

Abstract
Cyclophosphamide treatment on a medium-dose, intermittent chemotherapy (MEDIC) schedule activates both innate and adaptive immunity leading to major regression of implanted gliomas. Here, we show that this MEDIC treatment regimen induces tumor cell autonomous type-I interferon signaling, followed by release of soluble factors that activate interferon-stimulated genes in both tumor cells and tumor-infiltrating immune cells. In cultured GL261 and CT-2A glioma cells, activated cyclophosphamide stimulated production and release of type-I interferons, leading to robust activation of downstream gene targets. Antibody against the type-I interferon receptor IFNAR1 blocked the cyclophosphamide-stimulated induction of these genes in both cultured glioma cells and implanted gliomas. Furthermore, IFNAR1 antibody strongly inhibited the MEDIC cyclophosphamide-stimulated increases in tumor cell infiltration of macrophages, dendritic cells, B-cells, as well as natural killer cells and cytotoxic T-cells and their cytotoxic effectors. Finally, cyclophosphamide-treated dying glioma cells producing type-I interferons were an effective vaccine against drug-naïve glioma cells implanted in vivo. Thus, cyclophosphamide induces local, tumor cell-centric increases in type-I interferon signaling, which activates immunogenic cell death and is essential for the striking antitumor immune responses that MEDIC cyclophosphamide treatment elicits in these glioma models.
Keywords: metronomic chemotherapy, type-I interferon, IFNAR1, interferon-stimulated genes, 4-hydroperoxy-cyclophosphamide
1. Introduction
Many conventional clinical chemotherapy regimens are toxic to T cells, natural killer cells and dendritic cells, leading to immunosuppression [1, 2]. Further, certain cancers, including high-grade malignant gliomas, are in a highly immunosuppressive environment prior to chemotherapy [3, 4]. However, several cytotoxic anticancer drugs, including doxorubicin [5], oxaliplatin [6] and cyclophosphamide (CPA) [7, 8], can kill tumor cells by activating immunogenic tumor cell death (ICD), which may trigger robust antitumor immune responses [9, 10]. The hallmarks of chemotherapy-induced ICD include cell surface exposure of the endoplasmic reticulum protein calreticulin [11, 12], secretion of ATP [6], and release of the chromatin-binding protein HMGB1 [13]. Recent studies suggest that type-I interferon (type-I IFN) formation is also an important hallmark of ICD [14–16].
CPA is one of the most widely used alkylating agents for the treatment of hematologic and solid malignancies [17]. CPA has significant immune-modulatory activities, most notably its ability to suppress regulatory T cells and thereby counteract immunosuppression in the tumor microenvironment [18]. CPA is a liver cytochrome P450-activated alkylating agent prodrug that generally shows little or no activity in tumor cell culture studies due to the absence of P450 activity [19], but where its chemically activated derivative, 4-hydroperoxy-CPA (4HC), shows potent cytotoxic and immunomodulatory properties [20, 21]. Low-dose CPA treatment decreases splenic production of immunosuppressive cytokines and signaling molecules, such as IL-10, TGF-β and nitric oxide, restoring lymphoproliferative capacity [22] and can reduce myeloid-derived suppressor cells, both at the tumor site and in the peripheral blood and lymph nodes [23]. CPA can also induce hallmarks of ICD, including changes in the cell surface composition and release of soluble damage-associated molecular patterns [24], leading to the activation of tumor-specific immune responses [25–28]. The latter effects are most striking when CPA is given on an intermittent, metronomic schedule [25, 26, 29, 30], termed medium-dose, intermittent chemotherapy (MEDIC) [7].
MEDIC CPA treatment induces widespread transcriptional changes within the tumor environment, affecting both tumor cells and tumor-infiltrating immune cells critical for drug-stimulated antitumor immunity [21, 31]. In particular, studies in glioma models show that type-I interferon (IFN) signaling is an upstream regulator of the immunogenic gene responses activated by MEDIC CPA treatment [31], and a signature of type-I IFN activation is seen in peripheral blood cells of CPA-treated patients [32]. Tumors that are intrinsically sensitive to CPA cytotoxicity yet are unresponsive to MEDIC CPA-stimulated immune-based regression have been identified, and are largely deficient in these IFN-linked transcriptional changes [21], suggesting that type-I IFNs may contribute functionally to MEDIC CPA-induced tumor regression.
Type-I IFNs include IFNα proteins, which are encoded by 13 distinct but closely related genes in humans, and IFNβ, which is encoded by a single gene in humans and mice [33]. Type-I IFNs signal via the homodimeric cell surface receptor IFNAR1, which has a high affinity for IFNβ, or via IFNAR1—IFNAR2 heterodimers, which bind both IFNα and IFNβ [34]. Activation of these receptors leads to transcriptional activation of IFN-stimulated genes (ISGs), many of which elicit diverse immunostimulatory responses, including antitumor immune responses [35, 36]. CPA can stimulate IFNAR1-dependent proliferation of dendritic cells in tumor-bearing mice [37], and host cell IFNAR1 signaling may be important for CPA-induced immune cell infiltration and for control of metastatic tumor progression [38]. CPA can synergize with type-I IFNs to stimulate CD8 T-cell cross-priming by dendritic cells [37]. However, it is unknown whether type-I IFN production is induced in MEDIC CPA-responsive glioma cells in vivo, or whether type-I IFN signaling contributes functionally to the strong antitumor immune responses seen with this metronomic CPA treatment schedule.
Here, we examine these issues and characterize the effects of CPA on type-I IFN production and signaling in cultured glioma cells and in mouse models of glioma. We investigate CPA-induced changes in type-I IFN expression in both tumor cell-enriched (CD45-negative) and immune cell (CD45-positive) populations isolated from MEDIC CPA-treated GL261 gliomas. Furthermore, we investigate the contribution of type-I IFN signaling to CPA-induced immune cell infiltration associated with tumor regression, and we examine the antitumor vaccine activity of CPA-treated glioma cells undergoing ICD. Our findings establish that MEDIC CPA treatment induces tumor cell autonomous type-I IFN signaling that is essential for the robust immune cell infiltration that helps drive tumor regression in glioma models.
2. Materials and Methods
2.1. Glioma cell culture -
Mouse GL261 glioma cells were authenticated by and obtained from the Developmental Therapeutics Program Tumor Repository (National Cancer Institute, Frederick, MD). Mouse CT-2A glioma cells [39] were generously donated by Dr. Thomas Seyfried (Boston College, Boston, MA). Both glioma lines were grown at 37°C in a humidified, 5% CO2 atmosphere in RPMI 1640 culture medium containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin.
2.2. Drug treatments in cell culture -
Glioma cells were seeded in 6-well tissue culture plates 24 h prior to drug treatment. Cells were treated for 4 h with 10-20 μM 4-hydroperoxy-CPA (4HC; Santa Cruz Biotechnology, Inc), as specified in each study, then washed once and replenished with fresh, drug-free media. Cells were collected in TRIzol (Life Technologies, Grand Island, NY) to isolate RNA 24, 48, or 72 h after initiating 4HC treatment. The concentration 4HC was based on the EC50 value for 4HC cytotoxicity, 12.9 μM, measured in cultured GL261 cells in a 24 h growth inhibition assay (data not shown). The 4 h exposure time was chosen to mimic the exposure to active 4-hydroxy-CPA that occurs in mice in vivo, where CPA and 4-hydroxy-CPA show plasma half-lives of 20-40 min, and are essentially eliminated from circulation within 4 h [40, 41]. In some studies, GL261 cells were seeded in 150 mm culture dishes 24 h prior to treatment. Cells (70–80% confluent) were treated with 10 μM 4HC for 4 h, washed and then replenished with fresh culture medium (10 ml). Conditioned media from the 4HC-treated cells was collected 48 h later, passed through a 0.45 μm syringe filter to remove dead cells and debris, and then applied to fresh GL261 cells seeded in 6-well plates 24 h earlier. Cells were then treated with the conditioned media or with culture media containing 250 U/ml IFNβ1 (Recombinant Mouse IFNβ1, Cat. #581304, BioLegend, San Diego, CA) and collected in TRIzol for RNA isolation either 6, 12, 24, or 48 h later. In other experiments, GL261 cells or CT-2A cells were seeded in 6-well plates 24 h prior to treatment. Cells were treated for 4 h with 20 μM 4HC with or without anti-IFNAR1 antibody (10 μg/ml; clone MAR1-5A3, Cat. # BE0241, Bio-XCell, West Lebanon, NH). Cells were then washed and replenished with fresh, drug-free media containing 10 μg/ml anti-IFNAR1 antibody, then harvested in TRIzol for RNA isolation 44 h later.
