

Abstract
The high level of complexity underlying the heterogeneous pathophysiology of neuromuscular diseases is a fundamental limiting factor in understanding the role of physical activity in their onset and/or clinical evolution. To overcome this difficulty, it is essential to rely on and deep knowledge of the aetiology and on the physiological adaptations to physical exercise, in order to predict how they can impact on the clinical history of each disease. This paper illustrates the possible strategies of intervention in some neuromuscular disorders, through the analysis of their supposed pathogenic mechanisms. Nevertheless, no clear conclusions can be inferred so far.
Key words: muscle adaptation, exercise, neuromuscular disorders
Introduction
Translation of fundamental physiological aspects of skeletal muscle plasticity to neuromuscular diseases may help overcoming the high degree of complexity residing in the existence of a constellation of conditions that are very different in their pathophysiological origin.
In particular, open questions regard how to link physiological aspects of muscle adaptation to physical exercise, disuse, and aging to the pathophysiology of neuromuscular diseases and their evolution.
Considering the plastic nature of skeletal muscle tissue capable of a wide range of adaptations through qualitative and quantitative variations of cellular protein expression to load and nutrients, the physiological translation should pass through elements of intercorrelations between the cellular structure, from one side, and the its energy disposal, from the other, determined by its use or disuse/non-use, with the leitmotif of the aging process on the background, known to correlate with the arising of age-related diseases and consequent increased probability of death.
A fundamental observation from this point of view resides in the positive correlation between activity energy expenditure (AEE), unlike resting metabolic rate (RMR), and longevity, through significant delay of age-related diseases, such as cardiovascular, metabolic diseases, and cancer. As known, the subjects’ daily energy consumption includes resting metabolic rate, representing almost 50% of the total expenditure (EE), and AEE, that is the energy consumption due to exercise and daily activities, whereas the thermal effect of meals (TEM) represents a small and constant quote. Of course, the relative proportion of resting and activity expenditure is highly variable between subjects and is strictly linked to their daily habits. A really important finding that may help unraveling the role of energy consumption in longevity is the age-related decline of total energy consumption, reflected by changes in body mass (particularly from birth to 20y) and daily energy consumption (from 2550 kCal/day 18-19y to 2050 kCal/day over 60y WHO/FAO guidelines) (1).
In the large part of the population, the aging process adds to the generally observed reduction in the level of physical activity to determine a progressive decline in several physiological capacities. Therefore, in order to get insight into physiology of aging, it is of major importance to try normalizing this process over the level of physical activity. In other terms, it is mandatory to exclude the impact of physical activity to understand what aging per se is. In this sense, a precious help may derive from having a deep look at the ageing processin the so called master athletes (2), who have long lasting commitment to moderate to high intensity physical activity. In these subjects, it can be reasonably considered that the ageing process is, as much as possible, unlinked from inter-subjects variations in the basal level physical activity. That said, in master athlete performance inevitably declines with age and the major determinants of this loss are the reduction in the aerobic fitness (mirrored by VO2max decrease) (3) and the concomitant changes in the skeletal muscle functional and structural features, including variations in muscle phenotype (generally a fast-to-slow transition) (4). By comparing the aging process in sedentary subjects and master athletes, it is now clear that the maximum rate of oxygen consumption inevitably declines with age but this change is enormously conditioned by the level of physical activity maintained over time. In fact, an elder active subject may express much higher VO2max than a sedentary young (5). Therefore, being the total energy expenditure and maximal oxygen consumption largely dependent from the basal level of physical activity, from this point of view, a young may be considered elder and vice versa.
Considering the organ mass change over time and the relative contribution to total mass of each organ, the skeletal muscle appears as a fundamental contributor of this variation. This phenomenon is highly amplified with age by following an exponential, unlike linear, change due to ageing-associated muscle disuse and is predictable that an earlier exponential change may well represent the age-related variations in the skeletal muscle in presence of neuromuscular diseases, independently from their pathophysiology (6).
A fundamental regulator of skeletal muscle mass and its sensing of applied load and energy status is the so called mechanistic TOR, an atypical protein kinase essential for organism survival which plays the role of a catalytic subunit of two distinct dimer multiprotein complexes termed mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (7). In these complexes mTOR acts as a multieffector protein kinase. Four major inputs regulate mTORC1 by cooperating or antagonizing each other and include nutrients (amino acids and glucose), growth factors, energy status, and mechanical stress.
In particular, as chemical inhibitors of glycolysis suppress mTORC1 activity, the complex senses the cellular energy of the cell. This phenomenon suggests that changes in energy disposal may promote convergent upstream regulatory signals on mTORC1. As already known, glycolysis and mitochondrial respiration convert nutrients into energy, which is stored in the form of ATP. Glucose loss, inhibition of glycolysis, or mitochondrial respiration cause a significant reduction of the intracellular ATP levels that determines a change in the intracellular ADP/ATP and AMP/ATP ratios. This change is sensed by heterotrimeric complex AMP dependent protein kinase (AMPK) Under nutrient deprivation AMPK transmits stress signals to mTORC1(8). This process leads mTORC1 inhibition through direct and indirect mechanisms. Importantly, mechanical load and nutrients as branched chain amino acids (BCAA) (9), although first acting at different levels, seem to have a common target on the mTOR biosynthetic pathway controlling protein synthesis. For example, eccentric type exercise is able to activate downstream mTORC1 kinase effector (S6K1) responsible for translation initiation, through changes in the membrane level of phosphatidic acid (PA) (10, 11).
