Science; 11415-1), recombinant TGF-β1 (R&D Systems; 240-B-002), recombinant HGF (Gibco; PHG0324), recombinant EGF (Sigma; E9644), AG1478 (Sigma; 658552-5MG), SU11274 (Sigma; S9820-5MG), ruxolitinib (Selleckchem; S1378), and SB431542 (Fisher; 16-414-0). The drugs were reconstituted according to the manufacturers’ instructions and added to monolayer medium at the following concentrations: 25 U/ml (IFN-β), 5 ng/ml (TGF-β1), 100 ng/ml (EGF), 40 ng/ml (HGF), 1 μM (AG1478), 10 μM (ruxolitinib), 1 μM (SU11274), and either 5 μM or 10 μM (SB431542).
RNA extraction, RT-qPCR, and immunoblotting.
Total RNA was isolated using RNA-STAT 60 (TelTest, Inc.), digested with RNase-free DNase (Promega), phenol-chloroform extracted, and reverse transcribed using qScript (Quanta) as described previously (65). Quantitative PCR was performed using PerfeCTa SYBR Green SuperMix ROX (Quanta) on an Applied Biosystems StepOne Plus real-time PCR machine using the primers shown in Table 7. Protein lysates were prepared by adding 1× Cell Signaling lysis buffer, supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF), to cells and incubating them on ice for 5 min, followed by scraping, brief sonication, and clarification by centrifugation. SDS-PAGE and Western blotting were performed essentially as described previously (131), except SDS was excluded from the TGFBR2 gels. Blocking and antibody dilution were performed using Li-Cor Odyssey blocking buffer containing 0.1% Tween 20, and images were acquired using a Li-Cor Odyssey near-infrared imaging system. The antibodies used included IFN-κ (1:500; Abnova; H00056832-M01), phospho-STAT1 (Y701) (1:1,000; Cell Signaling; 7649 P), total STAT1 (1:1,000; Cell Signaling; 9172S), TGFBR2 (1:750; Fisher; PA5-35076), phospho-Smad3 (Ser423/425) (1:2,000; Abcam; ab52903), total JAK1 (1:500; Cell Signaling; 3332), total EGFR (1:2,500; Abcam; ab32562), total Smad2/3 (1:1,000; Cell Signaling; 5678), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (1:2,500; Santa Cruz; sc-47724), tubulin (1:1,000; NeoMarkers; MS581P), OAS2 (1:1,000; Santa Cruz; sc-271117), IFIT1 (1:1,000; Proteintech; 23247-1-AP), and HPV16-E7 (1:250; Cervimax; vs 13005). Images included in the figures are unsaturated.
siRNA transfection.
The DharmaFeCT siRNA protocol was followed. Briefly, half a million keratinocytes with or without an HPV genome were plated and incubated with E medium plus 5% fetal bovine serum (FBS) for 12 h on 6-well plates. siRNA was diluted to 35 nM in serum-free medium (Opti-MEM; Fisher). DharmaFeCT transfection reagent was separately diluted in Opti-MEM. The siRNA and transfection reagents were incubated at room temperature for 5 min, followed by combining the solutions and incubating them at room temperature for 20 min; 1,600 μl of E medium plus 5% FBS and 200 μl of the siRNA mixture were simultaneously added to the cells. Ninety-six hours after transfection, the cells were harvested for RNA or Western analysis as described above.
Bisulfate analysis and MeDIP.
Total DNAs were isolated using phenol-chloroform extraction as described previously (21). Bisulfite treatment was performed with Zymo EZ DNA Direct kit methylation, followed by PCR, cloning, and sequencing as previously described (65). Bisulfite-specific primers previously established (the same PCR conditions described in reference 53) were used to amplify the IFN-κ promoter with AmpliTaq Gold 360 Master Mix, as previously performed (65).
For MeDIP analysis, we used a previously described protocol (102); 10 μg total cellular DNA was sonicated using a Bioruptor (Diagenode) to yield 200- to 600-bp fragments, and fragmentation was confirmed using agarose gel electrophoresis. The sonicated DNA (500 ng) was diluted in Tris-EDTA (TE) buffer and denatured at 95°C for 10 min, followed by chilling on ice. Immunoprecipitation (IP) buffer was added to a final concentration (1×) of 10 mM sodium phosphate, pH 7.0, 0.14 M NaCl, 0.05% Triton X-100, followed by control nonspecific polyclonal rabbit serum or 0.25 μg/μl 5-hmC-specific antibody (Active Motif; 39769). Samples were incubated for 2 h at 4°C. DNA-antibody complexes were captured using magnetic protein G beads and incubation with rotation for 2 h at 4°C, followed by three washes in 1× IP buffer for 5 min each time at 4°C. DNA was eluted in elution buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 0.5% SDS, 0.25 mg/ml proteinase K) for 3 h at 50°C and then purified using an UltraClean PCR cleanup kit (MoBio). qPCR was used to quantify the yield of immunoprecipitated DNA using the primers listed in Table 7.
