

Abstract
Aims
To characterize the population pharmacokinetics (PK) of sildenafil and its active metabolite, N‐desmethyl sildenafil (DMS), in premature infants.
Methods
We performed a multicentre, open‐label trial to characterize the PK of sildenafil in infants ≤28 weeks gestation and < 365 postnatal days (cohort 1) or < 32 weeks gestation and 3–42 postnatal days (cohort 2). In cohort 1, we obtained PK samples from infants receiving sildenafil as ordered per the local standard of care (intravenous [IV] or enteral). In cohort 2, we administered a single IV dose of sildenafil and performed PK sampling. We performed a population PK analysis and dose‐exposure simulations using the software NONMEM®.
Results
We enrolled 34 infants (cohort 1 n = 25; cohort 2 n = 9) and collected 109 plasma PK samples. Sildenafil was given enterally (0.42–2.09 mg/kg) in 24 infants in cohort 1 and via IV (0.125 or 0.25 mg/kg) in all infants in cohort 2. A 2‐compartment PK model for sildenafil and 1‐compartment model for DMS, with presystemic conversion of sildenafil to DMS, characterized the data well. Coadministration of fluconazole (n = 4), a CYP3A inhibitor, resulted in an estimated 59% decrease in sildenafil clearance. IV doses of 0.125, 0.5 and 1 mg/kg every 8 hours (in the absence of fluconazole) resulted in steady‐state maximum sildenafil concentrations that were generally within the range of those reported to inhibit phosphodiesterase type 5 activity in vitro.
Conclusions
We successfully characterized the PK of sildenafil and DMS in premature infants and applied the model to inform dosing for a follow‐up, phase II study.
Keywords: sildenafil, premature infants, pharmacokinetics
What is already known about the subject
Sildenafil is a type 5 phosphodiesterase inhibitor increasingly used off‐label in infants to treat pulmonary hypertension.
The population pharmacokinetics of sildenafil have been previously characterized in term infants, but limited data are available in premature infants.
What this study adds
This study provides information on the population pharmacokinetics of sildenafil in premature infants following both intravenous and enteral administration.
Confirming previous findings, and after accounting for body size, fluconazole, a cytochrome P450 3A inhibitor, was identified as a significant covariate for sildenafil clearance.
1. INTRODUCTION
Bronchopulmonary dysplasia (BPD) is a common pulmonary complication of premature birth.1, 2 Premature infants with BPD are at risk for developing pulmonary hypertension, which increases mortality.3, 4 Currently, there are no therapeutics approved by the US Food and Drug Administration for the prevention of BPD.5 In preclinical models, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4743, a type 5 phosphodiesterase (PDE5) inhibitor, reduces pulmonary inflammatory response and vascular remodeling, and has been shown to stimulate alveolarization.6, 7 Case reports and retrospective cohort studies of premature infants receiving sildenafil demonstrate successful resolution of pulmonary hypertension associated with BPD following treatment.8, 9 Prospective trials of sildenafil in infants are needed, but the first step is characterization of sildenafil pharmacokinetics (PK) in this population.
When administered to adults, the mean absolute oral bioavailability of sildenafil is approximately 41%; sildenafil displays linear protein binding (~96% bound), and is metabolized by cytochrome P450 3A4 (CYP3A4)/CYP3A5 (major) and CYP2C9 (minor) to N‐desmethyl sildenafil (DMS).10, 11, 12, 13, 14 DMS, the major circulating metabolite, has an in vitro potency that is 50% of the parent compound.12 The population PK (PopPK) of sildenafil have been previously studied in term neonates and infants with pulmonary hypertension.15, 16, 17 In 36 term neonates with persistent pulmonary hypertension of the newborn or hypoxaemia, sildenafil clearance (CL) increased with postnatal age (PNA) in the first 3 weeks of life, likely reflective of maturation in CYP3A4 function.15 When administered to 11 infants with pulmonary hypertension supported with extracorporeal membrane oxygenation, concomitant administration of fluconazole, a CYP3A inhibitor, was identified as a significant covariate that reduced sildenafil CL.16 A widely accepted sildenafil exposure target has not been established in infants; however, steady‐state maximum sildenafil concentrations (Cmax,ss,SIL) of 47, 140 and 373 ng/mL are expected to correlate with unbound concentrations found to inhibit PDE5 activity in vitro by approximately 53, 77 and 90%, respectively.18
We performed a phase I study to evaluate sildenafil PK in 2 cohorts of premature infants; the first group was receiving open‐label sildenafil for treatment of pulmonary hypertension and the second group received a single dose of intravenous (IV) sildenafil for the purpose of PK measurements for the research study. We then developed a PopPK model that we applied to inform dosing for the phase II study of sildenafil in premature infants at risk for BPD.
2. METHODS
2.1. Patient population
Clinical and PK data were collected as part of the Pediatric Trials Network (PTN) study titled Pharmacokinetics of Sildenafil in Premature Infants (http://ClinicalTrials.gov Identifier: NCT01670136). The study was a phase I, multicentre, open‐label trial designed to characterize the PK of sildenafil in premature infants. A total of 7 clinical trial sites were activated, but only 6 sites actually enrolled study participants. For cohort 1, the inclusion criteria were gestational age (GA) ≤28 weeks at birth and <1 year PNA receiving sildenafil as standard clinical care (most commonly for treatment of pulmonary hypertension associated with BPD). The only exclusion criterion was any condition that would make the infant, in the opinion of the investigator, unsuitable for the study. For cohort 2, the inclusion criteria were: GA <32 weeks; 3–42 days PNA; mechanical ventilation or nasal continuous positive airway pressure or high‐flow (≥1.5 L/min) nasal cannula or fraction of inspired oxygen >0.21; and IV line in place. The exclusion criteria were: previous exposure to sildenafil within 7 days prior to enrolment; currently on a vasopressor for hypotension; known sickle cell disease; history of allergic reactions to sildenafil; and aspartate aminotransferase >225 U/L or alanine aminotransferase >150 U/L. The first and last participants were enrolled on 21 February 2013 and 22 July 2016, respectively. The study protocol was reviewed and approved by an institutional review board for each participating clinical trial site, and informed consent was obtained from each participant's parent/legal guardian.
2.2. Drug dosing and sample collection
In cohort 1, infants received enteral (compounded suspension) or IV administration of sildenafil per standard of care. In cohort 2, infants received a single dose of sildenafil (0.125 or 0.25 mg/kg) administered via IV over 90 minutes, followed by a 30‐minute flush. When possible, PK samples for cohort 1 and cohort 2 were collected according to Table S1 and Table S2, respectively. For cohort 1, the compounding and dispensing of sildenafil was done by the local clinical or investigational pharmacy per sites' local policies. For cohort 2, a commercially available IV formulation was used.
2.3. Analytical methods
At least 200 μL of blood was collected in an ethylenediaminetetraacetic acid‐containing tube and was processed into plasma immediately prior to freezing at the study sites. Plasma PK samples were sent to the PTN central laboratory (OpAns, LLC, Durham, NC, USA) for storage and analysis. Sildenafil and DMS concentrations in plasma were quantified using a validated high‐performance liquid chromatography–tandem mass spectrometry assay. During method validation, accuracy for all runs at all levels were within ±15% of the theoretical value. Precision values for all runs at all levels did not exceed 15%. The lower limits of quantification for sildenafil and DMS were 0.1 and 0.2 ng/mL, respectively. Additional details of the bioanalytical method are provided in the Supporting Information and Table S3.
At least 200 μL of whole blood was collected for genetic analysis. The genetic analysis was performed by Children's Mercy Hospital (Kansas City, MO, USA). The CYP3A gene locus was genotyped for the single‐nucleotide polymorphisms (SNPs) listed in Table S4. All infants in the study were genotyped for these variants. Additional details of the genotyping are provided in the Supporting Information.
