

Abstract

Scaffold hopping-driven lead optimizations were performed based on our prior lead 7-methoxy-4-(2-methylquinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one (2a) by C-ring expansion and isometric replacement of the A/B-ring, successively, aimed at finding new potential alternative drug candidates with different scaffold(s), high antitumor activity, and other improved properties to replace prior, once promising drug candidates that failed in further studies. Two series of new compounds 7 (a–d) and 13 (a–j) were synthesized and evaluated for antitumor activity, leading to the discovery of three highly potent compounds 13c, 13d, and 13e with different scaffolds. They exhibited similar high antitumor activity with single digital low nanomolar GI50 values (4.6–9.6 nM) in cellular assays, comparable to lead 2a, clinical drug candidate CA-4, and paclitaxel in the same assays. Further biological evaluations identified new active compounds as tubulin polymerization inhibitors targeting the colchicine binding site. Moreover, 13d showed better aqueous solubility than 2a and a similar log P value.
Keywords: Tubulin polymerization inhibitors; scaffold hopping; lead optimization; N-aryl-3,4-dihydroquinoxalin-2(1H)-ones; colchicine binding site
During the past decade, numerous small molecules targeting the tubulin colchicine site (CS) have been intensely investigated.1,2 The natural tubulin inhibitor combretastatin A-4 (CA-4) and its phosphate prodrug CA-4P (Figure 1) have been advanced in clinical trials for cancer therapies,3,4 such as anaplastic thyroid cancer, ovarian cancer, and nonsmall cellular lung cancer (NSCLC), but so far, neither compound has been approved as a new drug by the FDA. Therefore, the discovery and development of new tubulin polymerization inhibitors with novel structures or unique binding to the colchicine site should continue to determine the optimal drug candidates.
Figure 1.

Colchicine, CA-4, and related clinical trial drug candidates.
In our prior studies on novel antitumor agents (Figure 2), two series of lead compounds N-aryl-1,2,3,4-tetrahydronquiolines (1)5,6 and 4-(quinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-ones (2)7 were successively discovered and identified as novel classes of tubulin polymerization inhibitors. Both series of leads showed high cytotoxicity in human tumor cells and inhibitory activity in tubulin assembly and colchicine binding assays. However, the 2-series leads generally showed higher antitumor activity than corresponding 1-series compounds. For example, 2a exhibited low nanomolar GI50 values of 1.9–3.2 nM against several human tumor cell lines and a low submicromolar IC50 value of 0.77 μM to inhibit tubulin polymerization; it was more potent than corresponding 1a (GI50 11–18 nM and IC50 1.0 μM, respectively) as well as positive control drug paclitaxel in the same assays. Further biologic studies indicated that active 2a(8) possesses high antitumor potency in vivo, extremely high antiproliferative activity in the NIH-NCI 60 human tumor cell line panel with low to subnanomolar GI50 values, and desirable drug-like properties. Mechanism studies identified 2a as a novel tumor-VDA targeting the tubulin-colchicine binding site. Therefore, compound 2a served as a promising drug candidate for further development. Meanwhile, our prior iterativite lead optimizations also revealed some structure–activity relationships (SARs) and correlative structure–property relationships (SPRs) of lead 1- and 2-series. Their scaffold structures include a fused aromatic heterocycle (A/B-ring), a p-methoxyphenyl ring (D-ring), and a cyclic N-linker built as a C-ring to connect two aryl rings (A/B and D). In comparison, the lactam C-ring in the 2-series is preferred compared to the tetrahydropyridine C-ring in the 1-series for enhanced antitumor activity, and a 10-membered aromatic heterocycle (such as quinazoline) is a favorable moiety compared with pyridinyl, purinyl, naphthalenyl, and isoquinolinyl moieties. In addition, the methoxy group on the phenyl ring (D-ring) is necessary for antitumor activity, whereas the 2-substituent on the A-ring could alter the antitumor activity and improve drug-like properties.
Figure 2.

Prior leads 1 and 2 and current scaffold-hopping optimization strategies.
