

Abstract
Aims
To characterize the population pharmacokinetics (PK) and pharmacodynamics (PD) of the once‐weekly dipeptidyl peptidase‐4 (DPP‐4) inhibitor omarigliptin in healthy subjects and patients with type 2 diabetes mellitus, and use these models to support the dosing recommendation for patient labelling including patients with renal impairment.
Methods
PK and PD were assessed from a total of 9827 omarigliptin concentrations collected from 1387 healthy subjects and patients participating in Phase 1, 2 and 3 studies examining single‐ or multiple‐dose weekly administration of omarigliptin at doses ranging from 0.25 to 400 mg. Population PK and PD analyses were performed using nonlinear mixed effect modelling.
Results
A semi‐mechanistic 2‐compartment model with linear unbound clearance and concentration‐dependent binding of omarigliptin to the DPP‐4 enzyme in both the central and peripheral compartments adequately described omarigliptin PK. Key covariates on omarigliptin PK included reduced unbound clearance with renal impairment. A direct effect sigmoid maximum inhibitory efficacy model adequately described the relationship between omarigliptin plasma concentrations and DPP‐4 inhibition. These models supported the current Japan label instructions that the approved omarigliptin 25‐mg once‐weekly dose be halved in patients with severe renal impairment and in those with end‐stage renal disease. Also, if patients missed a dose, the next dose of omarigliptin should be taken as soon as remembered up to and including the day before the next scheduled dose. No other clinically important covariates were identified.
Conclusion
The models in the present analysis adequately described PK and PD characteristics of omarigliptin and supported the dosing and administration section of the omarigliptin label.
Keywords: dipeptidyl peptidase‐4 inhibitor, omarigliptin, population pharmacokinetics, population pharmacodynamics, type 2 diabetes mellitus
What is already known about this subject
Omarigliptin is a potent oral once‐weekly dipeptidyl peptidase‐4 inhibitor.
Omarigliptin 25 mg once weekly is approved in Japan for the treatment of type 2 diabetes mellitus.
Omarigliptin has a long terminal phase; this does not contribute significantly to the drug's exposure, with minimal accumulation occurring after multiple administration of the approved dose.
What this study adds
This is the first report to describe the pharmacokinetic and pharmacodynamic profiles of omarigliptin using models based on the drug's target (dipeptidyl peptidase‐4 enzyme) binding characteristics.
Except for renal function, no clinically important covariates were identified.
Simulations supported dosing in overall population, including those with renal impairment, and instructions for handling missed dosage.
1. INTRODUCTION
Omarigliptin is a potent oral dipeptidyl peptidase‐4 (DPP‐4) inhibitor with a long plasma half‐life, enabling once‐weekly (q.w.) dosing.1, 2 Omarigliptin (25‐and 12.5‐mg tablets) is approved in Japan as a q.w. DPP‐4 inhibitor for the treatment of adults with type 2 diabetes mellitus (T2DM). In a 12‐week, Phase 2b dose‐range finding study of omarigliptin, the dose‐dependent 2‐hour postmeal glucose reduction, fasting plasma glucose reduction, HbA1c reduction, and safety profile were similar to that of the once‐daily DPP‐4 inhibitor sitagliptin.3 Omarigliptin is rapidly absorbed with apparent high bioavailability.2, 4 The pharmacokinetic (PK) profile of omarigliptin is biphasic with a long terminal half‐life (>100 h).2 The long terminal phase does not contribute significantly to drug exposure for q.w. dosing with the 25‐mg dose, with minimal accumulation occurring after multiple dose administration.2 Following its rapid absorption, omarigliptin undergoes saturable plasma protein binding, with the unbound percentage ranging from ~25% at a plasma concentration of 1 nM to ~76% at 1 μM and higher, and with nearly‐constant plasma protein binding with plasma concentrations ≥50 nM.4, 5 At the approved 25‐mg q.w. dose, for the majority of the dosing duration, omarigliptin plasma concentrations are in the nearly‐constant plasma protein binding range (≥50 nM).4, 5 The concentration‐dependent plasma protein binding of omarigliptin was shown to be attributable to saturable binding of the drug to plasma DPP‐4.4 This protein‐binding behaviour also explains the observed nonlinear PK observed with lower omarigliptin doses and approximate dose‐proportional PK observed with higher clinical omarigliptin doses ranging from 10 to 100 mg.2
The PK and pharmacodynamic (PD) characterization of omarigliptin is based on data from Phase 1, Phase 2b and Japan‐specific and multinational Phase 3 studies, including 1 Phase 3 study in a specific population of patients with renal impairment. The objectives of the analyses described in this paper were to characterize the PK and PD profiles of omarigliptin using a population modelling approach, and to leverage these models to examine the appropriateness of the approved 25 mg q.w. dose in the overall T2DM patient population as well as to evaluate whether dose adjustment is necessary for omarigliptin in population subgroups such as patients with varying degrees of renal impairment. We also explored the potential effect of concurrent medication with anti‐hypertensive drugs and demographic factors (age, weight, race, sex) on omarigliptin exposures. These assessments supported the dosing recommendations of omarigliptin in the type 2 diabetes overall population including those with renal impairment in the approved product labelling. Finally, these models were also used to evaluate the impact of variance in adherence to treatment regimen on treatment effect.
2. METHODS
2.1. Studies
Table 1 summarizes the omarigliptin studies included in the population PK and PD analyses reported in this paper. For population PK analysis, data from 13 Phase 1 studies (Studies 001, 002, 003, 004, 005, 007, 009, 010, 017, 030, 031, 036 and 037), 1 Phase 2b study (Study 006) and 4 Phase 3 studies (Studies 011, 015, 019 and 020) with omarigliptin were pooled. For each of the 4 Phase 3 studies, only data from the base study (and not the extension) were included in the analyses. For PK‐PD analyses of DPP‐4 activity, all available data on DPP‐4 inhibition from 6 Phase 1 studies (Studies 001, 002, 004, 005, 009 and 031) and 1 Phase 2b study (Study 006) with omarigliptin were pooled.
Table 1.
List of omarigliptin studies supporting the final pharmacokinetic and pharmacodynamic (DPP‐4) models
| Study no. | Phase | n | Description | Final analysis | ||
|---|---|---|---|---|---|---|
| PK | DPP‐4 activity | |||||
| Overall | Renal impairment | |||||
| 001 | 1 | 24 | Single rising dose study in healthy young male subjects; omarigliptin 0.5, 1.5, 5, 12.5, 25, 50, 100, 200 and 400 mg | X | X | |
| 002 | 1 | 32 | Multiple rising dose study in healthy young male subjects; omarigliptin 10, 25, 50 and 100 mg | X | X | |
| 003 | 1 | 32 | Single‐dose study in healthy elderly male/female subjects, healthy obese young male/female subjects, and healthy young female subjects; omarigliptin 10 mg | X | ||
| 004 | 1b | 32 | Multiple‐dose study in healthy obese subjects and obese patients with type 2 diabetes; omarigliptin 50 mg q.w. for 4 weeks | X | X | |
| 005 | 1 | Part 1: 16 Part 2: 32 | Single‐ and multiple‐rising dose study in healthy Japanese male subjects; Part 1 (single‐dose): omarigliptin 5, 10, 25, 50, and 100 mg; Part 2 (multiple‐dose): omarigliptin 1, 10, 25 and 50 mg | X | X | |
| 007 | 1 | 6 | Mass balance study in healthy subjects; omarigliptin 25 mg C14 oral solution administered in the fasted state | X | ||
| 009 | 1 | 48 | PK study in patients with varying degrees of renal impairment vs healthy matched controls; single dose omarigliptin 3 mg | X | X | X |
| 010 | 1 | 60 | QT study in healthy subjects; 3‐period cross‐over; moxifloxacin 400 mg as positive control; omarigliptin 25, 175 mg | X | ||
| 017 | 1 | ~36 | Open label study in healthy postmenopausal or oophorectomized female subjects on oral contraceptive; omarigliptin 25 mg q.w. | X | ||
| 030 | 1 | ~12 | Open label study in healthy subjects on metformin; single dose omarigliptin 25 mg | X | ||
| 031 | 1 | 16 | Open‐label, single‐dose study in healthy subjects and patients with moderate hepatic impairment 25 mg | X | X | |
| 036 | 1 | ~12 | Food effect study in healthy subjects; cross‐over; omarigliptin 25 mg tablet in fed vs fasted | X | ||
| 037 | 1 | ~14 | Bioequivalence study in healthy subjects; cross‐over; omarigliptin 25 mg tablet vs capsule | X | ||
| 006 | 2b | 640 (base study) | Global dose‐range finding study with extension in patients with type 2 diabetes; base study: placebo or omarigliptin 0.25, 1, 3, 10 and 25 mg q.w. for 12 weeks | X | X | |
| 011 | 3 | ~400 (base study) | Global placebo‐controlled monotherapy study with extension in patients with type 2 diabetes; base study: placebo or omarigliptin 25 mg q.w. for 24 weeks | X | ||
| 015 | 3 | ~568 total/~300 with PK | Placebo‐controlled study of add‐on of omarigliptin to another oral antidiabetic agent in Japanese patients with type 2 diabetes; placebo or omarigliptin 25 mg q.w. | X | ||
| 019 | 3 | ~210 | Placebo‐controlled monotherapy study in patients with varying degrees of renal impairment; omarigliptin 12.5 mg q.w. | X | X | |
| 020 | 3 | ~410 total/~100 with PK (base study) | Placebo‐ and active (sitagliptin)‐controlled monotherapy study in Japanese patients with type 2 diabetes; base study: omarigliptin 25 mg q.w., sitagliptin 100 mg q.d. or placebo (2:2:1 ratio) for 24 weeks; PK samples taken randomly from first 100 patients to target (~40 omarigliptin‐treated patients) | X | ||
DPP‐4 = dipeptidyl peptidase‐4; PK = pharmacokinetic; q.d. = once daily; q.w. = once weekly
X denotes that study was included in respective analysis.
