

Abstract
Aims
Fremanezumab is a fully humanized IgG2Δa/κ monoclonal antibody specific for calcitonin gene‐related peptide developed and approved for the preventive treatment of migraine in adults. The population pharmacokinetics (PK) of fremanezumab were characterized in healthy subjects and patients with chronic migraine and episodic migraine, including the effects of intrinsic and extrinsic factors on PK variability.
Methods
Nonlinear mixed effects modelling was performed using NONMEM with data from 7 phase 1–3 clinical trials evaluating selected intravenous and subcutaneous dose regimens. The influence of covariates on fremanezumab PK was assessed and model evaluation was performed through visual predictive checks.
Results
A 2‐compartment model with first‐order absorption and elimination described the PK data well. Typical values for fremanezumab central clearance (0.0902 L/d) and central distribution volume (1.88 L) for a 71‐kg subject were consistent with previously reported values for IgG antibodies. Higher body weight was associated with increased central clearance and distribution volume. Effects of other covariates (age, albumin, renal function, sex, race, injection site, and acute, analgesic and preventive medication use for migraine) were not found to statistically significantly influence fremanezumab PK. There was no indication of reduced exposure in participants with positive anti‐drug antibody status or with mild to moderate hepatic impairment. Absolute bioavailability was estimated at 0.658.
Conclusions
A comprehensive population PK model was developed for fremanezumab following intravenous and subcutaneous administration in healthy subjects and patients with chronic migraine or episodic migraine, which will be used to further evaluate exposure–response relationships for efficacy and safety endpoints.
Keywords: fremanezumab, population pharmacokinetics, migraine
What is already known about this subject
Fremanezumab plasma concentration–time profiles and exposure parameters were similar in Japanese and Caucasian healthy subjects following single dose subcutaneous administration of 225, 675 and 900 mg.
The long half‐life of fremanezumab (30 days) supports monthly or quarterly administration.
What this study adds
The population pharmacokinetics of fremanezumab after intravenous and subcutaneous administration was characterized in over 2500 subjects (including patients with chronic migraine or episodic migraine) using a 2‐compartment model with first‐order absorption and elimination.
Weight was the only significant covariate predictive of variability in fremanezumab pharmacokinetics; higher weight was associated with lower exposure.
1. INTRODUCTION
Calcitonin gene‐related protein (CGRP) is a well‐studied neuropeptide and vasodilator that plays an important role in the pathogenesis of migraine, both centrally and peripherally.1, 2 Increased CGRP levels have been detected in the external jugular vein during an acute migraine attack,3 consistent with previous reports that intravenous (IV) CGRP administration induces migraine‐like headaches in most individuals with migraine, but not healthy control subjects.4 Small molecule CGRP receptor antagonists have demonstrated efficacy in acute migraine5, 6, 7 and monoclonal antibodies (mAbs) that target CGRP or its receptor have demonstrated efficacy as preventive treatment in episodic migraine (EM) and chronic migraine (CM) studies.8, 9
Fremanezumab, a fully humanized IgG2Δa/κ mAb, selectively targets CGRP and has been approved by the US Food and Drug Administration for the preventive treatment of migraine in adults.8 Fremanezumab binds to the CGRP ligand and blocks both the α‐ and β‐CGRP isoforms from binding to the CGRP receptor.8 Introduction of 2 mutations into the constant region of the fremanezumab heavy chain limits antibody effector functions, thereby preventing fremanezumab from stimulating antibody‐dependent cell‐mediated cytotoxicity and triggering complement‐mediated lysis.10
During the development of fremanezumab, pharmacometric modelling and simulation were used extensively to support development‐related decision‐making and are described in this manuscript. A population pharmacokinetic (PopPK) model was iteratively developed using a 3‐staged approach. Initially, it was developed using intensive data from healthy subjects in 2 phase 1 studies following IV and subcutaneous (SC) administration,11 then adding informative data from the long‐term safety (LTS) phase 3 study in EM and CM migraine patients after SC administration.12 Finally, it was further refined by adding trough samples collected after SC administration from 2 phase 2b13, 14, 15 and 3 phase 3 trials (2 pivotal studies and the LTS study.12, 16, 17 The final PopPK model provided a basis for prediction of subject‐specific exposure estimates to support future assessment of exposure–response relationships for efficacy and safety endpoints. This manuscript represents the first published PopPK analysis for a therapeutic mAb targeting the CGRP pathway.
2. METHODS
2.1. Data
Data from 7 clinical studies evaluating IV and SC administration of fremanezumab in healthy subjects and patients with EM or CM were pooled for analysis (Table 1). The 2 phase 1 studies included full‐profile blood sampling for determination of fremanezumab concentrations over 90 or 225 days, whereas a sparse sampling strategy (monthly predose trough samples) was implemented in the phase 2b and 3 studies (2 pivotal studies and 1 LTS study). Two additional nontrough samples were obtained from patients enrolled in the phase 3 LTS study who did not roll over from 1 of the 2 double‐blind phase 3 trials. Fremanezumab was administered subcutaneously, except for 1 phase 1 study that included both IV and SC administration.
Table 1.