Proteomics array.
The Phospho-Explorer array (Full Moon Biosystems; catalog no. PEX100) was used according to the manufacturer’s instructions. Uninfected HFKs, episomal HPV16-containing cells, and episomal E5 Stop cells were seeded at a concentration of 3 million cells in a 10-cm dish overnight. The following day, two plates for each cell line were washed 3 times in cold phosphate-buffered saline (PBS), followed by harvesting protein using the Full Moon Biosystems kit extraction buffer with PMSF added to inhibit proteases. The protein samples were biotinylated and incubated with the array slides. A GenePix Pro 6.0 array scanner was used to scan the array chips. Two independent experiments were performed, with different donor backgrounds in each. Spot intensities were normalized to the negative controls and then to the average intensity of the GAPDH spots for each array. Normalized intensities were used to calculate ratios between the various treatment groups. P values were calculated based on the log2 value of the intensity for each spot. A >2-fold change in the ratio with a significant P value (<0.05) was used as a cutoff. For each group comparison, we used DAVID to perform an in-depth statistical analysis of protein lists to determine their gene ontology (75, 76).
Statistics.
The significance of wild-type versus E5 Stop integration rates was determined using Fisher’s exact test. Significance in other experiments was calculated using Welch’s unequal variances t test. RT-qPCR analysis included at least 3 technical replicates of ≥3 biological experiments, and each immunoblot densitometry analysis included at least 1 technical replicate of ≥6 biological experiments. Western quantification included ≥4 biological replicates.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Center for Molecular and Tumor Virology for helpful discussions. We thank Rona Scott for the gift of NOKs, Arjun Yusufji for technical assistance, and Liudmila Chesnokova for assistance in processing the phosphoproteomics array.
This work was supported by grants from the National Institute of Allergy and Infectious Diseases (R01AI118904); the National Institute of General Medical Sciences (P30GM110703); the National Heart, Blood, and Lung Institute (HL141155); and the Feist-Weiller Cancer Center.
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health or the Feist-Weiller Cancer Center.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Munoz N, Castellsague X, de Gonzalez AB, Gissmann L. 2006. HPV in the etiology of human cancer. Vaccine 24(Suppl 3):1–10. doi: 10.1016/j.vaccine.2006.05.115. [DOI] [PubMed] [Google Scholar]
- 2.Cheetham D, Smith J, Wilson C, Munday PE, Coleman DV. 1984. Clinical significance of human papillomavirus infection of the uterine cervix in the development of cervical intraepithelial neoplasia. Br J Vener Dis 60:182–185. doi: 10.1136/sti.60.3.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coleman HN, Moscicki AB, Farhat SN, Gupta SK, Wang X, Nakagawa M. 2012. CD8 T-cell responses in incident and prevalent human papillomavirus types 16 and 18 infections. ISRN Obstet Gynecol 2012:854237. doi: 10.5402/2012/854237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moscicki AB, Schiffman M, Burchell A, Albero G, Giuliano AR, Goodman MT, Kjaer SK, Palefsky J. 2012. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 30(Suppl 5):F24–F33. doi: 10.1016/j.vaccine.2012.05.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowy DR, Kirnbauer R, Schiller JT. 1994. Genital human papillomavirus infection. Proc Natl Acad Sci U S A 91:2436–2440. doi: 10.1073/pnas.91.7.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doorbar J. 2006. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci 110:525–541. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- 7.Stanley MA, Pett MR, Coleman N. 2007. HPV: from infection to cancer. Biochem Soc Trans 35:1456–1460. doi: 10.1042/BST0351456. [DOI] [PubMed] [Google Scholar]
- 8.Nardelli B, Zaritskaya L, Semenuk M, Cho YH, LaFleur DW, Shah D, Ullrich S, Girolomoni G, Albanesi C, Moore PA. 2002. Regulatory effect of IFN-kappa, a novel type I IFN, on cytokine production by cells of the innate immune system. J Immunol 169:4822–4830. doi: 10.4049/jimmunol.169.9.4822. [DOI] [PubMed] [Google Scholar]
- 9.Gielen V, Schmitt D, Thivolet J. 1988. HLA class I antigen (heavy and light chain) expression by Langerhans cells and keratinocytes of the normal human epidermis: ultrastructural quantitation using immunogold labelling procedure. Arch Dermatol Res 280:131–136. doi: 10.1007/bf00456841. [DOI] [PubMed] [Google Scholar]
- 10.Stanley MA. 2009. Immune responses to human papilloma viruses. Indian J Med Res 130:266–276. [PubMed] [Google Scholar]