2.3. Reverse transcription-quantitative PCR analysis -
Isolation of total RNA from cultured cells or frozen tumor tissue, reverse transcription, and quantitative polymerase chain reaction (RT-qPCR) analysis were performed as previously described [26]. Briefly, total RNA was extracted from cultured cells or homogenized tumor tissue using TRIzol followed by DNase I treatment (Promega, Madison, WI) and cDNA synthesis using an Applied Biosystems High-Capacity cDNA Reverse Transcription kit (Life Technologies). For cultured cells, cells were trypsinized, washed, resuspended directly in TRIzol and homogenized by pipetting the sample up and down ~10 times. RT-qPCR was performed using Power SYBR Green (Life Technologies) and gene-specific primers (Table S1), and processed on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Grand Island, NY). Primers were designed using Primer Express software (Applied Biosystems, Carlsbad, CA) and evaluated by Blat analysis to ensure gene specificity. Data were analyzed using the comparative CT (ΔΔCT) method and are presented as relative levels of each RNA compared to the RNA level in untreated tumors after normalization to the 18S RNA content of each sample.
2.4. Tumor xenograft studies -
Mice were housed and treated under protocols approved by the Boston University Institutional Animal Care and Use Committee. GL261 glioma cells (4 ×106) or CT-2A glioma cells (5 ×106) were implanted in 6-week old (20–23 g) male C57BL/6NTac mice (Taconic Farms, Germantown, NY) by subcutaneous injection into the posterior flanks in fresh RIPM 1640 medium without fetal bovine serum (0.2 ml per site) using a U-100 insulin syringe and a 28-gauge needle (BD Biosciences, Cat. 329461) (n= 9-11 tumors per group). Tumor areas (length x width) were measured twice weekly using Vernier calipers (VWR International, Cat. 62379-531) and used to calculate: tumor volume = (π/6) x (length x width)3/2. Average tumor volumes for each treatment group were set to a value of 100% on the first day of drug treatment (t = 0 days) to normalize the size differences across the untreated tumors in each group. Mice were treated with CPA-monohydrate (Cat. # C0768, Sigma-Aldrich, St. Louis, MO) by intraperitoneal injection every 6 days at a dose of 140 mg CPA/kg body weight per day. Signaling through the IFNα and IFNβ receptor, IFNAR1, was inhibited using anti-IFNAR1 antibody. Anti-mouse IFNAR1 antibody (clone MAR1-5A3) and anti-mouse IgG1 Isotype Control (clone MOPC-21, BioXCell Cat. # BE0083) were dissolved in sterile saline and administered by intraperitoneal injection on day 0 (i.e., the first day of CPA treatment) at a dose of 2.5 mg/mouse, and on days 2, 6, and 10 at a dose of 1 mg/mouse. Antibody treatments on day 0 and on day 6 were given 4 h after CPA treatment.
2.5. Tumor dissection and cell immunoisolation -
Tumors from mice treated with CPA or PBS (vehicle control) on treatment day 0, or on treatment days 0 and 6, were excised from individual mice on days 1, 3, 6, 9, or 12 after the first CPA injection (n = 8-11 tumors/group). Tumors were dissected into 1-2 mm pieces and placed in a Miltentyi Biotec C-tube with 5 ml complete medium (RPMI 1640 containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin) containing 200 U/ml collagenase IV (Worthington Biochemical Corp., Lakewood, NJ, Cat. # CLS-4) and 0.1 mg/ml DNase I (Calbiochem/EMD Millipore, Billerica, MA, Cat# 260913). The tubes were incubated at 37°C for 30 min under shaking and were vortexed every 10 min. Then, the tumor samples were processed on a GentleMACS Tissue Dissociator (Miltenyi Biotec, San Diego, CA) using the manufacturer’s program, m_imp_tumor_4. The cell suspension was filtered through a 70 μm filter (Corning® cell strainer, Cat# 431751) into a fresh 50 ml conical tube. The filter was washed with 10 ml MACS labeling buffer (filtered 1 x PBS, pH 7.2, 2 mM EDTA), centrifuged and re-suspended in labeling buffer. Immune cells were immuno-purified from the digested tumor samples using a miniMACS Separator and a CD45 Cell Isolation Kit and (both from Miltenyi Biotec, Bergisch Gladbach, Germany), which captures cells displaying the cell surface marker CD45. CD45-positive cells (immune cell-enriched population) and CD45-negative cells (tumor cell-enriched population)were resuspended in MACS labeling buffer and divided into two aliquots; one used for RNA isolation, and the second used for flow cytometry. FACSCalibur analysis typically showed a purity of >85% (CD45-positive cells) and 45 to 95% (CD45-negative cells) after resuspension in 200 μl cell staining buffer. DAPI staining was used to discriminate between live and dead cells.
2.6. Tumor tissue processing and immunohistochemistry -
T umors were collected on day 1, 3, or 6 after the first CPA treatment (day 0) or as indicated in each figure. Tumors were excised and frozen in liquid nitrogen for RNA isolation, tissue cryosectioning, and immunohistochemistry. A second portion of each tumor was fixed overnight in 4% paraformaldehyde, dehydrated in 70% ethanol and embedded in paraffin at the Maine Medical Center Research Institute (Scarborough, ME). Paraffin sections (4 μm) were stained with hematoxylin and anti-IFNβ1 antibody (Cat. # LS-B1361, LifeSpan Biosciences, Seattle WA) after dewaxing with tissue deparaffinization solution (EZ-DeWax; BioGenex, Fremont, CA). Endogenous peroxidase was blocked for 5 min using 3% hydrogen peroxide. Protein blocking was performed for 20 min using 2% or 5% normal animal serum from the same species in which the secondary antibody was raised. The slides were incubated overnight at 4°C with anti-IFNβ antibody (primary antibody), followed by washing and incubation for 45 to 60 min at room temperature at a dilution of 1:200 with goat anti-rabbit biotinylated secondary antibodies (Cat. # PI-1000, Vector Laboratories, Burlingame, CA). An avidin-biotin blocking kit (Cat. No. SP-2001; Vector Laboratories) was used for all incubations, followed by ABC signal amplification using Vectastain ABC peroxidase reagent (ABC Elite Kit, Vector Laboratories, Cat. # PK-4000). The slides were submerged for 5 min in diaminobenzidine substrate (ImmPACT® DAB Peroxidase (HRP) Substrate, Vector Laboratories, Cat. # SK-4105) and stained for 5 min with Harris Modified hematoxylin solution (Sigma; Cat. No. HHS128). The slides were washed twice with ultra-pure water for 2 min, incubated for 1 min with 5% acetic acid, then washed sequentially with ultra-pure water (1 min), Scott’s Tap Water (45 sec; Sigma; Cat. No. S5134), and ultra-pure water (2 min x 2 washes) using a BioGenex i6000 Autostainer. The stained tumor sections were visualized under an Olympus BX51 microscope. For each tumor, tiled images typically covering 4 to 15 non-overlapping fields were collected (depending on the tumor size), and at least three randomly selected regions were analyzed (i.e., 10-40 images). Images were captured by an Olympus DP25 digital camera using Olympus DP2-BSW software. To quantify the staining, images were converted into 8-bit image files, a background threshold was determined using ImageJ software, and images were then processed using an ImageJ software macro to quantify the area of positive staining.