Description of participants, dosing regimens, and pharmacokinetic sampling plans for studies included in the population pharmacokinetic analyses
| Study number/phase | Study title | Participants | Duration of dosing | Dosing regimen | Pharmacokinetic sampling |
|---|---|---|---|---|---|
| LBR‐101‐011/phase 1 | A randomized, placebo‐controlled, double‐blind, parallel group study assessing the safety, tolerability, and pharmacokinetics of 2 different doses of TEV‐48125 given IV and SC | 36 healthy male and female subjects (18–60 years)24 (6/cohort) received TEV‐48125; 12 (6/cohort) received placebo | Single IV (1‐h infusion) or SC dose | Six treatment groups:225 mg, 1‐h IV infusion (n = 6)900 mg, 1‐h IV infusion (n = 6)225 mg, SC (n = 6)900 mg, SC (n = 6)placebo, 1‐h IV infusion (n = 6)placebo SC (n = 6) | Predose (0 h), EOI (1 h), 6, and 12 h after the start of infusion/postdose, and on d 2, 4, 6, 7, 10, 15, 21, 30, 60, and 90 |
| TV48125‐PK‐10078/phase 1 | A double‐blind, placebo‐controlled study to assess the pharmacokinetics, safety, and tolerability of single doses SC administration of TEV‐48125 (doses up to 900 mg) in Japanese and Caucasian healthy subjects | 64 healthy subjects enrolled: 32 Japanese and 32 Caucasian, matched based on sex, age and body mass index48 received fremanezumab and 16 received placebo | Single SC dose | One cohort (cohort 1) with the following doses administered in the following allocation ratio:225 mg or placebo SC 4:2 (n = 6)675 mg or placebo SC 4:1 (n = 5)900 mg or placebo SC 4:1 (n = 5)three cohorts (cohorts 2, 3, and 4) with the following doses administered in a 1:1:1:1 allocation ratio (n = 16 per cohort):225 mg SC675 mg SC900 mg SCplacebo SC | Predose (0 h) and 4, 8, and 12 h after study drug administration on d 1 and on d 2 (24 and 36 h postdose), 3 (48 and 60 h post), 4 (72 and 84 h postdose), 5 (96 and 108 h postdose), 6 (120 postdose), 8 ± 1 d, 12 ± 1 d, 15 ± 1 d, 29 ± 2 d, 43 ± 2 d, 57 ± 2 d, 85 ± 2 d, 113 ± 2 d, 141 ± 2 d, 169 ± 2 d, 197 ± 3 d, and 225 ± 3 d |
| LBR‐101‐021/phase 2b | A multicentre, randomized, double‐blind, double‐dummy, placebo‐controlled, parallel group, multi‐dose study comparing the efficacy and safety of SC TEV‐48125 with placebo for the preventive treatment of chronic migraine | ~240 male and female subjects (18–65 years) with history of frequent migraines suggestive of chronic migraine~160 (80/cohort) received fremanezumab; 80 received placebo | 3 SC doses total: Once monthly for 3 mo | Three treatment groups administered 3 doses every 28 d:675 (loading dose)/225 mg, SC (n = 88)900 mg, SC (n = 88)placebo, SC (n = 89) | Predose on d 1, 29, 57, and a sample collected on d 85 (follow‐up) |
| LBR‐101‐022/phase 2b | A multicentre, randomized, double‐blind, placebo‐controlled, parallel‐group study comparing the efficacy and safety of 2 doses of SC TEV‐48125 with placebo for the preventive treatment of high frequency episodic migraine | ~300 male and female subjects (18–65 years) fulfilling the criteria for episodic migraine~200 (100/cohort) received fremanezumab; 100 received placebo | 3 SC doses total: Once monthly for 3 mo | Three treatment groups administered 3 doses every 28 d:225 mg, SC (n = 96)675 mg, SC (n = 97)placebo, SC (n = 104) | Predose on d 1, 29, 57, and a sample collected on d 85 (follow‐up) |
| TV48125‐CNS‐30049/phase 3 | A multicentre, randomized, double‐blind, placebo‐controlled, parallel‐group study comparing the efficacy and safety of 2 dose regimens of SC administration of TEV‐48125 vs placebo for the preventive treatment of chronic migraine | ~1020 male and female subjects (18–70 years) fulfilling the criteria for chronic migraine~680 (340/cohort) received fremanezumab; 340 received placebo | 3 SC doses total: Once monthly for 3 mo | Three treatment groups administered 3 doses every 28 d:675 mg, SC loading dose; 225 mg, SC next 2 doses (n = 379)675 mg, SC (n = 376)placebo, SC (n = 375) | Predose on d 1, 29, 57, and a sample collected on d 85 (follow‐up) |
| TV48125‐CNS‐30050/phase 3 | A multicentre, randomized, double‐blind, placebo‐controlled, parallel‐group study comparing the efficacy and safety of 2 dose regimens of SC administration of TEV‐48125 vs placebo for the preventive treatment of episodic migraine | ~768 male and female subjects (18–70 years) fulfilling the criteria for episodic migraine~512 (256/cohort) received fremanezumab; 256 received placebo | 3 SC doses total: Once monthly for 3 mo | Three treatment groups administered 3 doses every 28 d:225 mg, SC (n = 290)675 mg, SC first dose; placebo, SC next 2 doses (n = 291)placebo, SC (n = 294) | Predose on d 1, 29, 57, and a sample collected on d 85 (follow‐up) |
| TV48125‐CNS‐30051/phase 3 | A multicentre, randomized, double‐blind, parallel‐group study evaluating the long‐term safety, tolerability, and efficacy of SC administration of TEV‐48125 for the preventive treatment of migraine | ~1842 male and female subjects (18–70 years)~867 subjects fulfilling the criteria for chronic migraine and ~675 subjects fulfilling the criteria for episodic migraine from pivotal efficacy study (studies TV48125‐CNS‐30049 and TV48125‐CNS‐30050)~300 new subjects fulfilling the criteria for chronic or episodic migraines | 12 or 4 SC doses total: Once monthly for 12 mo or once every 3 mo for 12 mo, respectively | One cohort (cohort 1) for subjects with chronic migraine (n = 1017):675 mg SC (loading dose) and 225 mg SC once monthly for 11 mo (total 12 doses)675 mg SC once every 3 mo for 12 mo (total 4 doses) | For subjects rolling over from a previous pivotal study (TV48125‐CNS‐30049 and TV48125‐CNS‐30050):Predose on d 85, 169, 253, and 337 and a sample collected on d 534 (follow‐up) |
| One cohort (cohort 2) for subjects with episodic migraine (n = 825):225 mg dose SC every 28 d for 12 mo (total 12 doses)675 mg SC once every 3 mo for 12 mo (total 4 doses) | For subjects not rolling over from the previous pivotal study:Predose on d 1, 85, 169, 253, and 337 and a sample collected on d 534 (follow‐up);two additional visits after any dose of study drug: 3 to 10 d or 15 to 20 d after study drug administration |
EOI, end of infusion; IV, intravenous; SC, subcutaneous; TEV 48125, fremanezumab.
All studies were conducted in accordance with the principles of the Declaration of Helsinki. Clinical protocols were reviewed and approved for each site by institutional review boards and all study participants provided written informed consent prior to enrolment in the studies.
2.2. Bioanalysis
A sandwich enzyme‐linked immunosorbent assay procedure was used for measurement of plasma fremanezumab concentrations. The method was validated in the concentration range of 250 to 3500 ng/mL as assessed via accuracy and precision using fremanezumab spiked at different levels in the human plasma samples. The accuracy of the spiked samples varied between 98 and 104% of the nominal concentrations. The interassay precision varied from 3 to 7% and the intra‐assay from 3 to 5%. The lower limit of quantitation (LLOQ) value for the PK assay used in the analysis was 250 ng/mL.
2.3. Population PK modelling
NONMEM, v7.3, was used for all model development (first‐order conditional estimation with interaction method), simulations and evaluation procedures,18 and KIWI, v3 (or later), was used for model discrimination and data visualization.19
Figure 1 illustrates the PopPK model development process for fremanezumab. Exploratory graphical analysis of the concentration–time data obtained following IV administration of fremanezumab in the phase 1 bioavailability study exhibited a biphasic profile, suggesting a 2‐compartmental model; however, models based on data following SC administration indicated a 1‐compartmental model. Hence, the modelling was initiated using only the IV data fitted to a 2‐compartment model. Then, a systematic, stepwise approach to model building was implemented given the heterogeneous nature of the pooled dataset after SC administration. Data collected after SC administration were added to the analysis dataset in stages from most to least informative: (i) intensive SC data from the study including both IV and SC administration (used to estimate bioavailability); (ii) data from the other phase 1 study with SC administration to establish a model based on all intensive phase 1 data; (iii) data from the phase 3 study with additional nontrough sampling; and (iv) trough samples from the phase 2b and 3 studies.
