

Abstract
Background/Aims:
Diabetic cardiomyopathy (DCM) is characterized by structural and functional alterations that can lead to heart failure. Several mechanisms are known to be involved in the pathogenesis of DCM, however, the molecular mechanism that links inflammation to DCM is incompletely understood. To learn about this mechanism, we investigated the role of inflammatory serine proteases (ISPs) during the development of DCM.
Methods:
Eight weeks old mice with deletion of dipeptidyl peptidase I (DPPI), an enzyme involved in the maturation of major ISPs, and wild type (WT) mice controls were injected with streptozotocin (50 mg/kg for 5 days intraperitoneally) and studied after 4, 8, 16, and 20 week after induction of type 1 diabetes mellitus (T1DM). Induction of diabetes was followed by echocardiographic measurements, glycemic and hemoglobulin A1c profiling, immunoblot, qPCR, enzyme activity assays, and immunohistochemistry (IHC) analysis of DPPI, ISPs, and inflammatory markers. Fibrosis was determined from left ventricular heart by Serius Red staining and qPCR. Apoptosis was determined by TUNEL assay and immunoblot analysis.
Results:
In the diabetic WT mice, DPPI expression increased along with ISP activation, and DPPI accumulated abundantly in the left ventricle mainly from infiltrating neutrophils. In diabetic DPPI-knockout (DPPI-KO) mice, significantly decreased activation of ISPs, myocyte apoptosis, fibrosis, and cardiac function was improved compared to diabetic WT mice. In addition, DPPI-KO mice showed a decrease in overall inflammatory status mediated by diabetes induction which was manifested by decreased production of pro-inflammatory cytokines like TNF-α, IL-1β and IL-6.
Conclusion:
This study elucidates a novel role of ISPs in potentiating the immunological responses that lead to the pathogenesis of DCM in T1DM. To the best of our knowledge, this is the first study to report that DPPI expression and activation promotes the inflammation that enhances myocyte apoptosis and contributes to the adverse cardiac remodeling that subsequently leads to DCM.
Keywords: Apoptosis, Diabetic cardiomyopathy (DCM), Dipeptidyl peptidase I (DPPI), ER stress, Inflammatory serine protease (ISP)
Introduction
Diabetes mellitus continues to be among the most common chronic medical conditions in the world. Ongoing studies throughout the field attest to the alarming rate of increasing incidence of diabetes mellitus (DM) is increasing. Bewteen 2002 and 2012, incidence of T1DM increased by 1.8% and incidence of T2DM increased by 4.8% [1]. According to the Global Burden of Disease report published in 2015, the prevalence of DM rose from approximately 333 million people worldwide to 435 million in the ten-year period between 2005 and 2015 [2].
DM affects nearly every organ system in the human body and claims approximately 1.5 million lives a year [3]. The heart is particularly vulnerable to DM, with cardiovascular diseases (CVDs) accounting for nearly two-thirds of diabetic mortality [1]. CVDs in diabetes were traditionally attributed to advanced atherosclerosis, autonomic neuropathy, and cardiomyopathy. DCM was first described in 1972 after Rubler et al. found significant left ventricular (LV) hypertrophy in the post-mortem hearts of diabetic patients with minimal coronary artery disease [4]. Histopathological analysis confirmed significant fibrosis and narrowing of coronary arterioles.
The prevalence of DCM is estimated to occur in 30%−60% in individual with DM depending on the study [3]. It is a vastly underestimated, enigmatic disease thought to result from a combination of hyperglycemia, dyslipidemia, and inflammation. It alters the environment, metabolism, and function of myocardium and leads to diastolic dysfunction followed by systolic dysfunction. Various mechanisms have been postulated, but because of its complicated multifactorial nature, the pathophysiology remains largely unknown [1].
Recently, our lab has worked to prove that inflammation is central to the development of DCM [5]. We have shown that chronic inflammation increases leukocyte invasion and infiltration in the myocardium where they release ISPs that trigger cardiac dysfunction. Previously, we have shown that ISPs regulate numerous biological pathways and signaling cascades involved in cardiac remodeling [6, 7]. These findings suggest that ISPs play a critical role in the alteration of both the geometry and mechanical properties of the myocardium [8, 9].
ISPs represent a broad category of enzymes derived mainly from neutrophils (i.e., elastase, proteinase 3, cathepsin G, neutrophil serine protease 4) [10, 11], cytotoxic T cells (i.e., granzyme B), and mast cells (i.e., chymase, tryptase). These enzymes predominantly function in the killing of intracellular pathogens [12–15]. A study previously published from our lab identified cardiomyocytes as the targets of many of these ISPs [6, 7]. For ISPs to function, they must be first activated by DPPI, also known as cathepsin C, a lysosomal cysteinyl protease that is constitutively expressed in both humans and rodents. Based on studies involving DPPI-KO mice, modulation of the activity of DPPI appears to be a selective way of inhibiting ISP activation and the subsequent inflammatory cascade [16–21].
DPPI-KO mice have been used to study asthma, chronic obstructive pulmonary disease, sepsis, and abdominal aortic aneurysms, with the general consensus being that the DPPIKO mice demonstrate less inflammatory cell infiltration, activation, and production of inflammatory cytokines [19, 22–25]. To our knowledge, DPPI-KO mice have not been studied in the context of DCM. Our study aimed to recapitulate the elevated inflammatory response in a DCM mouse model and also to illustrate the critical role that ISPs play in the development of DCM using a DPPI-KO version of the DCM mouse model.
Materials and Methods
Materials
Phospho-p38Thr180/182, phospho-AKTSer473, Bcl-2, Bax, cleaved-caspase-3, and CHOP antibodies were purchased from Cell Signaling Technology (Danvers, MA). AKT and p38 antibodies were purchased from Santa Cruz Biotechnology (Dallas, Texas). GRP78 was purchased from Enzo Life Science Inc (Farmingdale, NY). GAPDH was purchased from Millipore (Billerica, MA). NIMP-R14, chymase, and granzyme B were purchased from Abcam (Cambridge, MA). DPPI was purchased from R&D (Minneapolis, MN). All other chemicals were from standard suppliers.