2.7. Vaccination study –
For each mouse to be vaccinated, GL261 cells (6 x 106 per mouse) were treated in culture with 10 μM 4HC for 24 h and then incubated in fresh RPMI 1640 medium containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin for an additional 48 h (4HC group). Alternatively, cells were treated with 1 μM doxorubicin for 36 h (DOX group). Cells were then harvested in 200 μl PBS containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin, and injected subcutaneously in the lower flanks of 6-week-old male C57BL/6 mice (n = 8-9 tumors/group). Fresh, untreated GL261 cells (1 x 106) were inoculated into the opposite flank 7 day later, and the tumor growth at that site was monitored weekly. A statistically significant delay in the appearance of tumors at the latter inoculation site was taken as an indication of antitumor vaccine activity.
2.8. Statistical analysis -
Data were analyzed using GraphPad Prism to determine statistical significance by one-way analysis of variance (ANOVA) (for 3 of more comparisons) or one tailed Student’s t-test (for 2 group comparisons). Significance is indicated in each figure by: *, p < 0.05; **,p < 0.01; ***, p < 0.001; and ****, p < 0.0001.
3. Results
3.1. MEDIC CPA treatment induces regression and immune cell infiltration in CT-2A gliomas -
CPA treatment on a 6-day repeating MEDIC schedule [7] induces complete, immune cell-dependent regression of GL261 tumors implanted in syngeneic C57BL/6 mice and activates long-term tumor specific immunity [26]. Here, we confirmed these responses when this treatment schedule was used in a second syngeneic glioma model, CT-2A [39]. CPA-induced regression of CT-2A tumors was more rapid than that of GL261 tumors (Fig. 1A). Tumor volumes declined to 30% of drug-free control tumors by day 6 after the first CPA treatment, followed by a further decline after the second CPA injection. Tumor regression was associated with significant increases in immune cell infiltration, as revealed by marker gene analysis in CT-2A whole tumors excised on treatment day 12. Large increases in macrophages (CD68 and F4/80), natural killer cells (Nkp46), cytotoxic T-cells (CD8a), B-cells (B220), dendritic cells (CD74) and cytotoxic effectors of natural killer and T-cells (perforin1 and granzyme B) were observed (Fig. 1B), similar to our findings in GL261 tumors using the same MEDIC treatment schedule [25, 26, 30]. Strong induction of the ISGs MX1, CXCL10 and CXCL11 was also observed; however, the expression of the type-I interferons IFNB1 and IFNA4 was not significantly changed at this time point (Fig. 1C).
Fig. 1 -. MEDIC CPA induces tumor regression, immune infiltration and ISG expression in CT-2A gliomas.

(A) Tumor growth curves of CT-2A and GL261 gliomas untreated (blue ovals) or treated with 2 cycles of MEDIC CPA (red squares) on days indicated by arrows along X-axis. Data shown are mean ± SEM normalized tumor volumes for n=9-19 (CT-2A) or n=7 (GL261) tumors per group. * p < 0.05 for CPA versus control, evaluated only on day 6 (one-tailed t-test). (B, C) RT-qPCR analysis of immune cell marker genes (B) and ISGs and interferons (C) in untreated CT-2A whole tumors (first bar in each pair) and in CPA-treated CT-2A whole tumors excised on day 12 (second bar) (mean ± SEM, n = 9-10 tumor per group). NS: not significant; *p < 0.05, **p < 0.01, ***p < 0.001 , ****p < 0.0001 (unpaired Student’s t-test) compared to untreated tumors.
3.2. MEDIC CPA activates glioma cell production of type-I interferons –
Interferons have been identified as upstream regulators of the widespread transcriptional changes that MEDIC CPA treatment induces in regressing gliomas [21, 31]; however, little or no interferon induction was seen at the time points examined, typically 6 days after the second CPA treatment on day 6 (i.e., treatment day 12) [26] (Fig. 1C), when robust immune cell infiltration and tumor regression are already in progress. We therefore examined earlier time points after drug treatment, using the GL261 glioma model. Analysis of tumors excised 3, 6 and 12 days after initiating CPA treatment revealed significant increases in expression of IFNB1, IFNA4 and the ISGs MX1 and CXCL11. Expression peaked on day 6 and then declined (Fig. 2A). In contrast, the cytotoxic effector granzyme B showed further increases from day 6 to day 12 in the same tumor samples (Fig. 2A).
Fig. 2 -. Cancer cell–autonomous type-I interferon signaling in MEDIC CPA-treated GL261 gliomas.
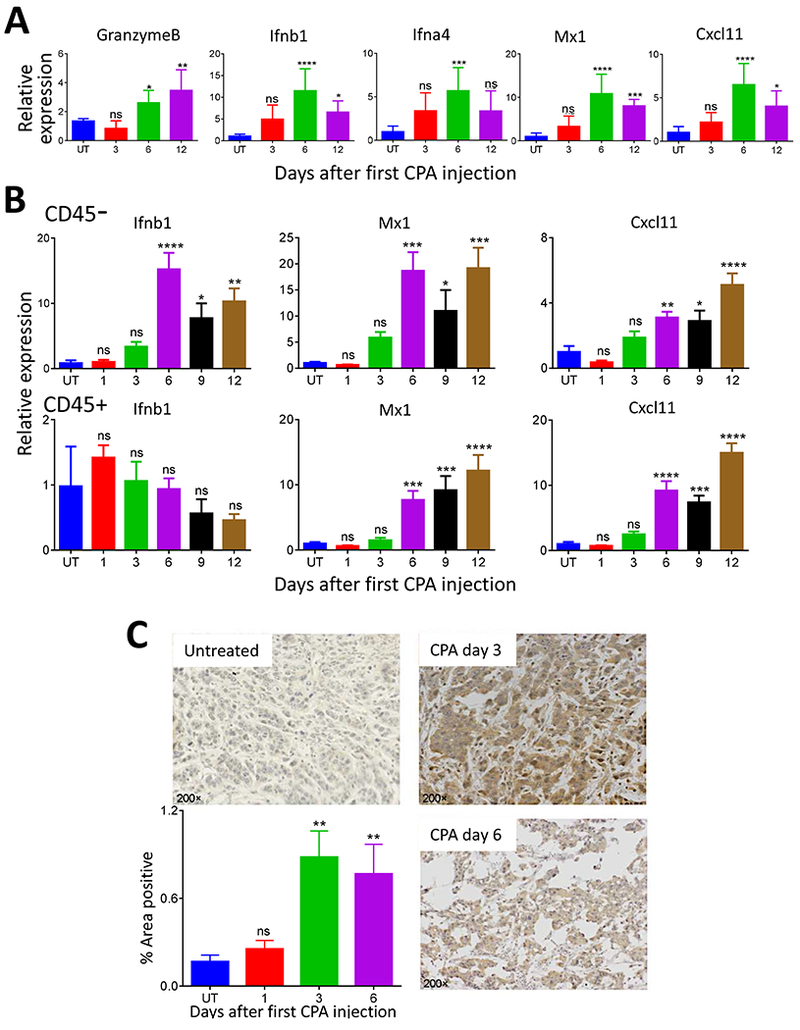
(A) RT-qPCR analysis of ISGs expressed in untreated GL216 gliomas and in GL261 gliomas excised on day 3, 6 or 12 after the first CPA treatment on day 0 (n = 7-8 tumors per group). Data are mean fold-change values ± SEM after normalization to the levels of 18S RNA, NS: not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001 (one-way ANOVA analysis) compared to untreated tumors. (B) Expression of ISGs in cell populations isolated from CPA-treated GL261 gliomas (n = 6-8 tumors per group) treated with PBS (untreated control, UT) or with CPA on days 0 and 6. Tumors were collected on the indicated days after first CPA treatment (X-axis values) and assayed for ISG induction by RT-qPCR after immuno-isolation of CD45-negative (first row) and CD45-positive cell populations (second row). IFNB1 was only induced in the tumor cell-enriched cell population, whereas the downstream ISG targets MX1 and CXCL11 were induced in both cell populations. Data are mean fold-change values ± SEM. NS: not significant; *p < 0.05, **p < 0.01, ***p < 0.001(unpaired Student’s t-test) compared to the untreated tumor. (C) Histochemical staining of the IFNβ protein in GL261 tumors on days 3 and 6 after CPA injection on day 0. Data are representative of n = 3-4 independent sections from each of n = 4-6 tumors per group. Image J quantification of immunostained IFNβ protein is shown on the bottom left. Error bars, mean ± SEM for n = 4-6 tumors per group, based on data averaged over 10 to 40 images per tumor. NS, not significant; *p < 0.05, **p < 0.01 (one-way ANOVA analysis) compared to untreated tumors.