Figure 1.
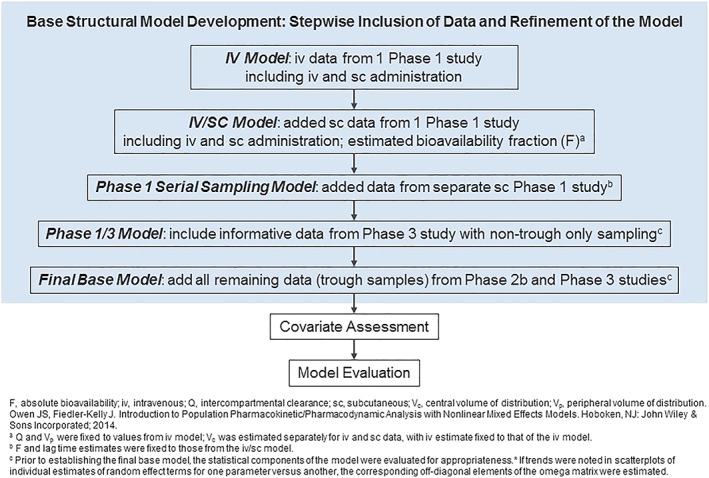
Flowchart of the fremanezumab population pharmacokinetic model development process
Between‐subject variability (BSV) in parameters was modelled using an exponential form. Residual variability (RV) was modelled using a proportional or additive plus proportional form, as appropriate.20
2.3.1. Covariate analysis
The influence of various intrinsic and extrinsic factors on the PK of fremanezumab was evaluated, including weight, age, race, sex, patient status, albumin, anti‐drug antibody (ADA) status, injection site, renal function category, hepatic function category, and use of preventive, acute or analgesic medications. Continuous covariates were centred about the population median value to improve the precision of the parameter estimates.20
Forward selection followed by backward elimination (α = 0.01 with decrease in BSV by ≥5% and α = 0.001, respectively; 1 df for χ2‐distribution) methods were used to assess covariate relationships. After backward elimination, the reduced multivariable model was then evaluated for any remaining biases in the BSV and RV error models and refined, as necessary.
2.3.2. Model evaluation
The adequacy of the final model was evaluated using a simulation‐based, prediction‐corrected visual predictive check (VPC) method.21, 22 Simulations were performed (n = 1000 replicates) using the final model and statistics of interest were calculated for comparison with observed data; e.g., the 5th, 50th (median) and 95th percentiles of the simulated and observed concentration distributions. These percentiles were plotted vs time, with the original observed dataset and percentiles based on the observed data overlaid to visually assess concordance between the model‐based simulated data and the observed data.
2.4. Simulations
Model‐based simulations were performed to evaluate exposures for the 2474 patients enrolled in the phase 2b or 3 trials, based on SC dosing regimens of fremanezumab administered in the phase 3 clinical trials: 225 mg SC monthly for 12 doses (with and without a starting dose of 675 mg) and 675 mg SC quarterly for 4 doses. Patient‐level measures of steady‐state fremanezumab exposure, including maximum drug concentration (Cmax,ss), average drug concentration (Cav,ss), and area under the fremanezumab drug concentration vs time curve (AUCss), were calculated based on the individual Bayesian estimates of model parameters from day 0 to day 28 or day 84 (for monthly or quarterly dosing regimens, respectively).
3. RESULTS
3.1. Exploratory data analysis
A total of 13 745 fremanezumab concentrations (2436 samples from phase 1 and 2b trials and 11 309 samples from phase 3 trials) collected from 2546 individuals (74 healthy subjects and 2474 patients) were used for the PopPK modelling. Concentrations below the LLOQ comprised 1.9% of all postdose samples and were excluded from the analysis dataset used for modelling.
The analysis population for the pooled data was primarily Caucasian (79.9%) and female (86.1%) with a median (range) age of 43 (18–71) years and median (range) body weight of 70.8 (43.5–131.8) kg. Approximately 20% of the PK samples were collected in the presence of preventive medications, 55% in the presence of acute medications and 9% in the presence of analgesics. The presence of ADAs was confirmed in only 0.7% of the samples collected. Supporting Table S1 provides a summary of the intrinsic and extrinsic factors by study phase.
Figure 2 displays a semilogarithmic scatterplot of dose‐normalized plasma fremanezumab concentrations vs time since the previous dose for the pooled phase 1/2b/3 data. The dose‐normalized concentration data generally show similar exposures between the phase 1, 2b and 3 data, with relatively greater variability evident in the sparse phase 2b/3 data.
Figure 2.

Dose‐normalized plasma fremanezumab concentrations vs time since previous dose. Conc, concentration; norm, normalized
3.2. Final fremanezumab population PK model
A 2‐compartment model with first‐order absorption and a lag time, first‐order elimination, an effect of weight on CL and central volume of distribution (Vc), a proportional RV model for the IV data and a combined additive and proportional RV model for the SC data (Figure 3) adequately fit the pooled phase 1/2b/3 data. The model included separate estimates of central volume for IV (Vc,IV) and SC (Vc,SC) administration. Parameters for the peripheral compartment (intercompartmental clearance [Q] and peripheral volume of distribution [Vp]) were estimated from the IV phase 1 data and fixed in subsequent stages. In addition, absolute bioavailability (F) and lag time were estimated based on both IV and SC phase 1 data and subsequently fixed for models including the entire pooled phase 1/2b/3 dataset. Table 2 provides the final parameter estimates and corresponding precisions for the final PopPK model of fremanezumab. The typical values (for a 71‐kg subject, the median weight in this population) for central CL and Vc,SC were 0.0902 L/d and 1.88 L, respectively, with an estimated F of 0.658 for SC administration. Based on the allometric relationships with weight, relative to a 71‐kg subject (the median weight in the population), a 51‐kg subject and a 101‐kg subject (i.e. 5th and 95th percentiles of body weight in the analysis dataset) are expected to have approximately 29% lower and 45% higher CL and 40% lower and 71% higher Vc, respectively.
Figure 3.