Experimental Animals
Eight-week-old C57BL/6 WT male mice were purchased from Jackson Laboratory (Bar Harbor, ME). DPPI-KO mice were provided by Dr. Pham (Washington University School of Medicine, St. Louis, MO) and genotyped as previously described [18–20, 26]. These animals were backcrossed to C57BL/6 mice for >10 generations, using the mutant allele as a marker for selection at each generation. All animals were housed at Thomas Jefferson University animal facility at 22 °C with a 12-h light/dark cycle and free access to standard rodent chow and tap water. The Institutional Animal Care Committee of Thomas Jefferson University approved all the animal protocols utilized in this study.
All animal experiments were conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health and approved by the American Physiological Society. Mice were divided into four groups: 1) STZ-treated WT mice; 2) WT-control mice; 3) STZ-treated DPPI-KO mice; and 4) DPPI-KO-control mice.
T1DM mouse model
STZ (Sigma-Aldich, St. Louis, MO), dissolved in 0.1 M sodium citrate (pH 4.5) was intraperitoneally injected (50 mg/Kg) to induce T1DM. Age matched controls received a vehicle injection (sodium citrate). Mice were injected for five consecutive days. This strategy minimizes nonspecific toxic effects of high-dose STZ and also provides a robust and consistent hyperglycaemic response [27]. T1DM was confirmed by a measurement of fasting blood glucose at greater than 250 mg/dL after anesthetization of animals using the inhalational anesthetic, isoflurane (2% for induction in 2L oxygen/min) [28]. Between 4–20 weeks after STZ injection, mice were sacrificed after they were anesthetized using an intraperitoneal injection of Ketamine (50 mg/kg) and Xylazine (10 mg/kg).
Plasma glycated hemoglobin (HbA1c)
Plasma HbA1c was measured using commercial assay kit following the protocol provided by manufacturer (Crystal Chem, Elk Grove Village, IL USA).
Echocardiography
Echocardiographic measurements were taken before and after STZ injection to determine the baseline heart function and ventricular dimensions in the experimental groups. Briefly, following light sedation using the inhalational anesthetic, isoflurane (2% for induction in 2L oxygen/min), an intraperitoneal injection of Ketamine (50 mg/kg) and Xylazine (10 mg/kg) will be administered to achieve semi-conscious sedation for 20 to 30 minutes. After a complete sedation, the animals received an echocardiogram. Mice were placed on a heated platform in the left lateral decubitus position for imaging. All hairs were removed from the thorax using a chemical depilatory agent, and aquasonic clear ultrasound gel (Parker Laboratories, Fairfield, NJ) was applied to the thorax surface to optimize the visibility of the cardiac chambers. Echocardiography was performed using Visualsonic Ultrasound System (Vevo770, Toronto, Canada) containing a 40 Mhz variable frequency probe. Standard long axis M-Mode and PW Doppler mode views were recorded when the mouse reached a target heart rate between 450 and 550 beats per minute. Posterior wall thickness (LVPWTd, LVPWTs) and left ventricular internal diameters (LVEDd, LVESs) were calculated and averaged from 4 consecutive contractions using manufacturer’s software. Functional calculations were acquired according to the guidelines of the American Society of Echocardiography. Heart rate, percent ejection fraction (%EF), percent fractional shortening (%FS), mitral peak velocity of early filling (E) to mitral peak velocity of late filling (A) E/A ratio were calculated [29].
Histopathology and immunohistochemistry (IHC)
Tissues were fixed in 10% formalin, embedded in paraffin, and sectioned at 6 μm thickness. Sections were stained for DPPI, NIMP-R14 (neutrophil), chymase (mast cells), and granzyme B (cytotoxic T cells) antibodies. Wheat germ agglutinin (WGA) staining was carried out to measure cardiomyocyte cell size. All images were captured in 400X magnification using bright field microscope (Nikon Eclipse 80i, NIS Elements version 4.3). The outline of myocytes was traced in the LV of each animal, using NIH Image J software to determine myocyte cross-sectional area. A value from each heart was calculated by the measurements of approximately 400–600 cells in a remote area from 5 randomly selected image areas in an individual heart. For evaluation of fibrosis, Picro-sirius red staining was performed using standard procedures. For immunostaining, paraffin heart sections were deparaffinized in xylene and re-hydrated. Antigen retrieval was achieved by boiling the slides in citrate solution for 12–15 minutes and then washing them with phosphate-buffered saline (PBS). After quenching endogenous tissue peroxidase activity with 3% H2O2 for 20 minutes, the slides were washed in PBS and blocked in PBS containing 5% bovine serum albumin (BSA) at room temperature for 30 minutes. Primary antibodies were applied overnight at 4°C in PBS containing 2% BSA. The samples were washed in PBS followed by incubation with Vectastain Elite ABC Kit (Avidin/Biotin/Horseradish Peroxidase-System, Vector Laboratories). The peroxidase reaction was visualized using 3, 3’-diaminobenzidine tetrahydrochloride (DAB) and slides were counterstained with hematoxylin. All images were captured in 400X magnification using bright field microscope (Nikon Eclipse 80i, NIS Elements version 4.3). Cells were counted using Image J (bundled with 64-bit Java 1.8.0_112) by adjusting to a lower and upper (Pixel) threshold (Image, Adjust, Threshold, Set or Auto). Then, using the analyze particle tool (Analyze, Analyze Particles), the number of spots corresponding the positive cells was counted automatically.
Terminal deoxynucleotidyl transferase (TdT) and tropomyosin immunolabeling
TdT assay was performed using the kit (Promega, Madison, WI) according to the manufacturer’s instructions. Five sections from each left ventricle (LV) cut perpendicularly to the major axis of the heart were sampled. TUNEL positive cells were counted throughout the LV and were expressed as percentage of the total number of nuclei as determined by DAPI (Molecular probes, Pittsburgh, PA). TUNEL costaining with tropomyosin was used to assess the percentage of apoptotic cardiomyocytes. All images were captured in 600X magnification using bright field microscope (Nikon Eclipse 80i, NIS Elements version 4.3). Quantification was done by ImageJ software (bundled with 64-bit Java 1.8.0_112).
Caspase-3 activity assay
Caspase-3 activity was measured with CaspACE assay system (Promega, Madison, WI) according to the manufacturer’s instructions. In brief, LV lysates were prepared by dounce homogenization in lysis buffer provided with the kit. The lysates were centrifuged at 12, 000 rpm for 20 minutes at 4 °C, and the supernatants containing 40 μg protein were used for caspase-3 assay. Caspase-3 activity was examined by measuring the rate of cleavage of fluorogenic conjugated substrate MCAVal-Asp-Gln-Met-Asp-Gly-TrpLys-(DNP)-NH2. The specificity of the assay was confirmed by addition of the specific caspase-3 inhibitor Z-DQMD-FMK in the reaction mixture at a concentration of 50 μM during the incubation.