As a part of our systematic lead optimizations to complement prior structural optimizations, scaffold hopping-driven lead opitimation based on lead 2a was recently carried out to explore the effects of the core structure on antitumor activity and possibly find new compounds with different scaffold(s) and high antitumor activity as new alternative drug candidates to replace once promising drug candidates that failed in further studies. Our scaffold-hopping strategies (Figure 2) focused successively on expansion of the C-ring, isometric replacement of the A/B ring, and introduction of additional substituents on the B-ring. The new compounds were tested for antiproliferative activity in a human tumor cell line (HTCL) panel, including A549 (lung carcinoma), MDA-MB-231 (triple-negative breast cancer), KB (identical to AV-3 as a cervical carcinoma HeLa derivative), multidrug-resistant (MDR) KB subline KB-VIN, and MCF-7 (estrogen receptor-positive breast cancer). Furthermore, we were also interested in determining whether new active compounds obtained from scaffold hopping and prior lead 2a share the same mechanism of action. Subsequently, new active compounds were selected for additional cell-based biological evaluations, such as impacts on cell cycle progression and microtubule formation. Thus, recent results about two series of new compounds 7 and 13 are reported herein, including chemical synthesis, antitumor activity in vitro, target identification, molecular modeling elucidation, and SAR analysis.
Based on the structure of lead 2a, we first tried to expand the lactam ring (C-ring) to investigate the effects of the lactam ring size on antitumor activity. With one, two, or three methylene (CH2) units inserted into the C-ring, related compounds 7a–c have an expanded 7-, 8-, or 9-membered lactam ring, respectively, whereas the amide group remains the same as in 2a. To compare the effect as a different amide type on the molecular activity, compound 7d was designed with a reversed amide group in a 7-membered lactam ring. As depicted in Scheme 1, compounds 7a–c were prepared from the intermediate 5(10) by a three-step reaction sequence: reduction of the nitro group by hydrogenation with Pd/C, acylation with 3-chloropropionyl chloride, 4-chlorobutyryl chloride, or 5-chlorovaleroyl chloride to produce corresponding amide compounds 6a–c, respectively, and subsequent intramolecular cyclization in N,N-dimethylacetamide (DMA) in the presence of K2CO3. The 7-membered lactam ring closure (7a) proceeded easily at room temperature, while the 8-membered (7b) and 9-membered (7c) ring closures required heating at 100 °C. In contrast, 7d was prepared by treating commercially available 2-amino-5-methoxy benzoic acid (4b) with 2-chloroethylamine to form amide 8, followed successively by a coupling reaction with 4-chloro-2-methylquinazole (3) and then the intramolecular cyclization described above.
Scheme 1. Preparation of 7a–c.

(i) i-PrOH, HCl, rt, 1 h; (ii) H2, Pd/C, EtOH/EtOAc, 50 psi, 2 h; (iii) carbonyl chloride, acetone, K2CO3, −5 °C–rt, 1 h; (iv) DMA, K2CO3, rt or 100 °C, 1 h; (v) 2-chloroethylamine, 1,1′-carbonyldiimidazole, Et3N, THF, rt, 3 h.
The new 7-series compounds were evaluated for antiproliferative activity in cellular assays against a HTCL panel as mentioned above in parallel with CA-4 as an experimental control. Their biologic activity in the cellular assay was determined by using the established sulforhidamine B (SRB) method as GI50 values and related data are summarized in Table 1. As the lactam size expanded from seven to eight to nine atoms (7a → 7b → 7c), the activity decreased. Compound 7a displayed sub-μM GI50 values (0.69–0.93 μM), 7b showed around 8-fold lower activity (GI50 5.5–10.5 μM) than 7a, and 7c was inactive with GI50 values greater than 10 μM. Also, compound 7d with a reversed amide group was much less potent (GI50 > 2 μM) than 7a, although both compounds have a 7-membered lactam ring. Finally, compared with the highly potent lead 2a (GI50 2.2–3.5 nM), all four new compounds with expanded lactam rings were less active. Thus, expansion of the 6-membered lactam C-ring decreased or abolished the molecular antitumor activity.
Table 1. Antiproliferative Activities of New Compounds 7a–7d in a Human Tumor Cell Line (HTCL) Panel.