Type-I interferons are produced by dendritic cells, but can also be formed by tumor cells [15, 42]. To determine the source of type-I interferon produced by the MEDIC CPA-treated gliomas, we used immunoaffinity beads to isolate two populations of cells from GL261 tumors: CD45-positive cells, which are primarily immune cells, and CD45-negative cells, which are largely devoid of immune cells and are tumor cell-enriched. Both cell populations were isolated from control tumors and from tumors collected 1, 3, 6, 9 and 12 days after initiating CPA treatment. IFNB1 was strongly induced in the tumor cell-enriched population but not in the immune cell (CD45-positive) population, as was first seen on treatment day 6 (Fig. 2B). In contrast, the ISGs MX1 and CXCL11 were strongly increased in both cell populations beginning on day 6. Tumor cell production of IFNβ protein was verified by immunohistochemistry, and was evident within 3 days of CPA treatment (Fig. 2C). The apparent delay until day 6 in the robust induction of IFNB1 mRNA might be explained by the instability of IFNB1 mRNA in induced cells [43, 44], coupled with the ability of IFNβ protein produced in initial, transient transcriptional responses to activate a feed-forward loop that leads to strong, sustained increases in IFNB1 expression [44, 45].
3.3. 4HC induces type-I interferon expression in glioma cells -
Given the tumor cell-centric induction of type-I interferons in the CPA-treated gliomas in vivo (Fig. 2), we examined the responsiveness of cultured glioma cells to CPA treatment. GL261 and CT-2A cells were treated with the activated CPA derivative 4HC (10 μM for 4 h), followed by incubation in drug-free medium for up to 72 h. 4HC induced the expression of IFNB1 RNA in both glioma lines (Fig. 3). In GL261 cells, small increases in IFNB1 were detected after 24 h, followed by much larger increases after 48 and 72 h. IFNA4 was also induced, but to a lower extent (Fig. 3A). Increases in expression of IFNB1, but not IFNA4, were seen in CT-2A cells (Fig. 3B). Furthermore, the increases in type-I interferons were accompanied by strong increases in many downstream ISGs after 48-72 h, with the strongest responses seen for MX1 and CXCL11 in both glioma lines (Fig. 3). These findings confirm that glioma cells are intrinsically responsive to induction of autonomous, tumor cell-centric type-I interferon signaling following CPA treatment.
Fig. 3 – Time course for induction of ISGs in glioma cell cultures treated with 4HC -.

GL261 (A) and CT-2A (B) cell cultures were treated with PBS or 4HC (20 μM for 4 h) and RNA was extracted 24-72 h after beginning 4HC treatment, as indicated, followed by assay for each of the indicated ISGs by RT-qPCR. Data shown are mean fold-change values ± SD of n = 3 replicate cultures, with intra-sample normalization to the levels of 18S RNA. Fold-change values sometimes varied between experiments (c.f., Fig. 5), in part due to variability in basal expression levels between cultures. *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001 (one-way ANOVA analysis) compared to untreated cells.
To investigate the possible involvement of soluble factors produced by the drug-treated glioma cells in type-I interferon signaling, conditioned medium was harvested from 4HC-treated GL261 cells and transferred to drug-naïve GL261 cells. MX1 and CXCL11 were both induced significantly in the recipient cells (Fig. 4A), indicating that soluble secreted factors, such as type-I interferons, contribute to ISG induction. Induction was maximal at 6 h then declined precipitously by 12 h. The same decline in response with time was seen when the cells were stimulated with exogenous IFNβ, indicating that the secreted IFNβ1 is unstable in culture medium. IFNB1 expression was also induced up to 5-fold by the conditioned medium, indicating there is a feed-forward loop for IFNB1 expression in these cells. However, given the instability of IFNβ in culture, this feed-forward response is unlikely to be a direct effect of IFNβ, as the increase in IFNB1 expression persisted through 48 h. Moreover, exogenous IFNβ was much less effective at increasing IFNB1 expression under conditions where it induced the ISGs MX1 and CXCL11 to very high levels (Fig. 4B).
Fig. 4 – Effects of 4HC conditioned medium and of exogenous IFNβ on IFNB1 and ISG expression in cultured GL261 cells –
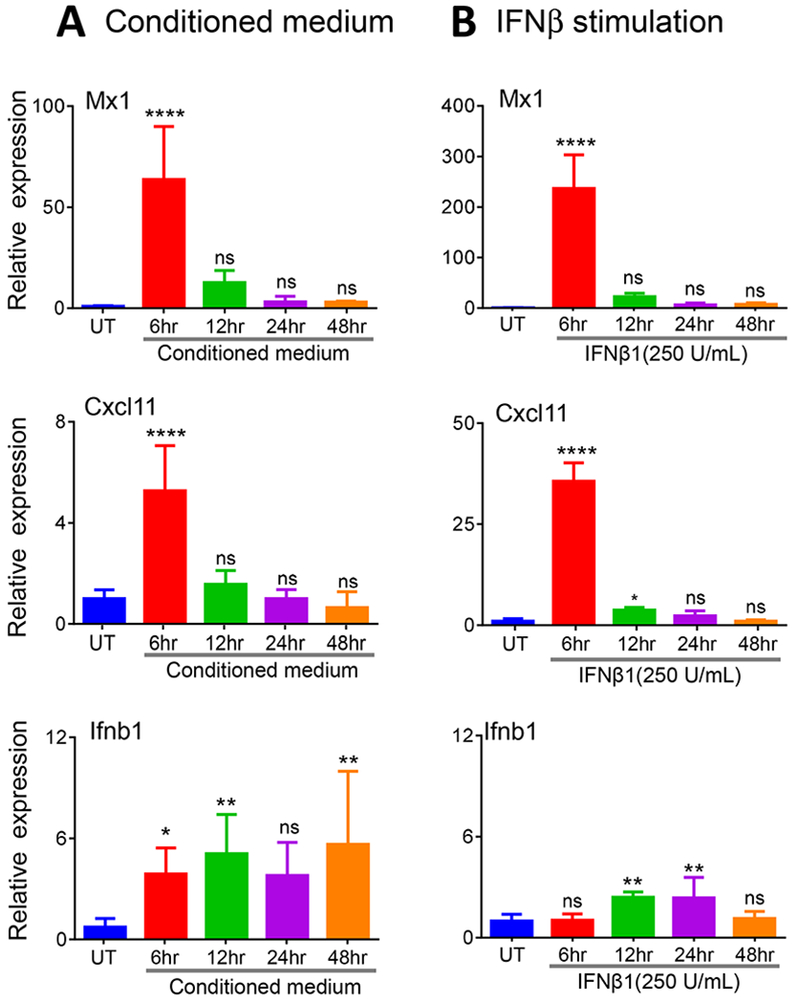
(A) Cultured GL261 cells were treated with 20 μM 4HC for 4 h, followed by incubation with drug-free medium for 44 h. The culture medium was then removed and placed on fresh, drug-naive GL261 cells, followed by incubation for times ranging from 6 to 48 h, as indicated. Gene expression was assayed by RT-qPCR analysis of RNA extracted from the recipient cells. (B) Cultured GL261 cells were treated with recombinant mouse IFNβ (250 U/ml) for the indicated times. Total RNA was extracted and analyzed as described in (A). Data shown are mean fold-change values ± SD for n= 3 replicate cultures, with intra-sample normalization to the 18S RNA level of each sample. NS, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way ANOVA analysis) compared to untreated cells.