- 11.Woodby B, Scott M, Bodily J. 2016. The interaction between human papillomaviruses and the stromal microenvironment. Prog Mol Biol Transl Sci 144:169–238. doi: 10.1016/bs.pmbts.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frazer IH. 2009. Interaction of human papillomaviruses with the host immune system: a well evolved relationship. Virology 384:410–414. doi: 10.1016/j.virol.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Palefsky JM, Minkoff H, Kalish LA, Levine A, Sacks HS, Garcia P, Young M, Melnick S, Miotti P, Burk R. 1999. Cervicovaginal human papillomavirus infection in human immunodeficiency virus-1 (HIV)-positive and high-risk HIV-negative women. J Natl Cancer Inst 91:226–236. doi: 10.1093/jnci/91.3.226. [DOI] [PubMed] [Google Scholar]
- 14.Serraino D, Carrieri P, Pradier C, Bidoli E, Dorrucci M, Ghetti E, Schiesari A, Zucconi R, Pezzotti P, Dellamonica P, Franceschi S, Rezza G. 1999. Risk of invasive cervical cancer among women with, or at risk for, HIV infection. Int J Cancer 82:334–337. doi: 10.1002/(sici)1097-0215(19990730)82:3<334::aid-ijc5>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 15.Scott M, Nakagawa M, Moscicki AB. 2001. Cell-mediated immune response to human papillomavirus infection. Clin Diagn Lab Immunol 8:209–220. doi: 10.1128/CDLI.8.2.209-220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mbulaiteye SM, Katabira ET, Wabinga H, Parkin DM, Virgo P, Ochai R, Workneh M, Coutinho A, Engels EA. 2005. Spectrum of cancers among HIV-infected persons in Africa: the Uganda AIDS-Cancer Registry Match Study. Int J Cancer doi: 10.1002/ijc.21443. [DOI] [PubMed] [Google Scholar]
- 17.Einstein MH, Kadish AS. 2004. Anogenital neoplasia in AIDS. Curr Opin Oncol 16:455–462. doi: 10.1097/00001622-200409000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Clarke B, Chetty R. 2002. Postmodern cancer: the role of human immunodeficiency virus in uterine cervical cancer. Mol Pathol 55:19–24. doi: 10.1136/mp.55.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellerbrock TV, Chiasson MA, Bush TJ, Sun XW, Sawo D, Brudney K, Wright TC Jr. 2000. Incidence of cervical squamous intraepithelial lesions in HIV-infected women. JAMA 283:1031–1037. doi: 10.1001/jama.283.8.1031. [DOI] [PubMed] [Google Scholar]
- 20.Bodily J, Laimins LA. 2011. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol 19:33–39. doi: 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fehrmann F, Klumpp DJ, Laimins LA. 2003. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol 77:2819–2831. doi: 10.1128/jvi.77.5.2819-2831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genther SM, Sterling S, Duensing S, Munger K, Sattler C, Lambert PF. 2003. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J Virol 77:2832–2842. doi: 10.1128/jvi.77.5.2832-2842.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crusius K, Auvinen E, Steuer B, Gaissert H, Alonso A. 1998. The human papillomavirus type 16 E5-protein modulates ligand-dependent activation of the EGF receptor family in the human epithelial cell line HaCaT. Exp Cell Res 241:76–83. doi: 10.1006/excr.1998.4024. [DOI] [PubMed] [Google Scholar]
- 24.Pim D, Collins M, Banks L. 1992. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 7:27–32. [PubMed] [Google Scholar]
- 25.Straight SW, Hinkle PM, Jewers RJ, McCance DJ. 1993. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J Virol 67:4521–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Straight SW, Herman B, McCance DJ. 1995. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J Virol 69:3185–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomakidi P, Cheng H, Kohl A, Komposch G, Alonso A. 2000. Modulation of the epidermal growth factor receptor by the human papillomavirus type 16 E5 protein in raft cultures of human keratinocytes. Eur J Cell Biol 79:407–412. doi: 10.1078/0171-9335-00060. [DOI] [PubMed] [Google Scholar]
- 28.Scott ML, Coleman DT, Kelly KC, Carroll JL, Woodby B, Songock WK, Cardelli JA, Bodily JM. 2018. Human papillomavirus type 16 E5-mediated upregulation of Met in human keratinocytes. Virology 519:1–11. doi: 10.1016/j.virol.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.French D, Belleudi F, Mauro MV, Mazzetta F, Raffa S, Fabiano V, Frega A, Torrisi MR. 2013. Expression of HPV16 E5 down-modulates the TGFbeta signaling pathway. Mol Cancer 12:38. doi: 10.1186/1476-4598-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munshi N, Yie Y, Merika M, Senger K, Lomvardas S, Agalioti T, Thanos D. 1999. The IFN-beta enhancer: a paradigm for understanding activation and repression of inducible gene expression. Cold Spring Harbor Symp Quant Biol 64:149–159. doi: 10.1101/sqb.1999.64.149. [DOI] [PubMed] [Google Scholar]