Single and multiple oral doses of omarigliptin ranging from 0.5 to 400 mg were given in the Phase 1 studies. Omarigliptin was given q.w. for 12 weeks in the Phase 2b Study 006 (0.25, 1, 3, 10 and 25 mg) and as 25 mg q.w. for 24 weeks in Phase 3 Studies 011, 015 and 020. In the Phase 3 Study 019, omarigliptin 25 mg q.w. was administered to patients with moderate renal impairment (estimated glomerular filtration rate [eGFR] ≥30–<60 mL/min/1.73 m2, while omarigliptin 12.5 mg q.w. was given to patients with severe renal impairment (eGFR ≥15–<30 mL/min/1.73 m2) and end‐stage renal disease (ESRD; eGFR <15 mL/min/1.73 m2).
Doses of omarigliptin were given as a liquid solution in Study 001 (cohorts 1 and 2) and as final market composition tablet formulation in Studies 036 and 037 (marketed formulation). In Studies 001 (cohort 3), 002, 003, 004, 005, 006, 009, 010 and 017, doses were given as a Phase 1/2b capsule formulation. In Studies 011, 015, 019, 020, 030, 031 and 037, doses were given as a Phase 1/3 capsule formulation. In Study 007, a carbon 14‐labeled omarigliptin dose in solution was administered.
2.2. PK and PD sampling strategy
Intensive sampling strategies for PK were used in Phase 1 Studies 001, 002, 003, 004, 005, 007, 009, 010, 017, 030, 031, 036 and 037, and sparse samples were collected in Phase 2b Study 006 and Phase 3 Studies 011, 015, 019 and 020.
Intensive sampling for DPP‐4 activity was used in Phase 1 Studies 001, 002, 004, 005, 009 and 031, and sparse sampling was used in Phase 2b Study 006 on Weeks 0 and 12.
2.3. PD response variables
The PD biomarker for modelling was plasma DPP‐4 activity. The levels of this biomarker were reported in mOD/min units. The measured DPP‐4 activity was modelled (as opposed to calculated percent DPP‐4 inhibition). The assay used to measure plasma DPP‐4 activity and the methods used to determine plasma DPP‐4 inhibition have been described previously.2
2.4. Population PK and PD analysis
The PK and PD analyses of omarigliptin were based on data from Phase 1, Phase 2b and Phase 3 studies. Population PK and PD models were developed and updated with new data from successive trials during the course of the programme. The structural components of the PK and PD models remained the same across all iterations with comparable values for parameter estimates (see supplementary material for details of the model).
The structural model for PK characterization was parameterized as an oral 2‐compartment model with each compartment having an additional binding compartment to describe the saturable binding of omarigliptin, as shown in Figure 1. This model characterized PK as a linear clearance system; however, the inclusion of saturable binding in plasma and tissues allowed adequate characterization of nonlinearity in plasma PK. An analysis of nonclinical experiments had concluded that the binding partner for saturable behaviour is probably DPP‐4 enzyme.4 In both of the additional binding compartments, binding of omarigliptin to the binding partner was considered to be reversible and competitive. Similar PK models with the feature of saturable protein binding have previously been successfully used to characterize apparent nonlinearities in pharmacokinetics for other DPP‐4 inhibitors, such as linagliptin.6
Figure 1.
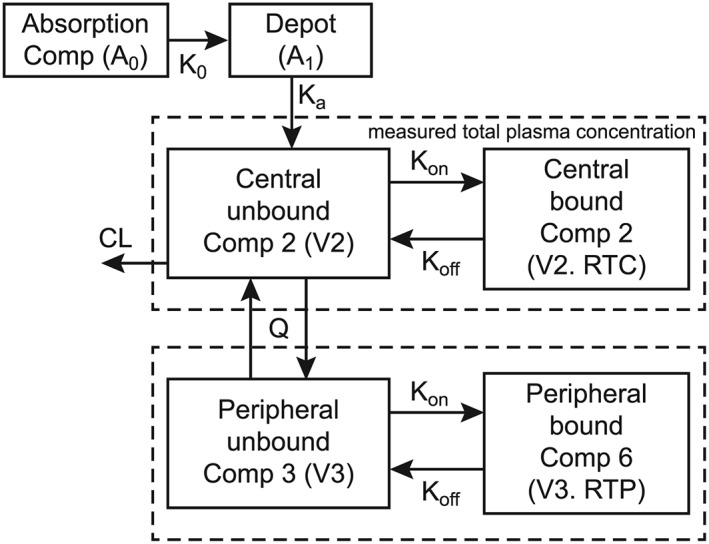
Schematic of final pharmacokinetic structural model. A0 = amount of drug in absorption compartment; K0 = zero‐order absorption, for which duration (D1) was estimated as the parameter; A1 = amount of drug in the transit compartment; CL = apparent clearance; Comp = compartment; Ka = first‐order absorption rate constant; Koff = dissociation rate constant; Kon = association rate constant; Q = intercompartmental clearance; RTC = central enzyme concentration; RTP = peripheral enzyme concentration; V2 = apparent central volume of distribution; V3 = apparent peripheral volume of distribution. Total plasma concentrations as measured by bioanalysis are represented by the sum of the concentrations in the plasma unbound and bound compartments
With respect to covariates, the base PK model included 2 covariate effects: body weight and eGFR. Body weight was assumed to influence the parameters of apparent unbound renal clearance (CL/F), apparent intercompartmental clearance (Q/F), apparent central and peripheral volumes of distribution (V2/F and V3/F, respectively) according to standard allometric relationships (body weight exponents were fixed at 0.75 and 1 for clearance‐ and volume‐of‐distribution‐related terms, respectively). Since the elimination of omarigliptin is primarily through renal elimination of intact parent drug, the effect of renal function (eGFR) on CL/F was also included as an additional covariate and this relationship was estimated based on available data. Additional demographic and clinical covariates were tested on CL/F and V2/F, including concomitant medications (for drugs that were coadministered for the entire duration of omarigliptin PK sampling and with usage frequency >5%), sex, race (white, Asian, black, other), ethnicity and patient status. For covariate evaluation, a process of forward selection followed by backward elimination was implemented, with a P‐value of <.05 for forward addition and a P‐value of <.001 for backward elimination.
The PK‐PD analysis used a sequential modelling approach, relating the individual‐predicted plasma concentrations from the final population PK model (with covariates) to observed DPP‐4 activity (assay value, not calculated inhibition) with a simple maximum inhibitory efficacy (Emax) model with intersubject variance on select PK‐PD parameters. The PK‐PD analysis evaluated the relationship between total omarigliptin plasma concentration and the DPP‐4 activity. The PD effect in this model was not assumed to be driven by the binding of unbound omarigliptin plasma concentration to the binding partner in the saturable compartment of the PK model. It has been shown previously for linagliptin, another DPP‐4 inhibitor, that both PK‐PD model structures yield similar results.6
Covariates evaluated in the analyses of DPP‐4 were race (white, Asian, black, other), ethnicity, age, sex and patient status. Similar to the PK model, covariate evaluation was based on forward selection followed by backward elimination.
