

Abstract

The redox noninnocence of the TAML scaffold in cobalt-TAML (tetra-amido macrocyclic ligand) complexes has been under debate since 2006. In this work, we demonstrate with a variety of spectroscopic measurements that the TAML backbone in the anionic complex [CoIII(TAMLred)]– is truly redox noninnocent and that one-electron oxidation affords [CoIII(TAMLsq)]. Multireference (CASSCF) calculations show that the electronic structure of [CoIII(TAMLsq)] is best described as an intermediate spin (S = 1) cobalt(III) center that is antiferromagnetically coupled to a ligand-centered radical, affording an overall doublet (S = 1/2) ground-state. Reaction of the cobalt(III)-TAML complexes with PhINNs as a nitrene precursor leads to TAML-centered oxidation and produces nitrene radical complexes without oxidation of the metal ion. The ligand redox state (TAMLred or TAMLsq) determines whether mono- or bis-nitrene radical complexes are formed. Reaction of [CoIII(TAMLsq)] or [CoIII(TAMLred)]– with PhINNs results in the formation of [CoIII(TAMLq)(N•Ns)] and [CoIII(TAMLq)(N•Ns)2]–, respectively. Herein, ligand-to-substrate single-electron transfer results in one-electron-reduced Fischer-type nitrene radicals (N•Ns–) that are intermediates in catalytic nitrene transfer to styrene. These nitrene radical species were characterized by EPR, XANES, and UV–vis spectroscopy, high-resolution mass spectrometry, magnetic moment measurements, and supporting CASSCF calculations.
Introduction
The use of base metals and redox noninnocent (or redox-active) ligands in radical-type carbene, oxo, and nitrene transfer reactions has evolved as a powerful tool for the direct functionalization of (unactivated) C–H bonds and olefins.1 The functionalized products of these reactions are motifs in pharmaceuticals and agrochemicals and are therefore highly valued.2 N-group transfer reactivity is an efficient way to afford the direct synthesis of secondary amines and aziridines, of which the synthesis otherwise typically requires harsh reaction conditions or multiple steps.3 Generation of the essential catalytic metal-nitrene intermediates has been achieved with second- and third-row transition metals (Ru,4 Rh,5 Pd,6 Ag7, and Au8) as well as more abundant base metals (Mn,9 Fe,10 Co,11 Ni12, and Cu13).
Our group, in collaboration with the Zhang group, has studied the formation and reactivity of nitrene adducts of cobalt(II)-porphyrin complexes, which are competent catalysts for a range of (enantioselective) amination and aziridination reactions.11a,11d−11j,14 The mononitrene species generated on cobalt upon reaction with an organic azide is most accurately described as a one-electron-reduced Fischer-type nitrene radical.14b This interesting electronic structure is the result of metal-to-substrate single-electron transfer (SET), wherein cobalt is oxidized from CoII to CoIII and the nitrene is reduced by one electron to produce a nitrene radical (N•R–) complex with single-electron population of the π symmetric Co–N antibonding orbital. Interestingly, the reaction of cobalt(II)-porphyrins with iminoiodinanes (PhINNs, Ns = nosyl) led to the formation of bis-nitrene radical species with two one-electron-reduced Fischer-type nitrenes, wherein the second nitrene is reduced via ligand-to-substrate SET. Intrigued by these nitrene-transfer catalysts, we became interested in the possibility of nitrene radical formation on square planar cobalt(III) platforms involving solely ligand-to-substrate single-electron transfer15 by studying systems containing redox-active ligands for which metal-to-substrate SET is difficult or even impossible.
When searching for suitable redox-active macrocyclic tetradentate ligand platforms that enforce a square planar coordination geometry around cobalt in an oxidation state higher than +II, we decided to investigate the tetra-amido macrocyclic ligand (TAML) platform designed by Collins’ group.16 The general structure of a TAML that met the aforementioned requirements is depicted in Scheme 1. Moreover, the potential redox noninnocence of TAML and related o-phenylenedicarboxamido complexes has been proposed in the literature, and for clarity, we will follow the nomenclature as presented in Scheme 1 for the fully reduced tetra-anion (red), mono-oxidized trianionic ligand-centered radical (sq), and fully oxidized dianion (q).16,17
Scheme 1. General Structure of the TAML Scaffold and the Potential Redox Noninnocence of the Backbone.

X1 = Cl, H, NO2, OMe. X2 = Cl, H. R = Et, Me, F.16
Iron complexes of these TAML activators have found widespread use in oxidation chemistry, and TAML complexes with Cr, Mn, Fe, Co, Ni and Cu have been reported with many variations of the TAML scaffold.16,18 Interestingly, ligand-centered oxidation of an [FeV(TAMLred)(NTs)]− complex was shown to afford [FeV(TAMLsq)(NTs)], which is a more active nitrene transfer species toward activated C–H bonds (bond dissociation energy between 75 and 80 kcal mol–1) and thioanisole than the reduced analogue.19 A similar trend was observed for a manganese-imido complex, wherein [MnV(TAMLred)(NMes)]− (Mes = mesityl) proved to be unreactive and metal-centered oxidized complex [MnVI(TAMLred)(NMes)] could be used for hydrogen atom transfer reactions and nitrene transfer to thioanisole.20 Apparently, the redox activity of the TAML ligand varies from complex to complex, depending on the metal and other ligands, and both metal- and ligand-centered redox processes can be used to influence the nitrene-transfer reactivity.
Specific [CoIII(TAMLred)]− complexes21 have been used for electrochemical water oxidation22,23 and oxygen reduction,24 cycloaddition of CO2 to epoxides,25 electrochemical sensing of H2O2,26 oxo transfer to C–H bonds,27 and electron-transfer reactions.28 However, to the best of our knowledge, no nitrene transfer reactions or stoichiometric reactions leading to the formation of Co(TAML)-based imido- or nitrene-complexes have been reported to date. Moreover, contrary to chemistry with iron, the existence of TAML-centered redox processes in cobalt complexes is still under debate (Figure 1).
Figure 1.

Interpretation of the ligand (non)innocence in cobalt-TAML complexes in chronological order. HFI = hyperfine interaction.
Collins et al.21 reported the synthesis and characterization of an anionic [CoIII(TAMLred)]− complex with a diamidophenyl backbone in 1991. The anionic parent complex was characterized as a triplet with an S = 1 Co center and a fully reduced o-phenylenedicarboxamido ligand. The oxidation of this complex afforded a neutral S = 1/2 system for which crystallographic bond metrics indicated single-electron oxidation of the ligand and electron paramagnetic resonance (EPR) data hinted at a cobalt-centered radical (Figure 1). This data was interpreted in 1998 as corresponding to an S = 1 cobalt(III) center antiferromagnetically coupled to a ligand-centered radical ([CoIII(TAMLsq)]).28 Ghosh et al.29 reported an elaborate density functional theory (DFT) study on the ligand noninnocence of multiple variations of the TAML backbone and suggested that the electronic structure of [CoIII(TAMLsq)] is better described as [CoIV(TAMLred)] (Figure 1). Their assignment was based on the Mulliken spin density, which was solely localized on cobalt. Collins and co-workers18 critically reinterpreted these spin densities as being evidence of an S = 1 CoIII center. It should be noted that multireference post-Hartree–Fock methods were not accessible at the time, and possible broken-symmetry solutions were apparently not explored. As such, optional antiferromagnetic coupling between an S = 1 Co center and a ligand-centered radical could have remained hidden in the applied DFT calculations.
Innocent behavior of the TAML scaffold was claimed in an electrochemical study reported in 201430 as well as in the characterization of Lewis acid-stabilized oxo-complex [CoIV(TAMLred)(O)].27 The TAMLred and CoIV oxidation states in an Sc3+-bound [CoIV(TAMLred)(O)]2– complex were based on UV–vis, EPR, XANES (X-ray absorption near edge spectroscopy), and EXAFS (extended X-ray absorption fine structure) studies, in combination with DFT-calculated Mulliken spin densities.27 On the contrary, TAML-centered redox activity in [CoIII(TAMLq)(OH)] was claimed in 2018 on the basis of UV–vis, EPR, and XPS (X-ray photoelectron spectroscopy) studies.23
Given (i) the contrasting descriptions of ligand and cobalt oxidation states in [Co(TAML)] complexes, (ii) our interest in generating cobalt-nitrene radical intermediates via ligand-to-substrate SET, and (iii) the previous characterization of [Fe(TAML)(imido)], [Mn(TAML)(imido)], and [Co(TAML)(oxo)] complexes, we set out to answer the following research questions:
Is the ligand in [Co(TAML)] complexes redox noninnocent, and can the different assignments in the literature be reconciled? (See Figure 2A.)
Figure 2.

(A) Electronic structure questions regarding the redox noninnocence of the TAML scaffold. (B) Possible electronic structures of the targeted nitrene species. The ligand color coding is as presented in Scheme 1. (C) Main findings with the assignment of the TAML scaffold being redox noninnocent in the coordination sphere of cobalt and its influence on nitrene (radical) formation.
Can the [Co(TAML)] platform be used to generate (catalytically competent) cobalt-nitrene (radical) species, and what is the influence of the (ligand) oxidation state on the (electronic) structure of the targeted nitrene (radical) species? (See Figure 2B.)
In case the TAML ligand platform is indeed redox-active, can we use this feature for ligand-to-substrate SET to produce nitrene radical species at square planar cobalt(III) species? (See Figure 2B).
The main findings of the investigations presented in this article are summarized in Figure 2C.
Results and Discussion
Ligand-Centered Oxidation of [CoIII(TAMLred)]−
The parent [CoIII(TAMLred)]– complex was obtained according to an adapted literature procedure.21,31 After a five-step synthesis procedure to obtain the ligand (TAMLH4), coordination of CoII to the fully deprotonated ligand (generated using n-BuLi) and aerobic oxidation afforded Li[CoIII(TAMLred)] or PPh4[CoIII(TAMLred)] after salt metathesis with PPh4Cl (Scheme 2). Crystals suitable for singe crystal X-ray diffraction (XRD) analysis of TAMLH4 and PPh4[CoIII(TAMLred)] were grown by the vapor diffusion of pentane into concentrated THF solutions of the ligand or complex, respectively. The solid state structure of PPh4[CoIII(TAMLred)] displays a square planar geometry around cobalt and a noncoordinating THF molecule in the crystal lattice. As expected and in accordance with the literature,21 an analysis of the crystallographic bond metrics (Supporting Information) of the diamidophenyl ring in TAMLH4 and PPh4[CoIII(TAMLred)] supports the preservation of aromaticity upon coordination to cobalt, with the ligand being fully reduced ((TAMLred)4–) and the metal adopting the CoIII oxidation state. The effective magnetic moment of PPh4[CoIII(TAMLred)], as determined via the Evans’ method,32 indicated a triplet (S = 1) ground state (μeff = 2.94μB). This is in accordance with the literature and is expected for an intermediate-spin CoIII center with two parallel metal-centered unpaired electrons.21 The DFT-optimized structure of [CoIII(TAMLred)]– in the triplet state at the BP86/def2-TZVP level of theory is consistent with these observations, and the calculated bond metrics closely match the experimental bond lengths (SI).