Importantly, with age, mTORC1 possibly increases its basal level of expression and activation (12). This change appears to be paralleled by reduced responsiveness to stimuli (ie, mechanical load, BCAA), partly ascribable to increased expression of negative regulators. In a condition in which mTORC1 responsiveness is blunted, increased loss of muscle mass and concomitant increased insulin resistance appears to contribute to the arising of age-related diseases. Of importance, muscle-specific inactivation of mTOR leads to severe myopathy, resulting in premature death. In fact, mTOR-deficient muscles display metabolic changes including impaired oxidative metabolism, altered mitochondrial regulation, and glycogen accumulation associated with protein kinase B/Akt hyperactivation. Importantly, loss of mTOR exacerbates the myopathic features in both slow oxidative and fast glycolytic muscles. Moreover, mTOR loss leads to reduced muscle dystrophin content thus exerting a control of dystrophin transcription (13).
Physiological muscle adaptations to exercise
Muscle adaptations to exercise include morphofunctional modifications triggered by the repetition of muscular contraction bouts, leading to increased mitochondrial biosynthesis and angiogenesis, fibers hypertrophy and fundamental changes in cell metabolism, including increase lactate tolerance (Tab. 1). These adaptations follow modifications of the expression and the activation a series of subcellular signals and a series, still partially unknown, of pre- and post-transcriptional events. The primitive stimulus (exercise or training) with its quality (strength or endurance) are able to activate a large number of cellular events, in turn responsible for the activation, inhibition or modulation of cross linked signaling routes, implicated in the regulation of transcription and translation. At the center of these routes mTORC1 and peroxisome PGC1α are cross-linked master regulators of the plastic responses due to strength type and endurance type exercise respectively (Fig. 1).
Table 1.
Muscular adaptations to endurance and resistance type exercise. Arrows indicate: ↑ minimal change; ↑↑ moderate change; ↑↑↑ large change; ↓ decrease; ↔ no change. Modified with permission from Physical Activity. Giuseppe D’Antona. Poletto publisher 2019 (1st edition) (62).
| Muscular adaptation | Endurance exercise | Resistance exercise |
|---|---|---|
| Hypertrophy | ↔ | ↑↑↑ |
| Strength | ↔↓ | ↑↑↑ |
| Cross sectional area | ↔ | ↑↑↑ |
| Neural adaptation | ↔↑ | ↑↑↑ |
| Anaerobic capacity | ↑ | ↑↑ |
| Lactate tolerance | ↑↑ | ↔ ↑ |
| Myofibrillar protein synthesis | ↔ ↑ | ↑↑↑ |
| Mitochondrial biogenesis, mitochondrial density, oxidative capacity | ↑↑↑ | ↔↑ |
| Fatigue resistance | ↑↑↑ | ↔ ↑ |
| Capillarization | ↑↑↑ | ↔ |
Figure 1.
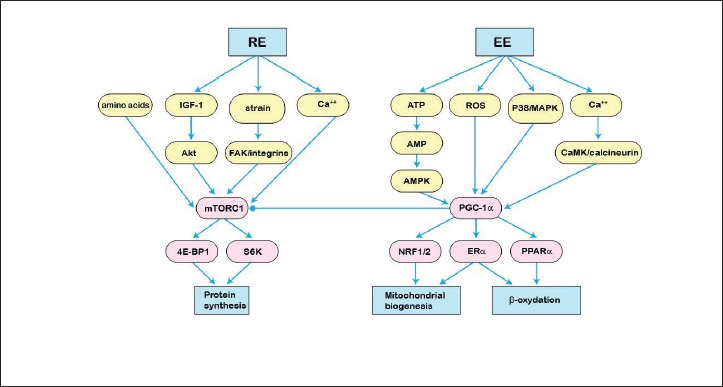
Schematic representation of the sub-cellular signals activated by the endurance (EE) and resistance (RE) exercises. In EE, the activation of the downstream pathways foresees modifications of the energetic availability (ATP), oxidative stress with production of reactive oxygen species (ROS), meccanostresses with p38 / MAPK activation or changes in cellular calcium content (ERα = alpha receptor for estrogens; PPARα = peroxisomal proliferator-activated receptor alpha), leading to PGC-1α mediated mitochondriogenesis and mitochondrial β-oxidation. In RE, Insulin Growth Factor 1 (IGF-1), amino acids, mechanical strain and calcium disposal converge on upstream signals activating mTORC1. Akt = protein kinase B; FAK = Focal Adhesion Kinase; S6K = ribosomal protein 6 kinase; 4E-BP1 = eukaryotic translation initiation factor 4E-binding protein 1. Modified with permission from: Physical Activity. Giuseppe D’Antona. Poletto publisher 2019 (1st edition) (62).
In particular, as regards mitochondrial biogenesis following endurance type exercise of sufficient duration (14), it is possible to identify two major governors of the process: AMPK and PGC1α AMPK regulator is the energy status of the cell defined by the AMP/ATP and Cr/PCr intracellular levels. An increased AMP/ATP ratio due to deprivation of glucose, oxidative stress and exercised induced ATP turnover, leads to acute or chronic AMPK activation. In the first case AMKP hypercactivation determines protein synthesis inhibition and increased lipid oxidation. In the latter it stimulates mitochondrial biogenesis. Endurance exercise is also associated with NAD+ fluctuations within the skeletal muscle fibers. NAD+ is a regulator of the deacetylasisirtuine family (SIRT). An increased NAD+/ NADH ratio due to acute exercise and fasting activates SIRT1 in the cytosol and SIRT3 in the mitochondria. The subsequent deacylation of transcription factors and mitochondrial enzymes is followed by a series of adaptations, which increase mitochondrial oxidative function. This adaptation is particularly significant in the presence of low load or in the recovery time from exercise. On the contrary if the load increases, the lactate production reduces the NAD+/ NADH ratio thus blunting the upregulation of mitochondrial function through this mechanism (15). Considering the final physiological effect of endurance type exercise on mitochondrial biogenesis and oxidative capacity, possible translation of this exercise paradigm regards spinal muscular atrophy.