3.4. Type-I interferon signaling is required for MEDIC CPA-induced anti-tumor immune responses -
Antibody to IFNAR1 was used to block interferon signaling in GL261 and CT-2A cells. IFNAR1 antibody had no effect on 4HC-stimulated expression of IFNB1 or IFNA4, as expected, but blocked induction of the ISGs in both glioma lines (Fig. 5). Thus, type-I interferon signaling via IFNAR is required for the induction of these ISGs by 4HC. Next, we used this antibody to determine the functional contribution of type-I interferon signaling to MEDIC CPA-induced immune cell infiltration in mice bearing GL261 gliomas. Tumor-bearing mice were given two 6-day cycles of CPA treatment, either alone or in combination with IFNAR1 antibody, which was given on the first day of CPA treatment (day 0), and again on days 2, 6, and 10 of the 12-day treatment schedule. IFNAR1 antibody did not slow the growth of the drug-free tumors, but had a small but significant inhibitory effect on CPA-stimulated tumor regression that was first seen after the second CPA treatment on day 6 (Fig. 6A). IFNAR1 antibody abolished the MEDIC CPA- stimulated increases in expression of MX1 and CXCL11 in the tumor compartment, consistent with its effects in cultured glioma cells. Most importantly, IFNAR1 antibody blocked the increases in tumor-infiltrating immune cells, as shown by RT-qPCR analysis of marker genes for macrophages (CD68), B-cells (B220), natural killer cells (Nkp46), and cytotoxic T-cells (CD8a), and for the cytotoxic effector granzyme B and perforin (Fig. 6B). Thus, immune cell infiltration stimulated by MEDIC CPA is fully dependent on type-I IFN signaling initiated by the tumor cell-centric production of type-I interferons.
Fig. 5 – IFNAR1 antibody inhibits 4HC-stimulated ISG induction in glioma cultures.

Gene expression data are shown for GL261 (A) and CT-2A (B) cells treated for 4 h with 4HC (20 μM) alone or in combination with IFNAR1 antibody (10 μg/ml), and then incubated for a further 44 h without 4HC, and with or without IFNAR1 antibody (arrows along x-axis), as indicated. Data shown are mean fold-change values ± SD for n= 3 replicate cultures, with intra-sample normalization to the levels of 18S RNA. NS, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way ANOVA analysis) for the indicated comparisons.
Fig. 6 – IFNAR1 antibody blocks ISG induction and immune cell infiltration in GL261 tumors.
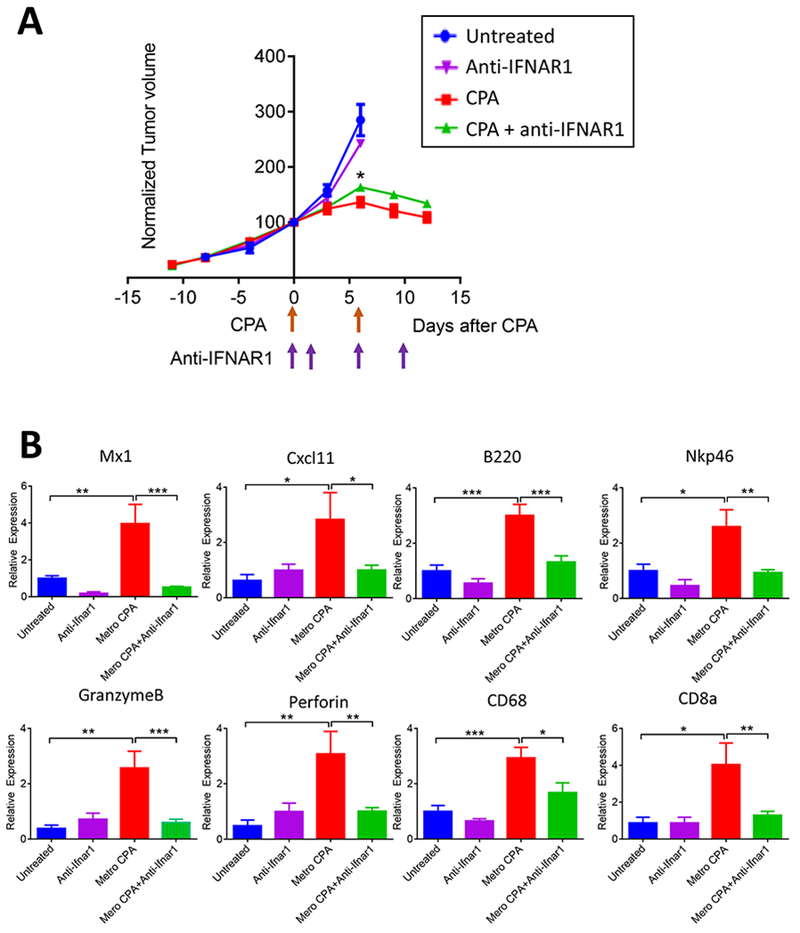
(A) Shown are growth curves of GL261 tumors untreated or treated with 2 cycles of MEDIC CPA (arrows along X-axis), in combination with a IFNAR1-inhibitory antibody or anti-mouse IgG1 isotype control, which were administered on days 0, 2, 6 and 10 (arrows along x-axis). Y-axis values are mean tumor volumes ± SEM for n=5-8 tumors/group, normalized to the volume on the first day of drug treatment (day 0 volume = 100). *p < 0.05 for normalized volume of CPA + IFNAR1 antibody versus CPA + anti-mouse IgG 1 isotype control, evaluated on day 6 (one-tailed t-test). (B) RT-qPCR analysis of immune cell marker genes showing the effects of CPA treatment or IFNAR1 antibody on immune cell infiltration in each of the four tumor groups whose volumes are shown in (A). Data shown are mean ± SEM values for n = 8-10 tumors per group for RNA extracted from tumors excised on day 6 (untreated and IFNAR1 antibody groups) or on day 12 (CPA + anti-mouse IgG1 isotype control, and CPA + IFNAR1 antibody groups). NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001 (one-way ANOVA analysis) for the indicated comparisons.
3.5. CPA-stimulated immunogenicity of GL261 glioma cells -
Tumor cells treated with doxorubicin, an established ICD drug, can vaccinate against a subsequent implantation of the same tumor type [13]. We used such a vaccination assay to investigate whether CPA treatment of GL261 cells, under conditions where the tumor cells produce and secrete type-I interferons, can similarly elicit an anti-GL261 immune response (Fig. 7A). Vaccination assays are considered the gold-standard for evaluating the ability of chemotherapy to cause bona fide ICD [46]. Cultured GL261 cells were treated with 4HC (10 μM 4HC for 24 h) and then changed to fresh complete medium for an additional 48 h. In parallel, as a positive control, GL261 cells were treated with doxorubicin (1 μM for 36 h), which also stimulated type-I IFN production and ISG activation (Fig. S1). The drug-treated tumor cells were injected into the left flank of mice (n=8-9 mice/group). One week later, fresh GL261 cells were implanted into the right flank of the vaccinated mice (Fig. 7A). Tumors formed within 25 days of implantation in 7 of 8 mice receiving PBS in place of the vaccine. In contrast, by day 70 after the tumor cell implantation, tumors were detected in only 4 of 9 mice vaccinated with the 4HC-pretreated GL261 cells (p = 0.0142, by Mantel-Cox test). Similarly, tumors formed in only 3 of 8 mice receiving with the doxorubicin-treated GL261 vaccine (p = 0.0040, by Mantel-Cox test) (Fig. 7B).
Fig. 7 – Drug-treated cultured GL261 cells can vaccinate against live GL261 tumor rechallenge in vivo.