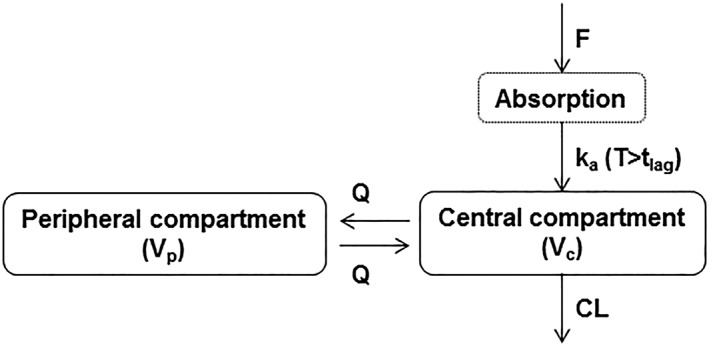
Schematic diagram of fremanezumab population pharmacokinetic model structure. CL, clearance; F, relative bioavailability; ka, first‐order absorption rate constant; Q, intercompartmental clearance; T, time; tlag, absorption lag time; Vc, central volume of distribution; Vp, peripheral volume of distribution
Table 2.
Parameter estimates and standard errors for the fremanezumab final population pharmacokinetic model
| Parameter | Final parameter estimate | Between‐subject variability | ||
|---|---|---|---|---|
| Typical value | %RSE | Magnitude | %RSE | |
| CL: Central clearance (L/d) | 0.0902 | 1.50 | 23.4%CVa , b | 4.60 |
| CL: Allometric exponent for weight (−)c | 1.05 | 4.33 | ||
| Vc,iv: Central volume of distribution IV (L)d | 2.98 | FIXED | NE | NA |
| Vc,SC: Central volume of distribution SC (L)e | 1.88 | 3.38 | 35.1%CVa , b , f | 19.9 |
| Vc: Allometric exponent for weight (−)d , e | 1.53 | 10.3 | ||
| ka: Absorption rate constant (/d) | 0.180 | 12.2 | 59.0%CVa , b , f | 15.8 |
| Q: Intercompartmental clearance (L/d) | 0.262 | FIXED | NE | NA |
| Vp: Peripheral volume of distribution (L) | 1.72 | FIXED | NE | NA |
| F1: Bioavailability | 0.658 | FIXED | NE | NA |
| ALAG1: Lag time (d) | 0.0803 | FIXED | NE | NA |
| Residual variability iv | 0.0467 | 32.6 | NA | NA |
| Residual variability SC proportional componentg | 0.0531 | 4.03 | ||
| Residual variability SC additive componentg | 0.204 | 25.6 | ||
| Minimum value of the objective function = 73303.561 | ||||
%CV, coefficient of variation expressed as a percent; IV, intravenous; NA, not applicable; NE, not estimated; %RSE, relative standard error expressed as a percent; SC, subcutaneous.
.
Bayesian shrinkage of η estimates was 10.6% for clearance, 37.1% for Vc, and 42.7% for ka.
Typical Value for CL = 0.0902 × (Weight/71)1.05
Typical Value for Vc,iv = 2.98 × (Weight/71)1.53
Typical Value for Vc,sc = 1.88 × (Weight/71)1.53
Phase 1 data (subcutaneous [SC]) and the patients with additional pharmacokinetic samples in Study TV48125‐CNS‐30051 were considered to have appropriate and informative data to contribute to the estimation of between‐subject variability (BSV) in the central volume of distribution (Vc) and absorption rate constant (ka). Thus, data from other patients were excluded from the estimation of BSV and calculation of shrinkage in the BSV of Vc and ka.
Epsilon shrinkage ranged from 3.1 to 9.0%.
All parameters were estimated well, with no high correlations (|r| > 0.9) between parameter estimates. The magnitude of estimated BSV was lower for CL (23.4%) and Vc (35.1%), compared to first‐order absorption rate constant (ka; 59.0%). To improve model stability, subjects with IV infusion administration or trough‐only sampling were not included in the estimation of BSV on ka or Vc; this was accomplished by fixing the individual values of ka or Vc for these subjects equal to the typical value. Estimated RV ranged from 93.3%CV at low individual predicted concentrations (values near the LLOQ) to 23.1% for fremanezumab predicted concentrations >10 μg/mL. Goodness‐of‐fit plots for the final PopPK model (Figure 4) indicate that the model provided a reasonable fit to the data following both IV and SC administration. Scatterplots of individual estimates of random effect terms of 1 parameter vs another, assessing the assumption of zero covariance, showed no apparent trends (data not shown). As such, no off‐diagonal omega matrix elements were estimated.
Figure 4.

Goodness‐of‐fit diagnostic plots for the fremanezumab final population pharmacokinetic model, including observed concentrations vs population predicted and individual predicted values; residuals and conditional weighted residuals vs population predicted values; conditional weighted residuals vs time; normalized prediction distribution errors (NPDE) values vs time since dosing; and individual weighted residuals vs individual predicted values. The red line is a reference line or the line of identity, the blue line is a smoothing spline. Conc, concentration
The effect of intrinsic and extrinsic factors (including dose) on specified PK parameters was evaluated. The inclusion of weight as a covariate was statistically significant and reduced the BSV in CL and Vc by 22 and 16%, respectively. No other covariate effects were found to be statistically significant predictors of fremanezumab PopPK.
Since data regarding injection site were not available from all subjects in the full dataset, the covariate effect of injection site on fremanezumab absorption was explored in a subpopulation analysis wherein only data deemed potentially informative to this covariate of interest were included. Patients from the LTS phase 3 study who had additional samples collected during days 3–10 or 15–20 were included in the analysis. Subjects were considered evaluable only if at each visit all injections for a given dose were within the same anatomic location (abdomen, thigh or upper arm), and the subsequent corresponding PK sample(s) was collected at an informative time (with respect to ka). Injection site was evaluated as a time‐varying covariate on ka, and all parameters (except ka) were fixed to the final model parameter estimates. Based on the prespecified criteria for covariate analysis (i.e. P < .01 and reduction in BSV in the parameter of interest of at least 5%), injection site was not considered a significant covariate on ka in this subpopulation analysis.
3.3. Evaluation of the model
Prediction‐corrected VPC was performed using the final PopPK model to ensure adequate model performance and assess the predictive capabilities of the model. The median and 90% prediction interval, derived from the simulated datasets, overlaid on the observed fremanezumab concentration data and corresponding percentiles for the intensive phase 1 IV and SC data and the sparse phase 2b/3 SC data are provided in Figure 5 and Figure 6, respectively. For the phase 1 data, a small degree of overprediction is apparent around the peak with some underprediction during the post‐absorptive phase of the profile. At almost all sampling time points for the phase 2b/3 data, the percentiles based on the observed data fall within the confidence interval around the corresponding simulation‐based percentile, indicating excellent predictive performance of the model for the data following SC administration in migraine patients.
Figure 5.