2.5. Predictive performance evaluation of final models
The final PK and PK‐PD models were qualified using a simulation‐based, visual predictive check methodology to assess concordance between the model‐based simulated data and the observed data, stratified by study and dose group. One thousand replicate simulations of the analysis dataset were performed. Statistics of interest were calculated from the simulated and observed data for comparison; for example, the 5th, 50th (median) and 95th percentiles of the distributions of simulated and observed plasma concentrations or PD biomarker (DPP‐4 activity) were compared. These percentiles were plotted vs time and overlaid on the original observed data and/or percentiles based on the observed data to visually assess concordance between the model‐based simulated data and the observed data.
2.6. Simulations to assess impact of covariates and missed dosing
Simulations were performed using the integrated population PK‐PD models to: (i) determine parameters characterizing the PK and PD profile of omarigliptin; (ii) provide support for the proposed 25 mg q.w. dose in the overall T2DM population; (iii) determine the dosing in specific population subgroups including those with renal impairment; and (iv) determine the appropriate dosing instructions for patients who miss their scheduled doses.
For objectives (i), (ii) and (iii), stochastic simulations of 5000 patients with T2DM administered each of 8 dosing regimens (omarigliptin 0, 0.25, 1, 3, 5, 10, 12.5 and 25 mg q.w.) were performed to predict the time to steady state, omarigliptin exposure, and the corresponding range of expected DPP‐4 responses. These 5000 virtual patients were created by resampling the demographic and laboratory data from Phase 2b Study 006 and Phase 3 Studies 011, 015 and 020. In addition, 1000 healthy virtual subjects were created by resampling covariate data from Phase 1 Studies 001, 002, 003, 004, 005, 007, 009, 010, 017, 030, 031, 036 and 037.
Final integrated population PK‐PD models were then used to simulate the PK and DPP‐4 outcomes for these patients at Weeks 12 and 24. The exposure measures of area under the concentration–time curve up to 168 hours (AUC0–168) and maximal observed drug plasma concentration (Cmax) were calculated using the log trapezoidal rule. Simulated exposure measures at steady state for the omarigliptin 25‐mg dose were used for the evaluation of the impact of baseline clinical or demographic factors (intrinsic and extrinsic factors) on PK. DPP‐4 inhibition profiles between healthy subjects and patients with T2DM were also compared.
Additional PK simulations were performed to understand the effect of renal impairment on the exposure of omarigliptin. Demographic and laboratory data from subjects enrolled in Studies 006, 009, 011, 015, 019 and 020 were used to obtain real patient data for moderate and severe renal impairment and ESRD. Patients with moderate and severe renal impairment and ESRD were re‐sampled with replacement to obtain 1200 virtual patients (800 with moderate renal impairment, 200 with severe renal impairment and 200 with ESRD) for PK simulation. Patients with normal renal function and mild renal impairment were obtained from the above described stochastic simulations of 5000 patients with T2DM. Each virtual patient was separately administered omarigliptin 12.5 and 25 mg q.w. for 24 weeks.
For objective (iv), simulations were performed for a population of 5000 virtual patients with T2DM for 3 different scenarios of missing doses to understand the effect of missing doses on the concentrations of omarigliptin as well as on DPP‐4 inhibition. These scenarios were selected to understand the impact of possible real‐world variations in dosing regimen on drug effect, and included the following: (i) administer dose 6 days late at steady‐state, i.e. normal weekly dosing week 1–12, week 13 dose 6 days late, followed by normal dosing in week 14 (week 13 and 14 doses administered on consecutive days); (ii) normal weekly dosing week 1–12, week 13 dose 6 days late followed by a new schedule for week 14, 7 days after the previous dose; and (iii) normal weekly dosing week 1–12, missed week 13 dose, followed by normal dosing in week 14. Scenario (i) represents the worst‐case scenario for the delay without completely skipping the dose.
2.7. Software
Data preparation was performed using SAS Version 9.2 or R Version 3.01. PK‐PD modelling was performed using NONMEM, Version 7, Level 1.2.i. All data analyses and presentations of data were performed using SAS Version 9.2 and KIWI Version 1.1 (Cognigen Corporation, Buffalo, NY, USA).
3. RESULTS
3.1. Data
A total of 9827 omarigliptin concentrations collected from 1387 individuals participating in Phase 1 (Studies 001, 002, 003, 004, 005, 007, 009, 010, 017, 030, 031, 036 and 037), Phase 2b (Study 006), and Phase 3 (Studies 011, 015, 019 and 020) studies examining single‐ or multiple‐dose administration of omarigliptin ranging from 0.25 to 400 mg, were used for the population PK analyses. The demographics and covariate characteristics of the healthy subjects and patients included in the population PK analysis are presented by study phase in Table 2. Overall, the baseline characteristics of the patients included in this analysis were representative of the general T2DM patient population.7
Table 2.
Demographic and covariate characteristics, by study phase, in the pharmacokinetic analysis of omarigliptin
| Characteristic | Phase I n = 327 | Phase IIb n = 552 | Phase III n = 508 | Overall n = 1387 |
|---|---|---|---|---|
| Age, y | 41.9 ± 15.5 | 55.1 ± 8.9 | 60.7 ± 10.2 | 54.1 ± 13.3 |
| BMI, kg/m2 | 26.4 ± 3.7 | 29.8 ± 5.2 | 27.8 ± 5.5 | 28.3 ± 5.2 |
| eGFR, mL/min/1.73 m2 | 96.3 ± 23.4 | 81.6 ± 15.4 | 73.2 ± 28.1 | 82.0 ± 24.3 |
| Body weight, kg | 74.5 ± 13.0 | 82.2 ± 17.6 | 75.6 ± 18.8 | 78.0 ± 17.5 |
| Race | ||||
| White | 109 (33.3) | 222 (40.2) | 146 (28.7) | 477 (34.4) |
| Black | 35 (10.7) | 22 (4.0) | 10 (2.0) | 67 (4.8) |
| Asian | 42 (12.8) | 150 (27.2) | 338 (66.5) | 530 (38.2) |
| Hispanic | 135 (41.3) | 146 (26.4) | 9 (1.8) | 290 (20.9) |
| Other | 6 (1.8) | 12 (2.2) | 5 (1.0) | 23 (1.7) |
| Sex | ||||
| Male | 199 (60.9) | 314 (56.9) | 323 (63.6) | 836 (60.3) |
| Female | 128 (39.1) | 238 (43.1) | 185 (36.4) | 551 (39.7) |
| Fed statea | ||||
| Fasted | 327 (92.6) | 552 (100) | 508 (100) | 1387 (98.2) |
| Fed | 26 (7.4) | 0 | 0 | 26 (1.8) |
| Formulationa | ||||
| Liquid solution | 16 (4.7) | 0 | 0 | 16 (1.1) |
| Phase I/IIb capsule | 254 (74.3) | 552 (100) | 0 | 806 (57.5) |
| FMC tablet | 29 (8.5) | 0 | 0 | 29 (2.1) |
| Phase I/III capsule | 43 (12.6) | 0 | 508 (100) | 551 (39.3) |
| Subjects | ||||
| Healthy subjects | 321 (98.2) | 0 | 0 | 321 (23.1) |
| Type 2 diabetic patients | 6 (1.8) | 552 (100) | 508 (100) | 1066 (76.9) |
| Renal function category | ||||
| Normal | 204 (62.4) | 137 (24.8) | 135 (26.6) | 476 (34.3) |
| Mild renal impairment | 111 (33.9) | 392 (71.0) | 244 (48.0) | 747 (53.9) |
| Moderate renal impairment | 6 (1.8) | 23 (4.2) | 76 (15.0) | 105 (7.6) |
| Severe renal impairment | 6 (1.8) | 0 | 33 (6.5) | 39 (2.8) |
| End‐stage renal disease | 0 | 0 | 20 (3.9) | 20 (1.4) |
Data are expressed as mean ± SD or n (%)
Subjects can contribute more than once based on study design.