Scheme 2. (A) Formation of Li[CoIII(TAMLred)] and PPh4[CoIII(TAMLred)] from TAMLH4, with Thermal Displacement Ellipsoid Plots (50% Probability Level) of TAMLH4 (B) and PPh4[CoIII(TAMLred)] (C). H Atoms (Except for NH) and Lattice Solvent (THF for PPh4[CoIII(TAMLred)]) Removed for Clarity.
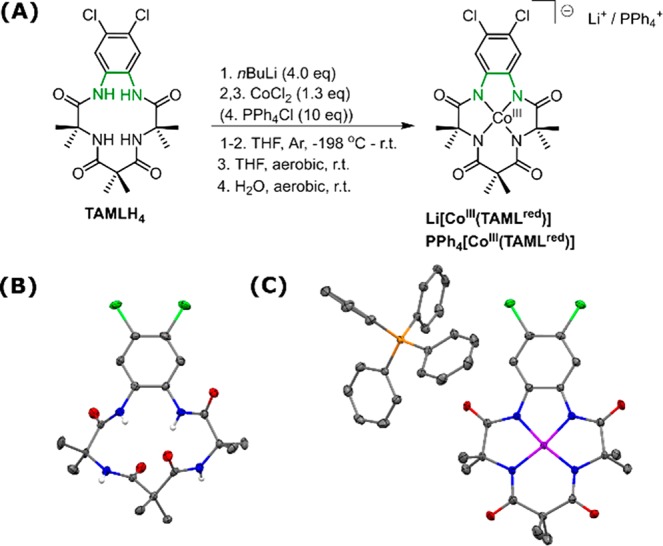
The electrochemical oxidation of PPh4[CoIII(TAMLred)] in CH2Cl2 (Scheme 3A) using cyclic voltammetry displays three fully reversible redox events at E1/2 = −1.18, +0.53, and +1.13 V vs Fc+/0, which are attributed to metal-centered reduction (CoIII/II) and two ligand-centered oxidations (TAMLred/sq and TAMLsq/q) respectively (vide infra).33 UV–vis spectroelectrochemical (UV–vis–SEC) monitoring of the oxidation event at +0.53 V vs Fc+/0 shows the disappearance of PPh4[CoIII(TAMLred)] (λmax = 510 nm) and the concomitant appearance of the characteristic absorption band of [CoIII(TAMLsq)] (λmax = 623 nm) with an isosbestic point at 545 nm (Scheme 3B).21,23,34,35 For clarity we already assigned the electronic structure of [CoIII(TAMLsq)] in the following descriptions. In the following sections we will further elaborate on the measurements and calculations leading to this assignment.
Scheme 3. (A) Cyclic Voltammogram of PPh4[CoIII(TAMLred)] in DCM (Details in the SI), (B) UV–vis–SEC Oxidation of PPh4[CoIII(TAMLred)] in DCM (Details in the SI), (C) Oxidation of [CoIII(TAMLred)]− to [CoIII(TAMLsq)] with (Thi)BF4, and (D) UV–vis Titration of PPh4[CoIII(TAMLred)] in DCM (0.15 mM) with Increasing Amounts of (Thi)BF4.
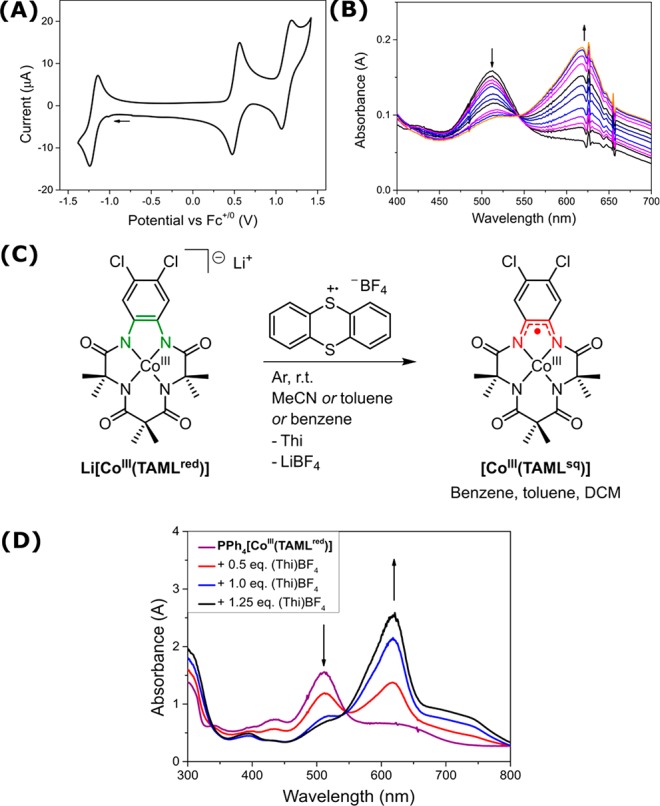
Chemical oxidation of TAML complexes with ceric ammonium nitrate ((NH4)2[Ce(NO3)6]) typically requires excess oxidant and large volumes of solvent to extract the product.21 For purple-colored PPh4[CoIII(TAMLred)] and Li[CoIII(TAMLred)], oxidation with a stoichiometric amount of thianthrenium tetrafluoroborate ((Thi)BF4) (Eo1/2 = 0.86 V vs Fc+/0)36 cleanly afforded the blue-colored [CoIII(TAMLsq)] complex (Scheme 3C). A UV–vis titration gave data identical to that obtained from UV–vis–SEC monitoring of the oxidation event at +0.53 V vs Fc+/0 (Scheme 3D).
The effective magnetic moment of [CoIII(TAMLsq)] (μeff = 1.88μB, Evans’ method) was found to be consistent with an overall net doublet (S = 1/2) ground state. Room-temperature (r.t.) X-band EPR studies in CH2Cl2 or toluene reveal a signal characteristic of a net S = 1/2 system with unpaired electron density on cobalt (giso = 2.22) (Figure 3A). EPR measurements at 10 K in toluene glass showed a rhombic signal with gx = 2.03, gy = 2.16, gz = 2.54, and partially unresolved cobalt hyperfine interactions (HFIs) (Figure 3B). The inclusion of 59Co (I = 7/2 nucleus) HFIs (ACox = 5.0 MHz, ACoy = 50.0 MHz, ACoz = 20.0 MHz) is necessary for an accurate simulation of the spectrum. The DFT-calculated cobalt HFIs are overestimated (B3LYP/def2-TZVP: gx = 2.04, gy = 2.25, gz = 2.26, ACox = 166.3 MHz, ACoy = 199.8 MHz, and ACoz = 641.3 MHz), which we attribute to the erroneous description of multireference systems with DFT methods (vide infra). Interestingly, the isotropic X-band EPR spectrum measured in MeCN (Figure 3C) revealed an eight-line pattern at giso = 2.00 attributed to hyperfine coupling with cobalt (ACoiso = 36.0 MHz) in [CoIII(TAMLsq)(MeCN)], which is in excellent agreement with the DFT-calculated parameters (B3LYP/def2-TZVP: giso = 2.00, ACoiso = 34.2 MHz). Notably, this species has a single-reference doublet electronic structure with the unpaired electron residing in a cobalt-ligand π* orbital (strongly delocalized over cobalt and the ligand; see the SI).
Figure 3.
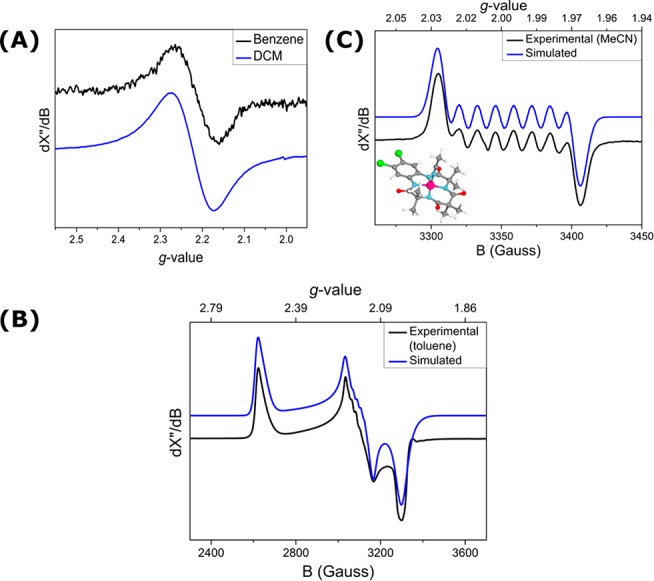
(A) X-band EPR spectrum of [CoIII(TAMLsq)] in benzene at r.t. (black line: microwave freq. 9.390167 GHz, mod. amp. 4 G, and power 2.518 mW) and CH2Cl2 (blue line: microwave freq. 9.3966 GHz, mod. amp. 5 G, and power 2.000 mW) with giso = 2.22. (B) Experimental (black) and simulated (blue) X-band EPR spectrum of [CoIII(TAMLsq)] in toluene at 10 K. Microwave freq. 9.365984 GHz, mod. amp. 4 G, and power 2.000 mW. Simulation parameters: gx = 2.03, gy = 2.16, gz = 2.54, ACox = 5.0 MHz, ACoy = 50.0 MHz, ACoz = 20.0 MHz, linear A strain −0.018 (z direction), and quadratic A strain −18 (x direction) and −2 (y direction). (C) Experimental (black) and simulated (blue) X-band EPR spectra of [CoIII(TAMLsq)(MeCN)] at r.t. in MeCN and the DFT (BP86/def2-TZVP/disp3)-optimized structure. Microwave freq. 9.3886 GHz, mod. amp. 3 G, and power 0.7962 mW. Simulated (calculated; B3LYP/def2-TZVP)) parameters: giso = 2.00 (2.00) and ACoiso = 36.0 (34.2) MHz.
The cobalt oxidation state of the four-coordinate complexes was further investigated using Co K-edge X-ray absorption near edge spectroscopy analysis. The Co K-edge XANES spectra of PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)] in toluene are compared in Figure 4. The edge position was 7721 eV for both complexes. Both spectra are identical, which is in line with the same oxidation state (+III) and similar coordination geometry of cobalt in the two complexes. The +III oxidation state of cobalt was already found in PPh4[CoIII(TAMLred)] (according to XRD-derived bond metric analysis, vide supra), and the observed edge position is equal to a related [CoIII(TAML)]− complex.27 The shoulder at approximately 7715 eV in the Co K-edge XANES spectra is typical for square planar Co complexes, including square planar Co-porphyrin complexes and a related cobalt-TAML complex.14b,27 The main edge feature arises primarily from 1s → 4p electron transitions, whereas the feature at 7715 eV is commonly assigned to 1s → 4pz and ligand-to-metal charge transfer (LMCT) shakedown transitions.37
Figure 4.