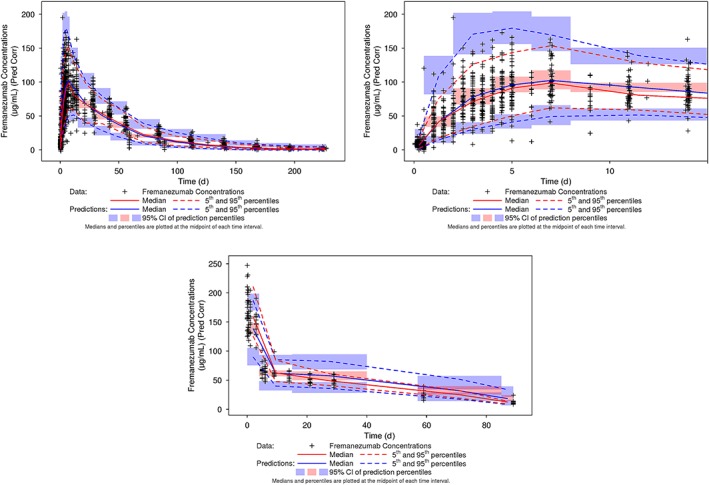
Prediction‐corrected visual predictive check for the fremanezumab final population pharmacokinetic model: Phase 1 serial sampling data in healthy subjects. CI, confidence interval; IV, intravenous; Pred Corr, prediction‐corrected; SC, subcutaneous
Figure 6.
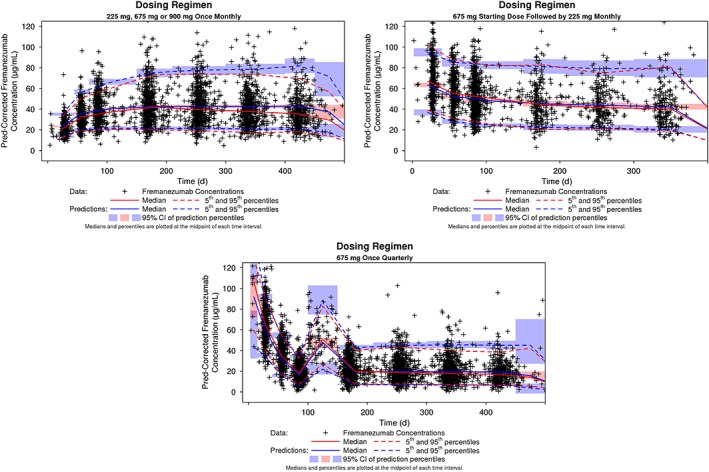
Prediction‐corrected visual predictive check for the fremanezumab final population pharmacokinetic model: Phase 2b and phase 3 data in migraine patients. CI, confidence interval; Pred, prediction
3.4. Simulations
Figure 7 illustrates the median (90% prediction interval) simulated fremanezumab concentration–time profiles for the SC dose regimens used in the phase 3 clinical trials over 12 months.
Figure 7.
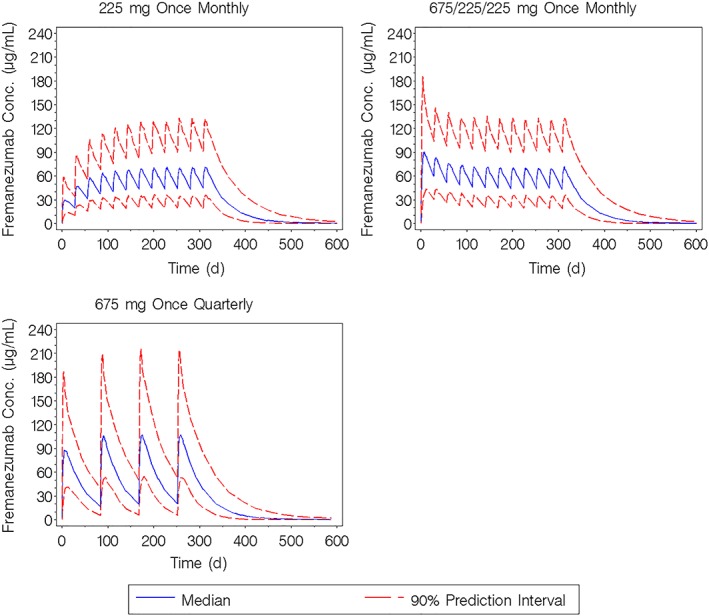
Simulated fremanezumab concentration–time profiles for the subcutaneous dose regimens used in the phase 3 clinical trials administered over 12 months. Conc, concentration
Boxplots of exposure measures vs select patient covariates were examined to determine if these covariates exhibited a clinically relevant impact on fremanezumab steady‐state exposure. Figure 8 provides representative boxplots of exposures (particularly, Cav,ss(0–28d) and Cav,ss(0–84d)) vs quartiles of body weight following administration of 225 mg SC once monthly and 675 mg SC once quarterly. Across the range of body weight (the only covariate found to be significant in the model), a decrease in fremanezumab exposure was evident across the quartiles of increasing body weight. The findings were consistent across additional exposure measures, including Cmax,ss(0–28d), Cmax,ss(0–84d), AUCss(0–28d), and AUCss(0–84d).
Figure 8.

Boxplots of model‐predicted Cav,ss(0–28d) and Cav,ss(0–84d) by quartiles of body weight in patients: 225 mg subcutaneous once monthly for 12 doses (left) and 675 mg subcutaneous once quarterly for 4 doses (right). Cav,ss(0–28d), average fremanezumab plasma concentration from 0 to 28 days at steady state; Cav,ss(0–84d), average fremanezumab plasma concentration from 0 to 84 days at steady state
Supporting Figure S1 and Figure S2 provide boxplots of fremanezumab Cav,ss following administration of 225 mg SC once monthly vs intrinsic and extrinsic factors evaluated as potential predictors of variability in PK. Figure S1 presents fremanezumab exposure vs sex, acute medication usage (ergotamine/triptan), and renal function category. For these factors, trends are evident for marginally higher exposures in females, patients receiving acute medications and patients with impaired renal function (as compared to males, patients not receiving acute medications and patients with normal renal function, respectively). However, despite these trends, none of these factors were found to be statistically significant covariates in the PopPK analysis, and overlap in the range of exposures is observed across groups. Similar results were observed for 675 mg quarterly doses (data not shown).
Supporting Figure S2 presents boxplots of fremanezumab Cav,ss following administration of 225 mg SC once monthly vs age quartile, race, liver function category, analgesic medication usage, preventive medication usage, ADA status and patient status. The results demonstrate that exposures are consistent across the sub‐groups representing each of these factors and are consistent with the PopPK analysis findings as there was no statistically significant effect of these factors on fremanezumab PK. Similar results were observed for 675 mg quarterly doses (data not shown).