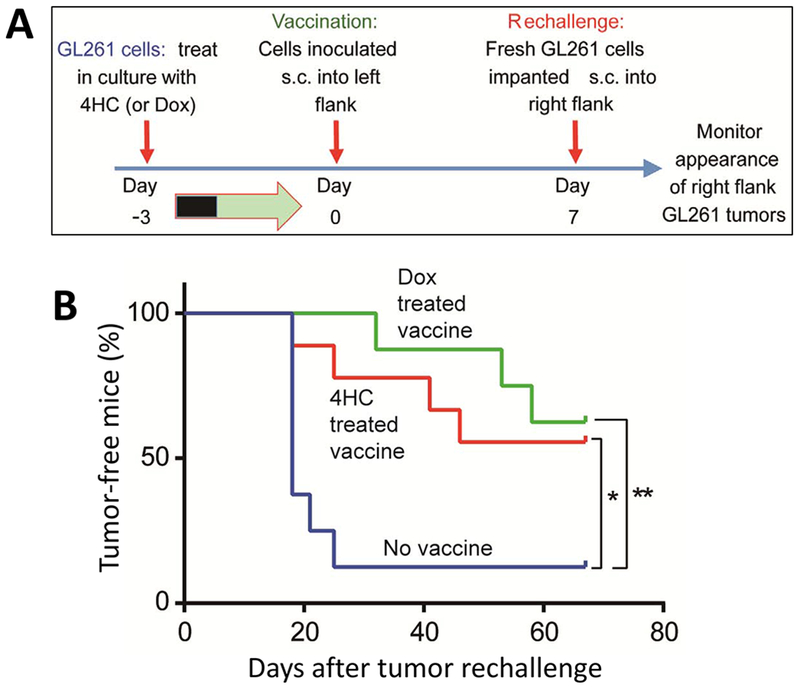
(A) Anti-tumor vaccine experimental design. (B) 4HC or doxorubicin (Dox)-treated GL261 cells were inoculated into untreated mice. Control mice received an equivalent volume of PBS as a vehicle control. Shown is the time course of tumor appearance in mice implanted with untreated GL261 cells 1 week later (day 0). Data is based on n = 8-9 mice per group. Significance of the incidence of tumor-free mice was determined using the Kaplan-Meier method, with *p < 0.05, **p < 0.01, compared to the control vaccine group.
4. Discussion
There is growing evidence that the success of conventional cancer therapies, including chemotherapeutics and radiotherapy, is dependent on type-I IFN signaling. Here, we investigated the role of type-I IFN production and signaling in the striking anti-tumor immune responses that are induced in both syngeneic and xenograft glioma models when CPA is given on a 6-day repeating metronomic treatment schedule, termed MEDIC [7]. We showed that MEDIC CPA induces robust type-I interferon signaling, and that this involves tumor cell autonomous production of type-I IFNs. Furthermore, although type-I IFN production was induced in tumor cells but not in the tumor infiltrating immune cells, both the tumor cells and the tumor-infiltrating immune cells showed strong ISG responses to the increase in IFNs in the tumor microenvironment. Most importantly, we found that type-I IFN signaling is required for MEDIC CPA-stimulated immune cell infiltration in vivo, as revealed by the inhibitory effects of anti-IFNAR1 antibody on both innate and adoptive immune cell infiltration, and on intratumoral formation of their cytotoxic effectors. MEDIC CPA-induced regression in these models is a combined response to immune cell infiltration and direct CPA cytotoxicity [26], and accordingly, we observed a significant, albeit partial decrease in tumor regression when type-I IFN signaling was blocked in the drug-treated tumors.
ICD is a major mechanism responsible for increasing tumor cell immunogenicity by anticancer agents such as doxorubicin [47, 48]. Although many clinical and preclinical studies have shown that CPA can stimulate anti-tumor immune responses, it is not entirely clear whether CPA can actually induce ICD in tumors in vivo [18]. Two gold standard criteria have been used to establish whether a given anticancer drug induces bona fide ICD in tumors in vivo [46]. First, ICD drugs stimulate the recruitment and infiltration of both innate and adaptive immune effector cells to the tumor microenvironment, which may synergize with drug-induced tumor cell cytotoxicity to increase the overall anti-tumor efficacy; and second, drugs that induce ICD show strong vaccine activity in vivo, that is, they elicit an immune response which protects against a subsequent challenge with live, drug naive tumor cells [46]. Supporting the first criteria for ICD, we previously established that MEDIC CPA scheduling ablates large implanted GL261 gliomas by activating both innate and adaptive antitumor immunity [25, 26], a finding confirmed here in a second syngeneic mouse glioma model, CT-2A. Further, we now show that cultured GL261 glioma cells treated with the chemically activated CPA derivative 4HC, under conditions where a robust type-I IFN response is induced, have significant anti-GL261 vaccine activity in vivo. Taken together, our findings establish that CPA induces bona fide ICD in vivo. Type-I IFN production is a critical hallmark of this response, with tumor regression induced by MEDIC CPA being at least partially dependent on this ICD pathway.
Transcriptional profiling has shown that type-I IFN signaling is an upstream regulator of the innate and adaptive immune responses required for MEDIC CPA-induced regression of glioma xenografts [21, 31]. Our finding here that MEDIC CPA directly stimulates glioma cells to produce type-I IFNs is consistent with recent studies of other anticancer therapies, including anthracyclines, radiotherapy and DNA demethylation agents [14]. The precise mechanisms whereby type-I IFN transcription is induced by CPA and other cytotoxic anticancer drugs are largely unknown, but could involve the sensing of cytosolic nucleic acids produced by the dying glioma cells by pathways involving STING (cytosolic DNA sensor) and RIG-1/MAVS (double-stranded RNA sensor) [49, 50]. Anthracyclines can stimulate the formation of double-stranded RNA by dying cancer cells, which may contribute to a type-I IFN-driven immune response [15]. Similarly, radiotherapy activates the binding of the cytoplasmic sensor RIG-I to the small nuclear RNAs U1 and U2 and triggers type-I IFN production and signaling [42]. DNA demethylation agents can also increase cytoplasmic levels of double-stranded RNAs, leading to interferon production and activation of ISGs [51, 52]. Studies of the molecular mechanisms whereby MEDIC CPA activates type-I IFN transcription will need to account for the delay of several days between drug treatment and robust IFN gene activation, seen here both in cell culture and in MEDIC CPA-treated tumors in vivo. Such studies may lead to the discovery of mechanism-based response markers, which could facilitate preclinical and clinical studies designed to identify more efficacious metronomic drug schedules and treatment regimens and advance efforts to devise novel combination chemo-immunotherapies.
Our previous work [26] and the present study establish that MEDIC CPA treatment can induce major regression of syngeneic mouse gliomas by an immune response-dependent mechanism. However, the same CPA treatment regimen effected only modest growth delay and induced minimal or no immune responses in Lewis lung carcinoma and B16F10 melanoma models, despite the intrinsic sensitivity of the latter two tumor cell lines to CPA cytotoxicity [21]. Thus, the immune stimulatory actions of metronomic CPA can differ dramatically across tumor models and/or tumor types and, most likely, individual cancer patients as well. Notably, the MEDIC CPA immune unresponsive tumor models did not show an ISG transcriptional signature [21], suggesting that tumor cell production of type-I IFNs and their downstream ISGs could serve as biomarkers for an effective MEDIC CPA-induced antitumor immune response.
Finally, we note two potential limitations of this study: one is that the in vivo data were obtained in subcutaneous tumor models, which may be more susceptible to immunotherapy as compared to orthotopic tumors [53]. A second limitation is that we did not investigate IFN production by human glioma models. We previously reported that human U251 gliomas show a strong innate immune response to MEDIC CPA that enhances tumor regression in mouse xenografts [25, 31]; however, these models are likely less informative due to the absence of the adaptive immune system, which plays a critical role in MEDIC CPA-induced tumor ablation, as seen in syngeneic mouse models [26].