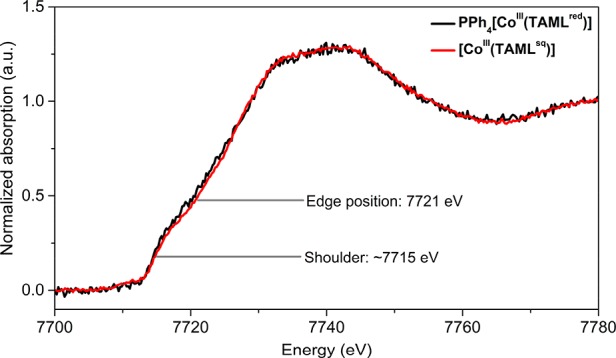
Co K-edge XANES analysis of PPh4[CoIII(TAMLred)] (black) and [CoIII(TAMLsq)] (red) in toluene.
In agreement with previous studies,27,29 DFT calculations with various GGA and hybrid functionals (BP86, B3LYP, PBE, and OPBE; see the SI for details) gave unsatisfactory results for the Co(TAML)-type complexes under investigation. An illustrative example of the problem encountered with DFT is found in the challenging description of the net-doublet ground state of the [CoIII(TAMLsq)] complex. Distinguishing between a genuine CoIV complex and a multireference electronic structure solution involving antiferromagnetic coupling between an S = 1 CoIII center and a TAML ligand-centered radical (as indicated by the B3LYP broken-symmetry DFT solution) is very difficult, if not impossible, when relying only on single-reference computational methods (such as DFT).38 We therefore decided to turn to multireference N-electron valence state perturbation theory (NEVPT2)-corrected complete active space self consistent field (CASSCF) calculations for a proper description of the electronic structures of the Co(TAML)-type complexes described in this article.39
CASSCF calculations were initiated on the anionic [CoIII(TAMLred)]– complex by the inclusion of all cobalt d orbitals and those ligand π orbitals (Lπ) that could have an interaction with cobalt. In the final CASSCF(14,13) calculation, all initial orbitals were preserved in the active space, except for the dxy orbital, which is uncorrelated (occupancy of 2.00).40 A selection of the most relevant active orbitals with their occupancies (in parentheses) is given in Figure 5A. Löwdin population analysis of the electronic configuration of the d shell gave (dxy)2.00(dz2)1.99(dyz)1.02(dxz)1.02, consistent with the assigned +III oxidation state of cobalt. Notably, the Lπ orbital at −0.268Eh has a weak bonding interaction with the dxz orbital and is fully filled (occupancy 1.91), consistent with the fully reduced oxidation state of the ligand.
Figure 5.
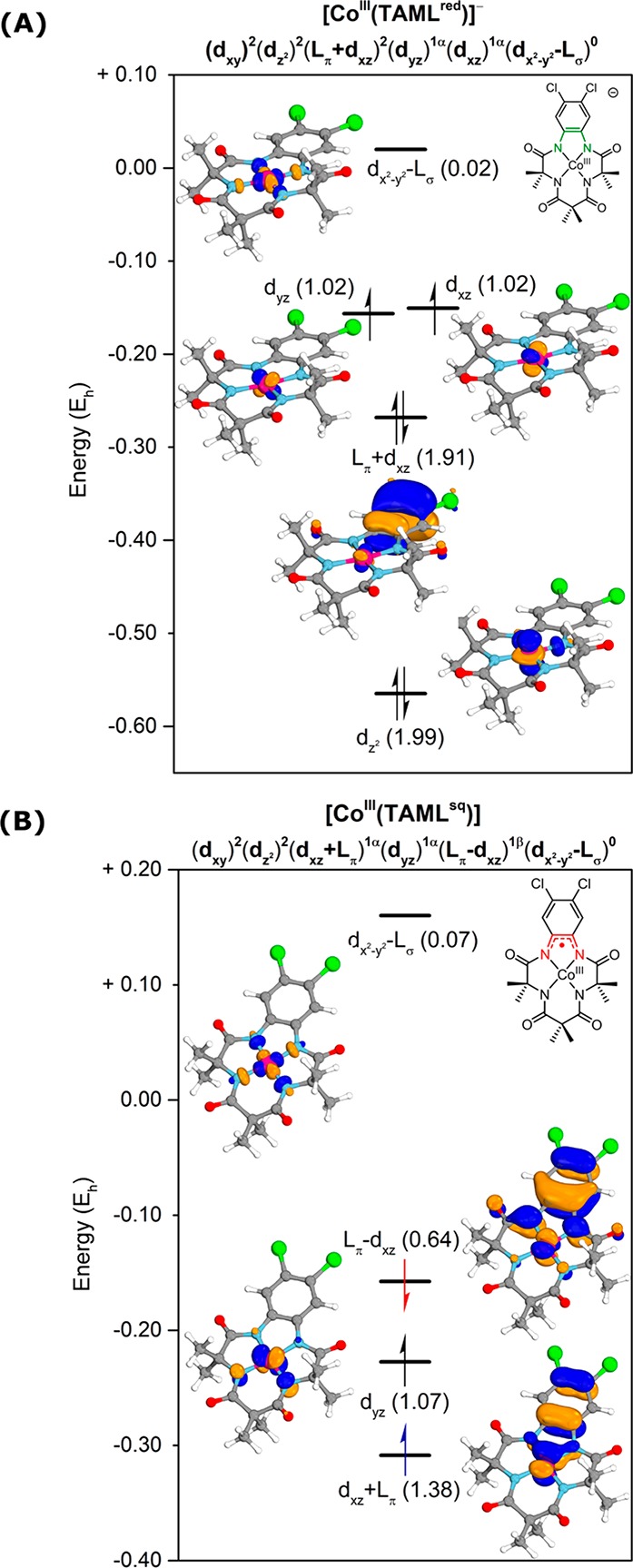
Relevant active orbitals and occupancies (in parentheses) of NEVPT2-corrected CASSCF(14,13) on [CoIII(TAMLred)]– (A) and CASSCF(13,12) on [CoIII(TAMLsq)] (B).
CASSCF(13,12) calculations on the neutral [CoIII(TAMLsq)] complex included a similar active space as for the parent anionic complex and revealed substantial multireference character. The uncorrelated dz2 and dxy orbitals (occupancy 2.00) were not preserved in the active space.40 The reduced charge on the complex causes increased stabilization of the cobalt d orbitals compared to the parent anionic complex, which increases overlap between the dxz and Lπ orbitals (Figure 5B). Because of this stabilization, the bonding and antibonding combinations of the dxz and Lπ orbitals are composed of substantial contributions from both dxz and Lπ. As a result of three orbitals being close in energy, significant population of the Lπ – dxz antibonding combination (occupancy 0.64) from the dxz + Lπ bonding combination (occupancy 1.38) occurs, while the dyz orbital is singly occupied (1.07). The net-doublet ground state of the neutral [CoIII(TAMLsq)] complex is thus best described as an S = 1 CoIII center that is antiferromagnetically coupled to an S = 1/2 TAML-centered radical, leading to a net-doublet system with a (dxy)2.00(dz2)2.00(dxz + Lπ)1.38(dyz)1.07(Lπ – dxz)0.64 electronic structure, in agreement with the early interpretation of Collins.28 Excitation energies derived from the CASSCF(13, 12) calculations revealed that the absorption band observed at λmax = 623 nm (Scheme 3B,D) is indeed characteristic of the ligand-centered radical. The corresponding calculated excitation (at 625 nm) is composed of ligand-centered Lπ → Lπ – dxz and metal-to-ligand (dxz + Lπ→ Lπ – dxz and dyz → Lπ – dxz) charge-transfer processes, with the ligand-centered radical orbital being the acceptor in all cases.
The combined data from magnetic moment measurements, EPR, UV–vis, and XANES spectroscopy, and NEVPT2-CASSCF calculations reveal that the oxidation of [CoIII(TAMLred)]– is ligand-centered, giving rise to the formation of [CoIII(TAMLsq)], wherein cobalt retains its +III oxidation state and its square planar coordination geometry.
Synthesis of [CoIII(TAMLq)(NNs)2]− and [CoIII(TAMLq)(NNs)] via Ligand-to-Substrate SET
With a proper understanding of their electronic structure, confirming that both complexes are square planar cobalt(III) species featuring a redox-active ligand but are in two different ligand oxidation states, we next set out to investigate nitrene formation at the anionic [CoIII(TAMLred)]– and neutral [CoIII(TAMLsq)] complexes. We were particularly interested in exploring the influence of the ligand oxidation state on the structure and overall composition of the targeted nitrene adducts.
The addition of 1 equiv of the nitrene precursor PhINNs to PPh4[CoIII(TAMLred)] in CH2Cl2 at r.t. led to a mixture of starting material, mononitrene adduct [Co(TAML)(NNs)]–, and trace amounts of bis-nitrene adduct [Co(TAML)(NNs)2]–, as revealed by negative-mode electrospray ionization high-resolution mass spectrometry (ESI-HRMS–) analysis. Upon addition of 10 equiv of PhINNs to PPh4[CoIII(TAMLred)] in CH2Cl2 or toluene at r.t., quantitative formation on bis-nitrene species [Co(TAML)(NNs)2]– was achieved on the basis of ESI-HRMS– and UV–vis analysis (Scheme 4 and Figure 6A,B).41 Although bis-nitrene formation was readily achieved for the anionic complex upon addition of excess PhINNs, the addition of 10 equiv of PhINNs to neutral complex [CoIII(TAMLsq)] in CH2Cl2 or toluene at r.t. led to the quantitative formation of only mononitrene species [Co(TAML)(NNs)], as shown by ESI-HRMS– and UV–vis analysis (Scheme 4 and Figure 6C,D). Also, the addition of alternative nitrene source PhINTs (10 equiv) to [CoIII(TAMLsq)] in CH2Cl2 or toluene at room temperature led to the formation of mononitrene complex [Co(TAML)(NTs)], according to ESI-HRMS– data. The effective magnetic moments of [Co(TAML)(NNs)2]– (2.75μB) and [Co(TAML)(NNs)] (1.53μB) are consistent with the formation of (net) triplet (S = 1) and doublet (S = 1/2) systems, respectively. For clarity, we already included the assigned oxidation states of the ligand and cobalt for anionic bis-nitrene ([CoIII(TAMLq)(NNs)2]–) and neutral mononitrene ([CoIII(TAMLq)(NNs)]) in Scheme 4 and Figure 6 and the following text. In the next sections, we will further elaborate on the measurements and calculations leading to these assignments.
Scheme 4. Synthesis of Bis-nitrene (Radical) Complex PPh4[CoIII(TAMLq)(NNs)2] and Mono-nitrene (Radical) Complex [CoIII(TAMLq)(NR)] (R = Ns, Ts) from PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)], Respectively.