The calculated model‐based median terminal half‐life was approximately 30 days (independent of dose or dose regimen). Based on predicted exposure levels in the phase 3 studies, fremanezumab concentrations approach steady state by 3 months of treatment and steady state is expected to be achieved by approximately 168 days (6 months) for the 225 mg monthly and 675 mg quarterly regimens (Figure 7). As shown in Table 3, comparison of the exposures following the final (12th) dose of the 225 mg monthly regimen to exposures following a single dose resulted in a median accumulation ratio of 2.43 and 2.38 for the area under the fremanezumab plasma concentration vs time curve from 0 to 28 days (AUC0–28d) and Cmax, respectively; for the 675 mg once‐quarterly dosing regimen, the exposures after dose 4 (final dose) compared to dose 1 resulted in a median accumulation ratio of 1.21 and 1.22 for area under the fremanezumab plasma concentration vs time curve from 0 to 84 days (AUC0–84d) and Cmax, respectively.
Table 3.
Summary of accumulation ratios: 225 mg subcutaneous once monthly and 675 mg subcutaneous once quarterly
| Dosing regimen | Percentile | ARAUC a | ARCmax b |
|---|---|---|---|
| 225 mg SC once monthly | 5th | 2.58 | 2.57 |
| 50th | 2.43 | 2.38 | |
| 95th | 2.55 | 2.23 | |
| 675 mg SC once quarterly | 5th | 1.18 | 1.30 |
| 50th | 1.21 | 1.22 | |
| 95th | 1.27 | 1.14 |
AR, accumulation ratio; AUC, area under the concentration–time curve; Cmax, maximum drug concentration.
AUC(0–28d, 225 mg 12th dose)/AUC(0–28d, 225 mg 1st dose) or AUC(0–84d, 675 mg 4th dose)/AUC(0–84, 675 mg 1st dose).
Cmax(225 mg 12th dose)/Cmax(225 mg 1st dose) or Cmax(675 mg 4th dose)/Cmax(675 mg 1st dose).
4. DISCUSSION
This manuscript describes the iterative PopPK model development for fremanezumab using a large pooled population of healthy subjects (n = 72) enrolled in 2 phase 1 studies and patients with EM or CM (n = 2474) enrolled in 2 phase 2b studies and 3 phase 3 studies. A 2‐compartment model with first‐order absorption and elimination and allometric weight scaling of CL and Vc (with estimated exponents) reasonably fit the phase 1/2b/3 data.
Despite the large pooled phase 1/2b/3 dataset, this modelling effort was somewhat limited by the designs of the studies included. Within the phase 1 healthy subject studies, no multiple‐dosing data were available, limiting knowledge of accumulation to steady‐state conditions and within‐subject variability in the healthy subject population. Furthermore, the phase 2b and pivotal 3 trials, which comprised the majority of the dataset, included exclusively sparse PK data collection, consisting entirely of trough, predose samples. However, additional information was obtained regarding absorption, peak concentration and the first elimination phase from 2 additional samples collected from a subset of patients in the LTS study at specified times following dosing.
The final model structure illustrates how the iterative approach maximized the use of informative data during model development to address the apparent discrepancy in compartmental structure for IV vs SC dosing. Exploratory plots demonstrated that the concentration–time profile following SC administration reflects a monophasic decrease after absorption, while the profile following IV administration reflects a biphasic process. In fact, when data following IV administration were not included in the dataset, the 2‐compartment model fit could not be supported.23 By fixing the parameters defining the peripheral compartment, both the IV and SC data were fit adequately and a precise estimate of bioavailability was obtained. Given that the apparent differences in fremanezumab elimination between IV and SC administration are probably due to the fact that the relatively slow absorption phase following SC administration masks the initial elimination phase observed following IV administration, rather than patient‐related differences, if IV data were available from patients with migraine (EM or CM), it would be expected that the estimates of Q and Vp would be similar to those observed in healthy volunteers. Furthermore, the effect of patient status, defined as healthy subjects vs patients with migraine, was evaluated as a covariate and found not to be a statistically significant predictor of variability in fremanezumab PK (see also Supporting Figure S2 illustrating the similarity in exposure between healthy subjects and patients).
In general, fixed and random effect parameters were estimated with good precision, and goodness‐of‐fit plots, as well as VPC diagnostics, indicated a good fit of the model to the data. The model characterized the overall PK data well and accurately captured the central tendency and shape of the multiple‐dose profile. After adjusting PK parameters for inclusion of IV data in the model (using the estimated F of 0.658), apparent clearance (CL/F) and total apparent volume (Vc/F + Vp/F) for a typical subject (weighing 71 kg) were 0.137 L/d and 5.47 L, respectively. These values are similar to those reported previously11 (mean CL/F values from 0.111 to 0.129 L/d and mean apparent volume of distribution during the terminal phase from 5.711 to 6.430 L) and are generally consistent with previously reported values for IgG antibodies.24, 25 Dose proportionality assessment was performed and suggested no changes in PK parameters with respect to dose.
As generally observed with mAbs, weight was a significant predictor of variability in PK for both CL and Vc; higher weight was associated with increased CL and Vc. The effects of other covariates evaluated (race, sex, albumin, age, renal function category, injection site, and acute, analgesic and preventive medications) were not found to statistically significantly influence the PK of fremanezumab. The lack of race as a significant covariate is consistent with a previous noncompartmental PK analysis comparing the single‐dose PK of fremanezumab in Japanese and matched Caucasian healthy subjects.11 Geometric mean ratios (i.e. Japanese/Caucasian ratios) for AUC and Cmax were within the default 0.80–1.25 no‐effect range across all doses, with similar median time of Cmax (range of 5–11 days) and mean half‐lives (range of 31–39 days) for both ethnicities. ADA status and liver function were evaluated on an exploratory basis due to limited sample size within the respective categories and the results showed no indication of these covariates having an impact on fremanezumab exposure.
Exploratory graphical evaluation and additional covariate analysis on a subset of the population was performed to evaluate the effect of injection site on ka and found no significant effect of injection site on variability in the rate of absorption. Due to the limited data available in the studies deemed to be informative to the estimation of F, the effect of injection site on F could not be formally evaluated. Although it is reported that injection site can have an impact on both the rate and the extent of absorption for mAbs, the effect of injection site on the extent of absorption is considered less likely.26, 27 Furthermore, since the lymphatic pathway may represent the primary mechanism for absorption of IgG‐type mAbs, it is possible that the inherent slow absorption makes the regional variation of lymph flow associated with different injection sites negligible in terms of an effect on F such that the extent of absorption does not differ significantly between dosing sites.28
Based on the model, fremanezumab exhibits linear, dose‐proportional kinetics with a calculated median terminal half‐life of approximately 30 days with fremanezumab concentrations approaching steady state by 3 months of treatment and steady‐state conditions are expected to be achieved by approximately 168 days (6 months) for the 225 mg SC monthly and 675 mg SC quarterly dosing regimens. This estimated half‐life is consistent with the typical range of half‐life values reported for other therapeutic mAbs.29, 30 In particular, for the mAbs that target the CGRP pathway, the reported half‐life is 28 days for erenumab and 27 days for galcanezumab.31, 32 Approximately 2‐fold accumulation based on AUC0–28 and Cmax is anticipated for the 225 mg SC once‐monthly dosing regimen, whereas, minimal accumulation is anticipated for the 675 mg SC once‐quarterly fremanezumab dosing regimen.