DPPI activity assay
Snap frozen LV-heart tissues were homogenized in ice-cold lysis buffer containing 100 mM acetate buffer (pH 5.5), 1 mM EDTA, 0.01% Brij-35, and 5 mM dithiothreitol (DTT). After centrifugation, supernatants containing 40 ug proteins were used for DPPI assay by measuring the rate of cleavage of fluorogenic conjugated substrate H-Gly-Arg-AMC (Bachem, Torrance, CA).
Cathepsin G, chymase, and granzyme activity assays
Snap frozen LV tissues were homogenized in ice-cold buffer containing 100 mM HEPES (pH 7.5), 1 M NaOH, 50 mM CaCl2, and 0.01% Igepal CA630 in the presence or absence of cathepsin G inhibitor. After centrifugation, supernatants containing 40 μg proteins were used for cathepsin G, chymase, and granzyme B activity assay by measuring the rate of cleavage of fluorogenic conjugated substrate Suc-Ala2-Pro-Phe-Amc (R&D Systems, Minneapolis, MN), SucLeu-Leu-Val-Tyr-AMC (Boston Biochem, Cambridge, MA), and Z-IleGlu-Thr-Asp-AFC (MilliporeSigma, Burlington, MA), respectively.
Immunoblot analysis
Extraction of proteins from heart tissue samples was performed as described previously [30]. Briefly, lysates were cleared by centrifugation at 12, 000 rpm and the supernatants were subjected to immunoblot analysis according to methods published previously. Each immunoblot panel in the figures represents results from a single blot exposed for a uniform duration with bands detected by LI-COR Odyssey imaging system (PCSH898; LI-COR, Inc., Nebraska USA).
Quantitative real-time (qRT) PCR
Total RNA were extracted from LV tissue with TRIZOL. cDNA was generated from 2μg of total RNA using High Capacity RNA-to-cDNA kit (Life Technology Grand Island, NY) according to the manufacturer’s instruction. qRT-PCR assays were performed using PerfeCT Super Green Super Mix (Quanta Biosciences, Beverly, MA) by using specific primers IL-6, TNF-α, ANP, BNP, β-MHC, Fibronectin, Collagen I, Collagen IV, TGF-β, CTGF, and MMP9. (Integrated DNA Technologies, Inc. Coralville, Iowa). Differences in expression were determined by the relative quantification method; the cycle threshold (CT) values of the target genes were first normalized to the CT values of endogenous control GAPDH.
Statistical analysis
All data are presented as mean ± SEM. Comparison of two groups was accomplished using an unpaired Student’s t test. One-way ANOVA followed by the Tukey post hoc test was used to compare multiple groups. Two-way ANOVA and subsequent Tukey test were performed to compare groups with different time points. A value of P < 0.05 was considered statistically significant.
Results
T1DM leads to increased DPPI expression and its activity
We examined DPPI expression and activity in the lef ventricular hearts of T1DM mice from 4 to 20 weeks post-STZ treatment. IHC revealed a steady increase in DPPI expression, with peak expression at 4 weeks post-STZ treatment (Fig. 1A & 1B). By comparison, age matched WT controls showed little to no expression of DPPI. At 4 weeks post-STZ treatment, the T1DM mice expressed an average of 18 ± 0.63 DPPI+ cells/mm2 compared to 1 ± 0.03 DPPI+ cells/mm2 in the WT controls. DPPI expression was also confirmed via immunoblotting at 4 weeks (Fig. 1D).
Fig. 1.
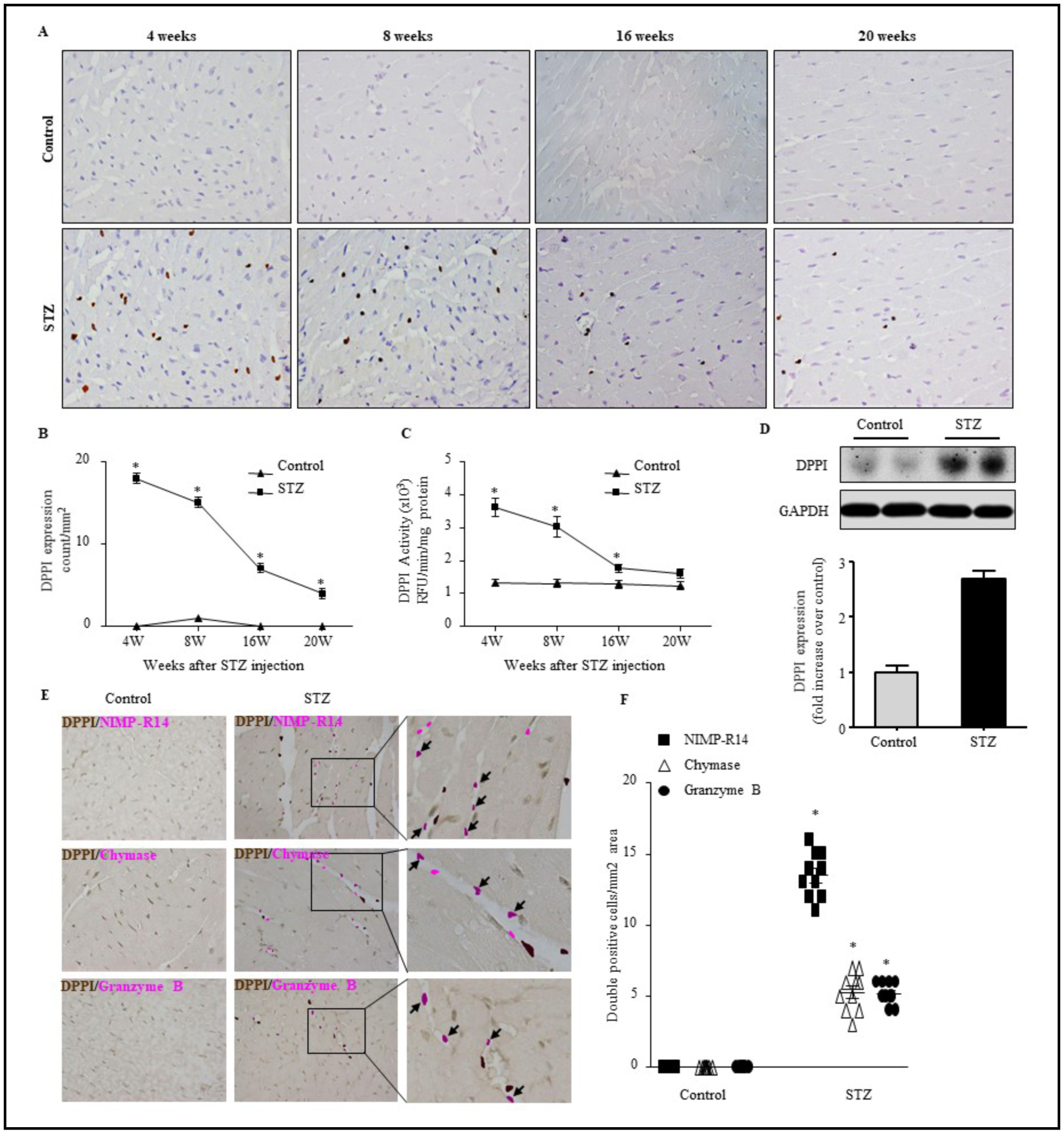