Figure 6.

(A) UV–vis spectrum of PPh4[CoIII(TAMLq)(NNs)2] (red) upon reaction of PPh4[CoIII(TAMLred)] (150 μM in CH2Cl2, black) with 10 equiv of PhINNs. (B) ESI-HRMS– spectrum (black) and simulated spectrum (red) for [CoIII(TAMLq)(NNs)2]–. (C) UV–vis spectrum of [CoIII(TAMLq)(NNs)] (blue), formed by the addition of 10 equiv of PhINNs to [CoIII(TAMLsq)] (78 μM in CH2Cl2, black). (D) ESI-HRMS– spectrum (black) and simulated spectrum (blue) for [CoIII(TAMLq)(NNs)].
As can be expected for an integer spin system, anionic bis-nitrene complex [CoIII(TAMLq)(NNs)2]– is X-band EPR silent at both r.t. and at 10 K. Neutral mononitrene complex [CoIII(TAMLq)(NNs)] displays an isotropic EPR signal (Figure 7A) at giso = 2.091 at r.t., showing well-resolved 59Co (ACoiso = 89.5 MHz) and poorly resolved (but necessary for accurate simulation) 14N (ANiso = 18.9 MHz) HFIs. The anisotropic low-temperature (20 K) EPR spectrum of [CoIII(TAMLq)(NNs)] recorded in toluene glass displays a slightly rhombic signal with small g anisotropy and multiple hyperfine coupling interactions, consistent with a net-doublet ground state (Figure 7B).42 The r.t. EPR spectrum of [CoIII(TAMLq)(NTs)] proved to be similar to that of [CoIII(TAMLq)(NNs)] (SI).
Figure 7.
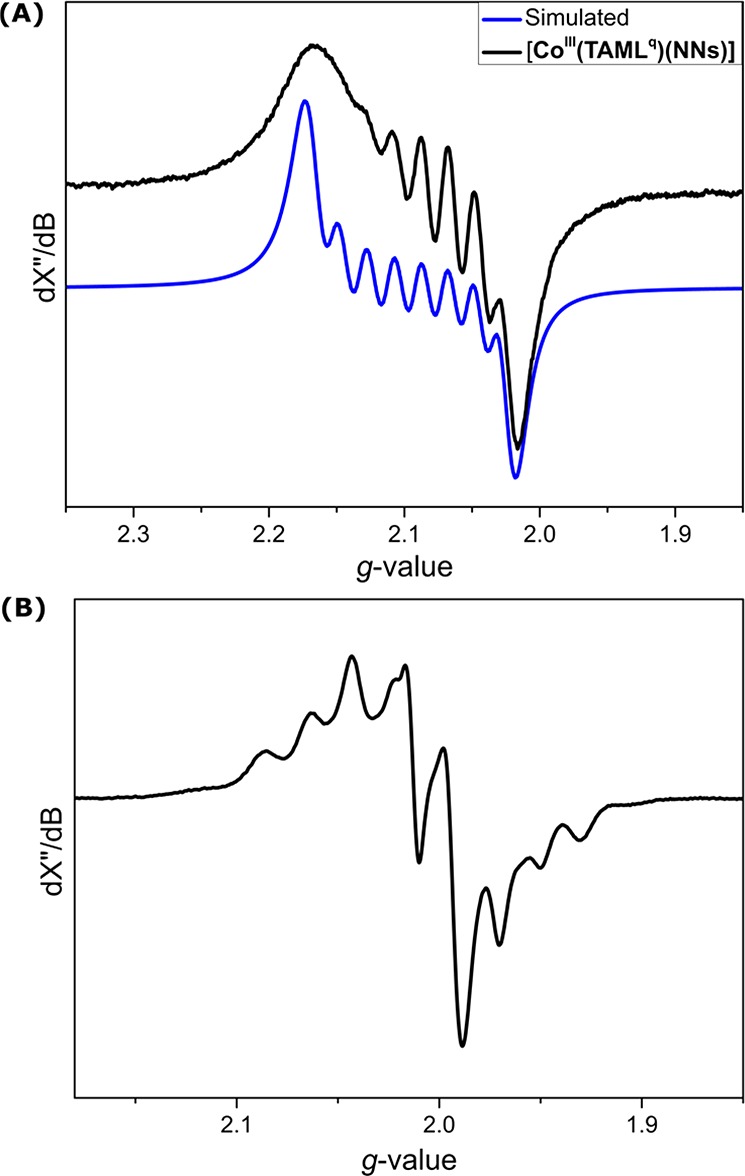
(A) Experimental (black) and simulated (blue) X-band EPR spectra of [CoIII(TAMLq)(NNs)] at r.t. in toluene. Microwave freq. 9.3716 GHz, mod. amp. 2.000 G, and power 6.325 mW. Simulated parameters: giso = 2.091, ACoiso = 89.5 (34.2) MHz, and ANiso = 18.9 MHz. (B) X-band EPR spectrum of [CoIII(TAMLq)(NNs)] in toluene glass at 20 K. Microwave freq. 9.376 GHz, mod. amp. 2.000 G, and power 6.325 mW.
The Co K-edge XANES spectra for PPh4[CoIII(TAMLq)(NNs)2] and [CoIII(TAMLq)(NNs)] are shown in Figure 8. As was observed for the parent complexes PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)], the edge position for both cobalt-nitrene complexes is detected at 7721 eV, suggesting that the cobalt centers in all four complexes have the same overall +III oxidation state. Interestingly, the intense shoulder absorption at 7715 eV observed in the spectra of PPh4[CoIII(TAMLq)(NNs)2] and [CoIII(TAMLq)(NNs)] (corresponding to 1s → 4pz + LMCT shakedown transitions characteristic of square planar cobalt complexes) is no longer visible in the nitrene adducts, thus suggesting that both complexes undergo changes in coordination number and/or geometry. This was also observed in related CoIII(porphyrin)-mono- and bis-nitrene complexes that displayed an octahedral coordination environment, with an axial coligand (NsNH2, NsNH–, H2O, or solvent) present in case of the mononitrene species.14b Moreover, an additional low-intensity pre-edge feature at 7711 eV is observed clearly for [CoIII(TAMLq)(NNs)] (inset in Figure 8). The pre-edge feature in the XANES spectrum of PPh4[CoIII(TAMLq)(NNs)2] is not well-resolved because of moderate data quality caused by low solubility of the complex. These pre-edge features arise from 1s → 3d transitions, and in centrosymmetric (i.e., square planar and octahedral) complexes, these transitions are weak because of quadrupole transitions.14b However, symmetry breaking enables 3d–4p hybridization of metal atomic orbitals, causing the pre-edge to gain intensity as a result of dipole-allowed transitions. It thus seems that [CoIII(TAMLq)(NNs)] bears an unidentified sixth coordinating coligand (octahedral coordination geometry) but is not fully centrosymmetric. However, similar low-intensity pre-edge features have been observed in a five-coordinate cobalt-TAML complex;27 therefore, square pyramidal coordination around cobalt cannot be fully excluded for [CoIII(TAMLq)(NNs)].
Figure 8.
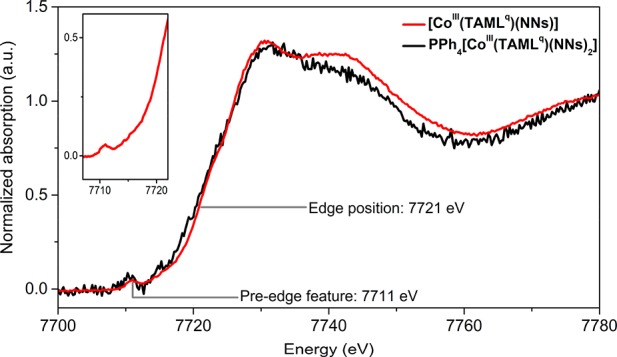
Co K-edge XANES analysis of PPh4[CoIII(TAMLq)(NNs)2] (black) and [CoIII(TAMLq)(NNs)] (red) in toluene. (Inset) Close-up of the pre-edge feature for [CoIII(TAMLsq)(NNs)].
Consistent with the above-mentioned experimental results, DFT calculations (BP86, def2-TZVP, disp3, and m4 grid) indicate that the formation of neutral mononitrene complex [CoIII(TAMLq)(NNs)] (S = 1/2; ΔGo298 K = −20.3 kcal mol–1) from [CoIII(TAMLsq)] (S = 1/2; reference point) is energetically more favorable than the formation of the neutral bis-nitrene adduct [Co(TAML)(NNs)2] (S = 1/2; ΔGo298 K = −14.5 kcal mol–1). However, the corresponding formation energies of the anionic mono- and bis-nitrene complexes [CoIII(TAMLsq)(NNs)]– (S = 1; ΔGo298 K = −27.9 kcal mol–1) and [CoIII(TAMLq)(NNs)2]– (S = 1; ΔGo298 K = −29.9 kcal mol–1) from [CoIII(TAMLred)]– (S = 1; reference point) are nearly equal (SI).
NEVPT2-corrected CASSCF calculations were performed to accurately describe the electronic structure of the nitrene species. All cobalt d orbitals, ligands Lπ, and nitrene-localized p orbitals were included in the active spaces. CASSCF(14,13) calculations on [CoIII(TAMLq)(NNs)2]– showed that the dxy orbital is not preserved in the active space (occupancy 2.00)40 and that the dz2 orbital forms bonding (nitrene-N1 and -N2 localized, occupancy 1.94) and antibonding (mostly dz2 localized, occupancy 0.07) combinations with the nitrene Npz orbitals. The dyz and dxz orbitals are both filled (occupancies 1.97 and 1.95, respectively), and Lπ–dxz (occupancy 0.10) is virtually empty. Given that the Lπ orbital was doubly filled in [CoIII(TAMLred)]– (vide supra), this implies that the formation of [CoIII(TAMLq)(NNs)2]– from [CoIII(TAMLred)]– is associated with the two-electron oxidation of the ligand. Interestingly, both nitrene nitrogen atoms bear a single unpaired electron in their Npy/Npx orbitals (both occupancies 1.00). The electronic structure is thus best described as (dxy)2.00(dyz)1.97(dxz)1.95(Npz1 + Npz + dz2)1.94(Npx2)1.00(Npy)1.00, consistent with a CoIII center, a fully oxidized TAML backbone (TAMLq), and two one-electron-reduced Fischer-type nitrene radical substrates (N•Ns–).43 Moreover, the cobalt(III) center has undergone a spin transition from intermediate spin in [CoIII(TAMLred)]– to low spin in [CoIII(TAMLq)(N•Ns–)2]– upon formation of the bis-nitrene radical species. As a result, the net total spin state does not change in the process and remains a triplet spin state (S = 1). The most relevant active orbitals and their occupation numbers are shown in Figure 9A. In addition, excitation energies derived from the CASSCF(14,13) calculation revealed that no intense absorption bands are expected in the 400–850 nm region (SI), consistent with the experimental spectrum depicted in Figure 6A.