| |
GI50 (nM) ± SDa |
|||||
|---|---|---|---|---|---|---|
| Compound | n | A549 | MDA-MB-231 | KB | KB-VIN | MCF-7 |
| 7a | 2 | 838 ± 2 | 95 ± 37 | 725 ± 22 | 696 ± 39 | 933 ± 10 |
| 7b | 3 | 5,900 ± 160 | 9,700 ± 1300 | 5,700 ± 100 | 5,556 ± 95 | 10,500 ± 600 |
| 7c | 4 | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 7d | 2 | >2,000 | >2,000 | >2,000 | >2,000 | >2,000 |
| 2ab | 1 | 3.2 ± 0.7 | NTc | 2.3 ± 0.5 | 2.2 ± 0.4 | NT |
| CA-4d | 5.5 ± 0.1 | 8.2 ± 0.0 | 3.6 ± 0.8 | 3.8 ± 1.1 | 84 ± 8 | |
The GI50 values are the concentrations corresponding to 50% cell growth inhibition and are expressed as the mean ± SD from the dose–response curves of at least three independent experiments.
Data published in ref (10).
NT: not tested.
Combretastatin A4 (CA-4) served as positive control in the same assays.
To better understand how different lactam ring size could affect the cytotoxic activity, a molecular modeling study was conducted by using the CDOCKER program in the Discovery Studio 3.0 software and the tubulin crystal structure (PDB code 1SA0).9,10 The lead compound 2a, which acts as a tubulin polymerization inhibitor binding at the colchicine binding site, and less active 7a were docked into the colchicine binding pocket on tubulin, and both compounds were superimposed with the original ligand DAMA-colchicine (Figure 1). The results (figure in the Supporting Information) indicated that the aromatic rings of the three compounds superimposed well in the binding site, but in contrast to 2a, the less active compound 7a (green stick) assumed a reversed orientation in the same binding pocket. Thus, an interaction with the key amino acid βCys241 was lost, which likely decreased affinity with the biological target. However, H-bonds were present between the carbonyl and NH in the lactam of 7a with αSer178 and αThr179, respectively, at the colchicine binding site of tubulin, like those formed by the side chain at the 7-position on the B-ring of DAMA-colchicine. Moreover, we tried to dock 7b into the colchicine-binding site but were unsuccessful using the same method under the same conditions. We postulated that a molecule with a large lactam ring containing more than six atoms might be too bulky to anchor into the colchicine-binding pocket due to twist conformation(s) of the C-ring. These modeling results supported our biological data and also confirmed our previous result that the six-membered lactam ring (C-ring) in 2 with a rigid planar conformation is very crucial for high antitumor activity.
Next, our scaffold hopping turned to isometric replacement of the A/B ring moiety (quinazoline), while the lactam C-ring in 2a was maintained, to investigate how A/B ring alterations as well as their substituents affected antitumor activity. We replaced the quinazoline with fused aromatic heterocycles with the same size and same topological shape but more or fewer nitrogen atoms, pyrido[2,3-d]pyrimidine (a), pyrido[3,4-b]pyrazine (b), 2-chloropyrido[3,2-d]pyrimidine (c), 2-methylquinoline (d), and multisubstituted quinazoline (e and f) (see Scheme 2). The new 4-aryl-7-methoxy-3,4-dihydroquinoxalin-2(1H)-ones (13a–j) were synthesized based on our previous synthetic methods as shown in Scheme 2. Aryl chloride reagents 10a–f, either easily synthesized (10a–b) or commercially available (10c–f), were individually coupled with 4-methoxy-2-nitroaniline (4a) in the presence of a catalytic amount of HCl or H2SO4 to afford corresponding N-aryl-4-methoxy-2-nitroanilines 11a–f, respectively. The nitro group of 11 was reduced by reaction with mild reagent zinc dust in acetic acid at room temperature. Without further purification, the intermediate amines were immediately treated with 2-chloroacetyl chloride to provide corresponding amides 12. Next, an intramolecular cyclization of 12 produced corresponding target compounds 13a–f with various aromatic A/B-heterocyclic ring moieties. However, reduction of 11b generated two products in a ratio of 4:1; one compound was the expected amine product (only nitro group reduced), and the other compound was an over-reduced amine-1,4-dihydropyrazine byproduct (both nitro group and pyrazine (B ring) reduced). The ultimate products 13b and 13g were separated after the following amidation and cyclization steps. Moreover, several multisubstituted quinazoline derivatives 13h, 13i, and 13j were prepared from 13f by converting the 2-chloro on the quinazoline to a 2-methoxy, methylamino, or 2-(3-hydroxypropyl)amino group, respectively. All synthesized new compounds were identified by 1H NMR and MS spectra.