Spinal muscular atrophy (SMA) is caused by homozygous deletion/mutations in the survival motor neuron 1(SMN1) gene with an estimated incidence of 1 in 11,000 live births. It is characterized by motoneurons degeneration in spinal cord and brainstem causing muscle wasting and weakness. In SMA clinical severity ranges from the extremely severe type 1 to the mildest type 3. Approximately 60% of affected infants have a type 1 SMA, with onset of symptoms at 6 months of age or younger, and a median life expectancy of less than 2 years without respiratory and nutritional support. New developmental motor milestones are rarely achieved after diagnosis. The mildest phenotype begins after the acquisition of autonomous ambulation, that is usually preserved till late if the onset was later than 3 ys of age, and gets lost about 10 yrs after the diagnosis if ambulation was acquired by 3 ys of age. Even an adult very mild form has been described, usually called type 4 characterized by adult onset and quite normal motor function. Between the extreme phenotypes there is a continuum with variable severity. The different phenotypes share the same molecular defects -a homozygous deletion or mutation in the survival motor neuron 1 which results in decreased expression of the survival motor neuron (SMN) protein and degeneration of motor neurons in the spinal cord and brain stem. A paralogous gene, survival motor neuron 2 (SMN2), also encodes the SMN protein; however, 90 to 95% of the translated protein is truncated and nonfunctional as a result of aberrant splicing. The more the SMN2 copies, the mildest the phenotype, as a general rule. Therefore, modulation of pre-messenger RNA (pre-mRNA) splicing of SMN2 to promote increased production of SMN protein has been shown to be an effective treatment strategy across the disease spectrum of spinal muscular atrophy. In addition to muscle weakness, fatigue is a common complaint in SMA. A possible explanation comes from SMA animal models and post-mortem studies, demonstrating abnormal development and maturation of the neuromuscular junction. Neuromuscular dysfunction has been found also in at least half of the patients with SMA. SMN has a role in myogenesis and normal muscle differentiation requires adequate levels of SMN, supporting the hypothesis that a delay in muscle maturation is one of the primary pathologic components of SMA. The association between normal myogenesis and increased oxidative metabolism has been demonstrated. Deregulated myogenesis and impaired mitochondrial biogenesis seem to be inversely proportional to SMN availability, being more prominent in muscle from patients with SMA-I than in muscle from patients with SMA-III. The reduced mitochondrial content makes SMA muscle unable to sustain muscle fiber maturation and contraction properly, contributing to patient weakness and hypotonia (16). Recent clinical observations reporting an incomplete response to aerobic exercise in patients with SMA also sustain this scenario (17). Considering its slow progression, SMA-III may benefit of endurance exercise training in terms of functional performance and exercise capacity through the amelioration of muscle fibers metabolic function and the interruption of further muscle functional and structural impairment induced by the vicious circle of inactivity (18). It has been shown that this exercise paradigm is able to determine positive effects on post-natal maturation of motor units and physical behavior in mouse models of SMA (19, 20). Although there are several inconsistencies regarding the human studies, particularly in terms of number of subjects enrolled, optimal training frequency, and possible adverse events (21), initial evidence seems to suggest a positive effect of endurance exercise type on maximal oxygen consumption (VO2max) in SMA-III (22). Despite these premises, considering the vulnerability of patients to exercise-induced fatigue, great future attention will be required to identify the correct dose of endurance exercise to be administered.
Another fundamental adaptation to exercise is related to calcium fluctuations within the sarcoplasm due to bouts repetition. As known, increased activity leads to calcium release at the sarcoplasmic reticulum through ryanodin receptors (RyR) and concomitant activation of calmodulin-dependent protein kinase (CaMK) signaling. Calpain 3 (CAPN3) is associated with the muscle triad through its interaction with RyR1. Following calcium release, phosphorylation of CaMK leads to the activation of p38 MAPK, which, in turn, stabilizes the transcriptional co-activator PGC1α, leading to increased transcription of muscle adaptation genes controlled by MEF2, PPAR and, possibly, other transcription factors. CaMK activation also phosphorylates transcriptional inhibitor HDAC, leading to its relocation from the nucleus to the cytoplasm, thus alleviating transcriptional repression. In the absence of CAPN3, as in calpain myopathy (limb girdle muscular dystrophy 2A), the levels of RyR and the amplitude of calcium release are blunted, leading to decreased activation of CaMK. As a result, both branches of downstream events are suppressed determining and the failure to up-regulate transcription of genes necessary for the adaptation to exercise arises.
Mutations in calpain 3 gene leads to autosomal recessive limb girdle muscular dystrophy 2A (LGMD2A), characterized by atrophy and weakness of proximal limb and girdle muscles. LGMD2A is one of the most common subtypes of LGMD worldwide, accounting for 15-40% of LGMD cases. Recently, a CAPN3 gene heterozygous deletion(c.643_663del21) has been associated with an autosomal dominant transmission pattern in thirteen unrelated European families (23). Clinical features may include a slowly progressive, symmetrical, limb-girdle weakness and selective muscle atrophy (e.g. hip adductors and extensors, and hamstring muscles), with an onset between the ages of 12 and 20. Scapular winging, scoliosis and joint contractures may also be observed. In general, ambulation loss occurs one to three decades after diagnosis; in fact,20% of LGMD2A patients may become wheelchair dependent before their thirties. Respiratory failure in calpainopathy is known to occur in patients with an advanced stage of the disease, particularly after ambulation loss. Early respiratory insufficiency requiring nocturnal non-invasive ventilation (NIV) in a 70-year-old ambulatory man with LGMD2A has recently been described (24). Most studies, with a few exceptions, have reported the lack of cardiac dysfunction in patients with calpainopathy (25). reported that cardiac function in 33 patients was normal on electrocardiogram and echocardiography, with the exception of 2 patients who had atrial fibrillation. In mice, calpain 3 transcripts are expressed during cardiogenesis, although its expression decreases as the organ matures. The absence in mature cardiomyocytes is a possible explanation for the absence of cardiomyopathy in the majority of patients. A few case reports have suggested cardiac involvement. For example, Okere et al. reported that a 23-year-old patient with calpainopathy had cardiomyopathy (26).