Figure 9.
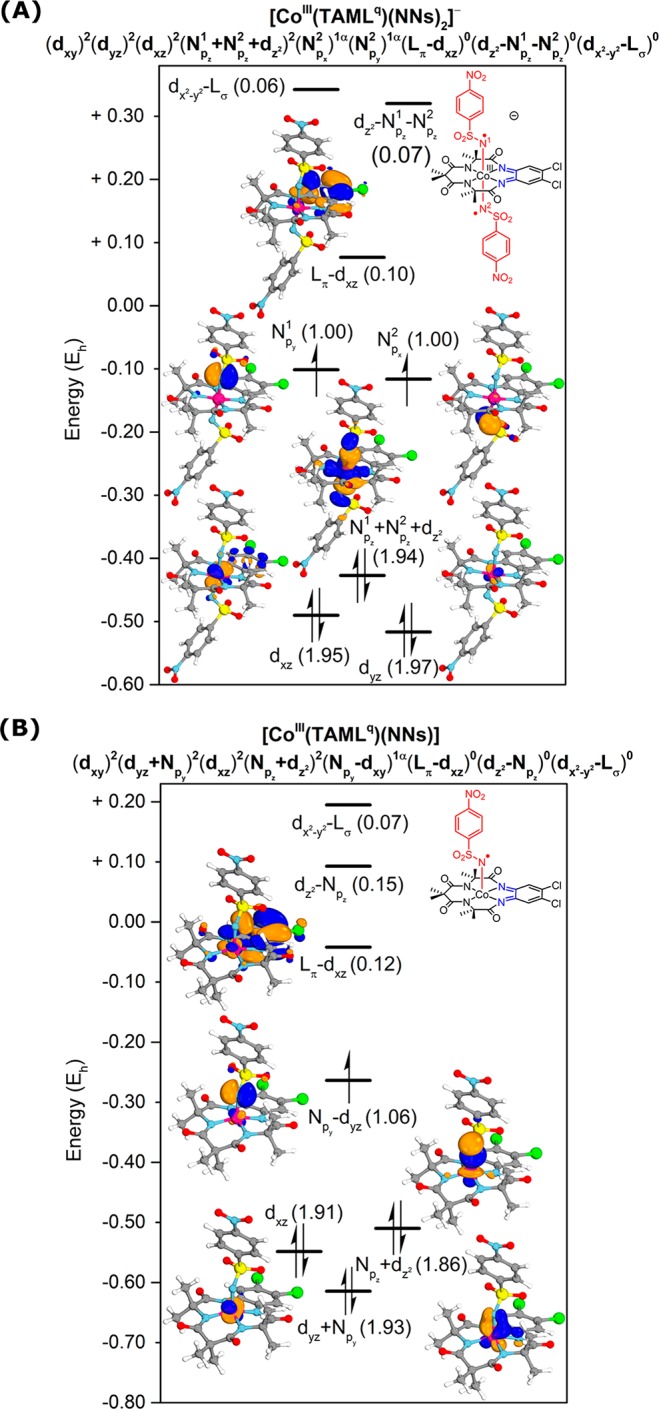
Most relevant active orbitals and occupancies (in parentheses) of NEVPT2-corrected CASSCF(14,13) ([CoIII(TAMLq)(NNs)2]–) (A) and CASSCF(13,12) ([CoIII(TAMLq)(NNs)] (B) calculations.
The complex bears some resemblance to the previously reported cobalt-porphyrin bis-nitrene ([CoIII(TPP•–)(N•Ns–)2])14 and ruthenium-porphyrin bis-imido ([RuVI(TPP)(NTs)2])44 complexes (TPP = tetraphenylporphyrin). The ruthenium bis-imido complex is formed exclusively via metal-centered oxidation processes. However, whereas in the cobalt-porphyrin complex double nitrene-radical formation is the result of combined metal-to-substrate and (porphyrin) ligand-to-substrate SET processes, the formation of [CoIII(TAMLq)(N•Ns–)2]– is an entirely (double) ligand-to-substrate single-electron-transfer process.
In a very similar fashion, CASSCF(13,12) calculations on [CoIII(TAMLq)(NNs)] reveal π (dyz+ Npy) and σ (dz2+ Npz) bonding interactions between cobalt and the nitrene, with occupations of 1.93 and 1.86 electrons, respectively.45 The dxz orbital is filled (occupancy 1.91), and the formerly half-filled Lπ–dxz orbital is now unoccupied (occupancy 0.12), indicating single-electron oxidation of the ligand (i.e., from TAMLsq to TAMLq). The single unpaired electron of the complex is mainly localized on the nitrene moiety (Npy–dyz, occupancy 1.06), again consistent with [CoIII(TAMLq)(N•Ns–)] being a Fischer-type nitrene radical complex with net π-bond order between cobalt and the nitrene of ∼0.5.43 As for the anionic bis-nitrene complex, the neutral mononitrene complex is generated via ligand-to-substrate SET. Once again, the cobalt(III) ion does not change its oxidation state in the process, but it does undergo a spin flip from intermediate spin in [CoIII(TAMLsq)] to low spin in [CoIII(TAMLq)(N•Ns–)]. The most relevant active orbitals and their occupations are shown in Figure 9B. Notably, neither [CoIII(TAMLq)(N•Ns–)2]– nor [CoIII(TAMLq)(N•Ns–)] has significant multireference character.
Interestingly, ligand-to-substrate SET combined with a metal-based spin flip effectively leads to a shift in the spin density from the metal to the nitrene nitrogen(s) in both the neutral mononitrene and the anionic bis-nitrene complexes, without the oxidation of cobalt and without changing the net total spin state of the complex. The redox events clearly occur on the TAML backbone (electron donor) and the nitrene (electron acceptor), wherein the former undergoes one-electron or two-electron oxidation to accommodate one or two nitrene radicals on the CoIII center.
Intrigued by the influence of the ligand oxidation state on the structure of the nitrene species, the mono- and bis-nitrene species were probed for catalytic nitrene transfer reactivity in the benchmark aziridination of styrene (Scheme 5).1a,3 A remarkable difference in the yield of aziridine product 1 was observed when using PPh4[CoIII(TAMLred)] (64%) or [CoIII(TAMLsq)] (35%) as the catalyst in nitrene transfer reactions from PhINNs to styrene, suggesting that the anionic bis-nitrene and neutral mononitrene exhibit markedly different activity and/or stability properties. A thorough investigation of the applicability and mechanisms of PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)] as aziridination catalysts is the subject of current investigations, which will be reported in due time. At this point it is worth mentioning that for cobalt-TAML complexes the reduced (anionic) [CoIII(TAMLred)]– species are apparently more effective nitrene-transfer catalysts than the corresponding oxidized (neutral) [CoIII(TAMLsq)] species, while for iron- and manganese-TAML complexes the reverse was observed.19,20
Scheme 5. Catalytic Aziridination of Styrene to Afford 1 Using PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)].

Conclusions
In this work we have conclusively shown that the ligand in Co(TAML) complexes is redox-active. The oxidation of [CoIII(TAMLred)]– using (Thi)BF4 cleanly affords [CoIII(TAMLsq)] via ligand-centered oxidation, with the electronic structure being best described as an intermediate spin (S = 1) cobalt(III) center that is antiferromagnetically coupled to a ligand-centered radical (S = 1/2).
Interestingly, cobalt-nitrene adducts of PPh4[CoIII(TAMLred)] and CoIII(TAMLsq) can be cleanly generated from PhINNs via ligand-to-substrate single-electron transfer to afford PPh4[CoIII(TAMLq)(NNs)2]– and [CoIII(TAMLq)(NNs)], respectively. CASSCF calculations revealed that both nitrene complexes are best described as one-electron-reduced Fischer-type nitrene radicals. The formation of a bis-nitrene adduct of PPh4[CoIII(TAMLred)] is attributed to the availability of two electrons within the reduced TAML framework for double ligand-to-substrate SET, whereas only one electron can be used for ligand-to-substrate SET on [CoIII(TAMLsq)], which therefore affords the mononitrene adduct. Intriguingly, in both cases the combination of ligand-to-substrate SET and a spin flip from intermediate spin (S = 1) to low spin (S = 0) at the cobalt(III) center effectively results in a shift of the spin density from the metal to the nitrene moieties, without the oxidation of cobalt and without changing the net total spin state of the complex.
Preliminary catalytic styrene aziridination reactions using PPh4[CoIII(TAMLred)] or [CoIII(TAMLsq)] as the catalyst reveal remarkable differences in activity/stability between the two systems. More elaborate studies on the underlying mechanisms, synthesis applicability, and differences between the two complexes in nitrene transfer catalysis will be reported in the near future.
Acknowledgments
Financial support from The Netherlands Organization for Scientific Research (NWO TOP-Grant 716.015.001 to B.d.B. and NWO VIDI grant 723.014.010 to M.T.) is gratefully acknowledged. We thank Ed Zuidinga for HRMS measurements and Lars Grooten for preliminary UV–vis studies. The authors thank the staff of beamline B18, Diamond Light Source (proposal number SP22432) in Didcot, U.K. for support and access to their facilities.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b11715.