BMI = body mass index; eGFR = estimated glomerular filtration rate; FMC = final market composition
The population PK‐PD analyses characterizing the inhibition of DPP‐4 as a function of omarigliptin plasma concentration were performed on data obtained from patients with available omarigliptin post hoc Bayesian PK parameters from Phase 1 (Studies 001, 002, 004, 005, 009, and 031) and Phase 2b (Study 006) studies as well as on data from patients who received placebo treatment in these studies. A total of 5435 DPP‐4 activity observations from 800 individuals were included in the PK‐PD DPP‐4 dataset for these analyses. The PK‐PD analysis population was composed of healthy subjects (22%) and patients with T2DM (78%).
3.2. Population PK analysis
The final PK model provided a good fit to the data for all studies, including the Phase 3 Studies 011, 015, 019 and 020. Population PK parameter estimates for omarigliptin are shown in Table 3. All fixed and random effect parameters were estimated with good precision (% standard error of the mean [%SEM] ≤26%; other than %SEM for association rate constant Kon, which was 54%). Type of formulation was found to have a minor effect on the initial (zero‐order) absorption rate; the duration of zero‐order absorption (D1) with the oral solution was negligible, and the duration for the Phase 1/2b capsule and final marketed tablet was decreased by 41% to approximately 0.5 hours when compared to the Phase 1/3 capsule formulation. The magnitude of the intersubject variability was large for D1, amount of binding partner in binding compartment linked with central compartment RTC, and absorption rate constant Ka (% coefficient of variation [%CV]: ≥42%), but was moderate for CL/F (27%) and RTP (30%). Eta‐shrinkage was moderate to high for all PK parameters, ranging from 17% for CL/F to 77% for RTP. Residual error variability for both the Phase 1 and Phase 2b/3 data was moderate (23% and 32%, respectively). In general, goodness‐of‐fit plots indicated an unbiased fit across the range of doses evaluated in these studies. Visual predictive check plots illustrated that both the central tendency as well as the magnitude of variability in concentrations were well described by the model (Figure 2).
Table 3.
Parameter estimates and standard errors for the final population pharmacokinetic model for omarigliptin
| Parameter | Final parameter estimate | Interindividual variability | ||
|---|---|---|---|---|
| Typical value | %SEM | Magnitude | %SEM | |
| D1: Zero‐order duration (h) | 0.88 | 5.1 | 75 %CV | 13 |
| FD1: Proportional shift in D1 when FED = 1 | 4.0 | 4.8 | ||
| F0D1: Fixed duration (~0 h) when FORM = 0 (OSF) | −1.0 | FIXED | ||
| F1D1: Proportional shift in D1 when FORM = 1 or 2 (DFC/tablet) | −0.41 | 5.9 | ||
| CL/F: Log apparent unbound renal clearance (L/h) | 1.1 | 1.2 | 27 %CV | 3.9 |
| eGFR: Power coefficient for eGFR on clearance | 0.44 | 3.3 | ||
| V2/F: Log apparent central volume of distribution (L) | 4.5 | 0.13 | NE | NA |
| Effect of patient status on V2 | −0.085 | 15 | ||
| V3/F: Log apparent peripheral volume of distribution (L) | 3.6 | 0.85 | ||
| Q/F: Log apparent intercompartmental clearance (L/h) | 3.3 | 1.7 | ||
| RTC: Log central enzyme concentration (nM) | 1.9 | 2.4 | 42 %CV | 9.4 |
| RTP: Log peripheral enzyme concentration (nM) | 4.0 | 1.7 | 30 %CV | 26 |
| KON: Log association rate constant (1/h × nM) | 0.61 | 54 | NE | NA |
| KEQ: Log equilibrium constant (nM) | 0.92 | 7.5 | ||
| KA: Absorption rate constant (1/h) | 1.9 | 5.7 | 71 %CV | 11 |
| Residual error for phase 1 dataa | 0.055 | 1.0 | NA | NA |
| Residual error for phase 2/3 dataa | 0.10 | 1.8 | NA | NA |
%CV = coefficient of variation expressed as a percentage; %SEM = standard error of the mean expressed as a percentage; DFC = dry‐filled capsule formulation; eGFR = estimated glomerular filtration rate; NA = not available; NE = not estimated; OSF = on‐site formulation
Residual variability (%CV) for Phase 1 data was √0.055 × 100 = 23% and for Phase 2/3 data was √0.10 × 100 = 32%.
Figure 2.
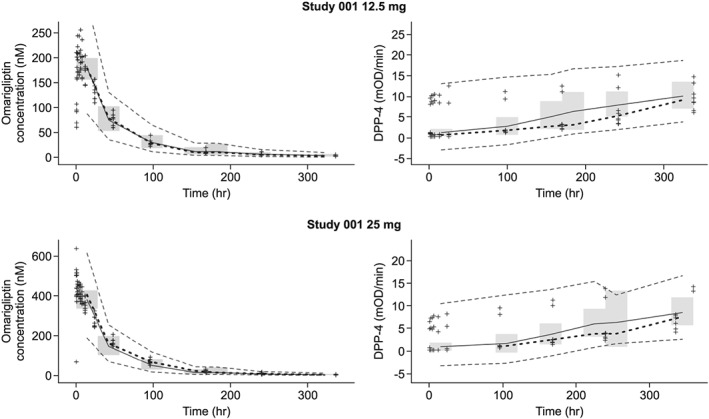
Representative visual predictive check plots for the final pharmacokinetic and pharmacokinetic–pharmacodynamic models. The median (solid grey line) and 90% prediction interval (dashed grey lines) were derived from the simulated datasets (n = 1000 replicates). These were overlaid on the observed omarigliptin plasma concentration or observed dipeptidyl peptidase‐4 (DPP‐4) activity vs time data (+ symbols) stratified by study and dose. The 95% confidence interval of the predicted median is shown as the shaded region. The median of the observed omarigliptin plasma concentration or observed DPP‐4 concentration vs time data is shown as a black dashed line
In addition to the effect of body weight on PK parameters captured as allometric exponents, the following additional covariates were found to be statistically significant for PK characterization: (i) impairment of renal function caused a reduction in unbound clearance, with an estimated power coefficient on eGFR of 0.44; (ii) the apparent volume of distribution of the central compartment was 9.1% higher in T2DM patients compared to healthy subjects; (iii) food delayed the duration of zero order absorption (D1 parameter) by approximately 4.4 hours, but without any marked differences in Cmax (~9% reduction in median Cmax). No statistically significant influence of race (white, Asian, black, other), ethnicity, age, sex or comedication was found for omarigliptin PK.
The simulations using the final model demonstrated that accumulation of AUC0–168, Cmax and Ctrough was minimal across dose groups and that near steady‐state exposures were reached after the first dose. This was consistent with the observed accumulation ratios (Week 3/Week 1) from early clinical studies for omarigliptin, ranging from 1.03 to 1.35 for AUC0–168, 0.87 to 1.36 for Cmax, and 0.97 to 1.24 for Ctrough.
3.3. Population PK/PD (DPP‐4 inhibition) analyses
The PK‐PD model was a sigmoid Emax model, which adequately described the relationship between omarigliptin concentrations and DPP‐4 activity. The maximal level of attainable DPP‐4 inhibition was fixed at 100%. Both fixed and random effect parameters were estimated with good precision (%SEM <30% for fixed effects and %SEM <40% for random effect terms). The magnitude of intersubject variability was moderate for both baseline DPP‐4 activity (%CV 22.5%) and omarigliptin plasma concentration, resulting in IC50 (%CV 37.3%; Table 4). Eta‐shrinkage was low for baseline DPP‐4 activity (11%) but higher for IC50 (46%), reflecting the expected increase in variability of the information content for individual subjects relative to the estimation of IC50. Residual variability was best described by a combined additive and proportional error model. With this error model structure, the residual variance ranged from >200% CV at the lowest DPP‐4 activity levels of 1 mOD/min (e.g. very high DPP‐4 inhibition) to only 8% CV at DPP‐4 activity levels of 40 mOD/min (e.g. baseline, 0% inhibition; Table 4). High residual variability at low DPP‐4 levels could be because of increased chance of an observation being censored at lower values. The visual predictive check plots (Figure 2) show that the prediction intervals from the simulated data covered the majority of the observed data, indicating that the magnitude of variability in observed DPP‐4 response was accurately characterized by the model.
Table 4.