STZ treatment increases leukocyte infiltration, DPPI expression and activation. LV sections from animals treated with citrate buffer or STZ for 4–20 weeks were assessed for anti-DPPI immunostaining (400X magnification with scale bars 50 μm) (A) and quatification (B), DPPI activity as determined by specific fluorogenic substrates (C), and DPPI immunoblot analysis (D). Double immunistaining with DPPI and NIMP-R14 (neutrophil), chymase (mast cells), or granzyme B (cytotoxic T cells) antibodies (400X magnification with scale bars 50 μm) (E) and quantification (F), respectively. n=5 for each group *=p< 0.05 vs control. One-way ANOVA followed by the Tukey post hoc test was used to compare multiple groups. Two-way ANOVA and subsequent Tukey test were performed to compare groups with different time points.
We also examined enzymatic activity in the WT controls and T1DM mice. We found that the enzymatic activity of DPPI was significantly increased in the myocardium of mice after diabetes induction as compared to controls, correlating with DPPI expression. DPPI expression was 3.6×103 ± 0.28×103 RFU/min/mg protein in the T1DM mice at 4 weeks post-STZ treatment compared to 1.3×103 ± 0.11×103 RFU/min/mg protein in the control (Fig. 1C).
Since DPPI expression and activation were both highest at 4-weeks post STZ, we chose this time point for rest of the studies.
Neutrophils are the predominant DPPI-expressing cells in T1DM heart
To identify which cells were expressing DPPI in the T1DM mice, we performed IHC for NIMP-R14 against neutrophils; chymase against mast cells; and granzyme B against cytotoxic T cells (CTL) in the heart (Fig. 1E). Interestingly, all three cell types were present in the T1DM mouse heart. Additionally, these cells colocalized with DPPI. Neutrophils constituted the largest DPPI-positive cell population among all leukocyte subtypes (Fig. 1E & 1F).
Downstream targets of DPPI demonstrate increased activity in T1DM heart
Serine proteases are the downstream targets of DPPI that are cleaved and activated by DPPI. These include and are not limited to cathepsin G (CG), chymase, and granzyme B, all of which were significantly increased in the present study in T1DM heart (Fig. 2B–2D) [19–21]. CG activity was higher in T1DM mice compared to age-matched control (1.7×103 ± 0.23 RFU/min/mg protein vs. 0.6×103 ± 0.12 RFU/min/mg protein). Chymase activity was higher in T1DM mice compared to control mice (7.6×103 ± 2.29 RFU/min/mg vs. 4.6×103 ± 1.24 RFU/min/mg protein). Granzyme B activity was higher in T1DM mice compared to control mice (3.9×103 ± 0.34 RFU/min/mg protein vs. 2.3×103 ± 0.30 RFU/min/mg protein) (Fig. 2B–2D).
Fig. 2.
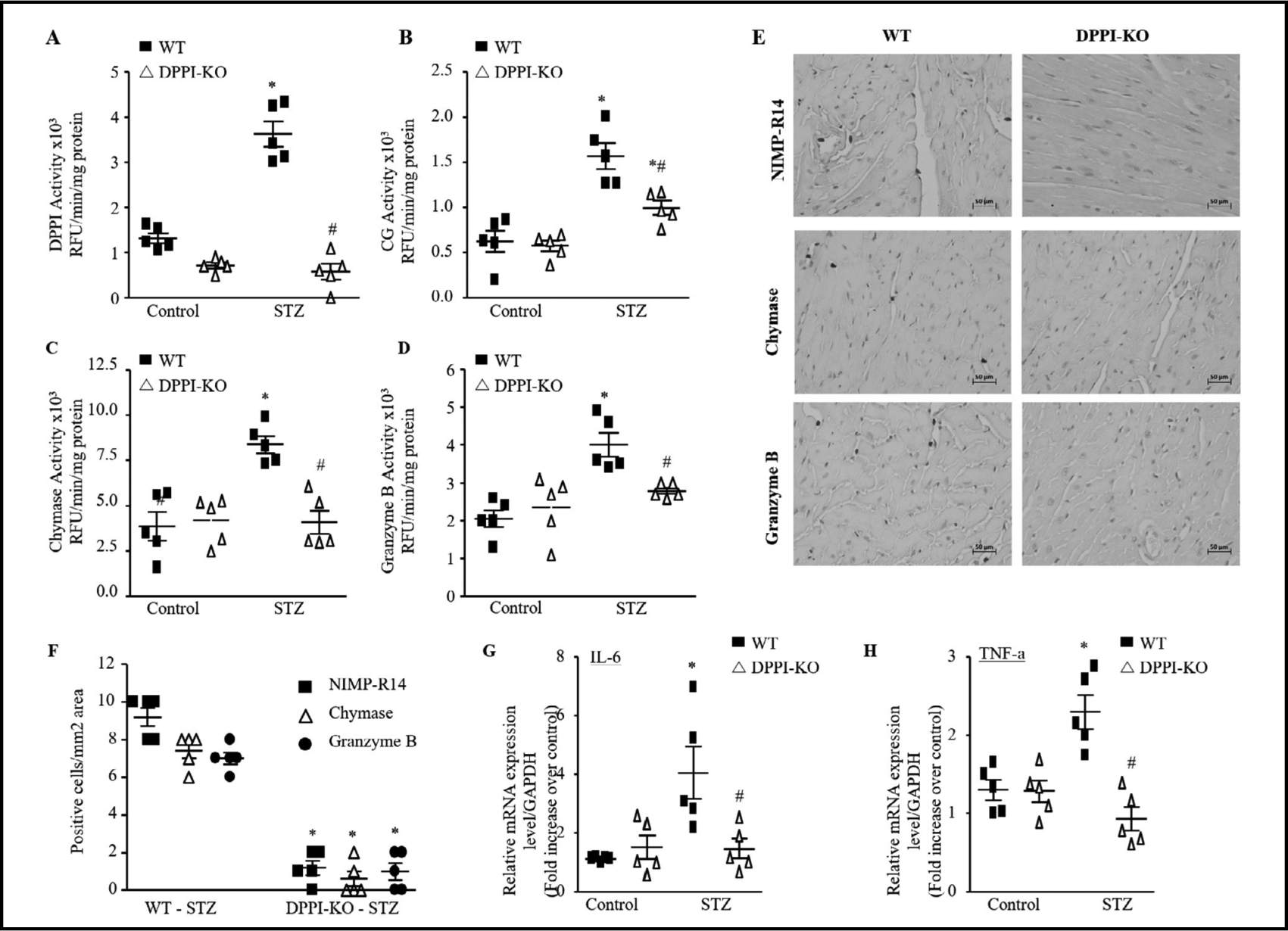
DPPI deletion reduces ISP activity and inflammation after diabetes induction. DPPI (A), cathespin G (CG) (B), chymase (C), and granzyme B (D) activity levels in the left ventricular heart as determined by enzymatic activity assay (n=5 for each group). (E) Representative NIMP-R14, chymase, and granzyme B immunstaining of paraffin-embedded heart sections of WT and DPPI subjected to diabetes induction (400X magnification with scale bars 50 μm). (F) Quantification of NIMP-R14, chymase, and granzyme B-positive cells (n=5 for each groups). Real-time qPCR analysis of IL-6 (G) and TNF-α (H) in controls and after diabetes induction in WT and DPPI-KO mice heart (n=5 for each groups). The data were normalized to GAPDH and were expressed as mean ± SEM. *P<0.05 vs. control, #P<0.05 vs. STZ-treated WT. One-way ANOVA followed by the Tukey post hoc test was used to compare multiple groups.