Considering the role of calpain and on the pathophysiology of calpainopathy, a working hypothesis in LGMD2A includes short duration bouts of endurance exercise. Notwithstanding these premises only pilot studies are available on the effects of training in LGMD2A, only focusing on resistance type exercise (27).
When the quality of training includes multi joint bouts heavy exercise, possibly with pyramiding increase of weight load and decrease in repetition number, the fundamental skeletal muscle adaptations consist in increased strength and hypertrophy appearance. It is widely known that, in this case, the hypertrophic plastic response of the muscle mostly depends on muscular damage due to the eccentric components of exercise. In fact, following eccentric contractions, a stereotyped response follows particularly in unaccustomed individuals including functional and biochemical features underpinning the structural alterations that the single muscle fibers sustain (Fig. 2). A fundamental role in muscle repair in presence of eccentric induce muscle damage is attributed to dysferlin.
Figure 2.
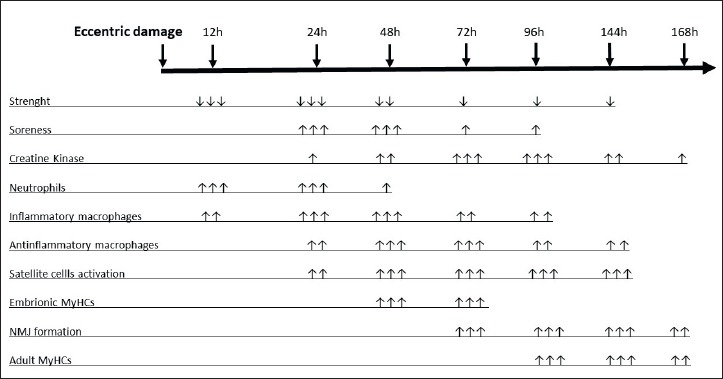
Stereotyped time course of muscular changes following eccentric exercise damage. MyHC = Myosin Heavy Chain; NMJ = neuromuscular junction; ↑ or ↓ = slight change; ↑↑ or ↓↓ = moderate change; ↑↑↑ or ↓↓↓ = large change.
DYSF gene, on Chromosome 2p13.2, encodes a protein called “dysferlin,” the muscle-specific member of a class of homologous proteins termed “ferlins”. Diseases related to mutation in DYSF Gene -Dysferlinopathies- are characterized by a selective and progressive involvement of proximal and/or distal muscles of the limb girdles, usually transmitted in autosomal recessive mode. The age at onset of muscle weakness varies widely (from congenital 14 to 73 years), but usually occurs in the teenage years or early adulthood (on average 15-27 years) (28). Serum creatine kinase (CK) levels are usually elevated (10-100 times normal values) from the early asymptomatic stage of the disease. HyperCKemia characterizes all clinical phenotypes of dysferlinopathy and is a hallmark of the disease. Three main phenotypes are usually associated with dysferlin gene mutations: limb girdle muscle dystrophy type 2B (LGMD2B), Miyoshi distal myopathy (MM) and distal myopathy with Anterior Tibialis onset (DMAT). Links between mutation type, location and phenotype are not straight forward. Dysferlin is responsible for plasma membrane repair, vesicle fusion and membrane trafficking. Not only does dysferlin widely exist in cell membranes of skeletal and cardiac muscles, it also exists in the membranes of non-myofiber cells, such as monocytes. The ferlin protein group to which dysferlin belongs all have Ca2+sensitive C2 domains. It has been demonstrated that an influx of Ca2+through the site of membrane injury triggers dysferlin-mediated membrane repair. Patients with mutations in the dysferlin gene often have impaired membrane resealing following mechanical or chemical stress, causing an influx of Ca2+. This is particularly relevant in muscle where mechanical stress due to muscle contraction is quite common and Ca2+ regulation is highly important. Furthermore, two reports have been published documenting mitochondrial abnormalities in patients with dysferlin mutations (29,30). The first report demonstrated accumulation of subsarcolemmal mitochondria in some patients’ muscle fibers including one patient with ragged red fibers and paracrystalline mitochondrial inclusions. More recently up to 10% of muscle fibers were reported to be cytochrome c oxidase (COX or complex IV) deficient, and some of these fibers have increased mtDNA copy number. In the same study, a reduced complex IV and complex III levels were observed in some patients, while others showed a significant downregulation of complex I and complex IV activities and only a mild reduction of complex III.