2.4. PopPK model development
Sildenafil and DMS plasma PK data collected following IV and enteral administration were fit simultaneously using the software NONMEM® (version 7.4.3, Icon Development Solutions, Ellicott City, MD, USA). Dose amounts were converted to micromoles using the molecular weight (474.6 g/mol for sildenafil and 460.6 g/mol for DMS), and sildenafil and DMS concentrations were converted to nanomolar units to facilitate simultaneous fitting of the parent and metabolite data. The first‐order conditional estimation method with interaction was used for all model runs. Prediction‐corrected visual predictive checks (pcVPCs) and a bootstrap analysis were performed with Perl‐speaks‐NONMEM (version 4.8.1).19, 20 Data visualization was performed using the ggplot2 (version 2.2.1) and lattice (version 0.20–34) packages in the software R (version 3.3.2, R Foundation for Statistical Computing, Vienna, Austria) and RStudio (version 1.0.136, RStudio, Boston, MA, USA).21, 22 A normalized prediction distribution error (NPDE) analysis was performed using the R package npde (version 2.0).23
One‐ and 2‐compartment PK models were explored for both sildenafil and DMS with assumed linear PK. Nonlinear elimination was also explored. Various absorption models for sildenafil were tested, including a zero‐order absorption, first‐order absorption, and use of a lag time. A hypothetical absorption compartment was also evaluated to incorporate presystemic conversion of sildenafil to DMS, as shown in Figure 1.24 When a hypothetical absorption compartment was used, the relative metabolite bioavailability (FDMS) was described according to Equation 1:
| (1) |
where TVF DMS is the typical value of the relative metabolite bioavailability (F DMS), which depended on the bioavailability of sildenafil (F SIL).24 In order to incorporate presystemic conversion of sildenafil to DMS, simultaneous enteral sildenafil and equimolar DMS dosing (scaled by the relative metabolite bioavailability) was assumed.24 In the absence of data for the metabolite administered IV, complete conversion of absorbed sildenafil to DMS was assumed in order to obtain an identifiable model (given that the relative metabolite bioavailability and extent of conversion of parent to metabolite were confounded).24 A logit transformation for FSIL and FDMS was used to constrain individual values between 0 and 1.
Figure 1.
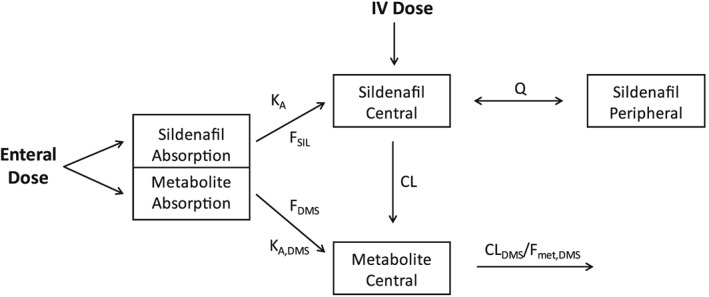
Final population pharmacokinetic model schematic for sildenafil (SIL) and its metabolite N‐desmethyl sildenafil (DMS). To incorporate presystemic conversion of SIL to DMS, simultaneous enteral SIL and equimolar DMS dosing (scaled by the relative metabolite bioavailability) was assumed.24 KA: SIL first‐order absorption rate constant; FSIL: SIL bioavailability; FDMS: relative DMS bioavailability; CL: SIL clearance (assuming complete conversion to DMS); VSIL,C: SIL central compartment volume of distribution; KA,DMS: DMS first‐order absorption rate constant; CLDMS/Fmet,DMS: DMS clearance; Fmet,DMS: fraction of SIL metabolized to DMS; Q: SIL intercompartmental clearance; and IV: intravenous
Interindividual variability was assessed for PK model parameters using an exponential relationship according to Equation 2:
| (2) |
where PARij denotes the estimate of parameter j in the ith individual; θ Pop,j is the population value for parameter j; and ηij denotes the deviation from the average population value for parameter j in the ith individual with mean zero and variance ω2.
Proportional, additive and combined (additive plus proportional) residual error models were explored. Separate residual error terms were estimated for sildenafil and DMS. The level 2 (L2) data item was used to estimate the covariance between residual error parameters for sildenafil and DMS.
Actual body weight (WT) was assumed to be a significant covariate for CL and volume of distribution (V) parameters, and was included in the base model. The last WT measurement taken prestudy dose was used and carried forward across all individual participant study records. The relationship between WT and PK parameters was scaled to a 70 kg standardized WT and modelled according to Equations 3 and 4:
| (3) |
| (4) |
where CL std and V std represent population estimates of CL and V in a 70 kg adult; WT i denotes body weight for the ith infant; and θWT,CL and θWT,V denote the exponents for the effect of weight on CL and V parameters, respectively. Estimation of these exponents was compared to using fixed values of 0.75 and 1 for θWT,CL and θWT,V, respectively. In addition, for CL parameters, we also evaluated use of an allometric equation with an exponent that changes with body weight.25 The body weight‐dependent exponent was modelled using an exponential function.25
After accounting for WT, additional covariates were tested for inclusion in the model. Determination of which covariates to test was based on physiological relevance, and by visual inspection of scatter and box plots (continuous and categorical variables, respectively) of the individual deviations from the population‐typical value PK parameters (ETAs) against covariates. The following covariates were explored: GA, PNA, postmenstrual age (PMA), serum creatinine, albumin, blood urea nitrogen, sex, ethnicity, genotype (Table S4), and concomitant medications considered individually (omeprazole, midazolam, caffeine, hydrocortisone, fentanyl, methadone, bosentan, erythromycin, fluconazole and phenobarbital).
With the exception of WT, other continuous covariates were normalized to the population median value as described in Equation 5, whereas for categorical covariates, a relationship as shown in Equation (6) was used. When evaluating the covariates GA, PNA and PMA, linear and sigmoidal relationships were also evaluated (Equations 7 and 8). For genotype groups, the covariate effect for the wild type group was assumed to be equal to 1, and then separate θcov,n estimates were obtained for the heterozygous variant and homozygous variant groups. If the θcov,n estimate for the heterozygous variant group was close to 1 or the model was unstable, only the covariate effect for the homozygous variant group was estimated and heterozygotes were treated as wild type study participants. Concomitant medications were evaluated as a binary variable, such that the effect of the concomitant medication (i.e. θcov,n) on CL was estimated when the concomitant medication was administered at the time of PK sampling.
| (5) |
| (6) |
| (7) |
| (8) |
where PAR ij denotes the estimate of parameter j in the ith individual; θ Pop,j is the population value for parameter j; covi denotes the individual covariate value; covm is the population median covariate value; θcov is a parameter that represents the covariate effect; θcov,n is a parameter that represents covariate effect of the nth category; TM 50 is the value of covi when 50% adult CL is reached; and HILL is a slope parameter.
For all continuous covariates, missing data values were imputed using the population median value. A forward inclusion (P < .05 and Δ objective function value [OFV] >3.8 points) and backward elimination (P < .001 and ΔOFV >10.8 points) approach was used to evaluate statistical significance.
2.5. PopPK model evaluation
During the PopPK model‐building process, successful minimization, diagnostic plots, pcVPCs, a numerical predictive check, plausibility and precision of parameter estimates, nonparametric bootstrapping, as well as OFV and shrinkage values were used to assess model appropriateness. Additional details regarding PopPK model evaluation are provided in the Supporting Information.
2.6. Dosing simulations
The final model was applied to inform dosing for a follow‐up phase II study. The following dosing regimens were evaluated in virtual premature infants (n = 1000) with age characteristics that were comparable to the phase II study population (≤29 weeks GA and 7–28 [inclusive] days PNA): 0.125, 0.5 and 1 mg/kg administered via IV every 8 hours; 0.25, 1 and 2 mg/kg administered enterally every 8 hours. For each virtual infant, concentrations were simulated every 0.5 hours from 0 to 192 hours after the first dose. A noncompartmental analysis was performed to determine the simulated Cmax,ss,SIL, and the area under the concentration (individual predicted concentrations) vs time curve from 0 to 24 hours (AUC0–24) for both sildenafil (AUC0–24,SIL) and DMS (AUC0–24,DMS) at steady‐state. Additional details regarding the dosing simulations are provided in the Supporting Information.