In conclusion, the PopPK model for fremanezumab well described the PK data observed in a large population of healthy subjects and patients with CM or EM following IV and SC administration of a wide range of doses and dosing intervals. As expected, this analysis confirmed the influence of weight on the PK parameters of fremanezumab, whereby higher body weight is associated with lower exposure. The prolonged absorption and long elimination half‐lives for fremanezumab supports the use of monthly or quarterly dosing regimens and the lack of significant influence of other intrinsic or extrinsic factors on fremanezumab exposure suggests no need for dose modification in special populations. Furthermore, the predictive performance of the model indicates its appropriateness in predicting subject‐specific estimates of exposure for use in the evaluation of relationships between exposure and safety and efficacy response.
COMPETING INTERESTS
Orit Cohen‐Barak, Michele Rasamoelisolo, Honglue Shen and Micha Levi are employees of Teva Pharmaceutical Industries Ltd. and own stock in Teva Pharmaceutical Industries Ltd.
Cognigen Corporation (a SimulationsPlus company) received financial support from Teva Pharmaceutical Industries Ltd. to perform these analyses. Jill B. Fiedler‐Kelly, Denise N. Morris and Elizabeth Ludwig were employed by Cognigen Corporation when the analyses were performed.
CONTRIBUTORS
J.B.F.‐K., D.N.M. and E.L. were responsible for performing the modeling and simulations, preparing the data visualizations, providing interpretation of the results, and drafting the manuscript. M.R. and H.S. were responsible for the bioanalytical assays used in these studies and reviewed the manuscript. O.C.‐B. and M.L. were responsible for providing strategic direction, clinical interpretation and oversight and review of these analyses as well as assisting with drafting and review of the manuscript.
Supporting information
TABLE S1
Summary statistics of demographic characteristics by phase, in the pharmacokinetic analysis of fremanezumab
FIGURE S1 Boxplots of model‐predicted Cav,ss(0–28d) by sex, acute medication usage and renal function category in patients: 225 mg subcutaneous once monthly for 12 doses. CAV,SS(0–28D), average fremanezumab plasma concentration from 0 to 28 days at steady state.
FIGURE S2 Boxplots of model‐predicted CAV,SS(0–28D) by adult age quartiles, race, liver function category, analgesic medication usage, preventive medication usage, ADA status, and patient status: 225 mg subcutaneous once monthly for 12 doses. boxplots of fremanezumab exposure vs ADA status considered all ADA‐positive samples whether treatment emergent or not. ADA, anti‐drug antibody; CAV,SS(0–28D), average fremanezumab plasma concentration from 0 to 28 days at steady state.
FIGURE S2 continued Boxplots of model‐predicted CAV,SS(0–28D) by adult age quartiles, race, liver function category, analgesic medication usage, preventive medication usage, ADA status, and patient status: 225 mg subcutaneous once monthly for 12 doses. boxplots of fremanezumab exposure vs ADA status considered all ADA‐positive samples whether treatment emergent or not. ADA, anti‐drug antibody; CAV,SS(0–28D), average fremanezumab plasma concentration from 0 to 28 days at steady state.
ACKNOWLEDGEMENTS
The authors would like to thank Pippa S. Loupe, PhD, (Global Research and Development Teva Pharmaceuticals Ltd.) for her valued assistance with this publication's development.
This study was funded by Teva Pharmaceuticals Ltd., Netanya, Israel.
Fiedler‐Kelly JB, Cohen‐Barak O, Morris DN, et al. Population pharmacokinetic modelling and simulation of fremanezumab in healthy subjects and patients with migraine. Br J Clin Pharmacol. 2019;85:2721–2733. 10.1111/bcp.14096
Morris, D. N., formerly employed at Cognigen Corporation
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to patient‐level data and related study documents including the study protocol and the statistical analysis plan. Patient‐level data will be de‐identified and study documents will be redacted to protect the privacy of trial participants and to protect commercially confidential information. Please email usmedinfo@tevapharm.com to make your request.
As this work involves the population pharmacokinetic modelling of the data collected from multiple clinical studies, the Principal Investigator of each trial is not included as an author on this paper. In this context, Orit Cohen‐Barak is the senior scientist who was responsible for this analysis.
REFERENCES
- 1. Eftekhari S, Edvinsson L. Possible sites of action of the new calcitonin gene‐related peptide receptor antagonists. Ther Adv Neurol Disord. 2010. Nov;3(6):369‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Olesen J. CGRP in migraine. Cephalalgia. 2011. Apr;31(5):638. [DOI] [PubMed] [Google Scholar]
- 3. Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990. Aug;28(2):183‐187. [DOI] [PubMed] [Google Scholar]
- 4. Hansen JM, Hauge AW, Olesen J, Ashina M. Calcitonin gene‐related peptide triggers migraine‐like attacks in patients with migraine with aura. Cephalalgia. 2010. Oct;30(10):1179‐1186. [DOI] [PubMed] [Google Scholar]
- 5. Olesen J, Diener HC, Husstedt IW, et al. Calcitonin gene‐related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004. Mar 11;350(11):1104‐1110. [DOI] [PubMed] [Google Scholar]
- 6. Ho TW, Ferrari MD, Dodick DW, et al. Efficacy and tolerability of MK‐0974 (telcagepant), a new oral antagonist of calcitonin gene‐related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo‐controlled, parallel‐treatment trial. Lancet. 2008. Dec 20;372(9656):2115‐2123. [DOI] [PubMed] [Google Scholar]
- 7. Hewitt DJ, Aurora SK, Dodick DW, Goadsby PJ, Ge YJ, Bachman R. Randomized controlled trial of the CGRP receptor antagonist MK‐3207 in the acute treatment of migraine. Cephalalgia. 2011. Apr;31(6):712‐722. [DOI] [PubMed] [Google Scholar]
- 8. Bigal ME, Rapoport AM, Silberstein SD, Walter S, Hargreaves RJ, Aycardi E. From LBR‐101 to fremanezumab for migraine. CNS Drugs. 2018. Nov;32(11):1025‐1037. [DOI] [PubMed] [Google Scholar]
- 9. Levin M, Silberstein SD, Gilbert R, et al. Basic considerations for the use of monoclonal antibodies in migraine. Headache. 2018. Nov;58(10):1689‐1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Armour KL, Clark MR, Hadley AG, Williamson LM. Recombinant human IgG molecules lacking Fcgamma receptor I binding and monocyte triggering activities. Eur J Immunol. 1999. Aug;29(8):2613‐2624. [DOI] [PubMed] [Google Scholar]
- 11. Cohen‐Barak O, Weiss S, Rasamoelisolo M, et al. A phase 1 study to assess the pharmacokinetics, safety, and tolerability of fremanezumab doses (225 mg, 675 mg and 900 mg) in Japanese and Caucasian healthy subjects. Cephalalgia. 2018. Nov;38(13):1960‐1971. [DOI] [PubMed] [Google Scholar]
- 12. Goadsby P, Yeung P, Blankenbiller T, Grozinski‐Wolff M, Yang R, Ma Y, et al. Fremanezumab long‐term efficacy and safety: interim results of a one‐year study. Poster presented at: American Headache Society; 2018. Jun 28‐Jul 01; San Francisco, California.