DPPI-KO attenuates STZ induced cardiac inflammation
The observed increase in DPPI expression in T1DM heart suggests that the enzyme plays a role in the development of DCM. To validate this observation, we subjected DPPI-KO mice to STZ injection in parallel with WT mice. A lack of DPPI activity was confirmed in DPPIKO mice both in the controls and STZ group (Fig. 2A). With IHC, we quantified neutrophil, mast cells, and CTL infiltration in STZ treated WT (WT-T1DM) and STZ treated-DPPI-KO (DPPI-KO-T1DM) mouse hearts. All three cell types were significantly decreased in DPPI-KO compared to WT (1.3 ± 0.33, 0.5 ± 0.34, 1.0 ± 0.37 versus 9.3 ± 0.42, 8.0 ± 0.29, 7.0 ± 0.29, respectively) (Fig. 2E & 2F). Examination of the same serine proteases that are activated by DPPI and increased in T1DM revealed a significant reduction in their activity in DPPI-KO group (Fig. 2B–2D). Previous studies have demonstrated that pro-inflammatory cytokines, such as interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-α), serve as chemotactic factors for leukocytes to diabetic cardiac tissue [31, 32]. T1DM cardiac tissue demonstrated a statistically significant increase in the expression of all these cytokines (1.5 ± 0.02 and 1.3 ± 0.07 fold increase, respectively). DPPI-KO-T1DM mice expressed these molecules at essentially the same levels as did the WT-control group (Fig. 2G & 2H).
DPPI deletion has no impact on fasting glucose levels in T1DM mice
Blood glucose levels in T1DM mice were significantly elevated compared to WT-control. Interestingly, blood glucose levels were not significantly different between WT-T1DM mice and DPPI-KO-T1DM mice (Fig. 3A). Hemoglobin A1c (HbA1c) also confirmed the induction of diabetes in both WT and DPPI-KO treated with STZ (Fig. 3B).
Fig. 3.

DPPI deletion improves the metabolic profile of diabetic mice. (A) Blood glucose levels. (B) Hemoglobulin A1c. Data are expressed as means ± SEM (n=10–15; only 5 animals are shown in each group).*=p< 0.05 vs control and #=p<0.05 vs STZ-treated WT. Oneway ANOVA followed by the Tukey post hoc test was used to compare multiple groups.
Cardiac functions in STZ induced T1DM model
We further investigated development of cardiomyopathy in STZ-induced diabetic animals by using echocardiography to measure cardiac functions that include LV ejection fraction (EF), fractional shortening (FS), LV end diastolic dimension (LVEDD), E/A ratio, LV chamber dilation (determined by the LV end diastolic dimension to LV wall thickness [LVEDD/PWT]) ratio, and LV end systolic dimension (LVESD). Induction of diabetes led to a significant reduction in LV EF, LV FS, and E/A ratio in the WT-T1DM compared to WT-controls (Fig. 4A–4C). At 4 weeks post induction of T1DM, average LV EF was 58.5 ± 1.96 in WT-T1DM mice compared to 67.2 ± 1.40 in WT-controls. Similarly, average LV FS was 30.9 ± 1.21 in WTT1DM mice compared to 36.7 ± 1.07 in WT-controls. Also the average E/A ratio in WT-T1DM mice was 0.9 ± 0.01 compared to 1.1 ± 0.01 in WT-controls (Fig. 4A–4C). The LVEDD, LVEDS, and LVEDD/PWT ratio also increased significantly in the WT-T1DM group compared to the WT-control group (Fig. 4D–4F).
Fig. 4.