X-ray structure of TAMLH4 (CIF)
X-ray structure of PPh4[CoIII(TAMLred)] (CIF)
Experimental details; synthesis procedures; relevant NMR, EPR, HRMS, XRD, UV–vis, electrochemical, and XANES data; geometries (xyz coordinates) of stationary points (DFT); and a description of the CASSCF calculations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a van Leest N. P.; Epping P. F. J.; van Vliet K. M.; Lankelma M.; van den Heuvel E. J.; Heijtbrink N.; Broersen R.; de Bruin B.. Single-Electron Elementary Steps in Homogeneous Organometallic Catalysis. Advances in Organometallic Chemistry; Elsevier: London, 2018; Vol. 70, pp 71–180. [Google Scholar]; b Broere D. L. J.; Plessius R.; van der Vlugt J. I. New avenues for ligand-mediated processes - expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem. Soc. Rev. 2015, 44, 6886–6915. 10.1039/C5CS00161G. [DOI] [PubMed] [Google Scholar]; c Chirila A.; Das B. G.; Kuijpers P. F.; Sinha V.; de Bruin B.. Application of Stimuli-Responsive and “Non-Innocent” Ligands in Base Metal Catalysis. In Non-Noble Metal Catalysis Molecular Approaches and Reactions; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018; pp 1–31. [Google Scholar]; d Lu H.; Zhang X. P. Catalytic C-H functionalization by metalloporphyrins: recent developments and future directions. Chem. Soc. Rev. 2011, 40, 1899–1909. 10.1039/C0CS00070A. [DOI] [PubMed] [Google Scholar]; e Lyaskovskyy V.; de Bruin B. Redox Non-innocent Ligands – Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. 10.1021/cs200660v. [DOI] [Google Scholar]
- Hili R.; Yudin A. K. Making carbon-nitrogen bonds in biological and chemical synthesis. Nat. Chem. Biol. 2006, 2, 284–287. 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- a Kuijpers P. F.; van der Vlugt J. I.; Schneider S.; de Bruin B. Nitrene Radical Intermediates in Catalytic Synthesis. Chem. - Eur. J. 2017, 23, 13819–13829. 10.1002/chem.201702537. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Park Y.; Kim Y.; Chang S. Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]; c Shin K.; Kim H.; Chang S. Transition-Metal-Catalyzed C–N Bond Forming Reactions Using Organic Azides as the Nitrogen Source: A Journey for the Mild and Versatile C–H Amination. Acc. Chem. Res. 2015, 48, 1040–1052. 10.1021/acs.accounts.5b00020. [DOI] [PubMed] [Google Scholar]
- a Intrieri D.; Carminati D. M.; Gallo E. Recent advances in C–H bond aminations catalyzed by ruthenium porphyrin complexes. J. Porphyrins Phthalocyanines 2016, 20, 190–203. 10.1142/S1088424616500383. [DOI] [Google Scholar]; b Zardi P.; Caselli A.; Macchi P.; Ferretti F.; Gallo E. Synthesis of Biologically Relevant Compounds by Ruthenium Porphyrin Catalyzed Amination of Benzylic C–H Bonds. Organometallics 2014, 33, 2210–2218. 10.1021/om500064d. [DOI] [Google Scholar]; c Fantauzzi S.; Gallo E.; Caselli A.; Ragaini F.; Casati N.; Macchi P.; Cenini S. The key intermediate in the amination of saturated C–H bonds: synthesis, X-ray characterization and catalytic activity of Ru(TPP)(NAr)2 (Ar = 3,5-(CF3)2C6H3). Chem. Commun. 2009, 3952–3954. 10.1039/b903238j. [DOI] [PubMed] [Google Scholar]
- a Guthikonda K.; When P. M.; Caliando B. J.; Du Bois J. Rh-catalyzed alkene oxidation: a highly efficient and selective process for preparing N-alkoxysulfonyl aziridines. Tetrahedron 2006, 62, 11331–11342. 10.1016/j.tet.2006.07.099. [DOI] [Google Scholar]; b Liang C.; Robert-Peillard F.; Fruit C.; Müller P.; Dodd R. H.; Dauban P. Efficient Diastereoselective Intermolecular Rhodium-Catalyzed C-H Amination. Angew. Chem., Int. Ed. 2006, 45, 4641–4644. 10.1002/anie.200601248. [DOI] [PubMed] [Google Scholar]; c Espino C. G.; When P. M.; Chow J.; Du Bois J. Synthesis of 1,3-Difunctionalized Amine Derivatives through Selective C–H Bond Oxidation. J. Am. Chem. Soc. 2001, 123, 6935–6936. 10.1021/ja011033x. [DOI] [Google Scholar]; d Roizen J. L.; Zalatan D. N.; De Bois J. Selective Intermolecular Amination of C-H Bonds at Tertiary Carbon Centers. Angew. Chem., Int. Ed. 2013, 52, 11343–11346. 10.1002/anie.201304238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Broere D. L. J.; de Bruin B.; Reek J. N. H.; Lutz M.; Dechert S.; van der Vlugt J. I. Intramolecular Redox-Active Ligand-to-Substrate Single-Electron Transfer: Radical Reactivity with a Palladium(II) Complex. J. Am. Chem. Soc. 2014, 136, 11574–11577. 10.1021/ja502164f. [DOI] [PubMed] [Google Scholar]; b Broere D. L. J.; van Leest N. P.; de Bruin B.; Siegler M. A.; van der Vlugt J. I. Reversible Redox Chemistry and Catalytic C(sp3)–H Amination Reactivity of a Paramagnetic Pd Complex Bearing a Redox-Active o-Aminophenol-Derived NNO Pincer Ligand. Inorg. Chem. 2016, 55, 8603–8611. 10.1021/acs.inorgchem.6b01192. [DOI] [PubMed] [Google Scholar]; c Okamoto K.; Oda T.; Kohigashi S.; Ohe K. Palladium-Catalyzed Decarboxylative Intramolecular Aziridination from 4H-Isoxazol-5-ones Leading to 1-Azabicyclo[3.1.0]hex-2-enes. Angew. Chem., Int. Ed. 2011, 50, 11470–11473. 10.1002/anie.201105153. [DOI] [PubMed] [Google Scholar]
- a Llaveria J.; Beltrán Á.; Díaz-Requejo M. M.; Matheu M. I.; Castillón S.; Pérez P. J. Efficient Silver-Catalyzed Regio- and Stereospecific Aziridination of Dienes. Angew. Chem., Int. Ed. 2010, 49, 7092–7095. 10.1002/anie.201003167. [DOI] [PubMed] [Google Scholar]; b Rigoli J. W.; Weatherly C. D.; Alderson J. M.; Vo B. T.; Schomaker J. M. Tunable, Chemoselective Amination via Silver Catalysis. J. Am. Chem. Soc. 2013, 135, 17238–17241. 10.1021/ja406654y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dolan N. S.; Scamp R. J.; Yang T.; Berry J. F.; Schomaker J. M. Catalyst-Controlled and Tunable, Chemoselective Silver-Catalyzed Intermolecular Nitrene Transfer: Experimental and Computational Studies. J. Am. Chem. Soc. 2016, 138, 14658–14667. 10.1021/jacs.6b07981. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Cui Y.; He C. Efficient Aziridination of Olefins Catalyzed by a Unique Disilver(I) Compound. J. Am. Chem. Soc. 2003, 125, 16202–16203. 10.1021/ja038668b. [DOI] [PubMed] [Google Scholar]
- Li Z.; Ding X.; He C. Nitrene Transfer Reactions Catalyzed by Gold Complexes. J. Org. Chem. 2006, 71, 5876–5880. 10.1021/jo060016t. [DOI] [PubMed] [Google Scholar]
- a Minakata S.; Ando T.; Nishimura M.; Ryu I.; Komatsu M. Novel Asymmetric and Stereospecific Aziridination of Alkenes with a Chiral Nitridomanganese Complex. Angew. Chem., Int. Ed. 1998, 37, 3392–3394. . [DOI] [PubMed] [Google Scholar]; b Nishikori H.; Katsuki T. Catalytic and highly enantioselective aziridination of styrene derivatives. Tetrahedron Lett. 1996, 37, 9245–9248. 10.1016/S0040-4039(96)02195-8. [DOI] [Google Scholar]; c Yang J.; Weinberg R.; Breslow R. The hydroxylation and amidation of equilenin acetate catalyzed by chloro[5,10,15,20-tetrakis(pentafluorophenyl)porphyrinato]manganese(III). Chem. Commun. 2000, 531–532. 10.1039/b000463o. [DOI] [Google Scholar]
- a Bagh B.; Broere D. L. J.; Sinha V.; Kuijpers P. F.; van Leest N. P.; de Bruin B.; Demeshko S.; Siegler M. A.; van der Vlugt J. I. Catalytic Synthesis of N-Heterocycles via Direct C(sp3)–H Amination Using an Air-Stable Iron(III) Species with a Redox-Active Ligand. J. Am. Chem. Soc. 2017, 139, 5117–5124. 10.1021/jacs.7b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Avenier F.; Latour J.-M. Catalytic aziridination of olefins and amidation of thioanisole by a non-heme iron complex. Chem. Commun. 2004, 1544–1545. 10.1039/b404561k. [DOI] [PubMed] [Google Scholar]; c Fingerhut A.; Serdyuk O. V.; Tsogoeva S. B. Non-heme iron catalysts for epoxidation and aziridination reactions of challenging terminal alkenes: towards sustainability. Green Chem. 2015, 17, 2042–2058. 10.1039/C4GC02413C. [DOI] [Google Scholar]; d Correa A.; Mancheño O. G.; Bolm C. Iron-catalysed carbon–heteroatom and heteroatom–heteroatom bond forming processes. Chem. Soc. Rev. 2008, 37, 1108–1117. 10.1039/b801794h. [DOI] [PubMed] [Google Scholar]; e Hennessy E. T.; Betley T. A. Complex N-Heterocycle Synthesis via Iron-Catalyzed, Direct C-H Bond Amination. Science 2013, 340, 591–595. 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]; f Iovan D. A.; Betley T. A. Characterization of Iron-Imido Species Relevant for N-Group Transfer Chemistry. J. Am. Chem. Soc. 2016, 138, 1983–1993. 10.1021/jacs.5b12582. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Patra R.; Coin G.; Castro L.; Dubourdeaux P.; Clémancey M.; Pécaut J.; Lebrun C.; Maldivi L. J.-M. Rational design of Fe catalysts for olefin aziridination through DFT-based mechanistic analysis. Catal. Sci. Technol. 2017, 7, 4388–4400. 10.1039/C7CY01283G. [DOI] [Google Scholar]; h Klotz K. L.; Slominksi L. M.; Hull A. V.; Gottsacker V. M.; Mas-Ballesté R.; Que L. Jr.; Halfen J. A. Non-heme iron(II) complexes are efficient olefin aziridination catalysts. Chem. Commun. 2007, 2063–2065. 10.1039/b700493a. [DOI] [PubMed] [Google Scholar]; i Klotz K. L.; Slominski L. M.; Riemer M. E.; Phillips J. A.; Halfen J. A. Mechanism of the Iron-Mediated Alkene Aziridination Reaction: Experimental and Computational Investigations. Inorg. Chem. 2009, 48, 801–803. 10.1021/ic8020244. [DOI] [PubMed] [Google Scholar]; j Chandrachud P. P.; Bass H. M.; Jenkins D. M. Synthesis of Fully Aliphatic Aziridines with a Macrocyclic Tetracarbene Iron Catalyst. Organometallics 2016, 35, 1652–1657. 10.1021/acs.organomet.6b00066. [DOI] [Google Scholar]; k Nakanishi M.; Salit A.-F.; Bolm C. Iron-Catalyzed Aziridination Reactions. Adv. Synth. Catal. 2008, 350, 1835–1840. 10.1002/adsc.200700519. [DOI] [Google Scholar]