Scheme 2. Preparation of 13a–j.

(i) i-PrOH, HCl, or H2SO4, rt–70 °C, 1 h–overnight; (ii) (a) Zn/CH3COOH, CH2Cl2, −5 °C; (b) 2-chloroacetyl chloride, acetone, K2CO3, −5 °C; (iii) DMA, K2CO3, rt, overnight or 100 °C for 1 h; (iv) see Supporting Information.
New compounds 13a–j exhibited a broad range of antiproliferative potency in the same cellular assays; the data are shown in Table 2. Although they have different A/B moieties, the most active compounds 13c (nitrogen at C-1, -3, -5), 13d (nitrogen at C-1), and 13e (nitrogen at C-1, -3) showed similar high antiproliferative activities with low single digit nM IC50 values (4.6–9.6 nM) in the tested HTCL panel, comparable to those of prior lead 2a, CA-4, and paclitaxel in the same assays, thus indicating that the A/B-ring system is changeable and favorable bioisosteres include 2-chloropyrido[3,2-d]pyrimidine, 2-methylquinoline, and 6-bromoquinazoline moieties. However, compounds 13a (nitrogen at C-1, -3, -8) (GI50 0.48–2.29 μM) and 13b (nitrogen at C-3, -5, -8) (GI50 0.61–0.92 μM) displayed 100-fold lower potency than 13c, even though each compound contains three N atoms within the A/B system. Therefore, we hypothesize that the molecular activity might be dependent on the N atom position and the nature of the aromatic heterocycle (A/B ring system). Notably, 13g with a 1,4-dihydropyrido[3,4-b]pyrazine was inactive (GI50 > 40 μM) in the cellular assays; thus, the loss of the aromatic character in the B-ring resulted in complete loss of molecular antitumor activity. Among several 6,7-dimethoxyquinazoline analogues, 2-chloro substituted 13f and 2-methylamino substituted 13i exhibited moderate antitumor activity with GI50 values of 45–82 nM and 51–84 nM, respectively, except 13i was less active against paclitaxel-resistant KBvin cells (GI50 507 nM). Otherwise, derivatives 13h with a 2-methoxy group and 13j with a longer side chain at the 2-position showed reduced potency (GI50 0.5–1.0 μM). The results from 13e–f and 13h–j confirmed our prior results that the type of 2-substituent on the A-ring of the quinazoline is related to the molecular antitumor activity. In addition, certain substituents on the B-ring of the quinazoline are tolerated and, thus, modifiable chemical space is available on the B-ring for further optimizations to improve drug-like properties without loss of potency.
Table 2. Antiproliferative Activities of New Compounds 13a–j against Human Tumor Cell Lines.
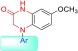

To identify the action mechanism of new active compounds, selected compounds 7a, 13a, 13c–f, and 13h–i were further investigated for effects on cell cycle progression in MDA-MB-231 cells11 in parallel with CA-4, a known tubulin polymerization inhibitor arresting cells in G2/M phase. As shown in Figure 3, the assay results demonstrated that all compounds induced cell cycle arrest in G2/M, like CA-4 as well as our previously reported CS agents. Thus, the new compounds most likely also target tubulin. Furthermore, immunocytochemistry studies were performed by immunofluorescence staining of MDA-MB-231 cells12 to confirm the effect of selected compound 13d on microtubule formation. Filamentous microtubules (Figure 4) were clearly seen in control cells, while no microtubules were observed in the cells treated with 13d or CA-4. These observations demonstrated that 13d inhibits tubulin polymerization in MDA-MB-231 cells. The aqueous solubility and log P value of 13d were measured according to methods described previously.13 The results at pH 7.4 were 10.9 μg/mL and 2.96, respectively; thus, compound 13d showed improved drug-like properties compared with 2a (8.28 μg/mL and 2.97, Figure 2). Thus, new compound 13d could be a potential alternate drug candidate for further development.
Figure 3.

Effects of compounds on cell cycle. MDA-MB-231 cells were treated for 24 h with vehicle (CTRL) or with the indicated compound at the concentration of 3 × IC50 values. DMSO was used as control. EtOH-fixed cells were stained with propidium iodide (PI) in the presence of RNase. PI-stained cells were analyzed by flow cytometer. The Y-axis represents the cell count, and the X-axis represents PI fluorescence on a linear scale.
Figure 4.