- 13. Cohen JM, Dodick DW, Yang R, et al. Fremanezumab as add‐on treatment for patients treated with other migraine preventive medicines. Headache. 2017. Oct;57(9):1375‐1384. [DOI] [PubMed] [Google Scholar]
- 14. Bigal ME, Dodick DW, Rapoport AM, et al. Safety, tolerability, and efficacy of TEV‐48125 for preventive treatment of high‐frequency episodic migraine: a multicentre, randomised, double‐blind, placebo‐controlled, phase 2b study. Lancet Neurol. 2015. Nov;14(11):1081‐1090. [DOI] [PubMed] [Google Scholar]
- 15. Bigal ME, Edvinsson L, Rapoport AM, et al. Safety, tolerability, and efficacy of TEV‐48125 for preventive treatment of chronic migraine: a multicentre, randomised, double‐blind, placebo‐controlled, phase 2b study. Lancet Neurol. 2015. Nov;14(11):1091‐1100. [DOI] [PubMed] [Google Scholar]
- 16. Dodick DW, Silberstein SD, Bigal ME, et al. Effect of fremanezumab compared with placebo for prevention of episodic migraine: a randomized clinical trial. JAMA. 2018. May 15;319(19):1999‐2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Silberstein SD, Dodick DW, Bigal ME, et al. Fremanezumab for the preventive treatment of chronic migraine. N Engl J Med. 2017. Nov 30;377(22):2113‐2122. [DOI] [PubMed] [Google Scholar]
- 18. Beal SL, Sheiner LB, Boeckmann AJ, Bauer RJ. (Eds). NONMEM 7.3.0 Users Guides. Hanover, MD: ICON Development Solutions; 1989. ‐2013. [Google Scholar]
- 19. KIWI [computer program]. Version 3. Buffalo, NY: Cognigen, a Simulations Plus Company; 2018.
- 20. Owen JS, Fiedler‐Kelly J. Introduction to population pharmacokinetic/pharmacodynamic analysis with nonlinear mixed effects models. Hoboken, NJ: John Wiley & Sons Incorporated; 2014. [Google Scholar]
- 21. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holford N. The visual predictive check: superiority to standard diagnostic (Rorschach) plots. Poster presented at: Population Approach Group in Europe (PAGE); 2005. Jun 16‐17; Pamplona, Spain.
- 23. Morris D, Fiedler‐Kelly J, Levi M, Cohen‐Barak O. Population pharmacokinetic modeling of fremanezumab in support of phase 3 development for patients with migraine. Poster presented at: Population Approach Group in Europe (PAGE); 2018. May 29‐Jun 01; Montreux, Switzerland.
- 24. Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093‐1095. [DOI] [PubMed] [Google Scholar]
- 25. Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010. Oct;49(10):633‐659. [DOI] [PubMed] [Google Scholar]
- 26. Gill KL, Machavaram KK, Rose RH, Chetty M. Potential sources of inter‐subject variability in monoclonal antibody pharmacokinetics. Clin Pharmacokinet. 2016. Jul;55(7):789‐805. [DOI] [PubMed] [Google Scholar]
- 27. Zhao L, Ji P, Li Z, Roy P, Sahajwalla CG. The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model. J Clin Pharmacol. 2013. Mar;53(3):314‐325. [DOI] [PubMed] [Google Scholar]
- 28. Xu Z, Wang Q, Zhuang Y, et al. Subcutaneous bioavailability of golimumab at 3 different injection sites in healthy subjects. J Clin Pharmacol. 2010. Mar;50(3):276‐284. [DOI] [PubMed] [Google Scholar]
- 29. Moek KL, Giesen D, Kok IC, et al. Theranostics using antibodies and antibody‐related therapeutics. J Nucl Med. 2017. Sep;58(Suppl 2):83S‐90S. [DOI] [PubMed] [Google Scholar]
- 30. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010. Aug;49(8):493‐507. [DOI] [PubMed] [Google Scholar]
- 31. Erenumab‐aooe (Aimovig) [product information]. Thousand Oaks, CA: Amgen Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761077s000lbl.pdf. Accessed January 6, 2019.
- 32. Galcanezumab‐gnlm (Emgality) [product information]. Indianapolis, IN: Eli Lilly and Company. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761063s000lbl.pdf. Accessed January 6, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1
Summary statistics of demographic characteristics by phase, in the pharmacokinetic analysis of fremanezumab
FIGURE S1 Boxplots of model‐predicted Cav,ss(0–28d) by sex, acute medication usage and renal function category in patients: 225 mg subcutaneous once monthly for 12 doses. CAV,SS(0–28D), average fremanezumab plasma concentration from 0 to 28 days at steady state.
FIGURE S2 Boxplots of model‐predicted CAV,SS(0–28D) by adult age quartiles, race, liver function category, analgesic medication usage, preventive medication usage, ADA status, and patient status: 225 mg subcutaneous once monthly for 12 doses. boxplots of fremanezumab exposure vs ADA status considered all ADA‐positive samples whether treatment emergent or not. ADA, anti‐drug antibody; CAV,SS(0–28D), average fremanezumab plasma concentration from 0 to 28 days at steady state.
FIGURE S2 continued Boxplots of model‐predicted CAV,SS(0–28D) by adult age quartiles, race, liver function category, analgesic medication usage, preventive medication usage, ADA status, and patient status: 225 mg subcutaneous once monthly for 12 doses. boxplots of fremanezumab exposure vs ADA status considered all ADA‐positive samples whether treatment emergent or not. ADA, anti‐drug antibody; CAV,SS(0–28D), average fremanezumab plasma concentration from 0 to 28 days at steady state.
Data Availability Statement
Qualified researchers may request access to patient‐level data and related study documents including the study protocol and the statistical analysis plan. Patient‐level data will be de‐identified and study documents will be redacted to protect the privacy of trial participants and to protect commercially confidential information. Please email usmedinfo@tevapharm.com to make your request.
As this work involves the population pharmacokinetic modelling of the data collected from multiple clinical studies, the Principal Investigator of each trial is not included as an author on this paper. In this context, Orit Cohen‐Barak is the senior scientist who was responsible for this analysis.