In 2016, Vincent et al. published a work regarding mitochondrial function in dysferlinopathies (30). The authors analyzed skeletal muscle biopsies for eight patients by quadruple immunofluorescent assay to assess oxidative phosphorylation protein abundance and Long-range PCR in single muscle fibers to look for presence of clonally expanded large-scale mitochondrial DNA rearrangements in patients’ skeletal muscle. They reported higher complex I and complexIV deficiency in patients than age matched controls but patients do not have rearrangements of the mtDNA. So, they hypothesized that respiratory chain deficiency could be the results of an increased cytosolic Ca2+ concentration (due to a membrane resealing defect) causing mitochondrial aberrations. Considering the crucial role of dysferlin in muscle repair to eccentric mechanical strain and possible translation regards dysferlin myopathies such as LGMD2B and Miyoshi myopathy due to dysferlin gene mutations. In fact, in dysferlinopathy it has been proposed that severity of the dystrophic process mainly depends on type of exercise (concentric/isometric and eccentric) instead of overload. Therefore, a working hypothesis in this neuromuscular disease include bouts of concentric exercise with abolishment of the eccentric components. To date, no studies have analyzed such a training paradigm in dysferlinopathy.
Another fundamental knowledge for translation regards muscle energy disposal and biochemical handling during rest and exercise. It is known that fluctuations in energy pool utilization mostly depends on duration and percentage of the maximal sustainable load that is the exercise intensity (Fig. 3).
Figure 3.
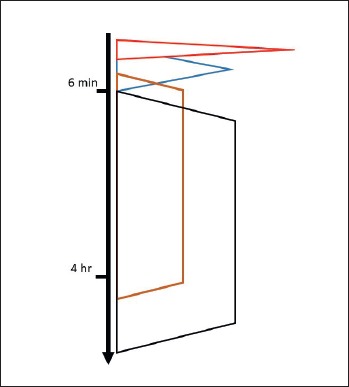
Schematic representation of the cellular energy handling versus time of exercise. From the top red: free ATP and phosphocreatine/creatine system, blue: anaerobic glycolysis (muscular glycogen), brown: aerobic oxidation (muscular glycogen, plasma glucose, liver glycogen), black: aerobic oxidation (adipose tissue triglycerides, plasma free fatty acids).
From this point of view, possible translation examples include mcArdle disease and CPT2 deficiency.
The myopathic form of carnitine palmitoyltransferase 2 (CPT2) deficiency, is an inherited metabolic disorder that affects mitochondrial oxidation of long chain fatty acids (LCFA).
CPT (carnitine palmitoyltransferase) II muscle deficiency is the most common form of muscle fatty acid metabolism disorders, inherited as autosomal recessive condition. It is one of the possible phenotypes related to mutation in the CPT system, consisting of two enzymes (CPT I and CPT II), involved in the transport of long-chain fatty acids into the mitochondrial compartment. The enzymes are located in the outer (CPT I) and inner mitochondrial membrane (CPT II) (31).
Three phenotypes of CPT II deficiency are known: a lethal neonatal form, a severe infantile hepatocardiomuscular form, and a mild myopathic form (32). It has also been reported an antenatal onset of CPT2 in a small subset of patients causing malformations including brain dysgenesis and neuronal migration defects (33). Muscle CPT II deficiency is the most frequent type of CPT II deficiency. Clinical features of the mild myopathic form are muscle weakness, myalgia, pain and rhabdomyolysis, possibly causing renal failure. Joshi et al. (2014) (34) showed that the manifestation of clinical symptoms occurred more frequently during infancy (one to 12 years old) than during adolescence (13-22 years old) and adulthood (> 22 years old). Trigger factors are prolonged exercise, fasting, infections, fever and exposure to cold (34).
In muscle CPT II deficiency, symptoms occur only intermittently. Lipid accumulation in muscle fibers can be detected (34). In approximately 90% of the patients the molecular basis is a p. S113L mutation in homozygous or heterozygous state with an allele frequency of 60-70% and more than 60 mostly private mutations (35). The normal protein content and enzyme activity allow a normal function of the CPT system in situations without stress on the fatty acid metabolism (36).
The biochemical consequences of the disease-causing mutations are still discussed controversially.
In former studies, CPT activities in muscles of patients with CPT II deficiency ranged from not detectable (38, 39) up to normal (41, 42). It is known that CPT I but not CPT II is sensitive to inhibition by malonyl-CoA. Trevisan et al. showed an almost complete inhibition of total CPT activity in patients by malonyl-CoA (43). So, it was inferred that the normal malonyl-CoA-insensitive CPT II activity is deficient. Due to the demonstration that total CPT activity is normal under optimal assay conditions but abnormal when inhibited by malonyl-CoA, palmitoylcarnitine, carnitine and Trition-X100 (non-ionic surfactant), the hypothesis raised of an abnormally regulated enzyme with a normal total CPT II concentration. CPT II with the S113L mutation, is most vulnerable to inhibition when it is most needed (44).
The study of Ørngreen and colleagues (45) about fuel utilization in CPT2 patients showed that they are unable to increase long-chain FAO during long-term, low-intensity cycle exercise, and carriers of single CPT2 gene mutations also can have impaired fat oxidation during exercise, which may explain milder symptoms of CPT II deficiency in these subjects. The authors demonstrated normal FAO at rest but impaired FAO during exercise in CPT II deficiency patients. Furthermore, they showed that the CPT II patients covered their energy deficit by carbohydrate metabolism, by enhanced muscle glycogenolysis (45).
