

Abstract
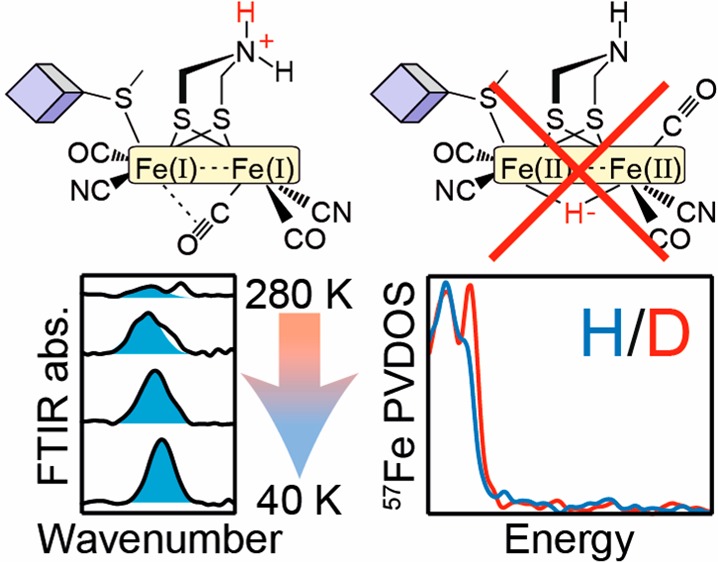
[FeFe] hydrogenases are extremely active H2-converting enzymes. Their mechanism remains highly controversial, in particular, the nature of the one-electron and two-electron reduced intermediates called HredH+ and HsredH+. In one model, the HredH+ and HsredH+ states contain a semibridging CO, while in the other model, the bridging CO is replaced by a bridging hydride. Using low-temperature IR spectroscopy and nuclear resonance vibrational spectroscopy, together with density functional theory calculations, we show that the bridging CO is retained in the HsredH+ and HredH+ states in the [FeFe] hydrogenases from Chlamydomonas reinhardtii and Desulfovibrio desulfuricans, respectively. Furthermore, there is no evidence for a bridging hydride in either state. These results agree with a model of the catalytic cycle in which the HredH+ and HsredH+ states are integral, catalytically competent components. We conclude that proton-coupled electron transfer between the two subclusters is crucial to catalysis and allows these enzymes to operate in a highly efficient and reversible manner.
Introduction
[FeFe] hydrogenases are highly active and efficient H2 conversion catalysts with turnover frequencies up to 10 000 s–1 for H2 production.1,2 The active site cofactor, the H-cluster, is composed of a unique [2Fe] subcluster ([2Fe]H) linked through a protein cysteine thiolate to a canonical [4Fe-4S] cluster ([4Fe-4S]H).3,4 The two iron ions are connected by a bridging CO and a 2-azapropane 1,3-dithiolate (ADT) ligand.5,6 Additionally, both irons are coordinated by terminal carbon monoxide (CO) and cyanide (CN–) ligands. The proximal iron (Fep), vicinal to [4Fe-4S]H, is coordinatively saturated, while the distal iron (Fed) features an open coordination site where substrates (H2 and/or H+) and inhibitors (such as CO and O2) bind.7,8 Some [FeFe] hydrogenases, including HydA1 from Chlamydomonas reinhardtii (CrHydA1), only contain the H-cluster.9,10 Other enzymes, including HydAB from Desulfovibrio desulfuricans (DdHydAB) and HydA1 from Clostridium acetobutylicum (CaHydA1 or CaI), contain additional iron–sulfur clusters (F-clusters) for exchanging electrons with redox partner proteins.11,12
The catalytic cycle of [FeFe] hydrogenases (Figure 1A) remains controversial in spite of intensive efforts by many research groups.13−15 Proton-coupled electron transfer (PCET) between the two subclusters of the H-cluster is believed to be essential for efficient and reversible catalysis.15,16 It has been demonstrated with pH-dependent FTIR spectroelectrochemistry that, for CrHydA1, two forms of the one-electron reduced state exist where the reducing equivalent is localized on either [4Fe-4S]H or [2Fe]H (Hred and HredH+, respectively).15 Electron transfer from [4Fe-4S]H to [2Fe]H is coupled to protonation of the bridgehead amine of the ADT ligand. This process was also shown to occur in DdHydAB, where it is enhanced by redox anticooperativity between [4Fe-4S]H and the F-cluster proximal to it.17 PCET between the two subclusters is also thought to play a role in formation of H2 from a terminal hydride-bound state, Hhyd,16,18 and activation of the oxygen-stable inactive state Hinact.19 PCET is disrupted in a sensory [FeFe] hydrogenase HydS from Thermotoga maritima(20) (TmHydS) or by an exchange of one of the cysteines ligating [4Fe-4S]H with histidine in CrHydA1.21
Figure 1.

(A) Putative catalytic mechanism for [FeFe] hydrogenase. The box represents the two Fe ions of the [2Fe]H subsite, while the cube represents the [4Fe-4S]H subsite. (B) The [2Fe]H subcluster is in a homovalent Fe(I)Fe(I) configuration with the bridging CO shifted to a semibridging position on Fed, and the ADT ligand in the protonated −NH2+– state, consistent with H(s)redH+. (C) The [2Fe]H subcluster is in a homovalent Fe(II)Fe(II) configuration with one of the CO ligands rotated into an apical position on Fed and a hydride in the bridging position. In the HredH+ state [4Fe-4S]H is oxidized to a 2+ state (green), and in HsredH+ it is reduced to a 1+ state (yellow).
An intriguing observation is that FTIR spectra of both the HredH+ state and the two-electron reduced state HsredH+ lack a peak corresponding to the bridging CO ligand.22−24 In agreement with this, the crystal structure of DdHydAB under reducing conditions revealed a shift in the bridging CO ligand to a semibridging position on Fed (Fed–μC distance of 1.69 Å compared with the Fep–μC distance of 2.4 Å, shown schematically in Figure 1B).5
In the past few years, a number of observations have suggested an alternative structure for both the HsredH+ and the HredH+ states. In 2014, Legér and co-workers demonstrated that CrHydA1 undergoes reversible low potential inactivation25 and attributed this conversion to the HsredH+ state. However, the time-scale required for the inactivation is on the order of minutes, while the HsredH+ state is routinely observed within seconds after reducing the enzyme.15,26 In the same year, Haumann and co-workers suggested that the HsredH+ (sred in their nomenclature, see supplementary discussion in the Supporting Information) state is best described as a bridging hydride based on site-selective X-ray absorption spectroscopy (XAS) and density functional theory (DFT) calculations (Figure 1C).27 Subsequently, the same group suggested that the HredH+ (Hred in their nomenclature) state contains a bridging hydride based on DFT simulations of FTIR26 and nuclear resonance vibrational spectra (NRVS).28 In H-cluster model systems bridging hydrides are more stable than isomeric terminal hydrides.29−32 This, and the fact that the the former would not be adjacent to the proton relaying ADT, implies that bridging hydrides would be catalytically inactive.33 As such, they have been proposed to serve a regulatory role.13,28
Despite the evidence in support of the bridging hydride structure of the HredH+ and HsredH+ states and their inactive nature,13,27,28 a few recent observations contradict this assignment. TmHydS can be poised in redox states with very similar FTIR spectra to the HsredH+ and HredH+ states but retaining the bridging CO.20 The same appears to be true for the highly active, reversible [FeFe] hydrogenase, CaI.14 Finally, a cysteine to histidine modification at [4Fe-4S]H in CrHydA1 increases the redox potential of [4Fe-4S]H, resulting in an enzyme that does not stably form the HredH+ state.21 If the HredH+ state is inactive, as has been proposed,13 then the C362H variant of CrHydA1 should show higher activity for H2 production than the wild-type enzyme. However, the C362H variant shows essentially no H2 production activity.