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.26
3. RESULTS
3.1. Patient population and PK data
A total of 34 infants were enrolled in this study (cohort 1 n = 25; cohort 2 n = 9). In cohort 1, all infants but 1 received sildenafil via enteral administration; in cohort 2, all infants received sildenafil via IV administration (Table 1). A total of 109 plasma PK samples were collected (Figure 2). There were 4 (3.7%) and 2 (1.8%) samples that were below the quantification limit for sildenafil and DMS, respectively, which were dropped from the analysis. The median (range) time after last dose for samples that were below the quantification limit was 28.5 (6.1–52.3) hours. The median (range) number of fresh plasma samples collected per infant was 3 (2–4). The median (range) dose was 0.25 (0.13–0.50) and 0.95 (0.42–2.09) mg/kg for IV and enteral dosing, respectively. Four infants, all in cohort 2, received concomitant fluconazole (2 for prophylaxis; 1 for empirical treatment of sepsis; and 1 had a positive endotracheal tube fungus culture). A summary of patient genotypes for the SNPs evaluated is provided in Table S5.
Table 1.
Clinical data
| Variablea | Cohort 1 (n = 25) | Cohort 2 (n = 9) | All (n = 34) |
|---|---|---|---|
| Dose (mg/kg)b | 0.95 [0.42–2.09] | 0.25 [0.13–0.25] | 0.79 [0.13–2.09] |
| Dose (mg/kg/d)b | 3.49 [1.25–8.38] | 0.25 [0.13–0.25] | 2.79 [0.13–8.38] |
| Samples per infantc | 3 [3–4] | 3 [2–4] | 3 [2–4] |
| Weight (kg) | 4.79 [1.36–8.06] | 0.75 [0.59–1.24] | 3.41 [0.59–8.06] |
| Birth weight (g) | 650 [450–1215] | 800 [425–980] | 666 [425–1215] |
| Gestational age (wk) | 25 [22–28] | 25 [23–27] | 25 [22–28] |
| Postnatal age (d) | 166 [52–279] | 18 [7–40] | 125.5 [7–279] |
| Postmenstrual age (wk) | 47.0 [31.3–62.7] | 27.4 [26.0–32.4] | 41.6 [26.0–62.7] |
| Serum creatinine (mg/dL)d | 0.2 [0.1–0.7] | 0.6 [0.4–1.4] | 0.3 [0.1–1.4] |
| Albumin (g/dL)c | 3.7 [1.8–4.3] | 2.8 [1.7–3.7] | 3.1 [1.7–4.3] |
| Fluconazole, n (%) | 0 | 4 (44%) | 4 (12%) |
| Formulation, n (%) | |||
| Enteral | 24 (96%) | 0 | 24 (71%) |
| Intravenous | 1 (4%) | 9 (100%) | 10 (29%) |
Descriptive statistics were calculated based on the value at the time of first record for each infant and reported as median [range] for continuous variables and n (%) for categorical variables. Fluconazole (%) was reported as the percentage of concomitant use of fluconazole in each group.
Doses (mg/kg and mg/kg/d) are calculated based on the first recorded dose.
Calculated from a total of 109 plasma pharmacokinetic samples collected, including 4 and 2 samples that were below the quantification limit for sildenafil and N‐desmethyl sildenafil, respectively.
Serum creatinine data were missing for 5 (15%) infants.
Albumin data were missing for 15 (44%) infants.
Figure 2.
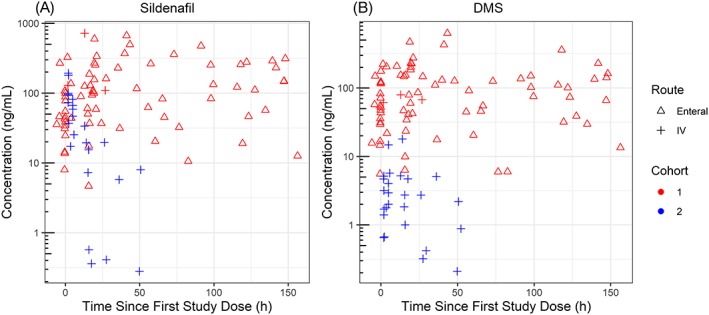
Sildenafil and N‐desmethyl sildenafil (DMS) plasma concentration vs time since first study dose data stratified by cohort and route of administration. Infants in cohort 1 had six standard of care sildenafil doses reported before any study doses. Cohort 2 infants received a single dose
3.2. PopPK model development and evaluation
A 2‐compartment model for sildenafil and a 1‐compartment model for DMS characterized the data well. Plots of ETAs vs covariates, and standard diagnostic plots, for the base model without WT as a covariate are provided as Figure S1 and Figure S2. Inclusion of WT as a covariate reduced the OFV by 39.5 points; therefore, WT was assumed to be a significant covariate for CL and V parameters, and was included in the base model. Use of a hypothetical absorption compartment to incorporate presystemic conversion of sildenafil to DMS reduced the OFV by 38.8 points and improved the fitting of the DMS data. Estimation of the allometric exponent for CL parameters resulted in a more plausible population estimate for CL; however, use of a body‐weight‐dependent exponent did not improve the data fitting. Estimation of the exponent for the effect of WT on volume of distribution parameters did not improve the data fitting, and thus this parameter was fixed to 1. Proportional residual error models were used for both sildenafil and DMS.
Fluconazole coadministration and the CYP3A5*3 (rs776746) polymorphism were found to result in a statistically significant reduction in the OFV during the forward inclusion step and were still retained in the model in the backward elimination. However, the interindividual variability in CL increased from 33.3 to 48.1% with the addition of CYP3A5*3 in the model. In addition, the model that included genotype as a covariate resulted in a high shrinkage value (>40%) for the random effect parameter characterizing the interindividual variability in CL, and the population estimate for CL was estimated to be much higher (67.9 L/h/70 kg) than previously reported values in adults. Lastly, upon bootstrap analysis, the covariate effect for CYP3A5*3 overlapped zero (2.5th and 97.5th percentiles ranged from a decreased to increased effect on CL, respectively). Therefore, fluconazole coadministration was the only covariate for CL included in our final model after accounting for WT.
Inclusion of fluconazole in the model resulted in a decrease in the interindividual variability in CL from 38.5 to 33.3%. Infants receiving fluconazole had a 59% reduction in CL. Random effect parameters used to characterize the interindividual variability were plotted against each other, and visualization of these data showed no apparent correlation. Standard diagnostic plots are shown in Figure 3 and Figure S3. Some model misspecification was noted for a limited number of samples >200 ng/mL (both sildenafil and DMS). Most of these samples were obtained from infants who received sildenafil via enteral administration. A zero‐order absorption model and other (additive and additive‐plus‐proportional) residual error models were also evaluated, but these did not significantly improve the misspecification. The pcVPCs for the final model are shown in Figure 4. Following enteral administration, the percent of observations that fell outside of the 90% prediction interval for sildenafil and DMS were 13.5 and 9.5%, respectively. Following IV administration, the percent of observations that fell outside of the 90% prediction interval for sildenafil and DMS were 3.7 and 10.3%, respectively. The final PopPK parameter estimates are shown in Table 2, and the final model NONMEM® control stream is provided in the Supporting Information. Individual empirical Bayesian estimates are presented in Table S6. Plots of the ETAs vs covariates for the final model are provided in Figure S4. NPDE analysis plots are provided in Figure S5.
Figure 3.
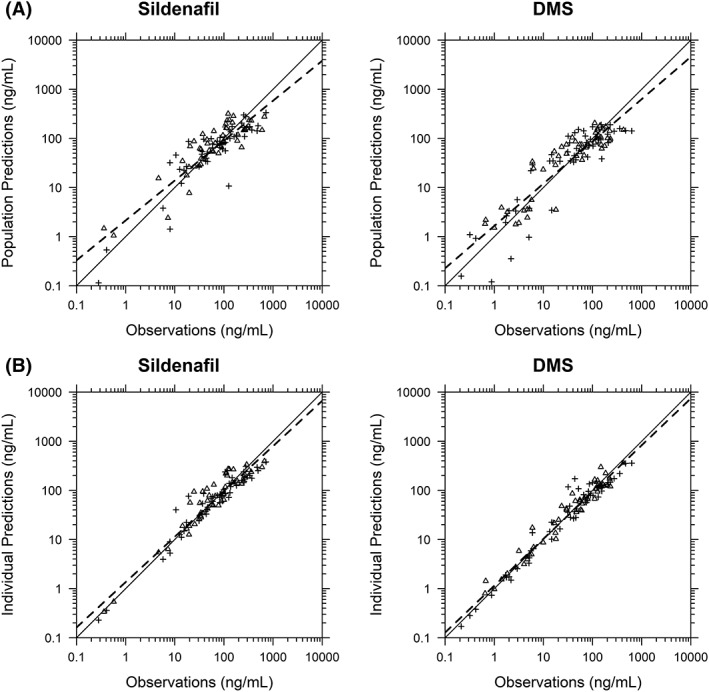
Sildenafil and N‐desmethyl sildenafil (DMS) observed vs population (A) and individual (B) predictions for the final population pharmacokinetic model. Triangles and cross symbols represent enteral and intravenous administration, respectively. The solid black line represents the line of unity. The dashed line represents the linear regression fit
Figure 4.