Parameter estimates and standard errors for the final pharmacokinetic–pharmacodynamic model for omarigliptin
| Parameter | Final parameter estimate | Interindividual variability | ||
|---|---|---|---|---|
| Typical value | %SEM | Magnitude | %SEM | |
| BSLN (mOD/min) | 20.1 | 0.911 | 22.5 %CV | 8.18 |
| BSLN: Asian race effect on BSLN DPP‐4 | 0.137 | 16.3 | ||
| BSLN: patient status effect on BSLN DPP‐4 | 0.0876 | 22.8 | ||
| BSLN: female sex effect on BSLN DPP‐4 | 0.0740 | 28.0 | ||
| IC50 (nM) | 3.83 | 2.65 | 37.3 %CV | 38.9 |
| Imax | 1.00 | FIXED | NE | NA |
| Hill coefficient | 1.10 | 2.34 | NE | NA |
| RVprop | 0.0616 | 22.9 | NA | NA |
| RVadd | 2.16 | 7.14 | ||
%CV, coefficient of variation expressed as a percentage; %SEM, standard error of the mean expressed as a percentage; BSLN, baseline; DPP, dipeptidyl peptidase‐4; IC50, concentration resulting in 50% of the maximum inhibition; Imax, maximum inhibition; NA, not applicable; NE, not estimated; RV, residual variability; RVadd, additive component of additive plus proportional RV; RVprop, proportional component of additive plus proportional RV.
The residual variability was calculated using the following equation: 100 × SQRT((2.16 × 2.16) + ((0.0616 × F)^2))/F, where F is the individual predicted value. The residual variability (%CV) ranged from 217 to 8.20% for F values (DPP‐4 activity levels) of 1–40 mOD/min, respectively
The omarigliptin plasma concentrations resulting in 50% (IC50) and 80% (IC80) of maximum DPP‐4 inhibition from the PK‐PD model were 3.83 and 13.6 nM, respectively. A scatterplot of the percent DPP‐4 inhibition vs omarigliptin plasma concentration for healthy subjects and T2DM patients administered omarigliptin 25 mg from Studies 001, 002 and 006, with simulated median and 5th and 95th percentiles overlaid, is shown in Figure 3.
Figure 3.
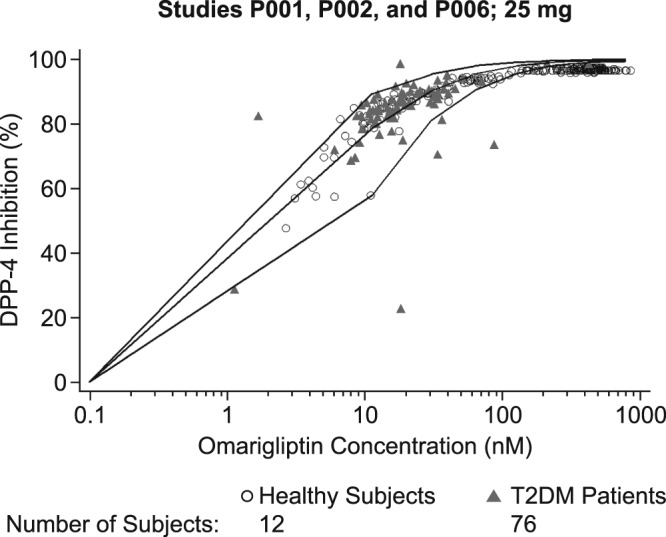
Scatterplot of percent dipeptidyl peptidase‐4 (DPP‐4) inhibition vs omarigliptin plasma concentration for subjects administered omarigliptin 25 mg once weekly, by patient status. The lines represent the simulated median as well as 5th and 95th percentiles for percent DPP‐4 inhibition. T2DM = type 2 diabetes mellitus
The following covariates were identified in the PD model: (i) Asian race, patient status, and male sex were all found to influence significantly the baseline DPP‐4 activity level, with higher baseline values predicted in Asian subjects compared to whites, in T2DM patients compared to healthy subjects, and in females compared to males; the predicted differences in baseline DPP‐4 activity across these subgroup comparisons were all <14%. (ii) No statistically significant influence of other races or ethnicity (white, Asian, black, Hispanic, other) or age was found for DPP‐4 inhibition with omarigliptin.
3.4. Support for 25 mg q.w. dose in T2DM patients
The predictions for DPP‐4 inhibition in T2DM patients showed that the systemic concentrations with the dose of 25 mg q.w. maintained DPP‐4 inhibition above 85% at the median level for the entire weekly dosing interval, and above 80% for most of the dosing interval in all patients (Figure 3). These results were consistent with the multiple‐dose study evaluating doses of 10–100 mg q.w. in which DPP‐4 inhibition of >80% was maintained up to 168 hours post dose at steady‐state (see supplementary material).2 These results were also consistent with the observed DPP‐4 inhibition of 80.7% for 25‐mg q.w. dose at week 12 in Phase 2 dose‐ranging study.3 The DPP‐4 inhibition above 80% is generally considered to be associated with full clinical response, supporting the adequacy of 25‐mg q.w. dose.
3.5. Impact of intrinsic and extrinsic factors on systemic exposure of omarigliptin and dosing recommendations
Effects of potential baseline clinical and demographic factors on PK were evaluated by simulation of patient subgroups to determine the need for dose adjustment for omarigliptin.
The subgroups examined for health status, race, age, sex and concomitant medication use did not have clinically meaningful impact on the PK profile for omarigliptin 25 mg q.w (Figure 4). Across the body weight range of approximately 40–180 kg, the predicted steady‐state AUC0–168 range remained generally within the margins of 0.5‐ and 2.0‐fold relative to the typical patient with T2DM. Calculated steady‐state AUC0–168 and Cmax values of omarigliptin were <20% different for comparison between T2DM patients compared with healthy subjects, Black/Asian/Hispanic compared with White race, obese compared with nonobese subjects, elderly (≥65 years) compared with nonelderly (<65 years) subjects, female compared with male subjects, subjects receiving concomitant amlodipine compared with subjects not on amlodipine, subjects receiving concomitant atorvastatin compared with subjects not on atorvastatin, and subjects receiving concomitant diuretics compared with subjects not on diuretics.
Figure 4.
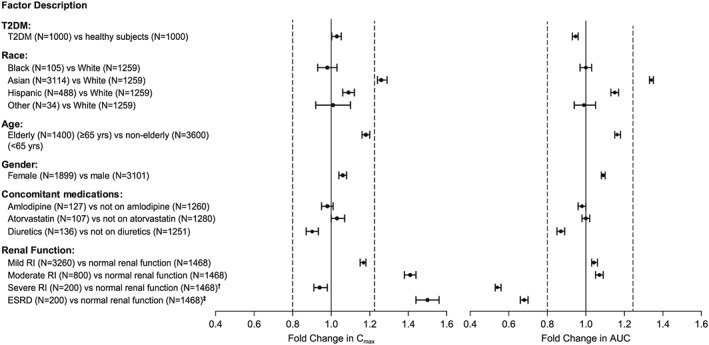
Effects of intrinsic and extrinsic factors on the pharmacokinetics of omarigliptin, based on simulations from the final population pharmacokinetic model. Horizontal axes show fold difference (geometric mean ratio [GMR] and 90% CI) relative to control in omarigliptin Cmax and AUC. Vertical dashed lines at 0.8 and 1.25 indicate typical lower and upper limits for bioequivalence comparison. Omarigliptin dose = 25 mg q.w. unless otherwise specified. †12.5 mg q.w. for severe RI group; 25 mg q.w. for normal renal function group. ‡12.5 mg q.w. for ESRD group; 25 mg q.w. for normal renal function group. T2DM = type 2 diabetes mellitus; RI = renal impairment; ESRD = end‐stage renal disease; AUC = area under the concentration–time curve; Cmax = maximal observed drug plasma concentration
Consistent with the disposition profile of omarigliptin, renal function was identified as a clinically meaningful intrinsic factor, with a clinically meaningful reduction in plasma clearance in patients with severe impairment and ESRD. The steady‐state AUC0–168 in patients with severe renal impairment and ESRD with the 25‐mg q.w. dose was 1.82‐fold and 2.94‐fold higher, respectively, compared to AUC observed at the 25‐mg q.w. dose in patients with normal renal function (Figure 4). A significant proportion (>35%) of patients with severe renal impairment and the majority of those with ESRD exceeded the 2‐fold exposure margin relative to exposure in normal renal function on the omarigliptin 25‐mg q.w. dose (Figure 5). Therefore, the recommended omarigliptin dose in severe renal impairment and ESRD was adjusted to 12.5 mg q.w. Simulations with this dose adjustment demonstrated steady‐state AUC0–168 in these subgroups comparable to that observed in subjects with normal renal function receiving an omarigliptin 25‐mg q.w. dose (exposures within 1.5‐fold range, see Figure 4). In patients with mild and moderate renal impairment, omarigliptin exposures for the 25‐mg q.w. dose were comparable to that observed in patients with normal renal function, with mean increases in steady‐state AUC0–168 of 1.17‐fold and 1.41‐fold, respectively (Figure 4 and Figure 5).