Clearly, new spectroscopic data are urgently required to resolve the involvement of the HredH+ and HsredH+ states. To this end, this paper described an investigation of the nature of the bridging ligand, specifically whether a (semi)bridging CO or a bridging H– ligand is present in the HredH+ and HsredH+ states. Our studies focused on the best characterized [FeFe] hydrogenases, CrHydA1 and DdHydAB. The former enzyme can be easily poised in the HsredH+ state,34 while the latter can be poised in the HredH+ state,22 both with almost complete purity. Crucially, low-temperature IR measurements reveal that both of these states feature bridging CO ligands. This result rules out the presence of a bridging hydride. Samples prepared in H2O and D2O show only subtle (<2 cm–1) differences in the CN– and CO peak positions. At the same time, NRVS studies on samples prepared in H2O and D2O show only very subtle isotope effects, with no evidence for a bridging hydride. DFT calculations give better agreement with a bridging CO structure with a protonated ADT ligand than a structure in which a hydride is bridged between the two Fe ions of the [2Fe]H subcluster. These findings have important consequences for the mechanism of H2 conversion by [FeFe] hydrogenases. Specifically, the presence of a bridging CO in the HredH+ and HsredH+ states favors their inclusion into the catalytic cycle as catalytically competent states.
Results and Discussion
In order to study the spectral properties of the HredH+ and HsredH+ states in more detail, we chose to apply variable-temperature FTIR spectroscopy. Initial experiments focused on sodium dithionite-reduced samples of CrHydA1 at pH 8, in which the HsredH+ state is highly populated.
Low-Temperature FTIR Reveals the Bridging CO in HsredH+
Figure 2 shows FTIR spectra of CrHydA1 reduced with 20 mM sodium dithionite (E ≈ −600 mV) taken at various temperatures (see Figure S1 for the full temperature range and Figure S2 for data before spline curve fitting to remove additional background). At 280 K it can be seen that CrHydA1 is predominantly in the HsredH+ state with terminal CO bands at 1882 and 1919 cm–1 and terminal CN– bands at 2026 and 2070 cm–1. Historically, an additional terminal CO band has been identified at 1954 cm–115,23,34,35 and assumed to be the fate of the missing bridging CO peak. However, the intensity of this peak varies dramatically between different reports and is unlikely to be associated with the HsredH+ state. Indeed, at 280 K we observed a low-intensity peak at 1954 cm–1 (colored yellow).
Figure 2.
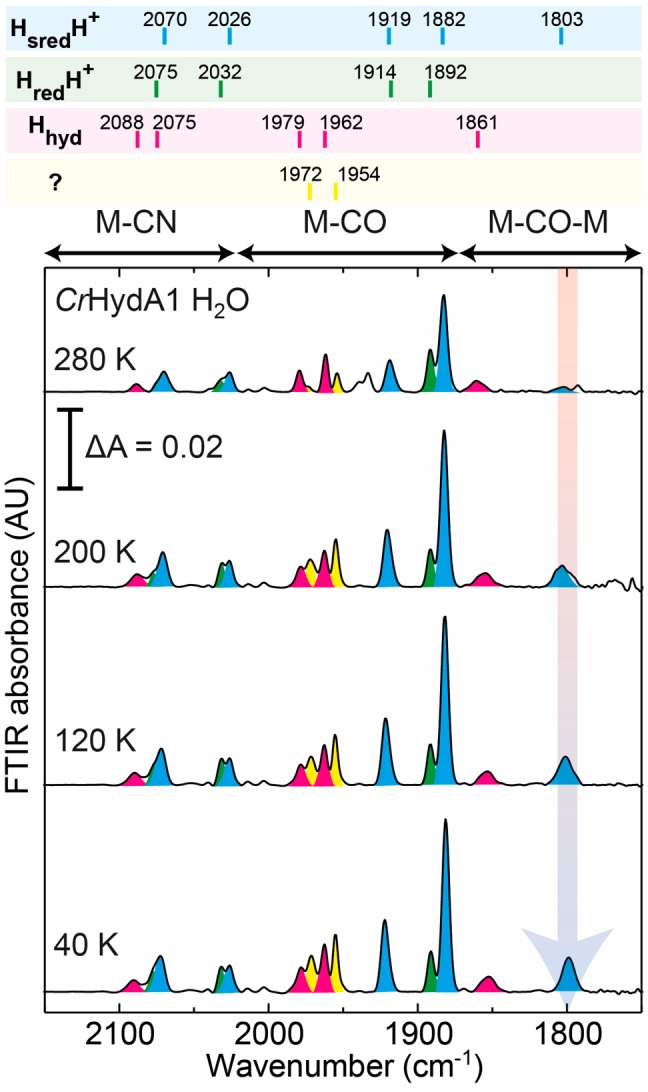
Variable-temperature FTIR spectra of CrHydA1 in H2O buffer (50 mM MES, 50 mM HEPES, 300 mM KCl, pH 8) reduced with 20 mM sodium dithionite. Peaks corresponding to the HsredH+ state are colored blue (60%). Smaller contributions from the HredH+ state (25%), the Hhyd state (10%), and an uncharacterized state similar to Hhyd (5%) are colored green, pink, and yellow, respectively.
In addition to the peaks from the HsredH+ state, we observe the most intense terminal CO peak from the HredH+ state at 1892 cm–1 and some evidence for the CN– bands of the same state as shoulders on the high-energy side of the HsredH+ CN– peaks. Very minor contributions from the Hox and Hred states can be identified from peaks at 1940 and 1934 cm–1, and a clear contribution from the Hhyd state can be observed as the pink peaks at 1861 (bridging CO) and 1962 and 1979 cm–1 (terminal CO). The highest energy CN– band from the Hhyd state is visible at 2088 cm–1, but the low-energy CN– band from Hhyd and the high-energy CN– bands from HredH+ and HsredH+ are overlapping in the 2070–2075 cm–1 region.
As the temperature is lowered, the majority of the peaks observed do not significantly change (small shifts in peak position and line width are shown in Table S1). However, one clear phenomenon can be observed: the appearance of a band at 1803 cm–1. We attribute this band to the bridging CO of the HsredH+ state, the dominant state observed at all temperatures. This assignment is supported by its similarity to a band of the HredH+ state observed for CaI.14 It is also clear that at lower temperature the peaks at 1972 and 1954 cm–1 have gained intensity. These changes also coincide with small changes to the Hhyd bridging CO peak (in the 1840–1870 cm–1 region, Figure S3). The peak is also clearly visible in spectra that have not yet been additionally background corrected with spline curve fitting (Figure S2).
The same experiment was performed in D2O and gave very similar results (Figures S4–S6). However, in D2O all the peaks of the HsredH+ state are shifted to slightly lower energies by 1–2 cm–1 (Figure S7), suggesting subtle changes to the electron density on the [2Fe]H subcluster or changes due to the vibrational coupling. These may be due to deuteration of the bridging ADT ligand, changes in hydrogen bond strength to [2Fe]H, or general changes in protein structure upon D2O exchange. The D2O sample also displayed much lower levels of additional states, yet retained similar intensity of the peak at 1803 cm–1, further supporting that this peak originates from the HsredH+ state. The lack of a 1954 cm–1 peak in the D2O spectra supports our suggestion that this peak does not originate from HsredH+.
Low-Temperature FTIR Reveals the Bridging CO in HredH+
FTIR measurements on the HredH+ state focused on the DdHydAB enzyme because CrHydA1-derived samples contain multiple states (Hox, HsredH+, and Hred). Conveniently, the DdHydAB enzyme, when reduced with 20 mM sodium dithionite at pH 8, produced almost exclusively the HredH+ state. This is because of redox-anticooperativity between the H-cluster and the proximal F-cluster, which lowers the intrinsic redox potential of [4Fe-4S]H.17 This effect destabilizes both Hred and HsredH+ in DdHydAB and favors HredH+.
At room temperature, peaks can be observed at 1894, 1916, 2041, and 2079 cm–1, corresponding to the terminal CO and CN– ligands (Figure 3; see also Figures S8 and S9). Small contributions can be observed from the Hox-CO state (2088, 2075, 2016, 1972, 1963, and 1810 cm–1) and some unknown states, including a large broad feature around 1984 cm–1. At lower temperatures the main change observed is the increase in intensity of the feature at 1810 cm–1, assigned to the bridging CO from the HredH+ state, with some additional subtle changes to peak positions and line widths (Table S1). The peak occurs at 7 cm–1 higher energy compared with the bridging CO in the HsredH+ state of CrHydA1, similar to the other CO and CN– peaks. The peak is, however, overlapping with the bridging CO from the Hox-CO state, which is already an observable contribution at high temperature.