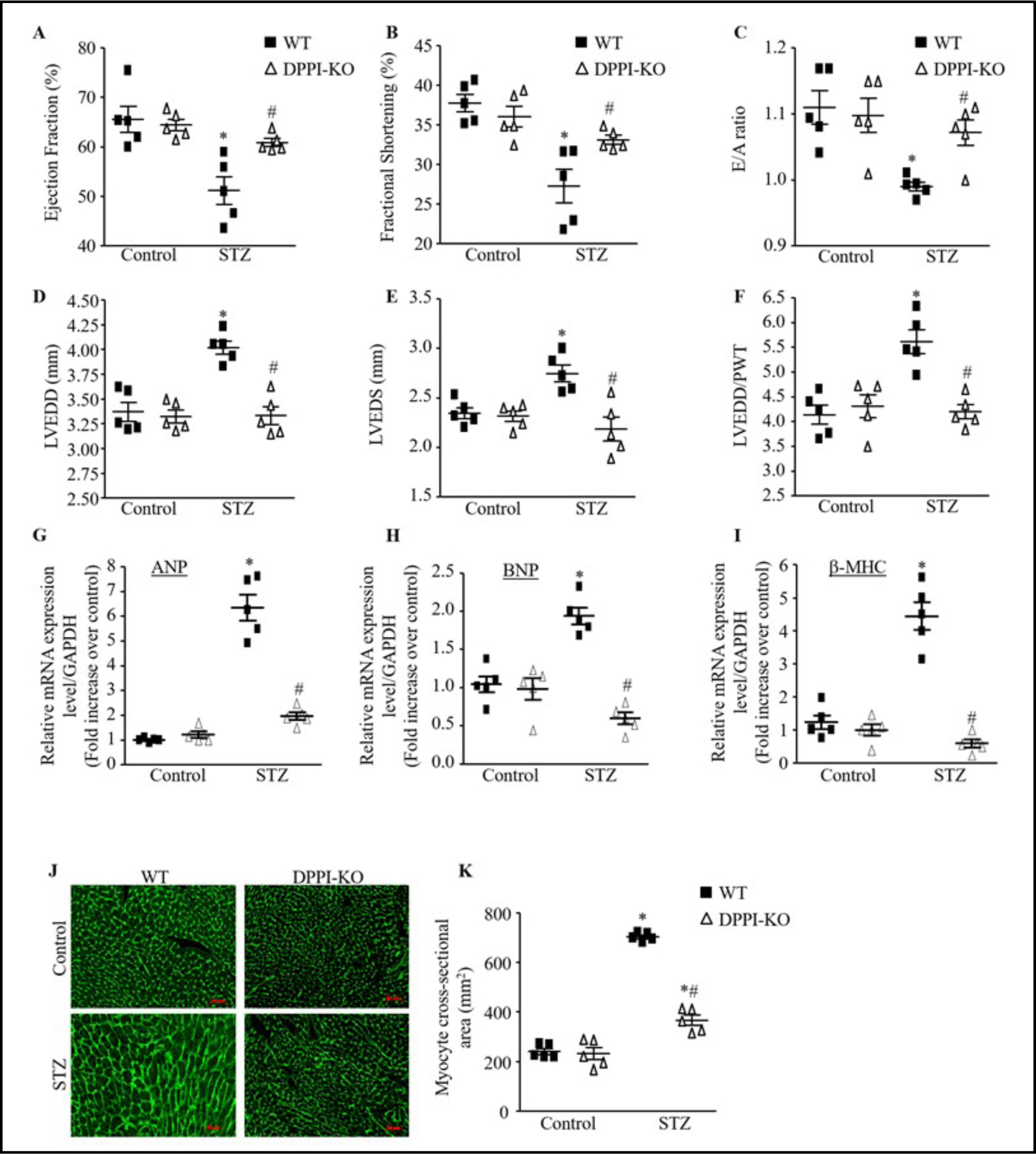
DPPI deletion improve cardiac function and remodeling induced by T1DM after STZ injection independently of hyperglycemia. (A-F) Echocardiography measurement of left ventricular (LV) ejection fraction (A), fractional shortening (B), E/A ratio (C), LV end diastolic (D) and systolic (E) dimension, and LV end diastolic dimension to wall thickness ration (LVEDD/PWT) ratio (F) in control and STZ-induced diabetic animals. (G-I) Expression of cardiac remodeling markers, atrial natriuretic peptide (ANP), B-type natriuretic peptite (BNP) hypertophic markers, and beta myosin heavy chain (β-MHC). (J) Representative images of WGA staining in the controls and diabetic hearts (400X magnification with scale bars 50 μm). (HK) Quantitative analysis of myocyte cross-sectional area. Data are expressed as means ± SEM (n=10–15; only 5 animals are shown in each group).*=p< 0.05 vs control and #=p<0.05 vs STZ-treated WT. One-way ANOVA followed by the Tukey post hoc test was used to compare multiple groups.
Development of cardiac hypertrophy in STZ induced T1DM mice
Cardiac hypertrophic markers were also analyzed after diabetes induction by qRT-PCR in these groups. Compared to WT-control, atrial natriuretic peptide (ANP) in the WT-T1DM demonstrated a significant increase of ~ 7 fold change (P<0.0001) (Fig. 4G); brain natriuretic peptide (BNP) showed a 2 fold increase (P<0.0001) (Fig. 4H); and beta-myosin heavy chain (β-MHC) demonstrated a ~ 4 fold change increase (P<0.0001) (Fig. 4I).
When myocyte cross-sectional area (CSA) was measured as an index of cardiac hypertrophy by means of WGA staining, we found that myocyte CSA was significantly increased in WT-T1DM mice compared to WT-control (Fig. 4J & 4K).
DPPI deletion improves cardiac function in T1DM
There were no significant differences between the baseline echocardiographic studies and measurements of hypertrophic markers in the WT and DPPI-KO- control mice (Fig. 4). Compared to WT-T1DM mice, DPPI-KO-T1DM mice demonstrated significantly increased LV EF (64.2 ± 1.06 vs. 58.5 ± 1.96, P<0.0446), LV FS (34.4 ± 0.86 vs. 30.9 ± 1.21, P<0.0351), and E/A ratio (1.1 ± 0.01 vs. 0.9 ± 0.00, P<0.0001) (Fig. 4A–4C). LVEDD, LVESD, and LVEDD/PWT were all 1.1–1.3 significantly decreased in DPPI-KO-T1DM mice compared to WT-T1DM mice (4D-4F).
DPPI-KO-T1DM expressed significantly decreased ANP, BNP, and β-MHC with expression levels near baseline levels in WT-control mice (Fig. 4G–4I). Myocyte CSA was also reduced in DPPI-KO-T1DM mice compared to WT-T1DM mice, to near baseline levels as in WT-control group (Fig. 4J & 4I).