Figure 5.
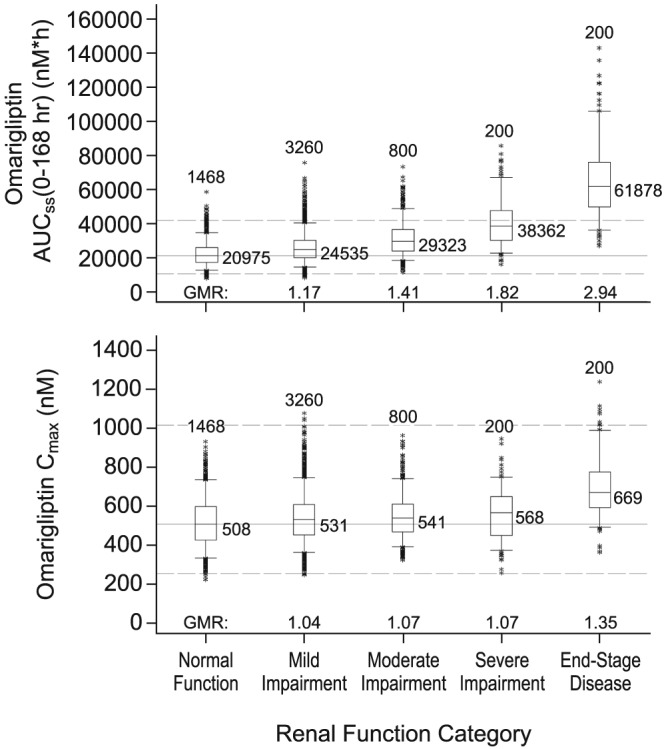
Effect of renal impairment on simulated steady‐state exposure with omarigliptin 25‐mg once‐weekly administration in patients with type 2 diabetes. Boxes are 25th, 50th and 75th percentiles. Whiskers are 5th to 95th percentiles. Asterisks show data points outside this range. The number of simulated patients is shown above each box. The median omarigliptin exposure is shown to the right of each box. The solid and dashed horizontal lines represent the median exposure and bounds of 0.5‐ and 2‐fold exposure for T2DM patients with normal renal function. AUCss = area under the concentration‐time curve from time 0–168 hours at steady‐state; Cmax = maximal observed drug plasma concentration; GMR = geometric mean ratio
With respect to the effect of baseline clinical and demographic factors on the PD profile of the approved omarigliptin 25‐mg q.w. clinical dose, as stated above, T2DM patients were found to have higher baseline DPP‐4 activity compared with healthy subjects by 9.4%. Despite this difference in baseline DPP‐4 activity, omarigliptin's DPP‐4 inhibition profile was not substantially altered between healthy subjects and patients with T2DM, as shown by the PK‐PD simulations in Figure 6.
Figure 6.
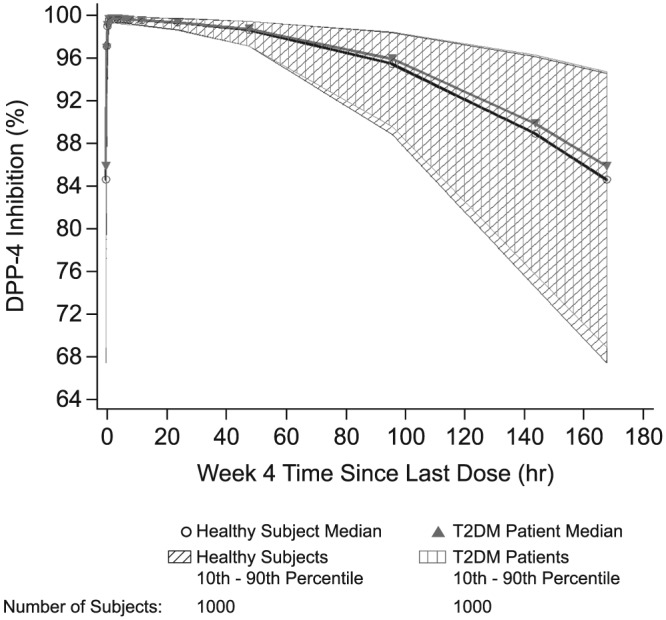
Population pharmacokinetic–pharmacodynamic simulation of dipeptidyl peptidase‐4 (DPP‐4) activity vs time in healthy subjects and patients with type 2 diabetes mellitus (T2DM). Data shown are simulated median and 80% CI for DPP‐4 inhibition profiles at week 4 based on stochastic simulations for 1000 healthy subjects and 1000 T2DM patients
3.6. Effect of missed doses on omarigliptin PK and PD
To evaluate the impact of missing a dose of omarigliptin and taking the missed dose later, omarigliptin Cmax and AUC0–168 at steady state and DPP‐4 inhibition were assessed for missed dose scenarios. PK exposures were approximately 50% greater when the missed dose of omarigliptin 25 mg was taken 6 days late and 1 day prior to the next regular dose (worst case scenario for the delay without completely skipping the dose) compared to the steady‐state Cmax and AUC0–168 for routine q.w. administration of omarigliptin 25 mg (Figure 7). Exposures much higher than these were well tolerated in an early clinical pharmacology multiple dose study, which tested doses up to 100 mg, and in a thorough QT study, which tested a dose of 175 mg.2, 8 The simulated DPP‐4 inhibition profiles showed that delaying or missing a scheduled omarigliptin dose may reduce DPP‐4 inhibition to <80%; however, DPP‐4 inhibition returned to levels generally associated with full clinical response (>80%) immediately after the next dose was administered (Figure 8). These results supported the labelling recommendation that omarigliptin dose could be taken any day up until the day prior to the next scheduled dose.
Figure 7.
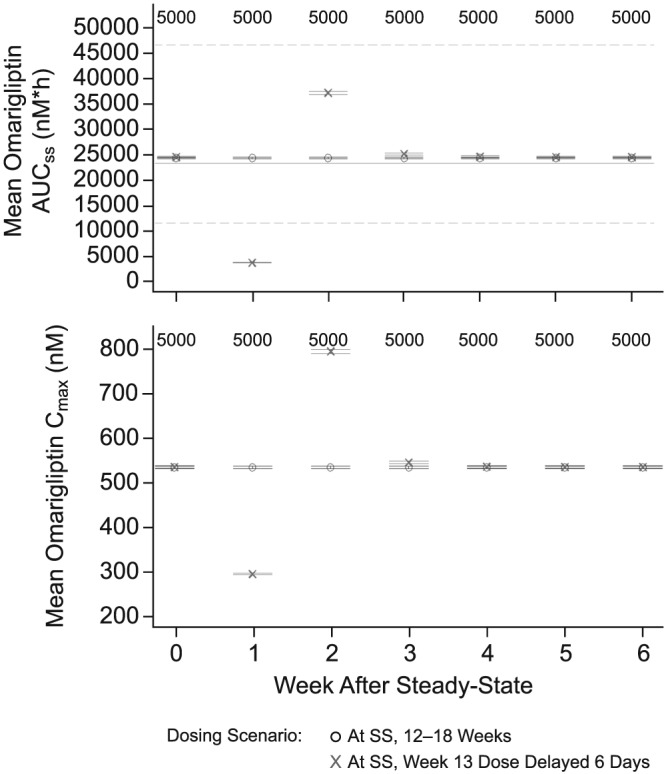
Simulated mean omarigliptin exposure measures vs week following 25‐mg once‐weekly dosing with a missed dose taken 6 days late (1 day prior to the next scheduled dose on the 13th week) compared to routine once‐weekly administration
Figure 8.