In conclusion, we show that MEDIC CPA treatment directly stimulates tumor cells but not tumor-infiltrating host immune cells to produce type-I IFNs in implanted gliomas. This tumor cell autonomous production of type-I IFNs, in turn, activates local ISG production by both tumor cells and host immune cells within the tumor microenvironment, and is sufficient to vaccinate mice from fresh glioma cell challenge. Finally, inhibition of type-I IFN signaling in CPA-treated gliomas in vivo resulted in substantial inhibition of immune cell infiltration and decreased the overall anti-tumor immune response to MEDIC CPA therapy. These findings highlight a novel mechanism for the antitumor actions of metronomic chemotherapy using MEDIC schedules.
Supplementary Material
Fig. S1 – Doxorubicin induction of type-I interferon signaling in cultured GL261 cells. GL261 cells were treated with PBS or doxorubicin (1 μM for 4 h), and RNA was extracted 4, 12, 24, 36, 48 and 72 h after beginning doxorubicin treatment, as indicated, followed by assay for each of the indicated ISGs by RT-qPCR. Data shown are mean fold-change values ± SD for n = 3 replicate cultures, with intra-sample normalization to the 18S RNA. NS, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001 (one-way ANOVA analysis) compared to PBS treated cells (control).
Highlights:
Cyclophosphamide induces tumor cell autonomous type-I interferon signaling
Abrogation of interferon signaling blocks drug-induced immune cell infiltration
Dying glioma cells producing interferons vaccinate against drug-naïve gliomas
Acknowledgments
Funding: This work was supported in part by the National Institutes of Health [grant number CA49248, to DJW] and by the National Natural Science Foundation of China [grant number 81872893, to BD].
Abbreviations:
- ANOVA
one-way analysis of variance
- CPA
cyclophosphamide
- ICD
immunogenic cell death
- IFN
interferon
- ISG
IFN-stimulated gene
- IFNAR1
interferon alpha and beta receptor subunit 1
- MEDIC
medium-dose, intermittent chemotherapy
- RT-qPCR
reverse transcription-quantitative polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement:
The authors declare no conflicts of interest
Financial disclosure: The authors have nothing to disclose.
References
- [1].Shurin MR, Naiditch H, Gutkin DW, Umansky V, Shurin GV, ChemoImmunoModulation: immune regulation by the antineoplastic chemotherapeutic agents, Curr Med Chem, 19 (2012) 1792–1803. [DOI] [PubMed] [Google Scholar]
- [2].Zitvogel L, Kepp O, Kroemer G, Immune parameters affecting the efficacy of chemotherapeutic regimens, Nat Rev Clin Oncol, 8 (2011) 151–160. [DOI] [PubMed] [Google Scholar]
- [3].Rolle CE, Sengupta S, Lesniak MS, Mechanisms of immune evasion by gliomas, Adv Exp Med Biol, 746 (2012) 53–76. [DOI] [PubMed] [Google Scholar]
- [4].Wainwright DA, Dey M, Chang A, Lesniak MS, Targeting Tregs in Malignant Brain Cancer: Overcoming IDO, Front Immunol, 4 (2013) 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Metivier D, Pichard E, Aucouturier P, Pierron G, Garrido C, Zitvogel L, Kroemer G, Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death, The Journal of experimental medicine, 202 (2005) 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Metivier D, Galluzzi L, Perfettini JL, Zitvogel L, Kroemer G, Molecular mechanisms of ATP secretion during immunogenic cell death, Cell Death Differ, 21 (2014) 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wu J, Waxman DJ, Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy, Cancer Lett, 419 (2018) 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Inoue H, Tani K, Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments, Cell Death Differ, 21 (2014) 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L, Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics, Oncoimmunology, 6 (2017) e1386829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li X, The inducers of immunogenic cell death for tumor immunotherapy, Tumori, 104 (2018) 1–8. [DOI] [PubMed] [Google Scholar]
- [11].Venkateswaran K, Verma A, Bhatt AN, Shrivastava A, Manda K, Raj HG, Prasad A, Len C, Parmar VS, Dwarakanath BS, Emerging Roles of Calreticulin in Cancer: Implications for Therapy, Curr Protein Pept Sci, 19 (2018) 344–357. [DOI] [PubMed] [Google Scholar]
- [12].Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G, Calreticulin exposure dictates the immunogenicity of cancer cell death, Nat Med, 13 (2007) 54–61. [DOI] [PubMed] [Google Scholar]
- [13].Bianchi ME, Crippa MP, Manfredi AA, Mezzapelle R, Rovere Querini P, Venereau E, High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair, Immunol Rev, 280 (2017) 74–82. [DOI] [PubMed] [Google Scholar]
- [14].Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G, Type I interferons in anticancer immunity, Nat Rev Immunol, 15 (2015) 405–414. [DOI] [PubMed] [Google Scholar]
- [15].Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, Fend L, Hannani D, Aymeric L, Ma Y, Niso-Santano M, Kepp O, Schultze JL, Tuting T, Belardelli F, Bracci L, La Sorsa V, Ziccheddu G, Sestili P, Urbani F, Delorenzi M, Lacroix-Triki M, Quidville V, Conforti R, Spano JP, Pusztai L, Poirier-Colame V, Delaloge S, Penault-Llorca F, Ladoire S, Arnould L, Cyrta J, Dessoliers MC, Eggermont A, Bianchi ME, Pittet M, Engblom C, Pfirschke C, Preville X, Uze G, Schreiber RD, Chow MT, Smyth MJ, Proietti E, Andre F,Kroemer G, Zitvogel L, Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy, Nat Med, 20 (2014) 1301–1309. [DOI] [PubMed] [Google Scholar]
- [16].Wang YJ, Fletcher R, Yu J, Zhang L, Immunogenic effects of chemotherapy-induced tumor cell death, Genes Dis, 5 (2018) 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Emadi A, Jones RJ, Brodsky RA, Cyclophosphamide and cancer: golden anniversary, Nat Rev Clin Oncol, 6 (2009) 638–647. [DOI] [PubMed] [Google Scholar]
- [18].Ahlmann M, Hempel G, The effect of cyclophosphamide on the immune system: implications for clinical cancer therapy, Cancer Chemother Pharmacol, 78 (2016) 661–671. [DOI] [PubMed] [Google Scholar]
- [19].Chang TK, Weber GF, Crespi CL, Waxman DJ, Differential activation of cyclophosphamide and ifosphamide by cytochromes P-450 2B and 3A in human liver microsomes, Cancer research, 53 (1993) 5629–5637. [PubMed] [Google Scholar]
- [20].Ozer H, Cowens JW, Colvin M, Nussbaum-Blumenson A, Sheedy D, In vitro effects of 4-hydroperoxycyclophosphamide on human immunoregulatory T subset function. I. Selective effects on lymphocyte function in T-B cell collaboration, The Journal of experimental medicine, 155 (1982) 276–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wu J, Jordan M, Waxman DJ, Metronomic cyclophosphamide activation of anti-tumor immunity: tumor model, mouse host, and drug schedule dependence of gene responses and their upstream regulators, BMC cancer, 16 (2016) 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Matar P, Rozados VR, Gervasoni SI, Scharovsky GO, Th2/Th1 switch induced by a single low dose of cyclophosphamide in a rat metastatic lymphoma model, Cancer Immunol Immunother, 50 (2002) 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang Z, Till B, Gao Q, Chemotherapeutic agent-mediated elimination of myeloid-derived suppressor cells, Oncoimmunology, 6 (2017) e1331807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, Sautes-Fridman C, Fucikova J, Galon J, Spisek R, Tartour E, Zitvogel L, Kroemer G, Galluzzi L, Trial Watch: Immunogenic cell death inducers for anticancer chemotherapy, Oncoimmunology, 4 (2015) e1008866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Doloff JC, Waxman DJ, VEGF receptor inhibitors block the ability of metronomically dosed cyclophosphamide to activate innate immunity-induced tumor regression, Cancer research, 72 (2012) 1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wu J, Waxman DJ, Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8(+) T-cell responses and immune memory, Oncoimmunology, 4 (2015) e1005521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ge Y, Domschke C, Stoiber N, Schott S, Heil J, Rom J, Blumenstein M, Thum J, Sohn C, Schneeweiss A, Beckhove P, Schuetz F, Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome, Cancer Immunol Immunother, 61 (2012) 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schiavoni G, Mattei F, Di Pucchio T, Santini SM, Bracci L, Belardelli F, Proietti E, Cyclophosphamide induces type I interferon and augments the number of CD44(hi) T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer, Blood, 95 (2000) 2024–2030. [PubMed] [Google Scholar]