DPPI deletion attenuates cardiac fibrosis in T1DM mice
We then studied collagen content in mice hearts because the deposition influences the passive mechanical properties of the myocardium, which then affects the cardiac performance [33–36]. Quantitative analysis of total collagen revealed the predicted increase in both interstitial and perivascular collagen deposition in WT-T1DM mice compared to WTcontrol mice (Fig. 5A). DPPI-KO-T1DM mice showed a significantly reduced degree of collagen deposition compared to WT-T1DM mice, with DPPI-KO decreasing collagen deposition levels to near baseline levels observed in WT-control mice (Fig. 5A).
Fig. 5.
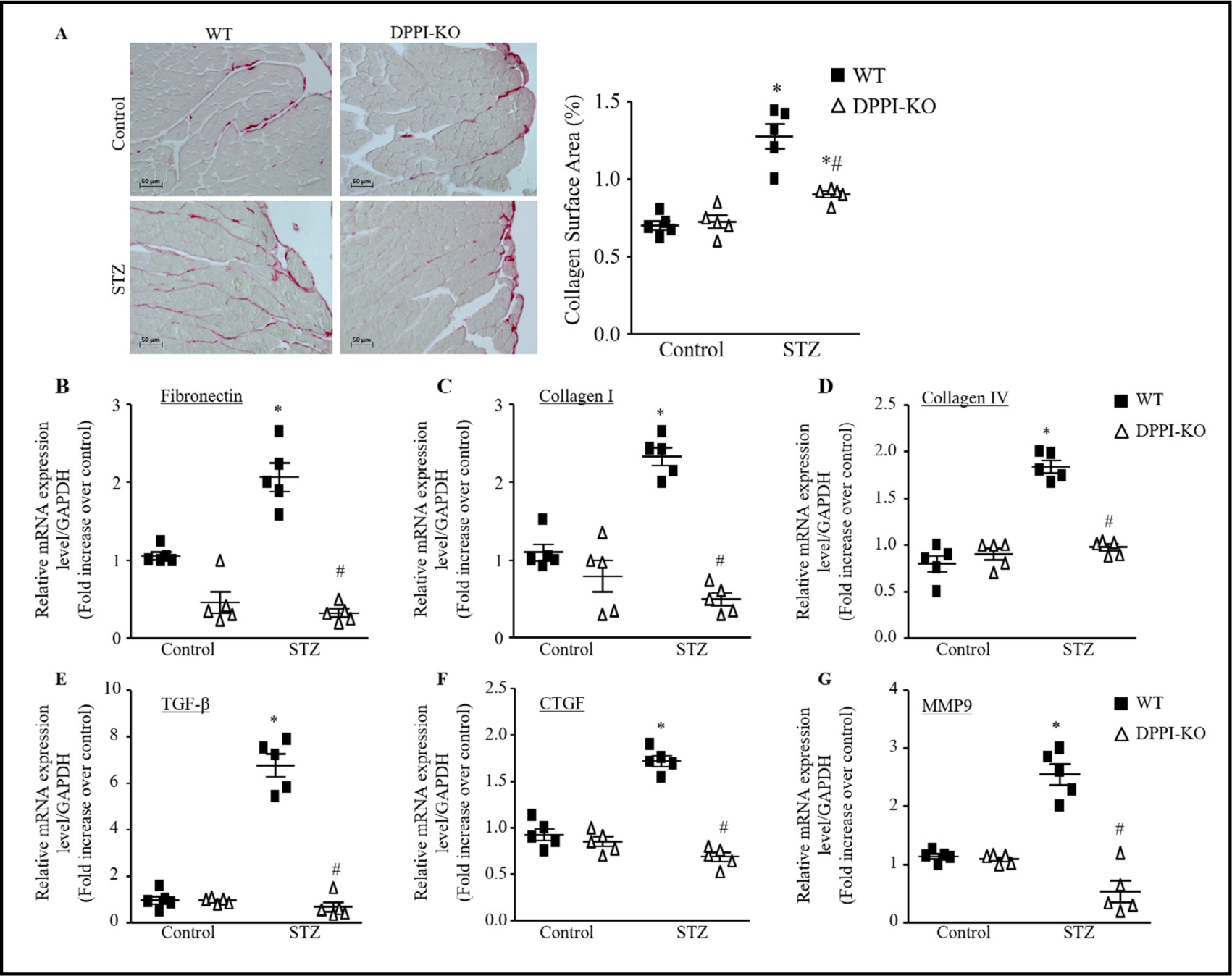
DPPI deletion attenuated fibrosis induced by T1DM. (A) Histological sections of left ventricular cardiac staining by picrosirius red and semiquantitative analysis was done by a computer imaging system (400X magnification with scale bars 50 μm). (B-G) cardiac mRNA expression of fibronectin (B), collagen I (C), collagen IV (D), TGF-β (E), connective tissue growth factor (CTGF) (F), and MMP9 (G). Data are presented as mean ± SEM. n=5 for each group. *=p< 0.05 vs control and #=p<0.05 vs STZ-treated WT. One-way ANOVA followed by the Tukey post hoc test was used to compare multiple groups.
To further validate the anti-fibrotic effect of DPPI deletion in STZ-induced T1DM at message level, we used qRT-PCR to evaluate the mRNA expression levels of the fibrotic genes (collagen, fibronectin), and profibrotic genes CTGF,TGF-β and MMP9. We found that fibronectin, collagen I, and collagen IV were significantly increased in WT-T1DM mice compared to WT-control group (Fig. 5B–5D). DPPI deletion significantly decreased fibronectin, collagen I, and collagen IV message levels compared to WT-T1DM mice (Fig. 5B–5D). T1DM cardiac tissue demonstrated marked increases in the expression of TGF-β and CTGF (Fig. 5E&5F). DPPI deletion significantly reduced this increase (Fig. 5E & 5F). MMP9 expression was also significantly decreased in the DPPI-KO-T1DM mice compared to the WTT1DM mice (Fig. 5G).