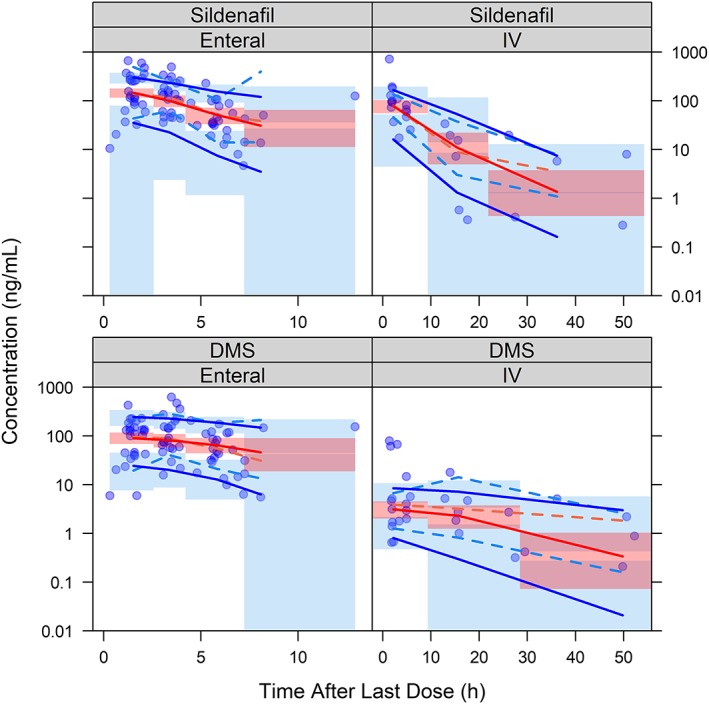
Prediction‐corrected visual predictive check for sildenafil and N‐desmethyl sildenafil (DMS) observations vs time after last dose, stratified by route of administration, and generated using the final population pharmacokinetic model. The dashed lines represent the 5th, 50th and 95th percentiles of the observed data. The solid lines represent the 5th, 50th and 95th percentiles of the predicted data. The shaded region represents the 90% confidence interval of the 5th, 50th and 95th percentiles of the predicted data. IV: intravenous
Table 2.
Final population pharmacokinetic model parameter estimates
| Parameterb | Estimate (RSE) | Bootstrap (n = 1000)a | ||
|---|---|---|---|---|
| 2.5th percentile | Median | 97.5th percentile | ||
| Structural model and covariate effect parameters | ||||
| KA (/h) | 1.59 (88.1%) | 0.27 | 1.09 | 4.52 |
| FSIL | 0.29 (33.0%) | 0.19 | 0.31 | 0.86 |
| CL (L/h/70 kg) | 27.8 (51.4%) | 10.9 | 35.5 | 322.8 |
| VSIL,C (L/70 kg) | 116.0 (16.1%) | 28.5 | 96.2 | 146.0 |
| VSIL,P (L/70 kg) | 28.1 (20.4%) | 17.2 | 34.1 | 186.2 |
| KA,DMS (/h) | 1.3 (49.4%) | 0.42 | 1.00 | 2.97 |
| TVFDMS | 1.29 (46.6%) | 0.55 | 1.42 | 11.59 |
| CLDMS/Fmet,DMS (L/h/70 kg) | 135 (62.4%) | 49.1 | 178.3 | 1363.3 |
| VDMS/Fmet,DMS (L/70 kg) | 1670.0 (22.2%) | 1029.1 | 1808.3 | 2819.5 |
| Q (L/h/70 kg) | 1.67 (74.3%) | 0.65 | 3.30 | 40.94 |
| Percentage CL reduction with fluconazole use (%) | 58.9 (19.1%) | 31.2 | 61.7 | 76.2 |
| Exponent (weight effect on clearance parameters)c | 0.96 (12.5%) | 0.75 | 1.03 | 1.48 |
| Interindividual variability (reported as %CV) d | ||||
| CL | 33.3 (45.5%) | 11.9 | 32.2 | 49.9 |
| VSIL,C | 26.8 (98.6%) | 0.27 | 24.1 | 73.9 |
| CLDMS/Fmet,DMS | 56.5 (27.3%) | 37.0 | 55.3 | 73.8 |
| VDMS/Fmet,DMS | 48.1 (48.1%) | 0.48 | 43.2 | 73.2 |
| Residual error (reported as %CV) e | ||||
| Proportional error for sildenafil (%) | 45.6 (24.2%) | 34.9 | 45.0 | 54.6 |
| Proportional error for DMS (%) | 40.7 (19.0%) | 32.7 | 40.8 | 49.2 |
| Covariance | ||||
| Covariance between residual error parameters | 0.12f (26.4%) | 0.07 | 0.13 | 0.20 |
RSE: relative standard error (expressed as a percentage); KA: sildenafil (SIL) first‐order absorption rate constant; FSIL: SIL bioavailability; CL: SIL clearance (assuming complete conversion to N‐desmethyl sildenafil [DMS]); VSIL,C: SIL central compartment volume of distribution; VSIL,P: SIL peripheral compartment volume of distribution; KA,DMS: DMS first‐order absorption rate constant; TVFDMS: typical value for the bioavailability of DMS; CLDMS/Fmet,DMS: DMS clearance; VDMS/Fmet,DMS: DMS volume of distribution; Fmet,DMS: fraction of SIL metabolized to DMS; Q: intercompartmental clearance for SIL.
83.4% of bootstrap runs resulted in a successful minimization.
KA (/h) = 1.59; FSIL = 0.29; CL (L/h) = 27.8*(WTi/70 kg)0.96*(0.411 [if infant received fluconazole])*exp(ηi,CL); VSIL,C (L) = 116*(WTi/70 kg)*exp(ηi,VSIL,C); VSIL,P (L) = 28.1*(WTi/70 kg); KA,DMS (/h) = 1.3; FDMS = TVFDMS*(1‐FSIL); CLDMS/Fmet,DMS (L/h) = 135*(WTi/70 kg)0.96*exp ; VDMS/Fmet,DMS (L) = 1670*(WTi/70 kg)*exp ; and Q (L/h) = 1.67*(WTi/70 kg), where WTi is the individual participant weight.
The same exponent is assumed for all SIL and DMS clearance parameters. The exponent for the weight effect on volume of distribution parameters is fixed to 1.
ETA shrinkage (%): CL: 10.5%; VSIL,C: 48.0%; CLDMS/Fmet,DMS: 6.43%; VDMS/Fmet,DMS: 38.2%.
EPS shrinkage (%): SIL: 11.7%; DMS: 12.0%.
Correlation coefficient: 0.65.
3.3. Dosing simulations
The effect of fluconazole on sildenafil's concentration vs time profile is shown in Figure 5. Simulated Cmax,ss,SIL following IV and enteral administration, with and without fluconazole, is shown in Figure 6. Simulated steady‐state AUC0–24 following enteral and IV dosing is presented in Table S7. Doses of 0.125, 0.5 and 1 mg/kg (in the absence of fluconazole) administered via IV every 8 hours resulted in simulated Cmax,ss,SIL values that generally exceeded the concentrations reported to inhibit PDE5 activity in vitro by approximately 53, 77 and 90%, respectively. Enteral doses of 0.25 and 2 mg/kg (in the absence of fluconazole) resulted in simulated Cmax,ss,SIL values that were generally slightly below the cut‐offs for in vitro inhibition by 53 and 90%, respectively, whereas 1 mg/kg generally resulted in exposures needed for 77% inhibition. When sildenafil was coadministered with fluconazole, IV dosing of 0.125, 0.5 or 1 mg/kg every 8 hours generally resulted in simulated Cmax,ss,SIL values that exceed cut‐offs for in vitro inhibition by 53, 77 and 90%, respectively; a similar trend was observed for enteral doses of 0.25, 1 or 2 mg/kg every 8 hours.