- 31.Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, Wathelet MG. 1998. Structure and function of the interferon-beta enhanceosome. Cold Spring Harbor Symp Quant Biol 63:609–620. doi: 10.1101/sqb.1998.63.609. [DOI] [PubMed] [Google Scholar]
- 32.Ford E, Thanos D. 2010. The transcriptional code of human IFN-beta gene expression. Biochim Biophys Acta 1799:328–336. doi: 10.1016/j.bbagrm.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. 2001. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 34.Iatropoulos MJ, Williams GM. 1996. Proliferation markers. Exp Toxicol Pathol 48:175–181. doi: 10.1016/S0940-2993(96)80039-X. [DOI] [PubMed] [Google Scholar]
- 35.Tay SS, Roediger B, Tong PL, Tikoo S, Weninger W. 2014. The skin-resident immune network. Curr Dermatol Rep 3:13–22. doi: 10.1007/s13671-013-0063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. 2009. Skin immune sentinels in health and disease. Nat Rev Immunol 9:679–691. doi: 10.1038/nri2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bos JD, Kapsenberg ML. 1993. The skin immune system: progress in cutaneous biology. Immunol Today 14:75–78. doi: 10.1016/0167-5699(93)90062-P. [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Amerio P, Sauder DN. 1999. Role of cytokines in epidermal Langerhans cell migration. J Leukoc Biol 66:33–39. doi: 10.1002/jlb.66.1.33. [DOI] [PubMed] [Google Scholar]
- 39.Goldschmidt MH, Kennedy JS, Kennedy DR, Yuan H, Holt DE, Casal ML, Traas AM, Mauldin EA, Moore PF, Henthorn PS, Hartnett BJ, Weinberg KI, Schlegel R, Felsburg PJ. 2006. Severe papillomavirus infection progressing to metastatic squamous cell carcinoma in bone marrow-transplanted X-linked SCID dogs. J Virol 80:6621–6628. doi: 10.1128/JVI.02571-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. 2007. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol 17:1140–1145. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 41.Williams IR, Kupper TS. 1996. Immunity at the surface: homeostatic mechanisms of the skin immune system. Life Sci 58:1485–1507. doi: 10.1016/0024-3205(96)00042-2. [DOI] [PubMed] [Google Scholar]
- 42.LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, Taylor K, Buergin M, Chinchilla D, Roshke V, Chen G, Ruben SM, Pitha PM, Coleman TA, Moore PA. 2001. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J Biol Chem 276:39765–39771. doi: 10.1074/jbc.M102502200. [DOI] [PubMed] [Google Scholar]
- 43.Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD. 2001. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J Virol 75:4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang YE, Laimins LA. 2000. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J Virol 74:4174–4182. doi: 10.1128/jvi.74.9.4174-4182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, Kelsey KT, Turek LP, Ahlquist P. 2007. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res 67:4605–4619. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wan F, Miao X, Quraishi I, Kennedy V, Creek KE, Pirisi L. 2008. Gene expression changes during HPV-mediated carcinogenesis: a comparison between an in vitro cell model and cervical cancer. Int J Cancer 123:32–40. doi: 10.1002/ijc.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nees M, Geoghegan JM, Munson P, Prabhu V, Liu Y, Androphy E, Woodworth CD. 2000. Human papillomavirus type 16 E6 and E7 proteins inhibit differentiation-dependent expression of transforming growth factor-beta2 in cervical keratinocytes. Cancer Res 60:4289–4298. [PubMed] [Google Scholar]
- 48.Thomas JT, Oh ST, Terhune SS, Laimins LA. 2001. Cellular changes induced by low-risk human papillomavirus type 11 in keratinocytes that stably maintain viral episomes. J Virol 75:7564–7571. doi: 10.1128/JVI.75.16.7564-7571.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cortés-Malagón EM, Bonilla-Delgado J, Díaz-Chávez J, Hidalgo-Miranda A, Romero-Cordoba S, Uren A, Celik H, McCormick M, Munguía-Moreno JA, Ibarra-Sierra E, Escobar-Herrera J, Lambert PF, Mendoza-Villanueva D, Bermudez-Cruz RM, Gariglio P. 2013. Gene expression profile regulated by the HPV16 E7 oncoprotein and estradiol in cervical tissue. Virology 447:155–165. doi: 10.1016/j.virol.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reiser J, Hurst J, Voges M, Krauss P, Munch P, Iftner T, Stubenrauch F. 2011. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J Virol 85:11372–11380. doi: 10.1128/JVI.05279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perea S, Lopezocejo O, Vongabain A, Arana M. 1997. Human papillomavirus type-16 (HPV-16) major transforming proteins functionally interact with interferon signaling mechanisms. Int J Oncol 11:169–173. doi: 10.3892/ijo.11.1.169. [DOI] [PubMed] [Google Scholar]