McArdle disease, or glycogen storage disease type 5 (GSD5), is an autosomal recessive disease, due to myophosphorylase deficiency and represents the commonest muscle glycogenosis (46). The typical clinical picture of McArdle disease consists of acute crises of early fatigue and contractures, occasionally accompanied by rhabdomyolysis and myoglobinuria usually triggered by muscle tasks that predominantly involve (aerobic/anaerobic) glycolysis for ATP production (47, 48). Disease time to onset is usually in the first two decades of life, but it can also occur in infancy with progressive weakness, hypotonia, respiratory distress, and early death (47, 49), and later in adult life, with atypical symptoms, such as pain and tenderness in the masticatory muscles when eating (50) or asymmetrical, slowly progressive, limb weakness and muscle wasting which may remain focal (51). The so-called ‘second wind’ phenomenon, that is marked improvement in tolerance to dynamic exercise (eg, bicycling at a constant, submaximal wattage) after 6-10 min of exertion, with subsequent disappearance of previous tachycardia, is a unique characteristic of patients with McArdle disease (52). Patients lack the enzyme required to mobilize glucose-1-phosphate from skeletal muscle glycogen myophosphorilase, the only isoform of glycogen phosphorylase expressed in skeletal-muscle tissue. Myophosphorylase catalyzes the breakdown of muscle glycogen into glucose-1-phosphate in muscle fibers, preventing the patients to obtain energy from their muscle glycogen stores (53). Muscle glycolysis is not totally impaired in these patients, because the muscle fibers of McArdle disease patients can still take up glucose from the blood and convert it into glucose-6-phosphate, which then enters glycolysis (53). In McArdle disease, due to the block in glycogenolysis, muscle oxidative phosphorylation (OXPHOS) capacity, is also impaired and skeletal muscle is unable to produce pyruvate (54), causing a marked decrease in skeletal-muscle capacity for ATP synthesis through OXPHOS, and in accumulation of ADP and Pi in muscle fibers. This, in turn, can potentially inhibit the myofibrillar ATPase, the sarcoplasmic reticulum Ca2_ ATPase (SERCA1) and the Na+-K+ ATPase reactions, leading to decreased contractility and premature fatigue (55-57). Deficient glycogen dependent ATP supply can result in further downregulation of Na+-K+ ATPase in the skeletal muscle fibers of patients and in impaired membrane excitability and muscle cramping (54). The causes for exercise rhabdomyolysis in these patients are not completely understood: either the mechanical stress imposed by large muscle glycogen stores (48), the downregulation of muscle Na+-K+pumps (55) (which are responsible for maintaining cellular volume and integrity), or increased oxidative stress (57) have been supposed to be involved. Furthermore, elevated Ca2+ levels in the sarcoplasm (owing to the above mentioned downregulation in SERCA1) might activate proteases, phospholipases, and other catabolic enzymes that cause structural damage (58), besides causing muscle fatigue and cramps, Haller and Vissing (59) studied the ‘second wind’ phenomenon. Either when occurring spontaneously, either when glucose induced, it was due to a substrate-dependent increase in muscle oxidative capacity. Also, by providing glycogen-derived pyruvate a small amount of residual myophophorylase activity normalizes the oxidative deficit of muscle phosphorylase deficiency and may eliminate the ‘second wind’. The spontaneous second wind seemed to be related to the lack of fuel that is critical for normal oxidative metabolism and makes muscle oxidative capacity dependent on the changing availability of extramuscular fonts, such as free fatty acids, due to the blocked glycogenolysis. Furthermore, patients with McArdle disease have been reported to be insulin resistant in terms of glucose uptake, glycogen synthase activation, and alterations in fuel oxidation. As a result, the ability of insulin to increase fat and carbohydrate oxidation is limited, and the findings suggest that skeletal muscle glycogen levels play an important role in the regulation of insulin-stimulated glycogen synthase activity (60). The onset of McArdle disease may be at the teenage years during which decreased sensitivity to insulin has been noted, so that there may well be limited transport and oxidation of extracellular glucose and nonesterified fatty acids resulting in reduced exercise tolerance. Insulin sensitivity decreases with age which may play a part in the late onset of some cases (61).
Considering the inefficient utilization of glycogen in McArdle disease and long chain fatty acids in CPT2 deficiency, a working hypothesis may be the application of repeated short duration bouts followed by appropriate rest in both cases. This approach may allow to gain the benefits of exercise through phosphocreatine consumption as the main energy substrate to fuel contraction in McArdle disease and through glycogen consumption in CPT2 deficiency in presence of a strict compliance to a dietetic regimen in which reduction of fat is adequately compensated by carbohydrates intake.
Preliminary results on the feasibility of this approach in ameliorating the muscle function in McArdle disease have been recently put forward by Santalla and collaborators (54).
Conclusions and perspectives
Without the deep knowledge of the physiological adaptations to exercise training, declined in terms of quality, intensity, frequency and duration, it is not possible to ascertain whether it is, or not, beneficial in neuromuscular diseases. This knowledge should be translated to pathophysiology, with the aim to find out the optimal matching between potential training adaptations and clinical evolution of the diseases. Without attempting this approach, feasibility of physical interventions will be far to be established.
Figures and tables
Footnotes
Conflict of interest
The Authors declare to have no conflict of interest.