Figure 3.
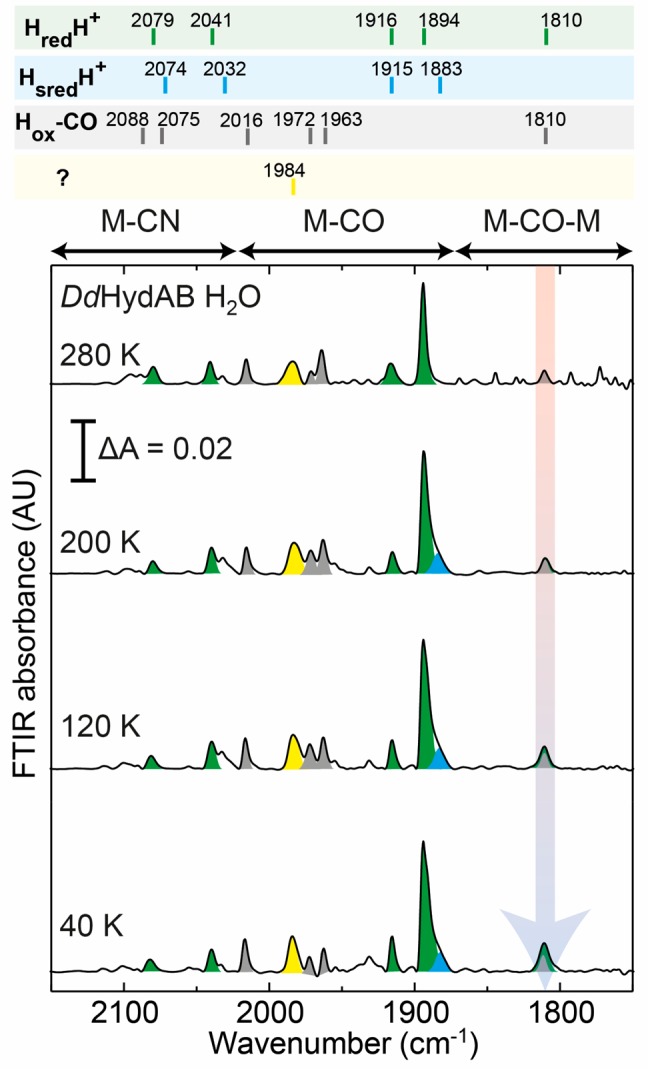
Variable-temperature FTIR spectra of DdHydAB in H2O buffer (50 mM MES, 50 mM HEPES, 300 mM KCl, pH 8) reduced with 20 mM sodium dithionite. The peaks corresponding to the HredH+ state are colored green. Small contributions from the HsredH+ state, the Hox-CO state, and an uncharacterized state are colored blue, gray, and yellow, respectively.
Samples prepared in D2O behaved similarly to those in H2O (Figures S10–S12), but with a smaller Hox-CO contribution. However, the more intense bridging CO peak at 1810 cm–1 further highlights that this peak originates from the HredH+ state. Again, only small shifts in the peaks of the HredH+ state were observed following D2O exchange, this time to higher energy (Figure S13).
To summarize, both the HsredH+ and HredH+ states of the [FeFe] hydrogenase appear to contain a bridging CO ligand. Meanwhile, none of the other peaks undergo particularly large shifts concomitant with the appearance of the bridging CO peak, excluding the possibility that a large conformational change occurs upon cooling the sample. Even if this were the case, the previous XAS and NRVS studies, which were used to propose the bridging hydride, were also performed on frozen samples under cryogenic conditions and should, therefore, contain a bridging CO.27,28 An IR H/D isotope effect, like that observed in the Hhyd state,16 which is well known to contain a terminal Fe hydride,18,36−38 could not be observed here. This further suggests that the HredH+ and HsredH+ states do not contain a bridging hydride.
DFT Calculations Reproduce Experimental FTIR Spectra of HredH+ and HsredH+ Using a Bridging CO Model
DFT calculations using bridging CO (μCO for HsredH+ and μCO+ for HredH+) and bridging hydride (μH for HsredH+ and μH+ for HredH+) models (Figure 4) were used to calculate FTIR spectra (Figure 5). These DFT models are compared in Figures S14–S17, with their additional characterization provided in Tables S2–S5. Notably, the models μCO and μCO+ produced vanishingly small spin populations at the two [2Fe]H Fe(I) sites, indicating the Fe(I)–Fe(I) metal–metal bonding as depicted in Figure 1B. The calculated bands for HsredH+ using the μCO model are at 2077 and 2009 cm–1 for the CN– ligands, 1940 and 1871 cm–1 for the terminal CO ligands, and 1801 cm–1 for the bridging CO ligand. The 1871 and 1801 cm–1 C–O modes display a minor degree of vibrational coupling (see Supporting Information for the normal mode animations). These values are quite close to the experimental values of 2070, 2026, 1919, 1882, and 1803 cm–1. While the C–O/N stretch vibrational energies of the terminal ligands assigned to Fep are somewhat overestimated, the energies of the terminal ligands assigned to Fed are underestimated (Table S4). Notably, the relative IR intensities of the observed bands were reproduced. Given the limitations of DFT, we find the agreement with the experiment to be very good. Importantly, while the calculations predict that the CO ligand retains its bridging character in the HsredH+ (and the HredH+) model, its carbon shifts 0.1 Å closer to Fed and 0.2 Å away from Fep when compared to, for example, the Hhyd state, as detailed in Table S2 and schematized in Figure 1B.
Figure 4.
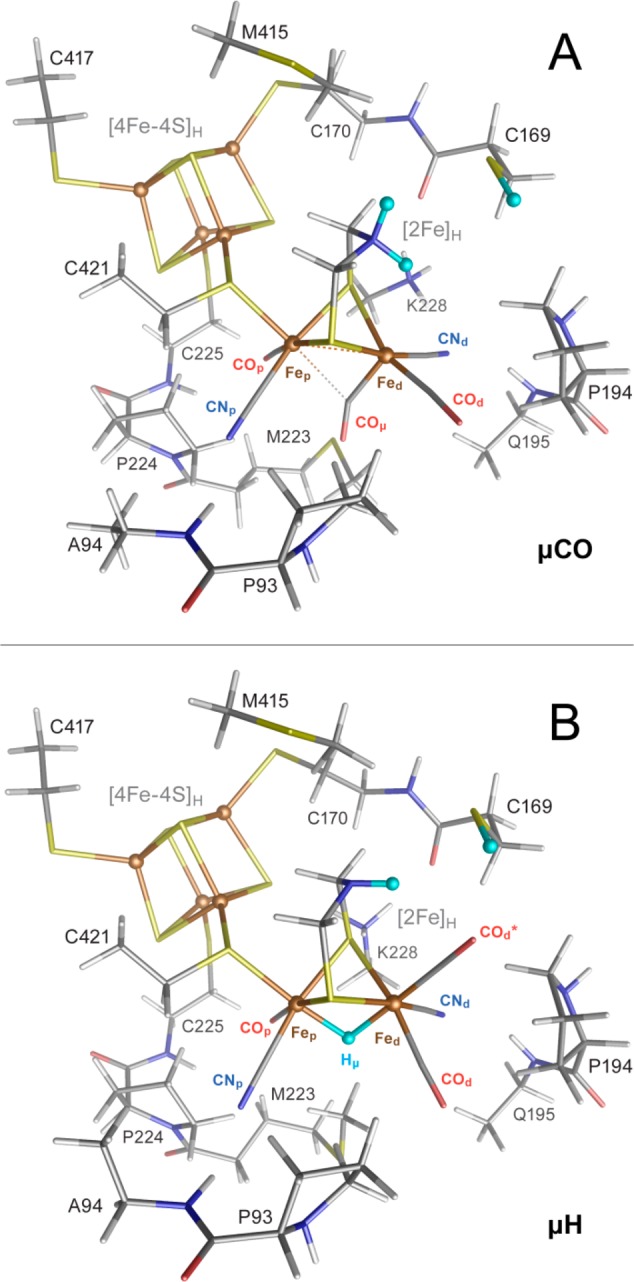
Optimized structures from DFT calculations on the μCO (A) and μH (B) models shown in tube representation. Additionally, in ball representation are indicated the Fe sites and the three H-to-D exchangeable protons (light blue). Single-letter amino acid labeling corresponds to the CrHydA1 enzyme sequence. Element colors are C (gray), H (white), N (blue), O (red), Fe (brown), and S (yellow). For extra details and DFT models of μCO+ and μH+, see Figures S14–S17.