Effect of 13d on microtubule formation. MDA-MB-231 cells were treated with compound 13d for 24 h at the concentration of 3 × IC50 values. DMSO or CS agent CA-4 was used as negative control or tubulin polymerization inhibitor, respectively. Cells were fixed in 4% PFA/PBS followed by labeling with antibody to α-tubulin (green) and DAPI (blue) for DNA. Stained cells were observed by confocal fluorescence microscopy, and images were represented as a projection of 15–20 confocal images.
In summary, our scaffold-hopping-driven optimization of prior lead 2a led to two series of new compounds, 4-(N-lactamphenyl)quinazolines (7a–d) and N-aryl-3,4-dihydroquinoxalin-2(1H)-ones (13a–j). Fourteen compounds were designed, synthesized, and evaluated for antitumor activity in cellular assays. Three new compounds 13c, 13d, and 13e with different scaffolds displayed extremely high antiproliferative activity with low single digit nanomolar GI50 values ranging from 4.6 to 9.4 nM, comparable to those of prior lead 2a, the clinical drug candidate CA-4, and paclitaxel in the same cellular assays. These three compounds were also efficient against paclitaxel-resistant KBvin cell growth with at least 340-fold higher potency than paclitaxel (1.96 μM). Based on further biological evaluations, such as the induction of cell cycle arrest at G2/M by inhibiting tubulin polymerization and immunofluorescence, we concluded that our new active compounds reported herein are CS agents. Current SAR studies revealed that (1) the six-membered lactam ring (C-ring) is a very crucial core-structure for highly potent antitumor activity, because its expansion substantially decreased antitumor potency; (2) aromatic heterocycles pyrido[3,2-d]pyrimidine, quinoline, and quinazoline are bioisoteres that are replaceable as the A/B ring system for high potency; (3) molecular antitumor activity is dependent on the nature of isometric A/B heterocycles, such as size, topologic shape, and the N-atom(s) position regardless of the numbers of N-atoms; and (4) introducing additional substituents on the B-ring, like the 6- and 6,7-positions of the quinazoline, can be tolerated to potentially improve drug-like properties. Additionally, drug-like property assessments indicated that the new active compound 13d has improved aqueous solubility and a similar log P value compared with 2a. Therefore, these new-scaffold lead compounds with high antitumor activity could be new starting points for further development of novel antitumor agents.
Acknowledgments
We wish to thank the Microscopy Service Laboratory (UNC-CH) for its expertise in the confocal microscopy studies. This investigation was supported by grant 81120108022 from the Natural Science Foundation of China (NSFC) awarded to L. Xie and NIH grant CA177584 from the National Cancer Institute awarded to K.-H. Lee. This study was also supported in part by the Eshelman Institute for Innovation awarded to M. Goto.
Glossary
Abbreviations
- CS
colchicine site
- HTCL
human tumor cell line
- PDB
protein database
- SAR
structure–activity relationship
- VDAs
vascular disrupting agents
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00352.
Synthetic experimental details, related spectroscopic data of intermediate and target compounds, and biological assay protocols (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Conceived project (L.X.), performed experiments (L.J., M.G., D.-Q.Z., M.C., P.-L.H., K.-P.L., X.H., L.X.), analyzed data (L.X., M.G.), and prepared manuscript (L.X., M.G., S.L.M.-N., K.-H.L.). All authors reviewed the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Perez-Perez M. J.; Priego E. M.; Bueno O.; Martins M. S.; Canela M. D.; Liekens S. Blocking Blood Flow to Solid Tumors by Destabilizing Tubulin: an Approach to Targeting Tumor Growth. J. Med. Chem. 2016, 59, 8685–8711. 10.1021/acs.jmedchem.6b00463. [DOI] [PubMed] [Google Scholar]