References
- 1.Human energy requirements. Report of a Joint FAO/WHO/UNU Expert Consultation Rome, 17-24 October 2001. [Google Scholar]
- 2.Geard D, Reaburn PRJ, Rebar AL, et al. Masters athletes: exemplars of successful aging? J Aging Phys Act 2017;25:490-500. [DOI] [PubMed] [Google Scholar]
- 3.Rogers MA, Hagberg JM, Martin WH, 3rd, et al. Decline in VO2max with aging in master athletes and sedentary men. J Appl Physiol (1985) 1990;68:2195-9. [DOI] [PubMed] [Google Scholar]
- 4.D’Antona G, Pellegrino MA, Adami R, et al. The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J Physiol 2003;552(Pt2):499-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trappe S. Master athletes. Int J Sport Nutr Exerc Metab 2001;11(Suppl):S196-207. [DOI] [PubMed] [Google Scholar]
- 6.Larsson L, Degens H, Li M, et al. Sarcopenia: aging-related loss of muscle mass and function. Physiol Rev 2019;99:427-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Antona G. mTOR, nutrition and ageing. Malavolta M, Mocchegiani E, Eds. Molecular basis of nutrition and ageing. New York, NY: Elsevier; 2016, pp. 141-53. [Google Scholar]
- 8.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115:577-90. [DOI] [PubMed] [Google Scholar]
- 9.D’Antona G, Nisoli E. mTor signaling as a target of amino acid treatment of the age related sarcopenia. Interdiscip Top Gerontol 2010;37:115-41. [DOI] [PubMed] [Google Scholar]
- 10.Lin SS, Liu YW. Mechanical stretch induces mTOR recruitment and activation at the phosphatidic acid-enriched macropinosome in muscle. Cell Front Cell Dev Biol 2019;8:7, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Neil TK, Duffy LR, Frey JW, et al. The role of phosphoinositide 3-kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J Physiol 2009;587(Pt 14):3691-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Risson V, Mazelin L, Roceri M, et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol 2009;187:859-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lundby C, Jacobs RA. Adaptations of skeletal muscle mitochondria to exercise training. Exp Physiol 2016;101:17-22. [DOI] [PubMed] [Google Scholar]
- 15.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 2013;17:162-84. [DOI] [PubMed] [Google Scholar]
- 16.Ripolone M, Ronchi D, Violano R, et al. Impaired muscle mitochondrial biogenesis and myogenesis in spinal muscular atrophy. JAMA Neurol 2015;72:666-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montes J, Garber CE, Kramer SS, et al. A randomized, controlled clinical trial of exercise in patients with spinal muscular atrophy: methods and baseline characteristics. J Neuromusc Dis 2014;1:151-61. [PubMed] [Google Scholar]
- 18.Abresch RT, Carter GT, Han JJ, et al. Exercise in neuromuscular diseases. Physical Med Rehab Clin North Am 2012;23:653-73. [DOI] [PubMed] [Google Scholar]
- 19.Biondi O, Grondard C, Lécolle S, et al. Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J Neurosci 2008;28:953-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grondard C, Biondi O, Armand AS, et al. Regular exercise prolongs survival in a type 2 spinal muscular atrophy model mouse. J Neurosci 2005;25:7615-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartels B, Montes J, van der Pol WL, et al. Physical exercise training for type 3 spinal muscular atrophy. Cochrane Database Syst Rev 2019;3:CD012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madsen KL, Hansen RS, Preisler N, et al. Training improves oxidative capacity, but not function, in spinal muscular atrophy type III. Muscle Nerve 2015;52:240-4. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Thompson JM, Niu Z, Tracy JA, et al. Autosomal dominant calpainopathy due to heterozygous CAPN3 C. Muscle Nerve 2018;57:679-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mori-Yoshimura M, Segawa K, Minami N, et al. Cardiopulmonary dysfunction in patients with limb-girdle muscular dystrophy 2A. Muscle Nerve 2017;55:465-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groen EJ, Charlton R, Barresi R, et al. Analysis of the UK diagnostic strategy for limb girdle muscular dystrophy 2A. 2007;130:3237-49. [DOI] [PubMed] [Google Scholar]
- 26.Okere A, Reddy SS, Gupta S, et al. A cardiomyopathy in a patient with limb girdle muscular dystrophy type 2A. Circ Heart Fail 2013;6:e12-3. [DOI] [PubMed] [Google Scholar]
- 27.Sveen ML, Andersen SP, Ingelsrud LH, et al. Resistance training in patients with Limb-girdle and Becker muscular dystrophies. Muscle Nerve 2013;47:163-9. [DOI] [PubMed] [Google Scholar]
- 28.Gayathri N, Alefia R, Nalini A, et al. Dysferlinopathy: spectrum of pathological changes in skeletal muscle tissue. Indian J Pathol Microbiol 2011;54:350-4. [DOI] [PubMed] [Google Scholar]
- 29.Liu F, Lou J, Zhao D, et al. Dysferlinopathy: mitochondrial abnormalities in human skeletal muscle. Int J Neurosci 2016;126:499-509. [DOI] [PubMed] [Google Scholar]
- 30.Vincent AE, Rosa HS, Alston CL, et al. Dysferlin mutations and mitochondrial dysfunction. Neuromuscul Disord 2016;26:782-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehmann D, Motlagh L, Robaa D, et al. Muscle Carnitine Palmitoyltransferase II deficiency: a review of enzymatic controversy and clinical features. Int J Mol Sci 2017;3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boemer F, Deberg M, Schoos R, et al. Diagnostic pitfall in antenatal manifestations of CPT II deficiency. Clin Genet 2016:89:193-7. [DOI] [PubMed] [Google Scholar]