- 52.DeCarlo CA, Severini A, Edler L, Escott NG, Lambert PF, Ulanova M, Zehbe I. 2010. IFN-kappa, a novel type I IFN, is undetectable in HPV-positive human cervical keratinocytes. Lab Invest 90:1482–1491. doi: 10.1038/labinvest.2010.95. [DOI] [PubMed] [Google Scholar]
- 53.Rincon-Orozco B, Halec G, Rosenberger S, Muschik D, Nindl I, Bachmann A, Ritter TM, Dondog B, Ly R, Bosch FX, Zawatzky R, Rösl F. 2009. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells. Cancer Res 69:8718–8725. doi: 10.1158/0008-5472.CAN-09-0550. [DOI] [PubMed] [Google Scholar]
- 54.Sunthamala N, Thierry F, Teissier S, Pientong C, Kongyingyoes B, Tangsiriwatthana T, Sangkomkamhang U, Ekalaksananan T. 2014. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-kappa transcription in keratinocytes. PLoS One 9:e91473. doi: 10.1371/journal.pone.0091473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lulli D, Carbone ML, Pastore S. 2016. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget 7:47777–47793. doi: 10.18632/oncotarget.10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Battcock SM, Collier TW, Zu D, Hirasawa K. 2006. Negative regulation of the alpha interferon-induced antiviral response by the Ras/Raf/MEK pathway. J Virol 80:4422–4430. doi: 10.1128/JVI.80.9.4422-4430.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Q, Gong R, Qu J, Zhou Y, Liu W, Chen M, Liu Y, Zhu Y, Wu J. 2012. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C virus replication via attenuation of the interferon-JAK-STAT pathway. J Virol 86:1544–1554. doi: 10.1128/JVI.00688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Komatsu Y, Christian SL, Ho N, Pongnopparat T, Licursi M, Hirasawa K. 2015. Oncogenic Ras inhibits IRF1 to promote viral oncolysis. Oncogene 34:3985–3993. doi: 10.1038/onc.2014.331. [DOI] [PubMed] [Google Scholar]
- 59.Takahara T, Fukuyama Y, Saito S, Ogino T, Miyajima N, Kohase M. 1999. Il-1, EGF, and HGF suppress the antiviral activity of interferon in primary monkey hepatic parenchymal cells. Jpn J Infect Dis 52:45–48. [PubMed] [Google Scholar]
- 60.Mascia F, Mariani V, Girolomoni G, Pastore S. 2003. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol 163:303–312. doi: 10.1016/S0002-9440(10)63654-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lupberger J, Duong FH, Fofana I, Zona L, Xiao F, Thumann C, Durand SC, Pessaux P, Zeisel MB, Heim MH, Baumert TF. 2013. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology 58:1225–1235. doi: 10.1002/hep.26404. [DOI] [PubMed] [Google Scholar]
- 62.Kalinowski A, Ueki I, Min-Oo G, Ballon-Landa E, Knoff D, Galen B, Lanier LL, Nadel JA, Koff JL. 2014. EGFR activation suppresses respiratory virus-induced IRF1-dependent CXCL10 production. Am J Physiol Lung Cell Mol Physiol 307:L186–L196. doi: 10.1152/ajplung.00368.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bedke N, Sammut D, Green B, Kehagia V, Dennison P, Jenkins G, Tatler A, Howarth PH, Holgate ST, Davies DE. 2012. Transforming growth factor-beta promotes rhinovirus replication in bronchial epithelial cells by suppressing the innate immune response. PLoS One 7:e44580. doi: 10.1371/journal.pone.0044580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qing J, Liu C, Choy L, Wu RY, Pagano JS, Derynck R. 2004. Transforming growth factor beta/Smad3 signaling regulates IRF-7 function and transcriptional activation of the beta interferon promoter. Mol Cell Biol 24:1411–1425. doi: 10.1128/mcb.24.3.1411-1425.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Woodby BL, Songock WK, Scott ML, Raikhy G, Bodily JM. 2018. Induction of interferon kappa in human papillomavirus 16 infection by transforming growth factor beta-induced promoter demethylation. J Virol 92:e01714-17. doi: 10.1128/JVI.01714-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yanai H, Negishi H, Taniguchi T. 2012. The IRF family of transcription factors: inception, impact and implications in oncogenesis. Oncoimmunology 1:1376–1386. doi: 10.4161/onci.22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu H, Zhang Y, Altomare D, Pena MM, Wan F, Pirisi L, Creek KE. 2014. Six1 promotes epithelial-mesenchymal transition and malignant conversion in human papillomavirus type 16-immortalized human keratinocytes. Carcinogenesis 35:1379–1388. doi: 10.1093/carcin/bgu050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bodily JM, Mehta KP, Cruz L, Meyers C, Laimins LA. 2011. The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J Virol 85:8852–8862. doi: 10.1128/JVI.00664-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dimaio D, Petti LM. 2013. The E5 proteins. Virology 445:99–114. doi: 10.1016/j.virol.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piboonniyom SO, Duensing S, Swilling NW, Hasskarl J, Hinds PW, Munger K. 2003. Abrogation of the retinoblastoma tumor suppressor checkpoint during keratinocyte immortalization is not sufficient for induction of centrosome-mediated genomic instability. Cancer Res 63:476–483. [PubMed] [Google Scholar]