Figure 5.

Experimental and DFT-calculated IR spectra of the HredH+ and HsredH+ states in H2O. The experimental spectra are the 40 K spectra from the HredH+ state of DdHydAB (Figure 3) and CrHydA1 in the HsredH+ state (Figure 2). The calculated μCO and μCO+ spectra were generated from DFT calculations on a bridging CO model (Figure 4A). The μH and μH+ spectra were generated from DFT calculations on a bridging hydride model (Figure 4B). The peaks corresponding to the HredH+ and HsredH+ states are colored green and blue, respectively. See Table S4 for comparison of experimental and calculated IR frequencies and assignment to the ligands on the proximal and distal Fe.
Calculations on a bridging hydride variant (μH) with two terminal CO ligands on Fed give radically different FTIR spectra with CN bands at 2088 and 2073 cm–1 and terminal CO bands at 1996 and 1929 cm–1. The high values compared with the bridging CO model are due to the fact that hydride formation leaves the two Fe ions formally in Fe(II) oxidation states (Figure 1C). The prediction of just two CO bands is due to an overlay of the terminal (at Fep and apical at Fed) C–O modes at ∼1929 cm–1, which become significantly mixed in the absence of the bridging CO ligand. The calculated spectra for the deuterated forms were only very subtly different from those of the protonated forms (Figure S18).
The calculated bands for HredH+ using a bridging CO (μCO+) model are at 2092 and 2007 cm–1 for the CN– ligands, 1940 and 1879 cm–1 for the terminal CO ligands, and 1822 cm–1 for the bridging CO ligand. These are quite similar to the calculated bands for HsredH+ but shifted to higher energy by up to 21 cm–1. The band positions, relative intensities, and most of the shifts relative to HsredH+ capture the trends observed in the experimental spectra. In both the μH+ vs μH and μCO+ vs μCO spectra comparisons, the IR band shifts are governed solely by the redox state of the [4Fe-4S]H subcluster, while the [2Fe]H subcluster harboring the CO/CN ligands remains in the same formal state. Our calculations indicate that, upon reduction of μCO+ to μCO, the [4Fe-4S]H fragment charge shifts by ∼0.9 units, and [2Fe]H obtains the remaining ∼0.1 units of the added electron density (Table S5). Calculations with a bridging hydride variant μH+ gave IR bands similar to those produced for the μH model. However, the vibrational energies of the terminal CO ligands are better separated in μH+ giving bands at 1940 and 1926 cm–1. Importantly, the bridging hydride models do not produce IR spectra remotely reminiscent of the experimental spectra.
The complete assignment of the bands for the HredH+ and HsredH+ states makes clear that the bridging CO does not move to a terminal apical position on Fed giving an FTIR peak around 1950–1960 cm–1. Therefore, the origin of the 1954 cm–1 peak in the reduced CrHydA1 and DdHydAB samples needs to be explained. We observed that the increase in intensity of the 1954 cm–1 peak in reduced CrHydA1 correlated with an increase in intensity of a peak at 1972 cm–1 and changes to the bridging CO peak associated with the Hhyd state. Therefore, we believe these peaks arise from an alternative form of the Hhyd state. A thorough assignment will require obtaining this state in higher purity, probably by performing cryogenic FTIR experiments on CrHydA1 samples under various reducing conditions. An important outcome of the present work is that samples measured at room temperature (e.g., with FTIR) cannot be directly compared with samples measured at cryogenic temperatures. A number of recent results will need to be revisited in light of this finding. Recently there have been numerous spectroscopic investigations into the Hhyd state, identifying it as a terminal hydride intermediate.16,18,36−39 However, IR measurements have never been performed at similarly low temperatures to those used for EPR, Mössbauer, and NRVS spectroscopies and could provide further insight.
NRVS and DFT Calculations Support a Bridging CO Structure for the H-Cluster in HsredH+ and HredH+
To test our assignment of the HredH+ and HsredH+ states, NRVS measurements were conducted on samples of CrHydA1 and DdHydAB. Samples were poised under identical conditions as for the FTIR samples, transferred to NRVS cells, and frozen in liquid nitrogen. To ensure high sensitivity, the [2Fe]H subsite was selectively labeled with 57Fe.
Figure 6 (and Figure S19) shows NRVS spectra for CrHydA1 and DdHydAB in the HsredH+ and HredH+ states, respectively, under H2/H2O (blue traces) and D2/D2O (red traces). Spectra measured in H2O and D2O in both enzymes look very similar. Importantly, at high energy (600–800 cm–1) no peaks can be observed in the H2O samples. Previous studies on the terminal hydride Hhyd state of the [FeFe] hydrogenase18,37,38 and the bridging hydride Ni-R state of the [NiFe] hydrogenase,40 as well as either terminal or bridging hydride containing diiron model complexes,41 show distinct peaks in this region. For CrHydA1 with an oxadithiolate (ODT) bridge the peaks at 670 and 727 cm–1 were assigned to bending motions of the terminal hydride correspondingly out-of-plane and in-plane, with respect to the pseudosymmetry plane of the [2Fe]H subcluster, respectively.38CrHydA1 with the native ADT cofactor showed the same pair of peaks but shifted to higher energy (675 and 744 cm–1), as did DdHydAB (675 and 747 cm–1, see Figure S20).18 In the C169S variant of CrHydA1, the in-plane peak is shifted to even higher energy (772 cm–1), while the out-of-plane peak is barely perturbed by the amino acid substitution (673 cm–1).37 For the [NiFe] hydrogenase in the Ni-R state a peak was observed at 675 cm–1,40 assigned to wagging motion of the bridging hydride. For a diiron model complex synthesized with a hydride in either terminal or bridging positions, Fe–H bending and Fe–H–Fe wagging NRVS intensities were observed in the 720–800 and 670–700 cm–1 regions.41 Upon H-to-D isotope exchange, these previously identified iron-hydride vibrations show large (up to ∼200 cm–1) shifts to lower energies, where they additionally perturb high-intensity Fe-CO bands.42 The fact that no such H2O vs D2O NRVS signatures are presently observed in either the HsredH+ or HredH+ samples directly contradicts the idea that Fe–H bonds exist in these states.
Figure 6.

NRVS of CrHydA1 in the HsredH+ state (A and B) in H2O buffer (50 mM MES, 50 mM HEPES, 300 mM KCl, pH 8) (blue traces) and D2O buffer (50 mM MES, 50 mM HEPES, 300 mM KCl, pD 8) (red traces) reduced with 20 mM sodium dithionite. NRVS of DdHydAB in the HredH+ state (C and D) in H2O buffer (50 mM MES, 50 mM HEPES, 300 mM KCl, pH 8) (blue traces) and D2O buffer (50 mM MES, 50 mM HEPES, 300 mM KCl, pD 8) (red traces) reduced with 20 mM sodium dithionite. B and D are expansions of the high-energy region from A and C, respectively. Error bars have been omitted for clarity. The regions of the spectrum corresponding to Fe-protein, Fe-[4Fe-4S]H, Fe-S, Fe-CN, Fe-CO, Fe-D, and Fe-H are indicated in A. Data including error bars are presented in Figure S19.