Figure 5.

Simulated sildenafil concentrations over 192 h following multiple intravenous (IV; A–C) or enteral (D–F) doses of sildenafil administered every 8 hours. Pink and teal shaded regions represent the 95% prediction intervals for virtual infants with and without fluconazole, respectively. Dashed and solid lines represent 2.5th, 50th and 97.5th percentiles for virtual infants with and without fluconazole, respectively. The black lines represent the 50th (median) percentile
Figure 6.
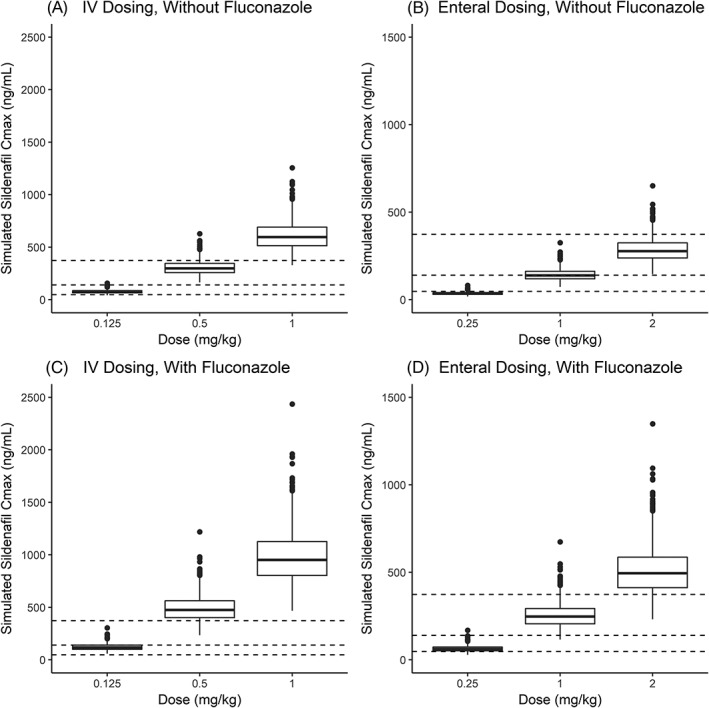
Box plots of simulated steady‐state maximum sildenafil concentrations (Cmax) following intravenous (IV; A, C) or enteral (B, D) doses of sildenafil administered every 8 h in virtual infants with (C, D) and without (A, B) fluconazole. The bottom and top of the box are the 25th and 75th percentile and the band in the middle of the box is the 50th percentile. The length of the box is the interquartile range (IQR). Upper whisker = 75th percentile +1.5 × IQR; lower whisker = 25th percentile – 1.5 × IQR. Dashed lines denote concentrations of 47, 140 and 373 ng/mL that are expected to correlate with unbound concentrations found to inhibit type 5 phosphodiesterase activity in vitro by approximately 53, 77 and 90%, respectively18
4. DISCUSSION
We developed a PopPK model for sildenafil to characterize its disposition and sources of variability by combining data from 2 separate cohorts of premature infants. Cohort 1 (n = 25) infants received open‐label, enteral sildenafil (IV administration in 1 participant) and had a median (range) PMA of 47.0 weeks (31.3–62.7); cohort 2 (n = 9) infants received a single dose of IV sildenafil at a much earlier PMA (27.4 weeks [26.0–32.4]). The sildenafil and DMS PK data collected in this study were reasonably well characterized using 2‐ and 1‐compartment models, respectively, with linear elimination. Previous paediatric PopPK model fitting and compartmental analyses for sildenafil similarly used either a 1‐ or 2‐compartment model with linear elimination for both molecules.15, 16, 17, 27, 28 Visual inspection of the sildenafil and DMS concentration vs time data suggested greater conversion to the metabolite following enteral administration, which led us to evaluate a model with presystemic conversion of sildenafil to DMS. This is in agreement with adult data suggesting presystemic conversion of sildenafil to DMS.29 CYP3A4 duodenal expression is known to increase with age,30, 31, 32 and the greater conversion to DMS in the infants who received sildenafil via enteral administration (cohort 1) may be supported by their greater PNA (median [range] 166 days [52–279]). In the absence of data for the metabolite administered IV, we assumed complete conversion of sildenafil to DMS because it was not possible to uniquely distinguish the extent of DMS's absorption from the conversion of sildenafil to DMS in the systemic circulation. However, even after accounting for presystemic conversion of sildenafil to DMS, there was still some notable model misspecification for concentrations >200 ng/mL (for both sildenafil and DMS). Models with nonlinear absorption or elimination were evaluated but were either not stable or did not result in improvements in the data fitting. The relatively small sample size and sparse sampling may have prevented us from explaining the pronounced variability in the absorption of the drug and metabolite.
In this study, the final population parameter estimates for sildenafil CL and volume of distribution (sum of central and peripheral compartment volumes), scaled to a 70‐kg standardized weight, were 27.8 L/h and 144.1 L, respectively. Our population estimate for CL (27.8 L/h/70 kg) is comparable to the model‐predicted typical value reported from a PopPK analysis of term infants (24.7 L/h/70 kg).15 Although the premature infants in our study were of a younger GA (mean [range] 25 weeks [22–28] vs mean 39.5 weeks [37–42]) when compared to this previous study, they were older in terms of PNA (mean 125.5 days [7–279] vs mean 1.4 days [0.5–3]).15 After correcting for bioavailability (29% in this study), the sildenafil CL estimate observed in our study (27.8/0.29 = 95.9 L/h) is higher than the CL/F estimate reported previously for infants (PNA 2–121 days; GA not reported) following capsule administration via nasogastric tube (62 L/h/70 kg).16 When compared to adult population estimates of CL/F (59 L/h),33 CL/F in our sample of premature infants was also higher. The oral bioavailability estimated for premature infants (29%) following suspension administration was lower than previously published values for adults receiving sildenafil tablets (41%), which may explain, at least in part, some of these differences in apparent CL.34 Sildenafil's volume of distribution (sum of central and peripheral volumes of distribution) in this study (144 L/70 kg) was lower than that reported in another study of term infants (456 L/70 kg), but not dissimilar from adult estimates (105–150 L).15, 34 We compared use of fixed or estimated allometric exponents for CL and V parameters, and found that estimation of the exponents for CL parameters improved the data fit and resulted in a value (0.96) >0.75. This may be because maturation in CL using GA, PNA or PMA was not found to result in a statistically significant reduction in the OFV following a forward inclusion–backward elimination covariate selection approach, and thus only body‐size differences were accounted for in CL predictions. In addition, our study's relatively small sample size and narrow age and weight range, may have affected our estimation of this allometric exponent.35
Concomitant fluconazole use was found to be a significant covariate that explains interindividual variability in sildenafil CL, which is consistent with another infant PopPK study of sildenafil.16 Fluconazole was administered to 2 infants for prophylaxis and 2 infants for treatment (1 empirical treatment for sepsis and 1 had a positive endotracheal tube culture), both in cohort 2. Fluconazole is a CYP3A inhibitor, and thus its use may result in a reduction in the conversion of sildenafil to DMS.36 In our study, the CL reduction with fluconazole use was 59%, which is slightly greater than that previously reported (47%).16 It is possible that this covariate may be explaining variability resulting from another patient variable (e.g. age, critical illness). It is important to note that there were 4 (12% overall; 44% of cohort 2) infants who received concomitant fluconazole use, and all received it via IV administration. In addition, we did not have information about the fluconazole dose received and related severity of illness. CYP3A5*3 also reached statistical significance as a covariate, but was not included in the final model because it did not decrease the interindividual variability, resulted in high ETA shrinkage for CL and the bootstrap estimate for the covariate effect overlapped zero. A previous study demonstrated that the CYP3A5*3 genotype is a significant predictor of sildenafil PK in healthy men.37 Given that CYP3A5 expression has been reported to be generally independent of age in human prenatal and paediatric liver samples,38, 39, 40 and that sildenafil N‐demethylation is catalysed by CYP3A,10, 11, 12, 13, 14 we hypothesized that genetic variation leading to differences in CYP3A5 expression could potentially explain interindividual variability in sildenafil PK. However, the modest impact of CYP3A5*3 on sildenafil PK in our premature infant analyses may have been, at least in part, a result of the small overall sample size and the effect size of this covariate in this population.41 This study highlights the challenges associated with evaluating genotype–phenotype relationships in infants where various factors must be considered when interpreting the results, including: the impact of ontogeny on protein expression; the age characteristics of the study population; a study's sample size and the number of infants with the genetic variant(s) of interest; and the magnitude of the interindividual variability not explained by genetic variation.