- 71.Stepp WH, Meyers JM, McBride AA. 2013. Sp100 provides intrinsic immunity against human papillomavirus infection. mBio 4:e00845. doi: 10.1128/mBio.00845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stepp WH, Stamos JD, Khurana S, Warburton A, McBride AA. 2017. Sp100 colocalizes with HPV replication foci and restricts the productive stage of the infectious cycle. PLoS Pathog 13:e1006660. doi: 10.1371/journal.ppat.1006660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Terenzi F, Saikia P, Sen GC. 2008. Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J 27:3311–3321. doi: 10.1038/emboj.2008.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marie I, Hovanessian AG. 1992. The 69-kDa 2-5A synthetase is composed of two homologous and adjacent functional domains. J Biol Chem 267:9933–9939. [PubMed] [Google Scholar]
- 75.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. 2007. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Lempicki RA. 2007. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim SH, Juhnn YS, Kang S, Park SW, Sung MW, Bang YJ, Song YS. 2006. Human papillomavirus 16 E5 up-regulates the expression of vascular endothelial growth factor through the activation of epidermal growth factor receptor, MEK/ERK1,2 and PI3K/Akt. Cell Mol Life Sci 63:930–938. doi: 10.1007/s00018-005-5561-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xi R, Pan S, Chen X, Hui B, Zhang L, Fu S, Li X, Zhang X, Gong T, Guo J, Zhang X, Che S. 2016. HPV16 E6-E7 induces cancer stem-like cells phenotypes in esophageal squamous cell carcinoma through the activation of PI3K/Akt signaling pathway in vitro and in vivo. Oncotarget 7:57050–57065. doi: 10.18632/oncotarget.10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wei LH, Kuo ML, Chen CA, Chou CH, Cheng WF, Chang MC, Su JL, Hsieh CY. 2001. The anti-apoptotic role of interleukin-6 in human cervical cancer is mediated by up-regulation of Mcl-1 through a PI 3-K/Akt pathway. Oncogene 20:5799–5809. doi: 10.1038/sj.onc.1204733. [DOI] [PubMed] [Google Scholar]
- 80.Crusius K, Auvinen E, Alonso A. 1997. Enhancement of EGF- and PMA-mediated MAP kinase activation in cells expressing the human papillomavirus type 16 E5 protein. Oncogene 15:1437–1444. doi: 10.1038/sj.onc.1201312. [DOI] [PubMed] [Google Scholar]
- 81.Crusius K, Rodriguez I, Alonso A. 2000. The human papillomavirus type 16 E5 protein modulates ERK1/2 and p38 MAP kinase activation by an EGFR-independent process in stressed human keratinocytes. Virus Genes 20:65–69. doi: 10.1023/A:1008112207824. [DOI] [PubMed] [Google Scholar]
- 82.Wasson CW, Morgan EL, Muller M, Ross RL, Hartley M, Roberts S, Macdonald A. 2017. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 8:103581–103600. doi: 10.18632/oncotarget.21658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou Y, Zhang Q, Gao G, Zhang X, Liu Y, Yuan S, Wang X, Chen JJ. 2016. Role of WDHD1 in human papillomavirus-mediated oncogenesis identified by transcriptional profiling of e7-expressing cells. J Virol 90:6071–6084. doi: 10.1128/JVI.00513-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patel D, Huang SM, Baglia LA, McCance DJ. 1999. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J 18:5061–5072. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levan J, Vliet-Gregg PA, Robinson KL, Katzenellenbogen RA. 2017. Human papillomavirus type 16 E6 and NFX1-123 mislocalize immune signaling proteins and downregulate immune gene expression in keratinocytes. PLoS One 12:e0187514. doi: 10.1371/journal.pone.0187514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li H, Zhan T, Li C, Liu M, Wang QK. 2009. Repression of MHC class I transcription by HPV16E7 through interaction with a putative RXRbeta motif and NF-kappaB cytoplasmic sequestration. Biochem Biophys Res Commun 388:383–388. doi: 10.1016/j.bbrc.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 87.Spitkovsky D, Hehner SP, Hofmann TG, Moller A, Schmitz ML. 2002. The human papillomavirus oncoprotein E7 attenuates NF-kappa B activation by targeting the Ikappa B kinase complex. J Biol Chem 277:25576–25582. doi: 10.1074/jbc.M201884200. [DOI] [PubMed] [Google Scholar]