Figure 7A and B show an overlay of DFT calculations of the HsredH+ H2O spectra generated using both a bridging CO model (μCO) and a bridging hydride model (μH). The experimental spectra are best reproduced by the μCO model. Notably, the μH model predicts an NRVS-detectable Fe–H–Fe mode at 738 cm–1, which is not experimentally observed. The calculations indicate that this peak is above the level of noise in the experimental data (Figure S21). H-to-D isotopically labeled (as specified in Figure 4) models μCOD and μD were used to calculate the D2O sample spectra, and again the μCOD model gave better agreement to the experimental data. The μCO/μCOD models suggest only very minor changes upon H/D exchange (Figure S22A), while the μH/D models suggest much larger changes (Figure S22B), particularly in the Fe-CO region (400–650 cm–1), which are not experimentally observed. Likewise, DFT calculations on μCO+ and μH+ (now with an oxidized [4Fe-4S]H2+ subcluster) models were used to calculate the spectra of HredH+ (Figure 7C and D and Figure S23). The μCO+ model provided better agreement with experimental data than the μH+ model. The μH+ model predicts an NRVS-detectable Fe–H–Fe mode now at 716 cm–1; however, a corresponding band is not produced by the HredH+ sample. Additionally, the μCO+/μCOD+ models, similarly to μCO/μCOD, suggest only small changes upon H/D exchange (Figure S24A), while the μH+/μD+ models suggest larger changes (Figure S24B). Notably, the bridging hydride models, formally different only in the [4Fe-4S]H oxidation level ([4Fe-4S]H2+ in μH+ vs [4Fe-4S]H1+ in μH), display a substantial 22 cm–1 deviation in the Fe–H–Fe wagging mode position. This highlights the sensitivity of the iron-hydride bands in [FeFe] hydrogenase to even subtle changes in the electronic structure and environment, as previously noted.18,37 A supplementary discussion in the Supporting Information additionally compares the NRVS-observed and DFT-calculated 57Fe-PVDOS spectra of HredH+ and HsredH+ vs that of the earlier reported Hhyd state18 (Figure S25), adding consistency to the present results.
Figure 7.

DFT calculations of the NRVS spectra in the HsredH+ state (A and B) and HredH+ state (C and D). The experimental NRVS spectra of CrHydA1 in the HsredH+ state (A and B) and DdHydAB in the HredH+ state (C and D) are shown in blue, while the DFT-calculated spectra generated using bridging CO (μCO and μCO+) models (Figure 4A) are shown in black, and the DFT-calculated spectra generated using bridging hydride (μH and μH+) models (Figure 4B) are shown in green. B and D are expansions of the high-energy regions of A and C. Experimental error bars have been omitted for clarity. Data including error bars are presented in Figures S21 and S23. See Figure S20 for an alternative comparison of the DFT-calculated and NRVS-observed 57Fe-PVDOS spectra.
To summarize, FTIR and NRVS spectra coupled with DFT calculations show clear evidence for the presence of a bridging CO in the HredH+ and HsredH+ states of the [FeFe] hydrogenase. These data exclude the possibility of a bridging hydride in these states. Our DFT rationalization of the IR signature with the prominent 1800–1820 cm–1 feature appears to be satisfactory only when using the bridging CO H-cluster model. An absence of this low-frequency band in the IR reference may support a “rotated” isomer model having terminal CO ligands only, thus implying the bridging hydride.26−28 Furthermore, the previous analysis assumed that peaks in the 1950–1970 cm–1 region derive from the HredH+ and HsredH+ states. In contrast, we suggest that these peaks are attributed to an alternative Hhyd state, and instead we find a peak in the 1800–1820 cm–1 region, which we assign to the bridging CO in the HredH+ and HsredH+ states. We believe that if the present findings were accounted for in Haumann and co-workers’ model, they may find that a μCO model is in better agreement with experimental data than a μH model.
We contend that there are no strong data indicating that the HredH+ and HsredH+ states should be considered inactive. A number of observations hint that these states are, in fact, likely to be catalytically relevant. Under H2, CrHydA1 exists in a mixture of Hred, HredH+, and HsredH+ states,34 while DdHydAB is almost completely in the HredH+ state.22 Neither enzyme shows significant lag-phases during H2 oxidation43,44 or H2 production,2,10 suggesting that these states are catalytically active. Otherwise, the published activities of 1000–150 000 s–1 2,10,43,44 must represent the low activity of the inactive HredH+ and HsredH+ states, which seems unlikely. Formation of HredH+ and HsredH+ requires reducing conditions at neutral to low pH, which are the conditions used for optimal H2 production. DdHydAB has a turnover frequency of 10 000 s–1 for H2 production2 and 150 000 s–1 for H2 oxidation43 and forms almost exclusively the HredH+ state under reducing conditions at neutral pH.22 Thus, if HredH+ is essentially inactive, then the true activity of the DdHydAB should be much higher than 10 000 s–1 for H2 production and much higher than 150 000 s–1 for H2 oxidation, which also seems unlikely. Recently, time-resolved IR measurements of the rate of formation and decay of the various catalytic intermediates demonstrated the catalytic relevance of the HredH+ and HsredH+ states in CrHydA1.45
The reason that the bridging CO has been so difficult to observe in the HredH+ and HsredH+ states at room temperature is unclear. It may be related to flexibility of this ligand or coupling with other vibrational modes. Either of these two effects could potentially affect the oscillating dipole moment and thus the extinction coefficient for the IR transition. Since the local structure around the H-cluster may change slightly as the temperature is lowered, the bridging CO ligand could become more rigid, leading to a larger oscillating dipole moment and hence increased IR intensity. Notable in this regard is that the present DFT methodology for the normal-mode analysis and spectral simulations best corresponds to zero-point vibrations at absolute zero. Studies on CO-bound heme proteins have shown similar temperature-dependent peak broadening and have interpreted this as increased conformational flexibility at higher temperature, populating multiple conformational substates.46,47 In agreement with this, the bridging CO can still be observed in the Hred* and Hsred* states of TmHydS at room temperature.20 This thermostable [FeFe] hydrogenase may have decreased conformational flexibility around the H-cluster and thus fewer conformational substates.
Another interesting feature of the bridging CO ligands in the HredH+ and HsredH+ states is their relatively high vibrational energy. The IR bands from the terminal CO and CN– ligands generally shift to lower energy as [2Fe]H becomes more reduced, due to increased metal–ligand π back-donation. However, the bridging CO in the HredH+ state has a higher energy IR vibration (1810 cm–1) than the Hox state (1802 cm–1). This may also reflect structural changes to the bridging CO ligand upon [2Fe]H reduction that prevent the increased π back-donation. Further IR studies coupled with DFT calculations and studies on amino acid variants may provide answers to these questions.
Conclusions
This paper clarifies the structure of two controversial states invoked for the [FeFe] hydrogenases, a large family of highly active H2 conversion catalysts. Specifically, new spectroscopic and computational evidence is presented that both HredH+ and HsredH+ contain bridging CO ligands, the presence of which precludes bridging hydride ligands.13,26−28 We propose that the HredH+ and HsredH+ states are catalytically relevant intermediates. The results support the overall hypothesis that [FeFe] hydrogenases operate via terminal hydride-containing intermediates, which are rendered hydridic or acidic by proton-coupled electron transfer between the two halves of the H-cluster.15,17,21
Methods
Protein overproduction and artificial maturation were performed as described previously.48−50 (Et4N)2[Fe2(ADT)(CO)4(CN)2] ((Et4N)2[2Fe]ADT) and (Et4N)2[57Fe2(ADT)(CO)4(CN)2] ((Et4N)2[257Fe]ADT) were synthesized following published procedures.51 FTIR experiments were performed using a Bruker Vertex 80v FTIR spectrometer equipped with a nitrogen-cooled Bruker mercury cadmium telluride detector, liquid helium cooled OPTIA Optistat CF continuous flow cryostat system, liquid helium transfer line, and an Oxford Instruments ITC503 temperature controller. In an anaerobic chamber, samples (5 μL) of 1–2 mM CrHydA1 or DdHydAB in 50 mM MES, 50 mM HEPES, and 300 mM KCl, pH 8, containing 20 mM sodium dithionite were loaded between CaF2 windows separated by a 50 μM Teflon spacer and closed inside a brass holder with rubber rings. Spectra were collected in the double-sided, forward–backward mode with a resolution of 2 cm–1, an aperture setting of 1.5 mm, and a scan velocity of 20 Hz. Data were processed using home-written routines in the MATLAB environment.