- 33.Joshi PR, Deschauer M, Zierz S. Carnitine palmitoyltransferase II (CPT II) deficiency: genotype-phenotype analysis of 50 patients. J Neurol Sci 2014;338:107-11. [DOI] [PubMed] [Google Scholar]
- 34.Bonnefont JP, Djouadi F, Prip-Buus C, et al. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med 2004;25:495-520. [DOI] [PubMed] [Google Scholar]
- 35.Isackson PJ, Bennett MJ, Vladutiu GD. Identification of 16 new disease-causing mutations in the CPT2 gene resulting in carnitine palmitoyltransferase II deficiency. Mol Genet Metab 2006;89:323-31. [DOI] [PubMed] [Google Scholar]
- 36.Lehmann D, Zierz S. Normal protein content but abnormally inhibited enzyme activity in muscle carnitine-palmitoyltransferase II deficiency. J Neurol Sci 2014;339:183-8. [DOI] [PubMed] [Google Scholar]
- 37.DiMauro S, di Mauro PM. Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science 1973;182:929-31. [DOI] [PubMed] [Google Scholar]
- 38.Bank WJ, DiMauro S, Bonilla E, et al. A disorder of muscle lipid metabolism and myoglobinuria. Absence of carnitine palmityl transferase. N Engl J Med 1975;292:443-9. [DOI] [PubMed] [Google Scholar]
- 39.Layzer RB, Havel RJ, McIlroy MB. Partial deficiency of carnitine palmityltransferase: physiologic and biochemical consequences. Neurology 1980;30:627-33. [DOI] [PubMed] [Google Scholar]
- 40.Vladutiu GD, Saponara I, Conroy JM, et al. Immunoquantitation of carnitine palmitoyl transferase in skeletal muscle of 31 patients. Neuromuscul Disord 1992;2:249-59. [DOI] [PubMed] [Google Scholar]
- 41.Fanin M, Anichini A, Cassandrini D, et al. Allelic and phenotypic heterogeneity in 49 Italian patients with the muscle form of CPT-II deficiency. Clin Genet 2012;82:232-9. [DOI] [PubMed] [Google Scholar]
- 42.Trevisan CP, Angelini C, Freddo L, et al. Myoglobinuria and carnitine palmityltransferase (CPT) deficiency: studies with malonyl-CoA suggest absence of only CPT-II. Neurology 1984;34:353-6. [DOI] [PubMed] [Google Scholar]
- 43.Zierz S, Engel AG. Regulatory properties of a mutant carnitine palmitoyltransferase in human skeletal muscle. Eur J Biochem 1985;149:207-14. [DOI] [PubMed] [Google Scholar]
- 44.Motlagh L, Golbik R, Sippl W, et al. Stabilization of the thermolabile variant S113L of carnitine palmitoyltransferase II. Neurol Genet 2016;2:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ørngreen MC, Dunø M, Ejstrup R, et al. Fuel utilization in subjects with carnitinepalmitoyltransferase 2 gene mutations. Ann Neurol 2005;57:60-6. [DOI] [PubMed] [Google Scholar]
- 46.McArdle B. Myopathy due to a defect in muscle glycogen breakdown. Clin Sci 1951;10:13-35. [PubMed] [Google Scholar]
- 47.Gordon N. Glycogenosis type V or McArdle’s disease developmental. Med Child Neurol 2003;45:640-4. [DOI] [PubMed] [Google Scholar]
- 48.Di Mauro S. Muscle glycogenoses: an overview. Acta Myol 2007;26:35-41. [PMC free article] [PubMed] [Google Scholar]
- 49.DiMauro S, Hartlage PL. Fatal infantile form of muscle phosphorylase deficiency. Neurology 1978;28:1124-9. [DOI] [PubMed] [Google Scholar]
- 50.Martin H. Masticatory muscle symptoms in a patient with McArdle’s disease. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1996;81:554-6. [DOI] [PubMed] [Google Scholar]
- 51.Wolfe GI, Baker NS, Haller RG, et al. McArdle’s disease presenting with asymmetrical, late-onset arm weakness. Muscle Nerve 2000;23:641-5. [DOI] [PubMed] [Google Scholar]
- 52.Delaney NF, Sharma R, Tadvalkar L, et al. Metabolic profiles of exercise in patients with McArdle disease or mitochondrial myopathy. PNAS 2017;114:8402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lucia A, Nogales-Gadea G, Perez M, et al. McArdle disease: what do neurologists need to know? Nat Clin Pract Neurol 2008;4:568-77. [DOI] [PubMed] [Google Scholar]
- 54.Santalla A, Munguía-Izquierdo D, Brea-Alejo L, et al. Feasibility of resistance training in adult McArdle patients: clinical outcomes and muscle strength and mass benefits. Front Aging Neurosci 2014;6:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haller RG, Clausen T, Vissing J. Reduced levels of skeletal muscle Na_K-ATPase in McArdle disease. Neurology 1998;50:37-40. [DOI] [PubMed] [Google Scholar]
- 56.Zange J, Grehl T, Disselhorst-Klug C, et al. Breakdown of adenine nucleotide pool in fatiguing skeletal muscle in McArdle’s disease: a noninvasive 31P-MRS and EMG study. Muscle Nerve 2003;27:728-36. [DOI] [PubMed] [Google Scholar]
- 57.Kitaoka Y, Ogborn DI, Nilsson MI, et al. Oxidative stress and Nrf2 signaling in McArdle disease. Mol Genet Metab 2013;110:297-302. [DOI] [PubMed] [Google Scholar]
- 58.Russo PJ, Phillips JW, Seidler NW. The role of lipid peroxidation in McArdle’s disease: applications for treatment of other myopathies. Med Hypotheses 1992;39:147-51. [DOI] [PubMed] [Google Scholar]
- 59.Haller RG, Vissing J. Spontaneous “second wind” and glucose-induced second “second wind” in McArdle disease. Arch Neurol 2002;59:1395-1402. [DOI] [PubMed] [Google Scholar]
- 60.Nielsen JN, Vissing J, Wojttaszewski JFP, et al. Decreased insulin action in skeletal muscle from patients with McArdle’s disease. Am J Physiol Endocrinol Metab 2002;282:e1267-75. [DOI] [PubMed] [Google Scholar]
- 61.O Dorin RI, Field JC, Boyle PJ, et al. Insulin resistance limits glucose utilization and exercise tolerance in myophosphorylase deficiency and NIDDM. J Appl Physiol 1996;81:1273-8. [DOI] [PubMed] [Google Scholar]
- 62.D’Antona G. Physical activity. Poletto publisher 2019. (1st edition). [Google Scholar]