- 88.Vandermark ER, Deluca KA, Gardner CR, Marker DF, Schreiner CN, Strickland DA, Wilton KM, Mondal S, Woodworth CD. 2012. Human papillomavirus type 16 E6 and E7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology 425:53–60. doi: 10.1016/j.virol.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karim R, Meyers C, Backendorf C, Ludigs K, Offringa R, van Ommen GJ, Melief CJ, van der Burg SH, Boer JM. 2011. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS One 6:e17848. doi: 10.1371/journal.pone.0017848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tummers B, Goedemans R, Pelascini LP, Jordanova ES, van Esch EM, Meyers C, Melief CJ, Boer JM, van der Burg SH. 2015. The interferon-related developmental regulator 1 is used by human papillomavirus to suppress NFkappaB activation. Nat Commun 6:6537. doi: 10.1038/ncomms7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tummers B, Goedemans R, Jha V, Meyers C, Melief CJ, van der Burg SH, Boer JM. 2014. CD40-mediated amplification of local immunity by epithelial cells is impaired by HPV. J Invest Dermatol 134:2918–2927. doi: 10.1038/jid.2014.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Da Costa R, Bastos MM, Medeiros R, Oliveira PA. 2016. The NFkappaB signaling pathway in papillomavirus-induced lesions: friend or foe?. Anticancer Res 36:2073–2083. [PubMed] [Google Scholar]
- 93.Hennighausen L, Robinson GW. 2008. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev 22:711–721. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A. 2006. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev 17:173–188. doi: 10.1016/j.cytogfr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 95.Sanchez-Prieto R, Sanchez-Arevalo VJ, Servitja JM, Gutkind JS. 2002. Regulation of p73 by c-Abl through the p38 MAP kinase pathway. Oncogene 21:974–979. doi: 10.1038/sj.onc.1205134. [DOI] [PubMed] [Google Scholar]
- 96.Murray-Zmijewski F, Lane DP, Bourdon JC. 2006. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 13:962–972. doi: 10.1038/sj.cdd.4401914. [DOI] [PubMed] [Google Scholar]
- 97.Lulli D, Carbone ML, Pastore S. 2017. The MEK inhibitors trametinib and cobimetinib induce a type I interferon response in human keratinocytes. Int J Mol Sci 18:E2227. doi: 10.3390/ijms18102227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maiti A, Drohat AC. 2011. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem 286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hill PW, Amouroux R, Hajkova P. 2014. DNA demethylation, Tet proteins and 5-hydroxymethylcytosine in epigenetic reprogramming: an emerging complex story. Genomics 104:324–333. doi: 10.1016/j.ygeno.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 100.Huang J, Wang H, Xie X, Gao H, Guo G. 2010. Developmental changes in DNA methylation of pollen mother cells of David lily during meiotic prophase I. Mol Biol 44:853–858. [PubMed] [Google Scholar]
- 101.Mendonca A, Chang EH, Liu W, Yuan C. 2014. Hydroxymethylation of DNA influences nucleosomal conformation and stability in vitro. Biochim Biophys Acta 1839:1323–1329. doi: 10.1016/j.bbagrm.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 102.Thillainadesan G, Isovic M, Loney E, Andrews J, Tini M, Torchia J. 2008. Genome analysis identifies the p15ink4b tumor suppressor as a direct target of the ZNF217/CoREST complex. Mol Cell Biol 28:6066–6077. doi: 10.1128/MCB.00246-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Herdman MT, Pett MR, Roberts I, Alazawi WO, Teschendorff AE, Zhang XY, Stanley MA, Coleman N. 2006. Interferon-beta treatment of cervical keratinocytes naturally infected with human papillomavirus 16 episomes promotes rapid reduction in episome numbers and emergence of latent integrants. Carcinogenesis 27:2341–2353. doi: 10.1093/carcin/bgl172. [DOI] [PubMed] [Google Scholar]
- 104.Pett MR, Herdman MT, Palmer RD, Yeo GS, Shivji MK, Stanley MA, Coleman N. 2006. Selection of cervical keratinocytes containing integrated HPV16 associates with episome loss and an endogenous antiviral response. Proc Natl Acad Sci U S A 103:3822–3827. doi: 10.1073/pnas.0600078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hong S, Mehta KP, Laimins LA. 2011. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol 85:9486–9494. doi: 10.1128/JVI.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chang YE, Pena L, Sen GC, Park JK, Laimins LA. 2002. Long-term effect of interferon on keratinocytes that maintain human papillomavirus type 31. J Virol 76:8864–8874. doi: 10.1128/jvi.76.17.8864-8874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Alazawi W, Pett M, Arch B, Scott L, Freeman T, Stanley MA, Coleman N. 2002. Changes in cervical keratinocyte gene expression associated with integration of human papillomavirus 16. Cancer Res 62:6959–6965. [PubMed] [Google Scholar]
- 108.Lace MJ, Anson JR, Haugen TH, Dierdorff JM, Turek LP. 2015. Interferon treatment of human keratinocytes harboring extrachromosomal, persistent HPV-16 plasmid genomes induces de novo viral integration. Carcinogenesis 36:151–159. doi: 10.1093/carcin/bgu236. [DOI] [PubMed] [Google Scholar]