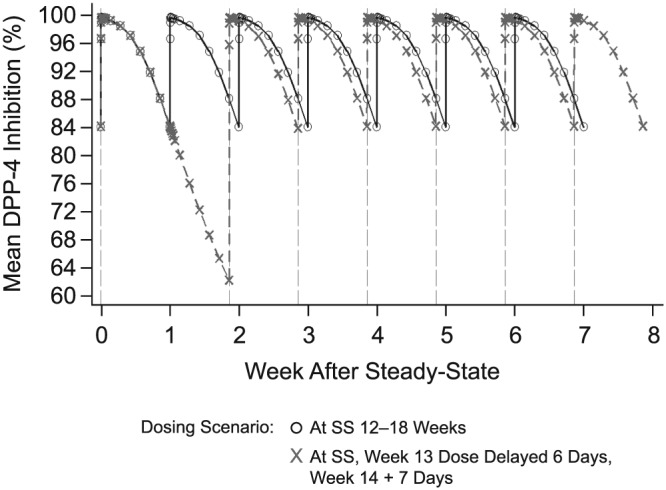
Simulated mean dipeptidyl peptidase‐4 (DPP‐4) inhibition vs week following omarigliptin 25‐mg once‐weekly dosing, with a missed dose taken 6 days late and the next dose taken 1 day later (new schedule)
4. DISCUSSION
This is the first manuscript to comprehensively characterize the PK and PD profiles of the once‐weekly DPP‐4 inhibitor omarigliptin using models based on the drug target (DPP‐4 enzyme) binding characteristics. This final PK model with saturable binding of omarigliptin to the DPP‐4 enzyme in plasma and tissues (central and peripheral compartments) and linear unbound clearance adequately described the nonlinear plasma PK observed with the lower omarigliptin doses investigated.2 This supports the conclusion that while omarigliptin has a concentration‐dependent volume of distribution and saturable protein binding, the clearance mechanism is constant.
PK‐PD analyses supported the dosing recommendation in the omarigliptin Japan label of 25 mg q.w. in T2DM patients with normal renal function and mild and moderate renal impairment, and 12.5 mg q.w. in patients with severe renal impairment and ESRD.5 Further, PK‐PD analyses also supported the labelling recommendations on missed dose in which patients are instructed that, if they miss a dose, they should take their missed dose as soon as they remember up to and including the day prior to the next scheduled dose and then take their next weekly dose on their regular day.
The covariate analysis showed that, except for the impact of renal function, no other covariate had a meaningful impact on PK exposures across the subgroups examined for race, body weight, age, concomitant medication use as well as formulations (see Figure 4 and supplementary material). Patient status was identified as a minor influential covariate on apparent volume of distribution of the central compartment, with this parameter being 9.1% higher in T2DM patients compared to healthy subjects. This finding may be related to differences in body composition and corresponding distribution in diabetics, which is not entirely captured by the inclusion of body weight and eGFR as covariates. The minor change in volume of distribution and corresponding slight reduction in Cmax was not considered to be clinically meaningful, given that Cmax for the omarigliptin 25‐mg dose was substantially above the concentrations required for maximum inhibition of DPP‐4. In addition, slight variations in the exposure of omarigliptin are not considered clinically meaningful for safety given that exposures much higher than those for the 25‐mg dose were well tolerated in early clinical studies, which tested doses up to 100 mg, and in a thorough QT study, which tested a dose of 175 mg.2, 8
These findings of covariate analysis are consistent with the observations from early clinical studies where no clinically meaningful differences were observed in PK and DPP‐4 activity based on age, sex or obesity. No clinically meaningful differences in formulations were confirmed in a dedicated bioequivalence study, which demonstrated comparability in PK exposures from the Phase 3 capsule and final market composition formulations, with bioequivalence criteria being met for all PK parameters Cmax, AUC0–168 and AUC extrapolated to infinity.
As expected based on elimination of omarigliptin by renal pathways, renal function was a significant covariate on apparent clearance. The findings about the impact of renal function on PK from this analysis are in general agreement with the observations from an earlier clinical pharmacology renal study with a lower, single 3‐mg dose of omarigliptin. In that study, a mean 1.56‐fold increase in AUC0–168 was observed in subjects with severe renal impairment and a mean 1.89‐ to 1.97‐fold increase in AUC0–168 was observed in ESRD subjects requiring dialysis. Analysis of dialysate samples from ESRD patients in this study had demonstrated that omarigliptin is not meaningfully removed by routine haemodialysis, regardless of whether it is dosed immediately after dialysis (~5% removal) or dialysis is initiated at the time of maximum plasma concentration (Tmax) for omarigliptin (~15% removal; data not shown).
It is worth noting that worsening renal function has notably less impact on the renal clearance of omarigliptin compared to that reported for other DPP‐4 inhibitors that are also primarily cleared by the kidney, namely, sitagliptin, alogliptin, as well as the only other approved once‐weekly DPP‐4 inhibitor, trelagliptin. Omarigliptin has up to a 2‐fold increase in mean exposures in severe renal function relative to normal renal function, while these other drugs have increases of 3‐fold or higher (Table 5). While the exact reason for this difference is not known, it could be possibly explained by differences in the overall contribution of renal clearance to total body clearance for each DPP‐4 inhibitor. Additionally, the renal excretion mechanism of omarigliptin appears to be passive filtration with net reabsorption by the tubules of the kidney, while other DPP‐4 inhibitors appear to be actively secreted by the tubules (Table 6). In the setting of renal impairment, active tubular secretion of sitagliptin, alogliptin and trelagliptin would be impaired with the reduction in GFR resulting in significant reduction of renal clearance. The reabsorption of omarigliptin per nephron would also be expected to decrease with renal impairment, as it is known that the fractional excretion of sodium increases and sodium/water reabsorption per nephron decreases with decline in GFR. However, possibly because of differences in renal clearance mechanisms, the resulting impact of decreasing GFR on renal clearance of omarigliptin is less compared to other DPP‐4 inhibitors (Table 6).
Table 5.
Ratios of reported areas under the concentration–time curve (AUCs) between various levels of renal impairment (mild, moderate, severe) for omarigliptin, sitagliptin, alogliptin and trelagliptin
| Level of renal impairment | Ratio of AUC (renal impaired/normal renal function) | |||
|---|---|---|---|---|
| Omarigliptin | Sitagliptin | Alogliptin | Trelagliptin | |
| Mild impairment | 1.17 | 1.1–1.6 | 1.2 | 1.56 |
| Moderate impairment | 1.41 | ~2 | ~2 | 2.06 |
| Severe impairment | 1.82 | ~4 | ~3–4 | 3.01 |
For omarigliptin, renal impairment was categorized using eGFR calculated using MDRD formula as follows: Normal ≥80 mL/min/1.73 m2; Mild ≥60 mL/min/1.73 m2, <80 mL/min/1.73 m2; Moderate ≥30 mL/min/1.73 m2; <60 mL/min/1.73 m2, Severe <30 mL/min/1.73 m2.
For sitagliptin, alogliptin and trelagliptin, renal impairment was categorized by creatinine clearance as follows: Normal >80 mL/min; Mild >50 mL/min, ≤80 mL/min; Moderate ≥30 mL/min, ≤50 mL/min; Severe <30 mL/min.
Table 6.
Comparison of renal clearance of dipeptidyl peptidase‐4 (DPP‐4) inhibitors that are eliminated through a renal pathway
| DPP‐4 inhibitor | CLr | fu | Clearance of unbound fraction (CLr/fu) | Inference | References |
|---|---|---|---|---|---|
| Sitagliptin 100 mg q.d. | 350 mL/min | 0.62 | 565 mL/min | Net active tubular secretion | 1, 4 |
| Alogliptin 25 mg q.d. | 160 mL/min | 0.80 | 200 mL/min | Net active tubular secretion | 2 |
| Trelagliptin 100 mg q.w. | 235 mL/min | ~0.75 | 313 mL/min | Net active tubular secretion | 3 |
| Omarigliptin 25 mg q.w. | ‐‐ | ‐‐ | 27–45 mL/min | Renal filtration with net reabsorption | 5 |
CLr = renal clearance; fu = fraction unbound; q.d. = once daily; q.w. = once weekly
The direct effect Emax model adequately described DPP‐4 inhibition profiles across the evaluated omarigliptin dose range. None of the tested covariates had any clinically meaningful impact on DPP‐4 inhibition with omarigliptin.
5. CONCLUSION
The population PK‐PD model adequately described the pharmacokinetic disposition and DPP‐4 inhibition profile of omarigliptin as well as supported dosing recommendations in the label for dose in the overall T2DM population, including those with renal impairment, and instructions for handling missed dosage.