There are no widely accepted exposure targets for sildenafil in the setting of BPD or lung disease in the first postnatal weeks likely to evolve to BPD. A previous PopPK analysis of enteral sildenafil in 11 infants with pulmonary hypertension supported with extracorporeal membrane oxygenation reported that all infants with a combined AUC0‐24 that included both sildenafil and DMS exposure (AUC0–24,SIL + DMS; sum of AUC0‐24, SIL and 50% of AUC0‐24, DMS) >2650 ng*h/mL survived, whereas 1 of the 4 who had an exposure lower than this cut‐off died of cardiac failure.16 In this previous study, the target was reached using enteral sildenafil dosing of 4.2 mg/kg/d, although wide variability was noted (10th and 90th percentiles for simulated AUC0–24,SIL + DMS were noted: 1000 and 8000 ng*h/mL). The median simulated exposures (AUC0–24,SIL + DMS) following enteral sildenafil dosing regimens using our final PopPK model were slightly higher, but within the range of simulated values reported in this previous publication (Table S7).16 When simulated Cmax,ss,SIL were compared to previous concentrations reported to inhibit PDE5 activity in vitro, in the absence of fluconazole coadministration, concentrations were generally within the range of those expected to lead to 53–90% inhibition in PDE5 activity. However, with coadministration of fluconazole, sildenafil concentrations generally exceeded 500 ng/mL following IV dosing of 1 mg/kg (or enteral dosing of 2 mg/kg) every 8 hours, which are greater than observed concentrations previously reported following sildenafil administration in infants.15, 42, 43 Additional studies are needed to characterize the relationship between sildenafil exposure and safety, particularly hypotension, for commonly used doses in this population.
This study it is not without limitations. First, even after explaining a significant amount of the interindividual variability in sildenafil CL by including fluconazole use as a covariate in the model and accounting for the impact of WT, misspecification of the observed data remained at high concentrations (>200 ng/mL) for both sildenafil and DMS. There are several potential reasons for this misspecification, including: sildenafil's absorption may have been inadequately characterized based on the modelling assumptions made to account for presystemic conversion of sildenafil to DMS using a hypothetical metabolite absorption compartment; the substantial inter‐ and intraindividual variability we observed in the sildenafil and DMS PK data; and the inability to account for covariates (other than WT and concomitant fluconazole use) that may explain interindividual variability in sildenafil disposition in premature infants. Although the precision of PopPK parameter estimates we observed was adequate, a larger study in this population may allow for further characterization of other relevant covariate effects and interindividual variability in sildenafil absorption and disposition. In addition, application of physiologically‐based pharmacokinetic modelling of sildenafil in premature infants provides an opportunity to mechanistically integrate drug parameters with physiological changes that occur in premature infants (e.g. age‐dependent changes in CYP3A expression), and further evaluate the interaction between sildenafil and fluconazole in infants. Second, there is no widely accepted exposure target for sildenafil in the setting of BPD, which limits the interpretation of our simulation results. Last, although we collected genotype data, SNPs in CYP3A4 and CYP3A7 did not meet the criteria for inclusion in our model, and we were not able to adequately characterize the impact of CYP3A5*3, possibly related to the relatively small sample size and the effect size of the SNPs evaluated in this patient population. Despite these limitations, this is the first study to characterize the PK of sildenafil in premature infants, and the data informed dosing for a follow‐up phase II study in this population (http://ClinicalTrials.gov Identifier: NCT03142568). For future studies in premature infants, our analysis demonstrates the value of collecting PK data following both IV and enteral drug administration, as well as data for relevant concomitant medications and the major circulating metabolite. In addition, it shows the importance of PK characterization in premature infants to inform dosing for a follow‐up phase II study.
COMPETING INTERESTS
D.G. receives support for research from the NICHD (5K23HD083465 and 1R01HD096435). M.M.L. receives support from the US government for work in paediatric pharmacology and trials (Food and Drug Administration R01FD005101, PI: Laughon; NHLBI 1R34HL124038, PI: Laughon; NICHD Pediatric Trials Network Government Contract HHSN267200700051C, PI: Benjamin). M.C‐W. receives support for research from the National Institutes of Health (1R01‐HD076676‐01A1), the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1TR001117), the National Institute of Allergy and Infectious Diseases (HHSN272201500006I and HHSN272201300017I), NICHD (HHSN275201000003I), the Biomedical Advanced Research and Development Authority (HHSO100201300009C), the nonprofit organization Thrasher Research Fund (http://www.thrasherresearch.org), and from industry for drug development in adults and children (http://www.dcri.duke.edu/research/coi.jsp). C.P.H. receives salary support for research from the NICHD (5K23HD090239), the US government for his work in paediatric and neonatal clinical pharmacology (Government Contract HHSN267200700051C, PI: Benjamin, under the Best Pharmaceuticals for Children Act), and industry for drug development in children. The remaining authors have nothing to disclose.
CONTRIBUTORS
D.G., M.M.L., P.B.S. and C.P.H. designed the research; D.G., M.M.L, P.B.S., S.G., N.A., A.A., G.M.S., C.D.H., D.S., G.M., B.B.P., R.G., M.M., M.‐C‐W., K.M. and C.P.H. performed the research; S.G. and D.G. analysed the data; and D.G. wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
DATA AVAILABILITY STATEMENT
To help expand the knowledge base for pediatric medicine, the Pediatric Trials Network is pleased to share data from its completed and published studies with interested investigators. For requests, please contact: PTN‐Program‐Manager@dm.duke.edu.
Supporting information
DATA S1 Supporting information
TABLE S1
Target pharmacokinetic sampling times for cohort 1.
TABLE S2 Target pharmacokinetic sampling times for cohort 2.
TABLE S3 Gradient mobile phase.
TABLE S4 Single nucleotide polymorphisms (SNPs) genotyped and assays used for each variant.
TABLE S5 Summary of genotypes.
TABLE S6 Empirical Bayesian pharmacokinetic parameter estimates stratified by cohort.
TABLE S7 Simulated steady‐state area under the concentration vs time curve from 0 to 24 hours (AUC0–24).
FIGURE S1 ETA vs covariates (weight, postnatal age [PNA], postmenstrual age [PMA] and gestational age [GA]) for both clearance (CL) and central volume of distribution (V2) for the base model without actual body weight as a covariate.
FIGURE S2 Sildenafil and N‐desmethyl sildenafil (DMS) observed vs population (A) and individual (B) predictions for the base population pharmacokinetic model without actual body weight included as a covariate. Triangles and cross symbols represent enteral and intravenous administration, respectively. The solid black line represents the line of unity. The dashed line represents the linear regression fit.
FIGURE S3 Conditional weight residuals (CWRES) vs time after last dose (A) and population predictions (B) for the final sildenafil population pharmacokinetic model.
FIGURE S4 ETAs vs covariates (weight, post‐natal age [PNA], post‐menstrual age [PMA], and gestational age [GA]) plots of the final model for both clearance (CL) and the central volume of distribution (V2).
FIGURE S5 Normalized prediction distribution error (NPDE) plots of sildenafil and DMS after enteral and intravenous administration. (Left panel) Histogram of the NPDE distribution plotted against the theoretical standard normal distribution [N (0,1)] overlaid in blue. (Middle panel) Scatterplot of NPDE vs population predicted concentrations. (Right panel) Scatterplot of NPDE vs time after last dose. In the middle and right panels, the solid red line depicts the median of the NPDE of the observations, and the shaded red area represents the 95% prediction interval for the median. In addition, for the middle and right panels, the solid blue lines represent the 2.5th and 97.5th percentiles of the NPDE for the observations, whereas the shaded blue area represents the 95% prediction interval for the corresponding percentiles. The dashed lines represent NPDE values of ± 1.96.