- a Kuijpers P. F.; Tiekink M. J.; Breukelaar W. B.; Broere D. L. J.; van Leest N. P.; van der Vlugt J. I.; Reek J. N. H.; de Bruin B. Cobalt-Porphyrin-Catalysed Intramolecular Ring-Closing C–H Amination of Aliphatic Azides: A Nitrene-Radical Approach to Saturated Heterocycles. Chem. - Eur. J. 2017, 23, 7945–7952. 10.1002/chem.201700358. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Goswami M.; Geuijen P.; Reek J. N. H.; de Bruin B. Application of [Co(Corrole)]- Complexes in Ring-Closing C-H Amination of Aliphatic Azides via Nitrene Radical Intermediates. Eur. J. Inorg. Chem. 2018, 2018, 617–626. 10.1002/ejic.201701343. [DOI] [Google Scholar]; c Baek Y.; Betley T. A. Catalytic C–H Amination Mediated by Dipyrrin Cobalt Imidos. J. Am. Chem. Soc. 2019, 141, 7797–7806. 10.1021/jacs.9b01262. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jin L.-M.; Lu H.; Cui Y.; Lizardi C. L.; Arzua T. N.; Wojtas L.; Cui X.; Zhang X. P. Selective radical amination of aldehydic C(sp2)–H bonds with fluoroaryl azides via Co(II)-based metalloradical catalysis: synthesis of N-fluoroaryl amides from aldehydes under neutral and nonoxidative conditions. Chem. Sci. 2014, 5, 2422–2427. 10.1039/C4SC00697F. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lu H.; Jiang H.; Wojtas L.; Zhang X. P. Angew. Chem., Int. Ed. 2010, 49, 10192–10196. 10.1002/anie.201005552. [DOI] [PubMed] [Google Scholar]; f Ruppel J. V.; Kamble R. M.; Zhang X. P. Selective Intramolecular C-H Amination through the Metalloradical Activation of Azides: Synthesis of 1,3-Diamines under Neutral and Nonoxidative Conditions. Org. Lett. 2007, 9, 4889–4892. 10.1021/ol702265h. [DOI] [PubMed] [Google Scholar]; g Jin L.-M.; Xu X.; Lu H.; Cui X.; Wojtas L.; Zhang X. P. Effective Synthesis of Chiral N-Fluoroaryl Aziridines through Enantioselective Aziridination of Alkenes with Fluoroaryl Azides. Angew. Chem., Int. Ed. 2013, 52, 5309–5313. 10.1002/anie.201209599. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Gao G.-Y.; Jones J. E.; Vyas R.; Harden J. D.; Zhang X. P. J. Cobalt-Catalyzed Aziridination with Diphenylphosphoryl Azide (DPPA): Direct Synthesis of N-Phosphorus-Substituted Aziridines from Alkenes. J. Org. Chem. 2006, 71, 6655–6658. 10.1021/jo0609226. [DOI] [PubMed] [Google Scholar]; i Subbarayan V.; Ruppel J. V.; Zhu S.; Perman J. A.; Zhang X. P. Highly asymmetric cobalt-catalyzed aziridination of alkenes with trichloroethoxysulfonyl azide (TcesN3). Chem. Commun. 2009, 4266–4268. 10.1039/b905727g. [DOI] [PubMed] [Google Scholar]; j Gao G.-Y.; Harden J. D.; Zhang X. P. Cobalt-Catalyzed Efficient Aziridination of Alkenes. Org. Lett. 2005, 7, 3191–3193. 10.1021/ol050896i. [DOI] [PubMed] [Google Scholar]; k Caselli A.; Gallo E.; Fantauzzi S.; Morlacchi S.; Ragaini F.; Cenini S. Allylic Amination and Aziridination of Olefins by Aryl Azides Catalyzed by CoII(tpp): A Synthetic and Mechanistic Study. Eur. J. Inorg. Chem. 2008, 2008, 3009–3019. 10.1002/ejic.200800156. [DOI] [Google Scholar]
- a Wiese S.; Aguila M. J. B.; Kogut E.; Warren T. H. β-Diketiminato Nickel Imides in Catalytic Nitrene Transfer to Isocyanides. Organometallics 2013, 32, 2300–2308. 10.1021/om300909n. [DOI] [Google Scholar]; b Iluc V. M.; Miller A. J. M.; Anderson J. S.; Monreal M. J.; Mehn M. P.; Hillhouse G. L. Synthesis and Characterization of Three-Coordinate Ni(III)-Imide Complexes. J. Am. Chem. Soc. 2011, 133, 13055–13063. 10.1021/ja2024993. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kogut E.; Wiencko H. L.; Zhang L.; Cordeau D. E.; Warren T. H. A Terminal Ni(III)–Imide with Diverse Reactivity Pathways. J. Am. Chem. Soc. 2005, 127, 11248–11249. 10.1021/ja0533186. [DOI] [PubMed] [Google Scholar]
- a van Vliet K. M.; Polak L. H.; Sieglet M. A.; van der Vlugt J. I.; Fonseca Guerra C.; de Bruin B. Efficient Copper-Catalyzed Multicomponent Synthesis of N-Acyl Amidines via Acyl Nitrenes. J. Am. Chem. Soc. 2019, 141, 15240–15249. 10.1021/jacs.9b07140. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Carsch K. M.; DiMucci I. M.; Iovan D. A.; Li A.; Zheng S.-L.; Titus C. J.; Lee S. J.; Irwin K. V.; Nordlund D.; Lancaster K. M.; Betley T. A. Synthesis of a copper-supported triplet nitrene complex pertinent to copper-catalyzed amination. Science 2019, 365, 1138–1143. 10.1126/science.aax4423. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Evans D. A.; Faul M. M.; Bilodeau M. T.; Anderson B. A.; Barnes D. M. Bis(oxazoline)-copper complexes as chiral catalysts for the enantioselective aziridination of olefins. J. Am. Chem. Soc. 1993, 115, 5328–5329. 10.1021/ja00065a068. [DOI] [Google Scholar]; d Li Z.; Quan R. W.; Jacobsen E. N. Mechanism of the (Diimine)copper-Catalyzed Asymmetric Aziridination of Alkenes. Nitrene Transfer via Ligand-Accelerated Catalysis. J. Am. Chem. Soc. 1995, 117, 5889–5890. 10.1021/ja00126a044. [DOI] [Google Scholar]; e Gephart R. T.; III; Warren T. H. Copper-Catalyzed sp3 C–H Amination. Organometallics 2012, 31, 7728–7752. 10.1021/om300840z. [DOI] [Google Scholar]; f Lebel H.; Parmentier M. Copper-catalyzed enantioselective aziridination of styrenes. Pure Appl. Chem. 2010, 82, 1827–1833. 10.1351/PAC-CON-09-12-12. [DOI] [Google Scholar]; g Mairena M. A.; Díaz-Requejo M. M.; Belderraín R. R.; Nicasio M. C.; Trofimenko S.; Pérez P. J. Copper-Homoscorpionate Complexes as Very Active Catalysts for the Olefin Aziridination Reaction. Organometallics 2004, 23, 253–256. 10.1021/om034158e. [DOI] [Google Scholar]; h Bagchi V.; Paraskevopoulou P.; Das P.; Chi L.; Wang Q.; Choudhury A.; Mathieson J. S.; Cronin L.; Pardue D. B.; Cundari T. R.; Mitrikas G.; Sanakis Y.; Stavropoulos R. A Versatile Tripodal Cu(I) Reagent for C–N Bond Construction via Nitrene-Transfer Chemistry: Catalytic Perspectives and Mechanistic Insights on C–H Aminations/Amidinations and Olefin Aziridinations. J. Am. Chem. Soc. 2014, 136, 11362–11381. 10.1021/ja503869j. [DOI] [PubMed] [Google Scholar]; i Ren Y.; Cheaib K.; Jacquet J.; Vezin H.; Fensterbank L.; Orio M.; Blanchard S.; Desage-El Murr M. Copper-Catalyzed Aziridination with Redox-Active Ligands: Molecular Spin Catalysis. Chem. - Eur. J. 2018, 24, 5086–5090. 10.1002/chem.201705649. [DOI] [PubMed] [Google Scholar]
- a Lyaskovskyy V.; Suarez A. I. O.; Lu H.; Jiang H.; Zhang X. P.; de Bruin B. Mechanism of Cobalt(II) Porphyrin-Catalyzed C–H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)–Nitrene Intermediates. J. Am. Chem. Soc. 2011, 133, 12264–12273. 10.1021/ja204800a. [DOI] [PubMed] [Google Scholar]; b Goswami M.; Lyaskobskyy V.; Domingos S. R.; Buma W. J.; Woutersen S.; Troeppner O.; Ivanović-Burmazović I.; Lu H.; Cui X.; Zhang X. P.; Reijerse E. J.; DeBeer S.; van Schooneveld M. M.; Pfaff F. F.; Ray K.; de Bruin B. Characterization of Porphyrin-Co(III)-‘Nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc. 2015, 137, 5468–5479. 10.1021/jacs.5b01197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vlugt J. I. Radical-Type Reactivity and Catalysis by Single-Electron Transfer to or from Redox-Active Ligands. Chem. - Eur. J. 2019, 25, 2651–2662. 10.1002/chem.201802606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T. J.; Ryabov A. D. Targeting of High-Valent Iron-TAML Activators at Hydrocarbons and Beyond. Chem. Rev. 2017, 117, 9140–9162. 10.1021/acs.chemrev.7b00034. [DOI] [PubMed] [Google Scholar]
- Dutta S. K.; Beckmann U.; Bill E.; Weyhermuller T.; Wieghardt K. 1,2-Bis(pyridine-2-carboxamido)benzenate(2−), (bpb)2-: A Noninnocent Ligand. Syntheses, Structures, and Mechanisms of Formation of [(n-Bu)4N][FeIV2(μ-N)(bpb)2(X)2] (X = CN-, N3-) and the Electronic Structures of [MIII(bpbox1)(CN)2] (M = Co, Fe). Inorg. Chem. 2000, 39, 3355–3364. 10.1021/ic0001107. [DOI] [PubMed] [Google Scholar]
- Popescu D.-L.; Chanda A.; Stadler M.; Tiago de Oliveira F.; Ryabov A. D.; Münck E.; Bominaar E. L.; Collins T. J. High-valent first-row transition-metal complexes of tetraamido (4N) and diamidodialkoxido or diamidophenolato (2N/2O) ligands: Synthesis, structure, and magnetochemistry. Coord. Chem. Rev. 2008, 252, 2050–2071. 10.1016/j.ccr.2007.11.006. [DOI] [Google Scholar]