- Kaur R.; Kaur G.; Gill R. K.; Soni R.; Bariwal J. Recent Developments in Tubulin Polymerization Inhibitors: an Overview. Eur. J. Med. Chem. 2014, 87, 89–124. 10.1016/j.ejmech.2014.09.051. [DOI] [PubMed] [Google Scholar]
- Zweifel M.; Jayson G. C.; Reed N. S.; Osborne R.; Hassan B.; Ledermann J.; Shreeves G.; Poupard L.; Lu S.-P.; Balkissoon J.; Chaplin D. J.; Rustin G. J. S. Phase II Trial of Combretastatin A4 Phosphate, Carboplatin, and Paclitaxel in Patients with Platinum-resistant Ovarian Cancer. Annals of Oncology 2011, 22 (9), 2036–2041. 10.1093/annonc/mdq708. [DOI] [PubMed] [Google Scholar]
- Arora S.; Gonzalez A. F.; Solanki K. Combretastatin A-4 and its Analogs in Cancer Therapy. Int. J. Pharm. Sci. Rev. Res. 2013, 22 (2), 168–174. [Google Scholar]
- Wang X. F.; Wang S. B.; Ohkoshi E.; Wang L. T.; Hamel E.; Qian K. D.; Morris-Natschke S. L.; Lee K. H.; Xie L. N-Aryl-6-methoxy-1,2,3,4-tetrahydroquinolines: a Novel Class of Antitumor Agents Targeting the Colchicine Site on Tubulin. Eur. J. Med. Chem. 2013, 67, 196–207. 10.1016/j.ejmech.2013.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. B.; Wang X. F.; Qin B.; Ohkoshi E.; Hsieh K. Y.; Hamel E.; Cui M. T.; Zhu D. Q.; Goto M.; Morris-Natschke S. L.; Lee K. H.; Xie L. Optimization of N-Aryl-6–methoxy-1,2,3,4-tetrahydroquinolines as Tubulin Polymerization Inhibitors. Bioorg. Med. Chem. 2015, 23, 5740–5747. 10.1016/j.bmc.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. F.; Guan F.; Ohkoshi E.; Guo W. J.; Wang L. L.; Zhu D. Q.; Wang S. B.; Wang L. T.; Hamel E.; Yang D. X.; Li L. N.; Qian K. D.; Morris-Natschke S. L.; Yuan S. J.; Lee K. H.; Xie L. Optimization of 4-(N-Cycloamino)phenylquinazolines as a Novel Class of Tubuline Polymerization Inhibitors Targeting the Colchicine Site. J. Med. Chem. 2014, 57, 1390–1402. 10.1021/jm4016526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M. T.; Jiang L.; Goto M.; Hsu P. L.; Li L.; Zhang Q.; Wei L.; Yuan S. J.; Hamel E.; Morris-Natschke S. L.; Lee K. H.; Xie L. In Vivo and Mechanistic Studies on Antitumor Lead 7-Methoxy-4-(2-methylquinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one and Its Modification as a Novel Class of Tubulin-Binding Tumor-Vascular Disrupting Agents. J. Med. Chem. 2017, 60, 5586–5598. 10.1021/acs.jmedchem.7b00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli R. B.G.; Gigant B.; Curmi P. A.; Jourdain I.; Lachkar S.; Sobel A.; Knossow M. Insight into Tubulin Regulation From a Complex with Colchicine and a Stathmin-like Domain. Nature 2004, 428, 198–202. 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- Dorléans A.; Gigant B.; Ravelli R. B. G.; Mailliet P.; Mikol V.; Knossow M. Variations in the Colchicine-binding Domain Provide Insight into the Structural Switch of Tubulin. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 13775–13779. 10.1073/pnas.0904223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa-Goto K.; Oda A.; Hamel E.; Ohkoshi E.; Lee K. H.; Goto M. Development of a Novel Class of Tubulin Inhibitor from Desmosdumotin B with a Hydroxylated Bicyclic B-Ring. J. Med. Chem. 2015, 58, 2378–2389. 10.1021/jm501859j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Goto M.; Oda A.; Hsu P. L.; Guo L. L.; Fu Y. H.; Morris-Natschke S. L.; Hamel E.; Lee K. H.; Hao X. J. Antiproliferative Aspidosperma-Type Monoterpenoid Indole Alkaloids from Bousigonia mekongensis Inhibit Tubulin Polymerization. Molecules 2019, 24, 1256–1265. 10.3390/molecules24071256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. Q.; Zhu L.; Qian K.; Qin B.; Huang L.; Chen C. H.; Lee K. H.; Xie L. Design, Synthesis, and Preclinical Evaluations of Novel 4-Substituted 1,5-Diarylanilines as Potent HIV-1 Non-nucleoside Reverse Transcriptase Inhibitor (NNRTI) Drug Candidates. J. Med. Chem. 2012, 55, 7219–7229. 10.1021/jm3007678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.