ACKNOWLEDGEMENTS
This work was funded under the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) contract HHSN275201000003I for the Pediatric Trials Network (Principal Investigator: Daniel K. Benjamin, Jr.). Phoenix® WinNonlin® was provided to the UNC Eshelman School of Pharmacy as part of the Center of Excellence program. The assay measuring sildenafil and DMS concentrations was developed by Robert M. Wurm, BS, at OpAns, LLC (Durham, NC, USA). The authors would like to acknowledge Andrea Gaedigk, PhD, from Children's Mercy Hospital for her contributions towards the genotyping analyses performed as part of this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
APPENDIX A.
A.1. The Best Pharmaceuticals for Children Act—Pediatric Trials Network Publication Committee
Gary Furda, Duke Clinical Research Institute, Durham, NC, USA; Daniel K. Benjamin, Jr., Duke Clinical Research Institute, Durham, NC, USA; Edmund Capparelli, University of California‐San Diego, San Diego, CA, USA; Gregory L. Kearns, Arkansas Children's Hospital Research Institute, Little Rock, AR, USA; Ian M. Paul, Penn State College of Medicine, Hershey, PA, USA; Jan Sullivan, University of Louisville, Louisville, KY, USA; Christoph P. Hornik, Duke Clinical Research Institute, Durham, NC, USA; Kelly Wade, Children's Hospital of Philadelphia, Philadelphia, PA, USA.
A.2. The Eunice Kennedy Shriver National Institute of Child Health and Human Development
David Siegel, Perdita Taylor‐Zapata, Anne Zajicek, Zhaoxia Ren, Ekaterini Tsilou and Alice Pagan.
A.3. The EMMES Corporation (Data Coordinating Center)
Ravinder Anand and Gina Simone.
A.4. Pediatric Trials Network Sildenafil Study Team, Principal Investigators, and Study Coordinators
Namasivayam Ambalavanan, MD (Principal Investigator [PI]), Tara E. McNair, RN, BSN (Study Coordinator [SC]), and Vivien Phillips, RN, BSN (SC), University of Alabama at Birmingham, Birmingham, AL, USA; Andrew Atz, MD (PI) and Kalyan Janakiram Choudhary, MSCR (SC), Medical University of South Carolina Children's Hospital, Charleston, SC, USA; Gregory M. Sokol, MD (PI), Susan Gunn, NNP‐BC, CCRC (SC), Dianne Herron, RN, CCRC (SC), and Lucy Smiley, BS, CCRC (SC), Riley Hospital for Children at Indiana University Health, Indiana University, Indianapolis, IN, USA; Chi D. Hornik, PharmD, BCPS, CPP (PI), C. Michael Cotten, MD (Investigator), and Lacey Andrews, BS (SC), Duke University Medical Center, Durham, NC, USA; Dan Stewart, MD (PI), Janice Sullivan, MD (Investigator), and Andrew Michael, RN, BSN (SC), University of Louisville Norton Children's Hospital, Louisville, KY, USA; Gratias Mundakel, MD (PI) and Subhatra Limbu, MBBS, MPH (SC), Kings County Hospital Center/SUNY Downstate Medical Center, Brooklyn, NY, USA; Brenda Poindexter, MD, MS (PI) and Sandra Wuertz, RN‐BSN, CCRP, CLC (SC), Cincinnati Children's Hospital Medical Center and the University of Cincinnati School of Medicine, Cincinnati, OH, USA.
Gonzalez D, Laughon MM, Smith PB, et al. Population pharmacokinetics of sildenafil in extremely premature infants. Br J Clin Pharmacol. 2019;85:2824–2837. 10.1111/bcp.14111
The authors confirm that the Principal Investigator for this paper is Matthew M. Laughon and that he had direct clinical responsibility for the patients.
REFERENCES
- 1. Khemani E, McElhinney DB, Rhein L, et al. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics. 2007;120(6):1260‐1269. [DOI] [PubMed] [Google Scholar]
- 2. Stoll BJ, Hansen NI, Bell EF, et al. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993‐2012. JAMA. 2015;314(10):1039‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berkelhamer SK, Mestan KK, Steinhorn RH. Pulmonary hypertension in bronchopulmonary dysplasia. Semin Perinatol. 2013;37(2):124‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bhat R, Salas AA, Foster C, Carlo WA, Ambalavanan N. Prospective analysis of pulmonary hypertension in extremely low birth weight infants. Pediatrics. 2012;129(3):e682‐e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beam KS, Aliaga S, Ahlfeld SK, Cohen‐Wolkowiez M, Smith PB, Laughon MM. A systematic review of randomized controlled trials for the prevention of bronchopulmonary dysplasia in infants. J Perinatol. 2014;34(9):705‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Visser YP, Walther FJ, Laghmani EH, Boersma H, van der Laarse A, Wagenaar GT. Sildenafil attenuates pulmonary inflammation and fibrin deposition, mortality and right ventricular hypertrophy in neonatal hyperoxic lung injury. Respir Res. 2009;10(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ladha F, Bonnet S, Eaton F, Hashimoto K, Korbutt G, Thébaud B. Sildenafil improves alveolar growth and pulmonary hypertension in hyperoxia‐induced lung injury. Am J Respir Crit Care Med. 2005;172(6):750‐756. [DOI] [PubMed] [Google Scholar]
- 8. Hon KL, Cheung KL, Siu KL, et al. Oral sildenafil for treatment of severe pulmonary hypertension in an infant. Biol Neonate. 2005;88(2):109‐112. [DOI] [PubMed] [Google Scholar]
- 9. Caputo S, Furcolo G, Rabuano R, et al. Severe pulmonary arterial hypertension in a very premature baby with bronchopulmonary dysplasia: normalization with long‐term sildenafil. J Cardiovasc Med (Hagerstown). 2010;11(9):704‐706. [DOI] [PubMed] [Google Scholar]
- 10. Ku HY, Ahn HJ, Seo KA, et al. The contributions of cytochrome P450 3A4 and 3A5 to the metabolism of the phosphodiesterase type 5 inhibitors sildenafil, udenafil, and vardenafil. Drug Metab Dispos. 2008;36(6):986‐990. [DOI] [PubMed] [Google Scholar]
- 11. Warrington JS, Shader RI, von Moltke LL, Greenblatt DJ. In vitro biotransformation of sildenafil (Viagra): identifiation of human cytochromes and potential drug interactions. Drug Metab Dispos. 2000;28(4):392‐397. [PubMed] [Google Scholar]
- 12. Pfizer, Inc . Revatio® package insert. Accessed via: http://labeling.pfizer.com/ShowLabeling.aspx?id=645.
- 13. Hyland R, Roe EG, Jones BC, Smith DA. Identification of the cytochrome P450 enzymes involved in the N‐demethylation of sildenafil. Br J Clin Pharmacol. 2001;51(3):239‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takahiro R, Nakamura S, Kohno H, et al. Contribution of CYP3A isoforms to dealkylation of PDE5 inhibitors: a comparison between sildenafil N‐demethylation and tadalafil demethylenation. Biol Pharm Bull. 2015;38(1):58‐65. [DOI] [PubMed] [Google Scholar]
- 15. Mukherjee A, Dombi T, Wittke B, Lalonde R. Population pharmacokinetics of sildenafil in term neonates: evidence of rapid maturation of metabolic clearance in the early postnatal period. Clin Pharmacol Ther. 2009;85(1):56‐63. [DOI] [PubMed] [Google Scholar]
- 16. Ahsman MJ, Witjes BC, Wildschut ED, et al. Sildenafil exposure in neonates with pulmonary hypertension after administration via a nasogastric tube. Arch Dis Child Fetal Neonatal Ed. 2010;95(2):F109‐F114. [DOI] [PubMed] [Google Scholar]
- 17. Watt S, Hayashi N, Harnisch L, Gao X. Population pharmacokinetics (PK) of sildenafil in paediatric and adult patients with pulmonary arterial hypertension (PAH). Poster presented at the European Society of Cardiology Congress 2010; August 28–September 1, 2010; Stockholm, Sweden.