NRVS spectra for [257Fe]H-CrHydA1 and [257Fe]H-DdHydAB in 50 mM MES, 50 mM HEPES, and 300 mM KCl, pH/D 8, containing 20 mM sodium dithionite were recorded at SPring-8 BL09XU and BL19LXU. BL09XU uses a Si(111) double crystal in a high heat load monochromator to produce 14.414 keV radiation with ∼1.0 eV resolution, followed by a high energy resolution monochromator [Ge(422)x2Si(975)] to increase the resolution to ∼0.8 meV. The beam flux was ∼2.5 × 109 photons/s, and the beam size was about 0.6 (height) × 1 (width) mm2. A 2 × 2 avalanche photodiode detector array was used to collect the delayed nuclear fluorescence and the Kδ fluorescence following nuclear excitation. The temperature at the base of the sample was maintained at 10 K with a closed cycle He cryostat. The Stokes/anti-Stokes imbalance derived real sample temperatures were 40–70 K. The setup at BL19LXU is similar to that at BL09XU except the average photo flux was ∼5.4 × 109 photons/s and a liquid He cryostat was used. NRVS spectral analysis was performed using the PHOENIX software package executed through Spectratools.52 Energy scale calibration was performed with a standard sample of [NEt4][57FeCl4] with a prominent peak at 380 cm–1. Scans were divided into segments with different data collection times. In general, 1 s per point (s/p) was used for −200 to 400 cm–1, 10 s/p for 400–600 cm–1, and 30 s/p for 600–800 cm–1. The scan ranges are all relative to the resonance energy.
The initial coordinates used for the DFT calculations were based on the 1.73 Å resolution 5BYQ X-ray data for semisynthetic Clostridium pasteurianum [FeFe] hydrogenase (CpI) maturated with the ODT variant of the [2Fe]H subcluster (CpIODT).53 A single serine side chain (S232 in CpI) had to be modified to alanine (A94 in CrHydA1). The approach used in the current work is analogous to our H-cluster DFT model construction described previously18,37 and denoted as L′ (Large prime). L′ corresponds to a molecular system including (i) a [2Fe]H subcluster, (ii) its immediate protein environment, and (iii) the [4Fe–4S]H subcluster as shown in Figures S14–S17. The structural optimizations and subsequent normal-mode calculations were performed using GAUSSIAN 09 Revision D.0154 based on the densities exported from single-point calculations performed by JAGUAR 9.4,55 which provided a high-quality initial guess. The PBE056,57 hybrid functional was employed in its unrestricted open-shell formulation. The LACV3P** basis set as implemented in JAGUAR was employed. For the first- and second-row elements, LACV3P** implies 6-311G** triple-ζ basis sets including polarization functions. For the Fe atoms, LACV3P** consists of a triple-ζ basis set for the outermost core and valence orbitals and the quasi-relativistic Los Alamos effective core potential for the innermost electrons. The molecular systems environment was considered using a self-consistent reaction field polarizable continuum model and integral equation formalism (IEF-PCM)58 as implemented in GAUSSIAN 09, with the static dielectric constant set to ε = 4.0 as often used for proteins, and the remaining IEF-PCM parameters at their default values for water. Furthermore, the computational scheme included two-body D3 empirical dispersion correction by Grimme et al. in its original formulation.59 The 57Fe-PVDOS and IR intensities were extracted from normal-mode outputs using an in-house program, Q-SPECTOR, successfully applied to simulate the [FeFe] hydrogenase NRVS spectra in previous works.18,37,38 To empirically account for the experimental line shape, the computed intensities were broadened by Lorentzian convolution with a full width at half-maximum (fwhm) = 14 cm–1 for 57Fe-PVDOS and by pseudo-Voigtian convolution with an fwhm = 8 cm–1 for IR. For 57Fe-PVDOS, empirical scaling by 0.94 was applied to the calculated frequencies in the >400 cm–1 region. For IR, the calculated CO/CN frequencies from the representative μCO+ and μCO models were fit linearly to the observed HredH+ and HsredH+ FTIR band positions, producing the 0.877 slope and +140.88 cm–1 offset parameters. For the H-cluster model as described above, H/D isotope exchange implied proton(s) of the ADT bridgehead, thiol proton of the adjacent cysteine (C169/178 in the CrHydA1/DdHydAB sequences, respectively), and the hydride if present, as indicated in Figure 4.
Acknowledgments
This work was supported by NIH GM-65440 (S.P.C.) and NIH GM-61153 (T.B.R.), by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC 2008/1 (UniSysCat) – 390540038 (V.P.), and by the Max Planck Society (J.A.B., W.L., and S.D.). S.D. and J.A.B. acknowledge funding from the DFG SPP 1927 “Iron–Sulfur for Life” project (Project Nos. DE 1877/1-1 and BI 2198/1-1). NRVS data collection was supported by a long-term proposal at BL09XU [2018B0141] and a general proposal at BLXU19 [2018B1379] at SPring-8. The authors thank Nina Breuer for help with enzyme preparation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b09745.
Author Contributions
¶ J.A.B. and V.P. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Lubitz W.; Ogata H.; Rüdiger O.; Reijerse E. Hydrogenases. Chem. Rev. 2014, 114 (8), 4081–4148. 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- Glick B. R.; Martin W. G.; Martin S. M. Purification and properties of the periplasmic hydrogenase from Desulfovibrio desulfuricans. Can. J. Microbiol. 1980, 26 (10), 1214–1223. 10.1139/m80-203. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Lanzilotta W. N.; Lemon B. J.; Seefeldt L. C. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 1998, 282 (5395), 1853–1858. 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- Nicolet Y.; Piras C.; Legrand P.; Hatchikian C. E.; Fontecilla-Camps J. C. Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Structure 1999, 7 (1), 13–23. 10.1016/S0969-2126(99)80005-7. [DOI] [PubMed] [Google Scholar]
- Nicolet Y.; de Lacey A. L.; Vernède X.; Fernandez V. M.; Hatchikian E. C.; Fontecilla-Camps J. C. Crystallographic and FTIR spectroscopic evidence of changes in Fe coordination upon reduction of the active site of the Fe-only hydrogenase from Desulfovibrio desulfuricans. J. Am. Chem. Soc. 2001, 123 (8), 1596–1601. 10.1021/ja0020963. [DOI] [PubMed] [Google Scholar]
- Silakov A.; Wenk B.; Reijerse E.; Lubitz W. 14N HYSCORE investigation of the H-cluster of [FeFe] hydrogenase: evidence for a nitrogen in the dithiol bridge. Phys. Chem. Chem. Phys. 2009, 11 (31), 6592–6599. 10.1039/b905841a. [DOI] [PubMed] [Google Scholar]
- Lemon B. J.; Peters J. W. Binding of exogenously added carbon monoxide at the active site of the iron-only hydrogenase (CpI) from Clostridium pasteurianum. Biochemistry 1999, 38 (40), 12969–73. 10.1021/bi9913193. [DOI] [PubMed] [Google Scholar]