- a Hong S.; Sutherlin K. D.; Vardhaman A. K.; Yan J. J.; Park S.; Lee Y.-M.; Jang S.; Lu X.; Ohta T.; Ogura T.; Solomon E. I.; Nam W. A Mononuclear Nonheme Iron(V)-Imido Complex. J. Am. Chem. Soc. 2017, 139, 8800–8803. 10.1021/jacs.7b04695. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hong S.; Lu X.; Lee Y.-M.; Seo M. S.; Ohta T.; Ogura T.; Clémancey M.; Maldivi P.; Latour J.-M.; Sarangi R.; Nam W. Achieving One-Electron Oxidation of a Mononuclear Nonheme Iron(V)-Imido Complex. J. Am. Chem. Soc. 2017, 139, 14372–14375. 10.1021/jacs.7b08161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; Xie J.; Lam W. W. Y.; Mab W.-L.; Mak C.-K.; Yiu S.-M.; Lee H. K.; Lau T.-C. Generation and Reactivity of a One-Electron-Oxidized Manganese(V) Imido Complex with a Tetraamido Macrocyclic Ligand. Chem. - Eur. J. 2019, 25, 12895–12899. 10.1002/chem.201902405. [DOI] [PubMed] [Google Scholar]
- Collins T. J.; Powell R. D.; Slebodnick C.; Uffelman E. S. Stable highly oxidizing cobalt complexes of macrocyclic ligands. J. Am. Chem. Soc. 1991, 113, 8419–8425. 10.1021/ja00022a033. [DOI] [Google Scholar]
- Das D.; Pattanayak S.; Singh K. K.; Garai B.; Gupta S. S. Electrocatalytic water oxidation by a molecular cobalt complex through a high valent cobalt oxo intermediate. Chem. Commun. 2016, 52, 11787–11790. 10.1039/C6CC05773J. [DOI] [PubMed] [Google Scholar]
- Du H.-Y.; Chen S.-C.; Su X.-J.; Jiao L.; Zhang M.-T. Redox-Active Ligand Assisted Multielectron Catalysis: A Case of CoIII Complex as Water Oxidation Catalyst. J. Am. Chem. Soc. 2018, 140, 1557–1565. 10.1021/jacs.8b00032. [DOI] [PubMed] [Google Scholar]
- Nasini U. B.; Gartia Y.; Ramidi P.; Kazi A.; Shaikh A. U.; Ghosh A. Oxygen reduction reaction catalyzed by cobalt(III) complexes of macrocyclic ligands supported on multiwalled carbon nanotubes. Chem. Phys. Lett. 2013, 566, 38–43. 10.1016/j.cplett.2013.02.046. [DOI] [Google Scholar]
- Ghosh A.; Ramidi P.; Pulla S.; Sullivan S. Z.; Collom S. L.; Gartia Y.; Munshi P.; Biris A. S.; Noll B. C.; Berry B. C. Cycloaddition of CO2 to Epoxides Using a Highly Active Co(III) Complex of Tetraamidomacrocyclic Ligand. Catal. Lett. 2010, 137, 1–7. 10.1007/s10562-010-0325-0. [DOI] [Google Scholar]
- Parnell C. M.; Watanabe F.; Nasini U. B.; Berry B. C.; Mitchell T.; Shaikh A. U.; Ghosh A. Electrochemical sensing of hydrogen peroxide using a cobalt(III) complex supported on carbonaceous nanomaterials. J. Electroanal. Chem. 2015, 740, 37–44. 10.1016/j.jelechem.2014.12.022. [DOI] [Google Scholar]
- Hong S.; Pfaff F. F.; Kwon E.; Wang Y.; Seo M.-S.; Bill E.; Ray K.; Nam W. Spectroscopic Capture and Reactivity of a Low-Spin Cobalt(IV)-Oxo Complex Stabilized by Binding Redox-Inactive Metal Ions. Angew. Chem., Int. Ed. 2014, 53, 10403–10407. 10.1002/anie.201405874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson R. E.; Gordon-Wylie S. W.; Woomer C. G.; Norman R. E.; Weintraub S. T.; Horwitz C. P.; Collins T. J. Electron-Transfer Oxidation by Phase-Separating Reagents. Inorg. Chem. 1998, 37, 4748–4750. 10.1021/ic9803661. [DOI] [PubMed] [Google Scholar]
- Conradie M. M.; Conradie J.; Gosh A. A DFT overview of high-valent iron, cobalt and nickel tetraamidomacrocyclic ligand (TAML) complexes: The end of innocence?. J. Inorg. Biochem. 2006, 100, 620–626. 10.1016/j.jinorgbio.2006.01.028. [DOI] [PubMed] [Google Scholar]
- Saavedra Díaz R. O.; Le Lagedec R.; Shen L. Q.; Ryabov A. D. In search for chelating TAMLs (tetraamido macrocyclic ligands) with peripheral bidentate donor centers: a cobalt(III) complex of the 3,3′-(2,2′-bipyridindiyl)-tailed TAML. J. Coord. Chem. 2014, 67, 3909–3919. 10.1080/00958972.2014.964224. [DOI] [Google Scholar]
- Ramidi P.; Sullivan S. Z.; Gartia Y.; Munshi P.; Griffin W. O.; Darsey J. A.; Biswas A.; Shaikh A. U.; Ghosh A. Catalytic Cyclic Carbonate Synthesis Using Epoxide and Carbon Dioxide: Combined Catalytic Effect of Both Cation and Anion of an Ionic CrV(O) Amido Macrocyclic Complex. Ind. Eng. Chem. Res. 2011, 50, 7800–7807. 10.1021/ie2003939. [DOI] [Google Scholar]
- a Evans D. F. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. 10.1039/jr9590002003. [DOI] [Google Scholar]; b Deutsch J. L.; Poling S. M. The determination of paramagnetic susceptibility by NMR: A physical chemistry experiment. J. Chem. Educ. 1969, 46, 167–168. 10.1021/ed046p167. [DOI] [Google Scholar]; c Piguet C. Paramagnetic Susceptibility by NMR: The ″Solvent Correction″ Removed for Large Paramagnetic Molecules. J. Chem. Educ. 1997, 74, 815–816. 10.1021/ed074p815. [DOI] [Google Scholar]; d Sur S. K. Measurement of magnetic susceptibility and magnetic moment of paramagnetic molecules in solution by high-field fourier transform NMR spectroscopy. J. Magn. Reson. 1989, 82, 169–173. 10.1016/0022-2364(89)90178-9. [DOI] [Google Scholar]; e Grant D. H. Paramagnetic Susceptibility by NMR: The ″Solvent Correction″ Reexamined. J. Chem. Educ. 1995, 72, 39. 10.1021/ed072p39. [DOI] [Google Scholar]; f Hoppe J. I. Effective magnetic moment. J. Chem. Educ. 1972, 49, 505. 10.1021/ed049p505. [DOI] [Google Scholar]; g Bain G. A.; Berry J. F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532–536. 10.1021/ed085p532. [DOI] [Google Scholar]
- Notably, a previous study (ref (23)) on the electrochemical behavior of Na[CoIII(TAMLred)] in MeCN displayed only one reversible oxidation event, whereas the second oxidation was found to be irreversible.
- Mak S.-T.; Wong W.-T.; Yam V. W.-W.; Lai T.-F.; Che C.-M. Cobalt(III) alkyl complexes of 1,2-bis(2-pyridinecarboxamido)benzene (H2bpb) and 4,5-dichloro-1,2-bis(2-pyridinecarboxamido)benzene (H2bpc) and X-ray crystal structures of [Co(bpc)(CH2CH2CMe = CH2)(H2O)] and [Co(bpb)Et(H2O)]. J. Chem. Soc., Dalton Trans. 1991, 1915–1922. 10.1039/DT9910001915. [DOI] [Google Scholar]
- CASSCF calculations demonstrated that the absorption band at 623 nm is dominated by ligand-centered π → π* and metal-to-ligand charge-transfer excitations. See the CASSCF section on [CoIII(TAMLsq)] for more details.
- a Connelly N. G.; Geiger W. E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]; b Boduszek B.; Shine H. J. Preparation of solid thianthrene cation radical tetrafluoroborate. J. Org. Chem. 1988, 53, 5142–4143. 10.1021/jo00256a042. [DOI] [Google Scholar]
- Lahanas N.; Kucheryavy P.; Lockard J. V. Spectroscopic Evidence for Room Temperature Interaction of Molecular Oxygen with Cobalt Porphyrin Linker Sites within a Metal–Organic Framework. Inorg. Chem. 2016, 55, 10110–10113. 10.1021/acs.inorgchem.6b01889. [DOI] [PubMed] [Google Scholar]
- Cramer C. J.Essentials of Computational Chemistry: Theories and Models, 2nd ed; John Wiley & Sons Ltd: West Sussex, U.K., 2004; pp 205–210. [Google Scholar]
- a Angeli C.; Cimiraglia R.; Evangelisti S.; Leininger T.; Malrieu J.-P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. 10.1063/1.1361246. [DOI] [Google Scholar]; b Angeli C.; Cimiraglia R.; Malrieu J.-P. N-electron valence state perturbation theory: a fast implementation of the strongly contracted variant. Chem. Phys. Lett. 2001, 350, 297–305. 10.1016/S0009-2614(01)01303-3. [DOI] [Google Scholar]; c Angeli C.; Cimiraglia R.; Malrieu J.-P. n-electron valence state perturbation theory: A spinless formulation and an efficient implementation of the strongly contracted and of the partially contracted variant. J. Chem. Phys. 2002, 117, 9138–9153. 10.1063/1.1515317. [DOI] [Google Scholar]
- We attempted to rotate the uncorrelated orbital(s) back into the active space. However, this was unsuccessful. Moreover, the inclusion of orbitals with occupancy 2.00 in the active space should be avoided.Aravena D.; Atanasov M.; Chilkuri V. G.; Guo Y.; Jung J.; Maganas D.; Mondal B.; Schapiro I.; Sivalingam K.; Ye S.; Neese F.. CASSCF Calculations in ORCA (4.2): A Tutorial Introduction. https://orcaforum.kofo.mpg.de/app.php/dlext/?cat=4 (accessed Sept 17, 2019).
- CASSCF calculations demonstrated that no intense characteristic absorption bands are expected in the 400–850 nm region. See the CASSCF section on [CoIII(TAMLq)(NNs)2]–.
- An accurate simulation of the experimental spectrum proved to be challenging and was unsuccessful.
- Olivos Suarez A. I.; Lyaskovskyy V.; Reek J. N. H.; van der Vlugt J. I.; de Bruin B. Nitrogen-Centred Ligand Radical Complexes; Classification, Spectroscopic Features, Reactivity and Catalytic Applications. Angew. Chem., Int. Ed. 2013, 52, 12510–12529. 10.1002/anie.201301487. [DOI] [PubMed] [Google Scholar]
- a Au S.-M.; Fung W.-H.; Cheng M.-C.; Che C.-M.; Peng S.-M. Synthesis, characterisation and reactivity of novel bis(tosyl)imidoruthenium(VI) porphyrin complexes; X-ray crystal structure of a tosylamidoruthenium(VI) porphyrin. Chem. Commun. 1997, 1655–1656. 10.1039/a702093g. [DOI] [Google Scholar]; b Au S.-M.; Huang J.-S.; Yu W.-Y.; Fung W.-H.; Che C.-M. Aziridination of Alkenes and Amidation of Alkanes by Bis(tosylimido)ruthenium(VI) Porphyrins. A Mechanistic Study. J. Am. Chem. Soc. 1999, 121, 9120–9132. 10.1021/ja9913481. [DOI] [Google Scholar]
- See the SI for a description of CASSCF-calculated UV–vis excitation energies and EPR g values.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.