- 18. Barst RJ, Ivy DD, Gaitan G, et al. A randomized, double‐blind, placebo‐controlled, dose‐ranging study of oral sildenafil citrate in treatment‐naive children with pulmonary arterial hypertension. Circulation. 2012;125(2):324‐334. [DOI] [PubMed] [Google Scholar]
- 19. Lindbom L, Ribbing J, Jonsson EN. Perl‐speaks‐NONMEM (PsN)‐‐a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85‐94. [DOI] [PubMed] [Google Scholar]
- 20. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2nd ed. New York: Springer‐Verlag; 2016. [Google Scholar]
- 22. Sarkar D. Lattice: Multivariate Data Visualization with R. 1st ed. New York: Springer; 2008. [Google Scholar]
- 23. Comets E, Brendel K, Mentré F. Computing normalised prediction distribution errors to evaluate nonlinear mixed‐effect models: the npde add‐on package for R. Comput Methods Programs Biomed. 2008;90(2):154‐166. [DOI] [PubMed] [Google Scholar]
- 24. Kerbusch T, Wählby U, Milligan PA, Karlsson MO. Population pharmacokinetic modelling of darifenacin and its hydroxylated metabolite using pooled data, incorporating saturable first‐pass metabolism, CYP2D6 genotype and formulation‐dependent bioavailability. Br J Clin Pharmacol. 2003;56(6):639‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ince I, de Wildt SN, Wang C, et al. A novel maturation function for clearance of the cytochrome P450 3A substrate midazolam from preterm neonates to adults. Clin Pharmacokinet. 2013;52(7):555‐565. [DOI] [PubMed] [Google Scholar]
- 26. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hill KD, Sampson MR, Li JS, Tunks RD, Schulman SR, Cohen‐Wolkowiez M. Pharmacokinetics of intravenous sildenafil in children with palliated single ventricle heart defects: effect of elevated hepatic pressures. Cardiol Young. 2016;26(2):354‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olguín HJ, Martínez HO, Pérez CF, et al. Pharmacokinetics of sildenafil in children with pulmonary arterial hypertension. World J Pediatr. 2017;13(6):588‐592. [DOI] [PubMed] [Google Scholar]
- 29. Muirhead GJ, Rance DJ, Walker DK, Wastall P. Comparative human pharmacokinetics and metabolism of single‐dose oral and intravenous sildenafil. Br J Clin Pharmacol. 2002;53(Suppl 1):13S‐20S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johnson TN, Tanner MS, Taylor CJ, Tucker GT. Enterocytic CYP3A4 in a paediatric population: developmental changes and the effect of coeliac disease and cystic fibrosis. Br J Clin Pharmacol. 2001;51(5):451‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fakhoury M, Litalien C, Medard Y, et al. Localization and mRNA expression of CYP3A and P‐glycoprotein in human duodenum as a function of age. Drug Metab Dispos. 2005;33(11):1603‐1607. [DOI] [PubMed] [Google Scholar]
- 32. Chen YT, Trzoss L, Yang D, Yan B. Ontogenic expression of human carboxylesterase‐2 and cytochrome P450 3A4 in liver and duodenum: postnatal surge and organ‐dependent regulation. Toxicology. 2015;330:55‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Milligan PA, Marshall SF, Karlsson MO. A population pharmacokinetic analysis of sildenafil citrate in patients with erectile dysfunction. Br J Clin Pharmacol. 2002;53(Suppl 1):45S‐52S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nichols DJ, Muirhead GJ, Harness JA. Pharmacokinetics of sildenafil after single oral doses in healthy male subjects: absolute bioavailability, food effects and dose proportionality. Br J Clin Pharmacol. 2002;53(Suppl 1):5S‐12S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sinha J, Al‐Sallami HS, Duffull SB. Choosing the allometric exponent in covariate model building. Clin Pharmacokinet. 2019;58(1):89‐100. [DOI] [PubMed] [Google Scholar]
- 36. Gibbs MA, Thummel KE, Shen DD, Kunze KL. Inhibition of cytochrome P‐450 3A (CYP3A) in human intestinal and liver microsomes: comparison of Ki values and impact of CYP3A5 expression. Drug Metab Dispos. 1999;27(2):180‐187. [PubMed] [Google Scholar]
- 37. Shon JH, Ku HY, Bae SY, et al. The disposition of three phosphodiesterase type 5 inhibitors, vardenafil, sildenafil, and udenafil, is differently influenced by the CYP3A5 genotype. Pharmacogenet Genomics. 2011;21(12):820‐828. [DOI] [PubMed] [Google Scholar]
- 38. Stevens JC, Hines RN, Gu C, et al. Developmental expression of the major human hepatic CYP3A enzymes. J Pharmacol Exp Ther. 2003;307(2):573‐582. [DOI] [PubMed] [Google Scholar]
- 39. Vyhlidal CA, Pearce RE, Gaedigk R, et al. Variability in expression of CYP3A5 in human fetal liver. Drug Metab Dispos. 2015;43(8):1286‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hines RN. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther. 2008;118(2):250‐267. [DOI] [PubMed] [Google Scholar]
- 41. Lagishetty CV, Coulter CV, Duffull SB. Design of pharmacokinetic studies for latent covariates. J Pharmacokinet Pharmacodyn. 2012;39(1):87‐97. [DOI] [PubMed] [Google Scholar]
- 42. Steinhorn RH, Kinsella JP, Pierce C, et al. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J Pediatr. 2009;155(6):841‐847.e1. [DOI] [PubMed] [Google Scholar]
- 43. Thakkar N, Gonzalez D, Cohen‐Wolkowiez M, et al. An opportunistic study evaluating pharmacokinetics of sildenafil for the treatment of pulmonary hypertension in infants. J Perinatol. 2016;36(9):744‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DATA S1 Supporting information
TABLE S1
Target pharmacokinetic sampling times for cohort 1.
TABLE S2 Target pharmacokinetic sampling times for cohort 2.
TABLE S3 Gradient mobile phase.
TABLE S4 Single nucleotide polymorphisms (SNPs) genotyped and assays used for each variant.
TABLE S5 Summary of genotypes.
TABLE S6 Empirical Bayesian pharmacokinetic parameter estimates stratified by cohort.
TABLE S7 Simulated steady‐state area under the concentration vs time curve from 0 to 24 hours (AUC0–24).
FIGURE S1 ETA vs covariates (weight, postnatal age [PNA], postmenstrual age [PMA] and gestational age [GA]) for both clearance (CL) and central volume of distribution (V2) for the base model without actual body weight as a covariate.
FIGURE S2 Sildenafil and N‐desmethyl sildenafil (DMS) observed vs population (A) and individual (B) predictions for the base population pharmacokinetic model without actual body weight included as a covariate. Triangles and cross symbols represent enteral and intravenous administration, respectively. The solid black line represents the line of unity. The dashed line represents the linear regression fit.
FIGURE S3 Conditional weight residuals (CWRES) vs time after last dose (A) and population predictions (B) for the final sildenafil population pharmacokinetic model.
FIGURE S4 ETAs vs covariates (weight, post‐natal age [PNA], post‐menstrual age [PMA], and gestational age [GA]) plots of the final model for both clearance (CL) and the central volume of distribution (V2).
FIGURE S5 Normalized prediction distribution error (NPDE) plots of sildenafil and DMS after enteral and intravenous administration. (Left panel) Histogram of the NPDE distribution plotted against the theoretical standard normal distribution [N (0,1)] overlaid in blue. (Middle panel) Scatterplot of NPDE vs population predicted concentrations. (Right panel) Scatterplot of NPDE vs time after last dose. In the middle and right panels, the solid red line depicts the median of the NPDE of the observations, and the shaded red area represents the 95% prediction interval for the median. In addition, for the middle and right panels, the solid blue lines represent the 2.5th and 97.5th percentiles of the NPDE for the observations, whereas the shaded blue area represents the 95% prediction interval for the corresponding percentiles. The dashed lines represent NPDE values of ± 1.96.
Data Availability Statement
To help expand the knowledge base for pediatric medicine, the Pediatric Trials Network is pleased to share data from its completed and published studies with interested investigators. For requests, please contact: PTN‐Program‐Manager@dm.duke.edu.