- 109.Doorbar J. 2018. Host control of human papillomavirus infection and disease. Best Pract Res Clin Obstet Gynaecol 47:27–41. doi: 10.1016/j.bpobgyn.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 110.Saikia P, Fensterl V, Sen GC. 2010. The inhibitory action of P56 on select functions of E1 mediates interferon’s effect on human papillomavirus DNA replication. J Virol 84:13036–13039. doi: 10.1128/JVI.01194-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Park JS, Kim EJ, Kwon HJ, Hwang ES, Namkoong SE, Um SJ. 2000. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem 275:6764–6769. doi: 10.1074/jbc.275.10.6764. [DOI] [PubMed] [Google Scholar]
- 112.Ronco LV, Karpova AY, Vidal M, Howley PM. 1998. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev 12:2061–2072. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Perea SE, Massimi P, Banks L. 2000. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int J Mol Med 5:661–666. doi: 10.3892/ijmm.5.6.661. [DOI] [PubMed] [Google Scholar]
- 114.Um SJ, Rhyu JW, Kim EJ, Jeon KC, Hwang ES, Park JS. 2002. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett 179:205–212. doi: 10.1016/s0304-3835(01)00871-0. [DOI] [PubMed] [Google Scholar]
- 115.Oldak M, Tolzmann L, Wnorowski A, Podgorska MJ, Silling S, Lin R, Hiscott J, Muller CS, Vogt T, Smola H, Smola S. 2011. Differential regulation of human papillomavirus type 8 by interferon regulatory factors 3 and 7. J Virol 85:178–188. doi: 10.1128/JVI.00998-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. 2006. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 117.Hynes NE, MacDonald G. 2009. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol 21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 118.Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, Ratzkin BJ, Yarden Y. 1996. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol 16:5276–5287. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gan HK, Walker F, Burgess AW, Rigopoulos A, Scott AM, Johns TG. 2007. The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor AG1478 increases the formation of inactive untethered EGFR dimers. Implications for combination therapy with monoclonal antibody 806. J Biol Chem 282:2840–2850. doi: 10.1074/jbc.M605136200. [DOI] [PubMed] [Google Scholar]
- 120.Chen SL, Lin ST, Tsai TC, Hsiao WC, Tsao YP. 2007. ErbB4 (JM-b/CYT-1)-induced expression and phosphorylation of c-Jun is abrogated by human papillomavirus type 16 E5 protein. Oncogene 26:42–53. doi: 10.1038/sj.onc.1209768. [DOI] [PubMed] [Google Scholar]
- 121.Bachman M, Uribe-Lewis S, Yang X, Williams M, Murrell A, Balasubramanian S. 2014. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat Chem 6:1049–1055. doi: 10.1038/nchem.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yee C, Krishnan-Hewlett I, Baker CC, Schlegel R, Howley PM. 1985. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am J Pathol 119:361–366. [PMC free article] [PubMed] [Google Scholar]
- 123.Shirasawa H, Tomita Y, Sekiya S, Takamizawa H, Simizu B. 1987. Integration and transcription of human papillomavirus type 16 and 18 sequences in cell lines derived from cervical carcinomas. J Gen Virol 68:583–591. doi: 10.1099/0022-1317-68-2-583. [DOI] [PubMed] [Google Scholar]
- 124.Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, Zur Hausen H. 1985. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 314:111–114. doi: 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- 125.Thierry F, Yaniv M. 1987. The BPV1-E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. EMBO J 6:3391–3397. doi: 10.1002/j.1460-2075.1987.tb02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. 1987. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol 61:962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jeon S, Allen-Hoffmann BL, Lambert PF. 1995. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol 69:2989–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Corden SA, Sant-Cassia LJ, Easton AJ, Morris AG. 1999. The integration of HPV-18 DNA in cervical carcinoma. Mol Pathol 52:275–282. doi: 10.1136/mp.52.5.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Collins SI, Constandinou-Williams C, Wen K, Young LS, Roberts S, Murray PG, Woodman CB. 2009. Disruption of the E2 gene is a common and early event in the natural history of cervical human papillomavirus infection: a longitudinal cohort study. Cancer Res 69:3828–3832. doi: 10.1158/0008-5472.CAN-08-3099. [DOI] [PubMed] [Google Scholar]
- 130.Wilson R, Laimins LA. 2005. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods Mol Med 119:157–169. doi: 10.1385/1-59259-982-6:157. [DOI] [PubMed] [Google Scholar]
- 131.Nakamura M, Bodily JM, Beglin M, Kyo S, Inoue M, Laimins LA. 2009. Hypoxia-specific stabilization of HIF-1alpha by human papillomaviruses. Virology 387:442–448. doi: 10.1016/j.virol.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.