COMPETING INTERESTS
L.J., A.S.Y.C., D.A.T., E.A.K. and E.L. are employees of and may hold stock or stock options in Merck & Co., Inc., Kenilworth, NJ, USA. J.H. and J.A.P. are employees of Cognigen Corporation, Buffalo, NY, USA, which was contracted by Merck & Co., Inc., Kenilworth, NJ, USA to conduct this study.
CONTRIBUTORS
All of the authors are responsible for the work described in this manuscript. All authors were involved in at least 1 of the following: study conception, study design, data acquisition, data analysis or interpretation of data. All authors were also involved in drafting the manuscript and/or revising it for important intellectual content. All authors provided final approval of the version to be published.
Supporting information
Table S1 Comparison of parameter estimates and standard errors for the final population pharmacokinetic model and the 2014 model for omarigliptin
Table S2 Effects of intrinsic and extrinsic factors on the pharmacokinetics of omarigliptin based on simulations from the final population pharmacokinetic model
Figure S1 Simulated mean (± standard deviation) plasma omarigliptin concentration vs time profiles for 25 mg after 3 weeks of dosing in healthy subjects and patients with type 2 diabetes
Figure S2
Figure S3 Boxplots of simulated omarigliptin exposure measures for healthy subjects and patients with type 2 diabetes administered 25 mg once weekly for 3 weeks
Figure S4 Boxplots of simulated omarigliptin exposure measures for race in patients with type 2 diabetes administered 25 mg once weekly
Figure S5 Impact of body weight on simulated steady‐state omarigliptin area under the concentration–time curve up to 168 hours in patients with type 2 diabetes administered 25 mg once weekly
Figure S6 Impact of age on simulated steady‐state omarigliptin area under the concentration–time curve up to 168 hours in patients with type 2 diabetes administered 25 mg once weekly
Figure S7 Boxplots of simulated omarigliptin exposure measures in nonelderly and elderly patients administered 25 mg once weekly
Figure S8 Impact of sex on simulated steady‐state omarigliptin exposure measures in patients with type 2 diabetes administered 25 mg once weekly
Figure S9 Impact of concomitant amlodipine use on simulated steady‐state omarigliptin exposure measures in patients with type 2 diabetes administered 25 mg once weekly
Figure S10 Impact of concomitant atorvastatin use on simulated steady‐state omarigliptin exposure measures in patients with type 2 diabetes administered 25 mg once weekly
Figure S11 Impact of concomitant diuretic use on simulated steady‐state omarigliptin exposure measures in patients with type 2 diabetes administered 25 mg once weekly
ACKNOWLEDGEMENTS
The authors wish to thank Shiyao Sherrie Xu (Merck & Co., Inc., Kenilworth, NJ, USA) for her contributions to the nonclinical PK data included in this report. The authors also wish to thank Alan Meehan (Merck & Co., Inc., Kenilworth, NJ, USA) for writing assistance as well as Jennifer Rotonda and Michele McColgan (both of Merck & Co., Inc., Kenilworth, NJ, USA) for assistance in preparing this paper for publication.
The studies included in this report were funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Jain L, Chain ASY, Tatosian DA, et al. Pharmacokinetic–pharmacodynamic (dipeptidyl peptidase‐4 inhibition) model to support dose rationale in diabetes patients, including those with renal impairment, for once‐weekly administered omarigliptin. Br J Clin Pharmacol. 2019;85:2759–2771. 10.1111/bcp.14103
Principal Investigator: The authors confirm that the PI for this paper is Lokesh Jain and that he had direct responsibility for the analysis.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Biftu T, Sinha‐Roy R, Chen P, et al. Omarigliptin (MK‐3102): a novel long‐acting DPP‐4 inhibitor for once‐weekly treatment of type 2 diabetes. J Med Chem. 2014;57(8):3205‐3212. [DOI] [PubMed] [Google Scholar]
- 2. Krishna R, Addy C, Tatosian D, et al. Pharmacokinetics and pharmacodynamics of omarigliptin, a once‐weekly dipeptidyl peptidase‐4 (DPP‐4) inhibitor, after single and multiple doses in healthy subjects. J Clin Pharmacol. 2016;56(12):1528‐1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sheu WH, Gantz I, Chen M, et al. Safety and efficacy of omarigliptin (MK‐3102), a novel once‐weekly DPP‐4 inhibitor for the treatment of patients with type 2 diabetes. Diabetes Care. 2015;38(11):2106‐2114. [DOI] [PubMed] [Google Scholar]
- 4. Xu S, Tatosian D, Mcintosh I, et al. Absorption, metabolism and excretion of [14C]omarigliptin, a once‐weekly DPP‐4 inhibitor, in humans. Xenobiotica. 2017;48(6):584‐591. 10.1080/00498254.2017.1346333 [DOI] [PubMed] [Google Scholar]
- 5. MARIZEV® (omarigliptin tablets) product circular. Avalaible at: http://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/170050_3969025F1022_1_05. Last accessed 13 October 2017.
- 6. Retlich S, Duval V, Graefe‐Mody U, Jaehde U, Staab A. Impact of target‐mediated drug disposition on linagliptin pharmacokinetics and DPP‐4 inhibition in type 2 diabetic patients. J Clin Pharmacol. 2010;50(8):873‐885. [DOI] [PubMed] [Google Scholar]
- 7. Litwak L, Goh S‐Y, Hussein Z, Malek R, Prusty V, Khamseh ME. Prevalence of diabetes complications in people with type 2 diabetes mellitus and its association with baseline characteristics in the multinational A1chieve study. Diabetol Metab Syndr. 2013;5(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tatosian DA, Cardillo Marricco N, Glasgow XS, et al. A thorough QTc study confirms early pharmacokinetics/QTc modeling: a supratherapeutic dose of omarigliptin, a once‐weekly DPP‐4 inhibitor, does not prolong the QTc interval. Clin Pharmacol Drug Dev. 2016;5(5):383‐392. [DOI] [PubMed] [Google Scholar]
- 9. JANUVIA® (sitagliptin) Tablets US Product Circular. 10 August 2017. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021995s040lbl.pdf. Accessed October 7, 2017.
- 10. NESINA (alogliptin) Tablets US Product Circular. 12 December 2016. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/022271s011lbl.pdf. Accessed October 7, 2017.
- 11. Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau Ministry of Health, Labour and Welfare, Japan. Report on the Deliberation Results for Zafatek (Trelagliptin Succinate) Tablets. February 5, 2015. Available at: https://www.pmda.go.jp/files/000213963.pdf. Accessed October 7, 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Comparison of parameter estimates and standard errors for the final population pharmacokinetic model and the 2014 model for omarigliptin
Table S2 Effects of intrinsic and extrinsic factors on the pharmacokinetics of omarigliptin based on simulations from the final population pharmacokinetic model
Figure S1 Simulated mean (± standard deviation) plasma omarigliptin concentration vs time profiles for 25 mg after 3 weeks of dosing in healthy subjects and patients with type 2 diabetes
Figure S2
Figure S3 Boxplots of simulated omarigliptin exposure measures for healthy subjects and patients with type 2 diabetes administered 25 mg once weekly for 3 weeks
Figure S4 Boxplots of simulated omarigliptin exposure measures for race in patients with type 2 diabetes administered 25 mg once weekly
Figure S5 Impact of body weight on simulated steady‐state omarigliptin area under the concentration–time curve up to 168 hours in patients with type 2 diabetes administered 25 mg once weekly
Figure S6 Impact of age on simulated steady‐state omarigliptin area under the concentration–time curve up to 168 hours in patients with type 2 diabetes administered 25 mg once weekly
Figure S7 Boxplots of simulated omarigliptin exposure measures in nonelderly and elderly patients administered 25 mg once weekly
Figure S8 Impact of sex on simulated steady‐state omarigliptin exposure measures in patients with type 2 diabetes administered 25 mg once weekly
Figure S9 Impact of concomitant amlodipine use on simulated steady‐state omarigliptin exposure measures in patients with type 2 diabetes administered 25 mg once weekly
Figure S10 Impact of concomitant atorvastatin use on simulated steady‐state omarigliptin exposure measures in patients with type 2 diabetes administered 25 mg once weekly
Figure S11 Impact of concomitant diuretic use on simulated steady‐state omarigliptin exposure measures in patients with type 2 diabetes administered 25 mg once weekly
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.