- Swanson K. D.; Ratzloff M. W.; Mulder D. W.; Artz J. H.; Ghose S.; Hoffman A.; White S.; Zadvornyy O. A.; Broderick J. B.; Bothner B.; King P. W.; Peters J. W. [FeFe]-hydrogenase oxygen inactivation is initiated at the H cluster 2Fe subcluster. J. Am. Chem. Soc. 2015, 137 (5), 1809–1816. 10.1021/ja510169s. [DOI] [PubMed] [Google Scholar]
- Happe T.; Naber J. D. Isolation, characterization and N-terminal amino acid sequence of hydrogenase from the green alga Chlamydomonas reinhardtii. Eur. J. Biochem. 1993, 214 (2), 475–481. 10.1111/j.1432-1033.1993.tb17944.x. [DOI] [PubMed] [Google Scholar]
- Kamp C.; Silakov A.; Winkler M.; Reijerse E. J.; Lubitz W.; Happe T. Isolation and first EPR characterization of the [FeFe]-hydrogenases from green algae. Biochim. Biophys. Acta, Bioenerg. 2008, 1777 (5), 410–416. 10.1016/j.bbabio.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Adams M. W.; Eccleston E.; Howard J. B. Iron-sulfur clusters of hydrogenase I and hydrogenase II of Clostridium pasteurianum. Proc. Natl. Acad. Sci. U. S. A. 1989, 86 (13), 4932–4936. 10.1073/pnas.86.13.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande H. J.; Dunham W. R.; Averill B.; Van Dijk C.; Sands R. H. Electron paramagnetic resonance and other properties of hydrogenases isolated from Desulfovibrio vulgaris (strain Hildenborough) and Megasphaera elsdenii. Eur. J. Biochem. 1983, 136 (1), 201–7. 10.1111/j.1432-1033.1983.tb07727.x. [DOI] [PubMed] [Google Scholar]
- Haumann M.; Stripp S. T. The molecular proceedings of biological hydrogen turnover. Acc. Chem. Res. 2018, 51 (8), 1755–1763. 10.1021/acs.accounts.8b00109. [DOI] [PubMed] [Google Scholar]
- Ratzloff M. W.; Artz J. H.; Mulder D. W.; Collins R. T.; Furtak T. E.; King P. W. CO-bridged H-cluster intermediates in the catalytic mechanism of [FeFe]-hydrogenase CaI. J. Am. Chem. Soc. 2018, 140 (24), 7623–7628. 10.1021/jacs.8b03072. [DOI] [PubMed] [Google Scholar]
- Sommer C.; Adamska-Venkatesh A.; Pawlak K.; Birrell J. A.; Rüdiger O.; Reijerse E. J.; Lubitz W. Proton coupled electronic rearrangement within the H-cluster as an essential step in the catalytic cycle of [FeFe] hydrogenases. J. Am. Chem. Soc. 2017, 139 (4), 1440–1443. 10.1021/jacs.6b12636. [DOI] [PubMed] [Google Scholar]
- Mulder D. W.; Ratzloff M. W.; Bruschi M.; Greco C.; Koonce E.; Peters J. W.; King P. W. Investigations on the role of proton-coupled electron transfer in hydrogen activation by [FeFe]-hydrogenase. J. Am. Chem. Soc. 2014, 136 (43), 15394–15402. 10.1021/ja508629m. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Maciá P.; Pawlak K.; Rüdiger O.; Reijerse E. J.; Lubitz W.; Birrell J. A. Intercluster redox coupling influences protonation at the H-cluster in [FeFe] hydrogenases. J. Am. Chem. Soc. 2017, 139 (42), 15122–15134. 10.1021/jacs.7b08193. [DOI] [PubMed] [Google Scholar]
- Pelmenschikov V.; Birrell J. A.; Pham C. C.; Mishra N.; Wang H.; Sommer C.; Reijerse E.; Richers C. P.; Tamasaku K.; Yoda Y.; Rauchfuss T. B.; Lubitz W.; Cramer S. P. Reaction coordinate leading to H2 production in [FeFe]-hydrogenase identified by nuclear resonance vibrational spectroscopy and density functional theory. J. Am. Chem. Soc. 2017, 139 (46), 16894–16902. 10.1021/jacs.7b09751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Maciá P.; Reijerse E. J.; van Gastel M.; DeBeer S.; Lubitz W.; Rüdiger O.; Birrell J. A. Sulfide protects [FeFe] hydrogenases from O2. J. Am. Chem. Soc. 2018, 140 (30), 9346–9350. 10.1021/jacs.8b04339. [DOI] [PubMed] [Google Scholar]
- Chongdar N.; Birrell J. A.; Pawlak K.; Sommer C.; Reijerse E. J.; Rüdiger O.; Lubitz W.; Ogata H. Unique spectroscopic properties of the H-cluster in a putative sensory [FeFe] hydrogenase. J. Am. Chem. Soc. 2018, 140 (3), 1057–1068. 10.1021/jacs.7b11287. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Maciá P.; Kertess L.; Burnik J.; Birrell J. A.; Hofmann E.; Lubitz W.; Happe T.; Rüdiger O. His-ligation to the [4Fe-4S] subcluster tunes the catalytic bias of [FeFe] hydrogenase. J. Am. Chem. Soc. 2019, 141 (1), 472–481. 10.1021/jacs.8b11149. [DOI] [PubMed] [Google Scholar]
- Pierik A. J.; Hulstein M.; Hagen W. R.; Albracht S. P. A low-spin iron with CN and CO as intrinsic ligands forms the core of the active site in [Fe]-hydrogenases. Eur. J. Biochem. 1998, 258 (2), 572–578. 10.1046/j.1432-1327.1998.2580572.x. [DOI] [PubMed] [Google Scholar]
- Roseboom W.; de Lacey A. L.; Fernandez V. M.; Hatchikian E. C.; Albracht S. P. The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. II. Redox properties, light sensitivity and CO-ligand exchange as observed by infrared spectroscopy. JBIC, J. Biol. Inorg. Chem. 2006, 11 (1), 102–118. 10.1007/s00775-005-0040-2. [DOI] [PubMed] [Google Scholar]
- Katz S.; Noth J.; Horch M.; Shafaat H. S.; Happe T.; Hildebrandt P.; Zebger I. Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution. Chem. Sci. 2016, 7 (11), 6746–6752. 10.1039/C6SC01098A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajj V.; Baffert C.; Sybirna K.; Meynial-Salles I.; Soucaille P.; Bottin H.; Fourmond V.; Léger C. FeFe hydrogenase reductive inactivation and implication for catalysis. Energy Environ. Sci. 2014, 7 (2), 715–719. 10.1039/C3EE42075B. [DOI] [Google Scholar]
- Mebs S.; Senger M.; Duan J.; Wittkamp F.; Apfel U. P.; Happe T.; Winkler M.; Stripp S. T.; Haumann M. Bridging hydride at reduced H-cluster species in [FeFe]-hydrogenases revealed by infrared spectroscopy, isotope editing, and quantum chemistry. J. Am. Chem. Soc. 2017, 139 (35), 12157–12160. 10.1021/jacs.7b07548. [DOI] [PubMed] [Google Scholar]
- Chernev P.; Lambertz C.; Brunje A.; Leidel N.; Sigfridsson K. G.; Kositzki R.; Hsieh C. H.; Yao S.; Schiwon R.; Driess M.; Limberg C.; Happe T.; Haumann M. Hydride binding to the active site of [FeFe]-hydrogenase. Inorg. Chem. 2014, 53 (22), 12164–12177. 10.1021/ic502047q. [DOI] [PubMed] [Google Scholar]
- Mebs S.; Duan J.; Wittkamp F.; Stripp S. T.; Happe T.; Apfel U. P.; Winkler M.; Haumann M. Differential protonation at the catalytic six-iron cofactor of [FeFe]-Hydrogenases revealed by 57Fe nuclear resonance X-ray scattering and quantum mechanics/molecular mechanics analyses. Inorg. Chem. 2019, 58 (6), 4000–4013. 10.1021/acs.inorgchem.9b00100. [DOI] [PubMed] [Google Scholar]
- Bruschi M.; Greco C.; Kaukonen M.; Fantucci P.; Ryde U.; de G. L. Influence of the [2Fe]H subcluster environment on the properties of key intermediates in the catalytic cycle of [FeFe] hydrogenases: hints for the rational design of synthetic catalysts. Angew. Chem., Int. Ed. 2009, 48 (19), 3503–3506. 10.1002/anie.200900494. [DOI] [PubMed] [Google Scholar]
- Filippi G.; Arrigoni F.; Bertini L.; De Gioia L.; Zampella G. DFT dissection of the reduction step in H2 catalytic production by [FeFe]-hydrogenase-inspired models: can the bridging hydride become more reactive than the terminal isomer?. Inorg. Chem. 2015, 54 (19), 9529–9542. 10.1021/acs.inorgchem.5b01495. [DOI] [PubMed] [Google Scholar]