- [29].Chen CS, Doloff JC, Waxman DJ, Intermittent metronomic drug schedule is essential for activating antitumor innate immunity and tumor xenograft regression, Neoplasia, 16 (2014) 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wu J, Waxman DJ, Metronomic cyclophosphamide schedule-dependence of innate immune cell recruitment and tumor regression in an implanted glioma model, Cancer Lett, 353 (2014) 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Doloff JC, Waxman DJ, Transcriptional profiling provides insights into metronomic cyclophosphamide-activated, innate immune-dependent regression of brain tumor xenografts, BMC cancer, 15 (2015) 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Moschella F, Torelli GF, Valentini M, Urbani F, Buccione C, Petrucci MT, Natalino F, Belardelli F, Foa R, Proietti E, Cyclophosphamide induces a type I interferon-associated sterile inflammatory response signature in cancer patients’ blood cells: implications for cancer chemoimmunotherapy, Clin Cancer Res, 19 (2013) 4249–4261. [DOI] [PubMed] [Google Scholar]
- [33].Trinchieri G, Type I interferon: friend or foe?, The Journal of experimental medicine, 207 (2010) 2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kaur S, Platanias LC, IFN-beta-specific signaling via a unique IFNAR1 interaction, Nat Immunol, 14 (2013) 884–885. [DOI] [PubMed] [Google Scholar]
- [35].Bracci L, Sistigu A, Proietti E, Moschella F, The added value of type I interferons to cytotoxic treatments of cancer, Cytokine Growth Factor Rev, 36 (2017) 89–97. [DOI] [PubMed] [Google Scholar]
- [36].Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I, Direct effects of type I interferons on cells of the immune system, Clin Cancer Res, 17 (2011) 2619–2627. [DOI] [PubMed] [Google Scholar]
- [37].Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, Sanchez M, Lorenzi S, D’Urso MT, Belardelli F, Belardelli F, Gabriele L, Proietti E, Bracci L Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis, Cancer research, 71 (2011) 768–778. [DOI] [PubMed] [Google Scholar]
- [38].Mokyr MB, Place AT, Artwohl JE, Valli VE, Importance of signaling via the IFN-alpha/beta receptor on host cells for the realization of the therapeutic benefits of cyclophosphamide for mice bearing a large MOPC-315 tumor, Cancer Immunol Immunother, 55 (2006) 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Seyfried TN, el-Abbadi M, Roy ML, Ganglioside distribution in murine neural tumors, Mol Chem Neuropathol, 17 (1992) 147–167. [DOI] [PubMed] [Google Scholar]
- [40].Pass GJ, Carrie D, Boylan M, Lorimore S, Wright E, Houston B, Henderson CJ, Wolf CR, Role of hepatic cytochrome p450s in the pharmacokinetics and toxicity of cyclophosphamide: studies with the hepatic cytochrome p450 reductase null mouse, Cancer research, 65 (2005) 4211–4217. [DOI] [PubMed] [Google Scholar]
- [41].Ma J, Chen CS, Blute T, Waxman DJ, Antiangiogenesis enhances intratumoral drug retention, Cancer research, 71 (2011) 2675–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ranoa DR, Parekh AD, Pitroda SP, Huang X, Darga T, Wong AC, Huang L, Andrade J, Staley JP, Satoh T, Akira S, Weichselbaum RR, Khodarev NN, Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs, Oncotarget, 7 (2016) 26496–26515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Paste M, Huez G, Kruys V, Deadenylation of interferon-beta mRNA is mediated by both the AU-rich element in the 3’-untranslated region and an instability sequence in the coding region, Eur J Biochem, 270 (2003) 1590–1597. [DOI] [PubMed] [Google Scholar]
- [44].Abe K, Ishigami T, Shyu AB, Ohno S, Umemura S, Yamashita A, Analysis of interferon-beta mRNA stability control after poly(I:C) stimulation using RNA metabolic labeling by ethynyluridine, Biochem Biophys Res Commun, 428 (2012) 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kawai T, Akira S, Toll-like receptor and RIG-I-like receptor signaling, Ann N Y Acad Sci, 1143 (2008) 1–20. [DOI] [PubMed] [Google Scholar]
- [46].Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, Bracci L, Breckpot K, Brough D, Buque A, Castro MG, Cirone M, Colombo MI, Cremer I, Demaria S, Dini L, Eliopoulos AG, Faggioni A, Formenti SC, Fucikova J, Gabriele L, Gaipl US, Galon J, Garg A, Ghiringhelli F, Giese NA, Guo ZS, Hemminki A, Herrmann M, Hodge JW, Holdenrieder S, Honeychurch J, Hu HM, Huang X, Illidge TM, Kono K, Korbelik M, Krysko DV, Loi S, Lowenstein PR, Lugli E, Ma Y, Madeo F, Manfredi AA, Martins I, Mavilio D, Menger L, Merendino N, Michaud M, Mignot G, Mossman KL, Multhoff G, Oehler R, Palombo F, Panaretakis T, Pol J, Proietti E, Ricci JE, Riganti C, Rovere-Querini P, Rubartelli A, Sistigu A, Smyth MJ, Sonnemann J, Spisek R, Stagg J, Sukkurwala AQ, Tartour E, Thorburn A, Thorne SH, Vandenabeele P, Velotti F, Workenhe ST, Yang H, Zong WX, Zitvogel L, Kroemer G, Galluzzi L, Consensus guidelines for the detection of immunogenic cell death, Oncoimmunology, 3 (2014) e955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kroemer G, Galluzzi L, Kepp O, Zitvogel L, Immunogenic cell death in cancer therapy, Annu Rev Immunol, 31 (2013) 51–72. [DOI] [PubMed] [Google Scholar]
- [48].Kepp O, Tesniere A, Schlemmer F, Michaud M, Senovilla L, Zitvogel L, Kroemer G, Immunogenic cell death modalities and their impact on cancer treatment, Apoptosis, 14 (2009) 364–375. [DOI] [PubMed] [Google Scholar]
- [49].Bose D, cGAS/STING Pathway in Cancer: Jekyll and Hyde Story of Cancer Immune Response, Int J Mol Sci, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Iurescia S, Fioretti D, Rinaldi M, Targeting Cytosolic Nucleic Acid-Sensing Pathways for Cancer Immunotherapies, Front Immunol, 9 (2018) 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, Makarov V, Budhu S, Slamon DJ, Wolchok JD, Pardoll DM, Beckmann MW, Zahnow CA, Merghoub T, Chan TA, Baylin SB, Strick R, Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses, Cell, 162 (2015) 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, O’Brien C, De Carvalho DD, DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts, Cell, 162 (2015) 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li HY, McSharry M, Bullock B, Nguyen TT, Kwak J, Poczobutt JM, Sippel TR, Heasley LE, Weiser-Evans MC, Clambey ET, Nemenoff RA, The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade, Cancer immunology research, 5 (2017) 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 – Doxorubicin induction of type-I interferon signaling in cultured GL261 cells. GL261 cells were treated with PBS or doxorubicin (1 μM for 4 h), and RNA was extracted 4, 12, 24, 36, 48 and 72 h after beginning doxorubicin treatment, as indicated, followed by assay for each of the indicated ISGs by RT-qPCR. Data shown are mean fold-change values ± SD for n = 3 replicate cultures, with intra-sample normalization to the 18S RNA. NS, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001 (one-way ANOVA analysis) compared to PBS treated cells (control).