- Zampella G.; Fantucci P.; Gioia L. D. Unveiling how stereoelectronic factors affect kinetics and thermodynamics of protonation regiochemistry in [FeFe] hydrogenase synthetic models: a DFT investigation. J. Am. Chem. Soc. 2009, 131 (31), 10909–10917. 10.1021/ja902727z. [DOI] [PubMed] [Google Scholar]
- Pullen S.; Maji S.; Stein M.; Ott S. Restricted rotation of an Fe(CO)2(PL3)-subunit in [FeFe]-hydrogenase active site mimics by intramolecular ligation. Dalton Trans. 2019, 48 (18), 5933–5939. 10.1039/C8DT05148H. [DOI] [PubMed] [Google Scholar]
- Finkelmann A. R.; Stiebritz M. T.; Reiher M. Inaccessibility of the μ-hydride species in [FeFe] hydrogenases. Chem. Sci. 2014, 5 (1), 215–221. 10.1039/C3SC51700D. [DOI] [Google Scholar]
- Adamska A.; Silakov A.; Lambertz C.; Rüdiger O.; Happe T.; Reijerse E.; Lubitz W. Identification and characterization of the “super-reduced” state of the H-cluster in [FeFe] hydrogenase: a new building block for the catalytic cycle?. Angew. Chem., Int. Ed. 2012, 51 (46), 11458–11462. 10.1002/anie.201204800. [DOI] [PubMed] [Google Scholar]
- Silakov A.; Kamp C.; Reijerse E.; Happe T.; Lubitz W. Spectroelectrochemical characterization of the active site of the [FeFe] hydrogenase HydA1 from Chlamydomonas reinhardtii. Biochemistry 2009, 48 (33), 7780–7786. 10.1021/bi9009105. [DOI] [PubMed] [Google Scholar]
- Mulder D. W.; Guo Y.; Ratzloff M. W.; King P. W. Identification of a catalytic iron-hydride at the H-Cluster of [FeFe]-hydrogenase. J. Am. Chem. Soc. 2017, 139 (1), 83–86. 10.1021/jacs.6b11409. [DOI] [PubMed] [Google Scholar]
- Pham C. C.; Mulder D. W.; Pelmenschikov V.; King P. W.; Ratzloff M. W.; Wang H.; Mishra N.; Alp E. E.; Zhao J.; Hu M. Y.; Tamasaku K.; Yoda Y.; Cramer S. P. Terminal hydride species in [FeFe]-hydrogenases are vibrationally coupled to the active site environment. Angew. Chem., Int. Ed. 2018, 57 (33), 10605–10609. 10.1002/anie.201805144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijerse E. J.; Pham C. C.; Pelmenschikov V.; Gilbert-Wilson R.; Adamska-Venkatesh A.; Siebel J. F.; Gee L. B.; Yoda Y.; Tamasaku K.; Lubitz W.; Rauchfuss T. B.; Cramer S. P. Direct observation of an iron-bound terminal hydride in [FeFe]-hydrogenase by nuclear resonance vibrational spectroscopy. J. Am. Chem. Soc. 2017, 139 (12), 4306–4309. 10.1021/jacs.7b00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler M.; Senger M.; Duan J.; Esselborn J.; Wittkamp F.; Hofmann E.; Apfel U. P.; Stripp S. T.; Happe T. Accumulating the hydride state in the catalytic cycle of [FeFe]-hydrogenases. Nat. Commun. 2017, 8, 16115. 10.1038/ncomms16115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H.; Krämer T.; Wang H.; Schilter D.; Pelmenschikov V.; van Gastel M.; Neese F.; Rauchfuss T. B.; Gee L. B.; Scott A. D.; Yoda Y.; Tanaka Y.; Lubitz W.; Cramer S. P. Hydride bridge in [NiFe]-hydrogenase observed by nuclear resonance vibrational spectroscopy. Nat. Commun. 2015, 6, 7890. 10.1038/ncomms8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M. R.; Gray D. L.; Richers C. P.; Wang W.; Zhao P.-H.; Rauchfuss T. B.; Pelmenschikov V.; Pham C. C.; Gee L. B.; Wang H.; Cramer S. P. Sterically stabilized terminal hydride of a diiron dithiolate. Inorg. Chem. 2018, 57 (4), 1988–2001. 10.1021/acs.inorgchem.7b02903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelmenschikov V.; Guo Y.; Wang H.; Cramer S. P.; Case D. A. Fe–H/D stretching and bending modes in nuclear resonant vibrational, Raman and infrared spectroscopies: Comparisons of density functional theory and experiment. Faraday Discuss. 2011, 148 (0), 409–420. 10.1039/C004367M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatchikian E. C.; Forget N.; Fernandez V. M.; Williams R.; Cammack R. Further characterization of the [Fe]-hydrogenase from Desulfovibrio desulfuricans ATCC 7757. Eur. J. Biochem. 1992, 209 (1), 357–365. 10.1111/j.1432-1033.1992.tb17297.x. [DOI] [PubMed] [Google Scholar]
- Siebel J. F.; Adamska-Venkatesh A.; Weber K.; Rumpel S.; Reijerse E.; Lubitz W. Hybrid [FeFe]-hydrogenases with modified active sites show remarkable residual enzymatic activity. Biochemistry 2015, 54 (7), 1474–1483. 10.1021/bi501391d. [DOI] [PubMed] [Google Scholar]
- Sanchez M. L. K.; Sommer C.; Reijerse E.; Birrell J. A.; Lubitz W.; Dyer R. B. Investigating the kinetic competency of CrHydA1 [FeFe] hydrogenase intermediate states via time-resolved infrared spectroscopy. J. Am. Chem. Soc. 2019, 141 (40), 16064–16070. 10.1021/jacs.9b08348. [DOI] [PubMed] [Google Scholar]
- Hong M. K.; Braunstein D.; Cowen B. R.; Frauenfelder H.; Iben I. E.; Mourant J. R.; Ormos P.; Scholl R.; Schulte A.; Steinbach P. J.; et al. Conformational substates and motions in myoglobin. External influences on structure and dynamics. Biophys. J. 1990, 58 (2), 429–436. 10.1016/S0006-3495(90)82388-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaposi A. D.; Vanderkooi J. M.; Stavrov S. S. Infrared absorption study of the heme pocket dynamics of carbonmonoxyheme proteins. Biophys. J. 2006, 91 (11), 4191–4200. 10.1529/biophysj.105.068254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; Grady-Smith C. S.; Bingham A. S.; George S. J.; Cramer S. P.; Swartz J. R. High-yield expression of heterologous [FeFe] hydrogenases in Escherichia coli. PLoS One 2010, 5 (11), e15491 10.1371/journal.pone.0015491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell J. A.; Wrede K.; Pawlak K.; Rodríguez-Maciá P.; Rüdiger O.; Reijerse E. J.; Lubitz W. Artificial maturation of the highly active heterodimeric [FeFe] hydrogenase from Desulfovibrio desulfuricans ATCC 7757. Isr. J. Chem. 2016, 56 (9–10), 852–863. 10.1002/ijch.201600035. [DOI] [Google Scholar]
- Esselborn J.; Lambertz C.; Adamska-Venkatesh A.; Simmons T.; Berggren G.; Noth J.; Siebel J.; Hemschemeier A.; Artero V.; Reijerse E.; Fontecave M.; Lubitz W.; Happe T. Spontaneous activation of [FeFe]-hydrogenases by an inorganic [2Fe] active site mimic. Nat. Chem. Biol. 2013, 9 (10), 607–609. 10.1038/nchembio.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Rauchfuss T. B. Iron carbonyl sulfides, formaldehyde, and amines condense to give the proposed azadithiolate cofactor of the Fe-only hydrogenases. J. Am. Chem. Soc. 2002, 124 (5), 726–727. 10.1021/ja016964n. [DOI] [PubMed] [Google Scholar]
- Sturhahn W. CONUSS and PHOENIX: Evaluation of nuclear resonant scattering data. Hyperfine Interact. 2000, 125 (1), 149–172. 10.1023/A:1012681503686. [DOI] [Google Scholar]
- Esselborn J.; Muraki N.; Klein K.; Engelbrecht V.; Metzler-Nolte N.; Apfel U. P.; Hofmann E.; Kurisu G.; Happe T. A structural view of synthetic cofactor integration into [FeFe]-hydrogenases. Chem. Sci. 2016, 7 (2), 959–968. 10.1039/C5SC03397G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J. T. G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, 2009.
- Jaguar, version 9.4; Schrodinger, Inc.: New York, NY, 2016.
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110 (13), 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105 (8), 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.