DPPI deletion inhibits cardiomyocyte apoptosis induced by T1DM
We used TUNEL staining to assess the degree of apoptotic cell death in cardiac tissue. DPPI-KO-T1DM mice had a significantly reduced number of TUNEL-positive myocytes when compared to WT-T1DM mice (Fig. 6A). Cardiac caspase-3 activity was also reduced in the DPPI-KO-T1DM group compared to the WT-T1DM group (Fig. 6B). T1DM-WT mice had significantly elevated expression of pro-apoptotic proteins Bax (100%, P-0.05) (Fig. 6C & 6D) and cleaved caspase-3 (70%, p<0.05) (Fig. 6C & 6F) compared to WT-control. Antiapoptotic Bcl-2 levels were decreased in WT-T1DM mice compared to WT-control group (Fig. 6C & 6E). Cardiac cleaved caspase-3 expression and Bax were all significantly decreased in DPPI-KO-T1DM mice compared to WT-T1DM mice (Fig. 6C, 6D, & 6F).
Fig. 6.
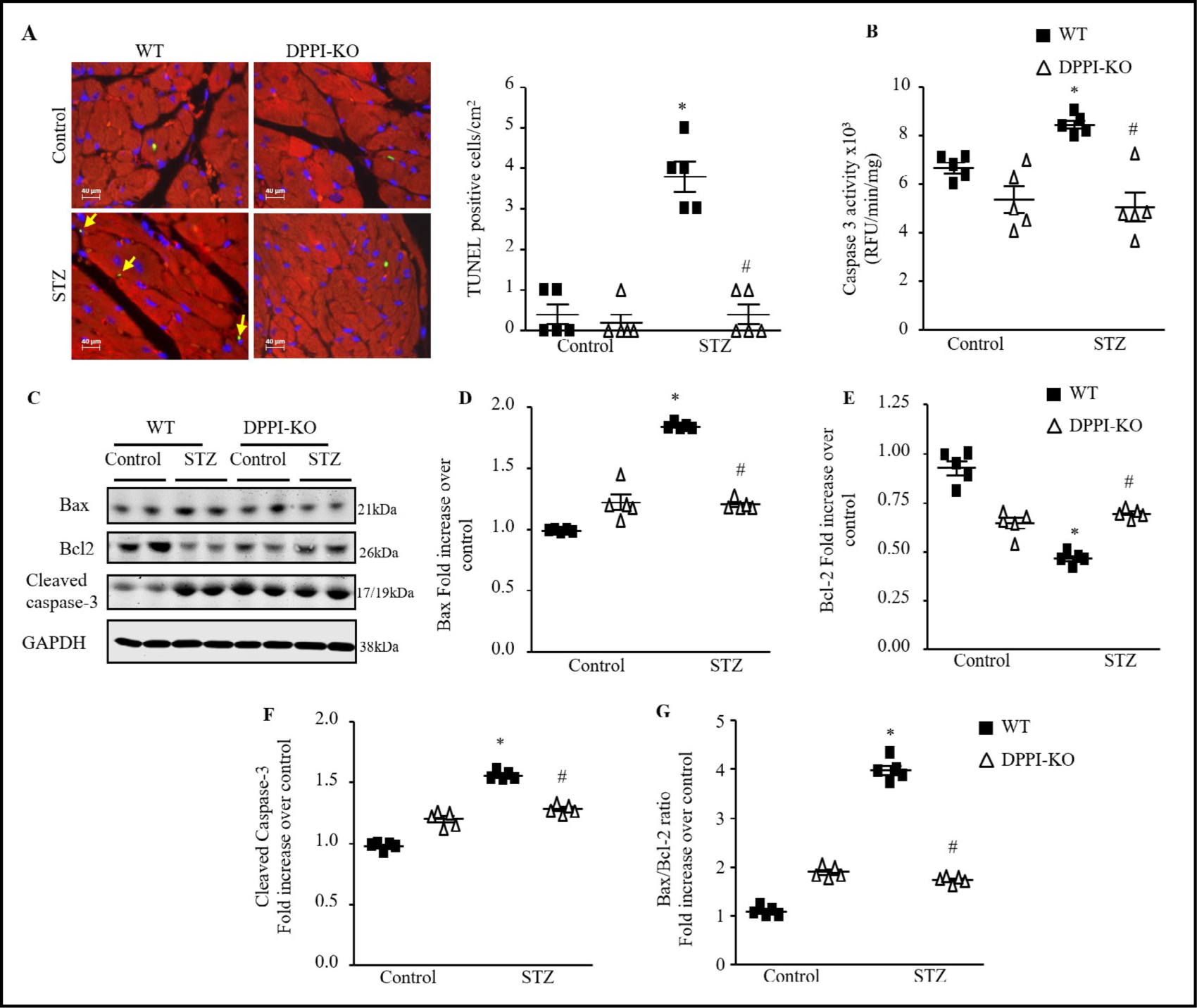
Myocardial cell apoptosis assessed by TUNEL staining and cardiac caspase-3 protein expression before and after diabetes induction. (A) Left ventricular (LV) tissue sections were assessed for apoptosis with the use of the terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assay (green), tropomyosin (red), and DAPI (4′,6-diamidino-2-phenylindole; blue) staining and the number of TUNELpositive myocytes was expressed as a percentage of total nuclei detected by DAPI staining (600X magnification with scale bars 40 μm). (B) Quantification of caspase-3 activity in LV with the use of caspase-3–specific fluorogenic substrate. RFU indicates relative fluorescence units. Immunoblot analysis and quantification of cardiac expression of Bax (C & D), Bcl-2 (C & E) and cleaved caspase-3 (C & F). (G) Bax/Bcl-2 ratio. Data are presented as mean ± SEM. n=5 for each group. *=p< 0.05 vs control and #=p<0.05 vs STZ-treated WT. Oneway ANOVA followed by the Tukey post hoc test was used to compare multiple groups.
Analysis of the Bax/Bcl-2 ratio, an indicator of activation of the mitochondria-mediated apoptotic pathway, showed a significant increase in WT-T1DM mice compared to WT-control (Fig. 6G). The Bax/Bcl-2 ratio was significantly decreased in DPPI-KO-T1DM group compared to the WT-T1DM group.
To better understand how DPPI modulates apoptosis, we conducted a throughput analysis of pathways involved in the regulation of apoptosis and inflammation. We measured the expression of p38 and AKT to identify the impact of DPPI-KO on upstream apoptotic signaling in the myocardium. In T1DM cardiac tissue, the p38 phosphorylation was activated, however no significant changes have been found with AKT phosphorylation. With DPPI-KOT1DM mice, expression of p38 phosphorylation was significantly normalized back to the baseline in WT-control mice (Fig. 7A & 7B).
Fig. 7.
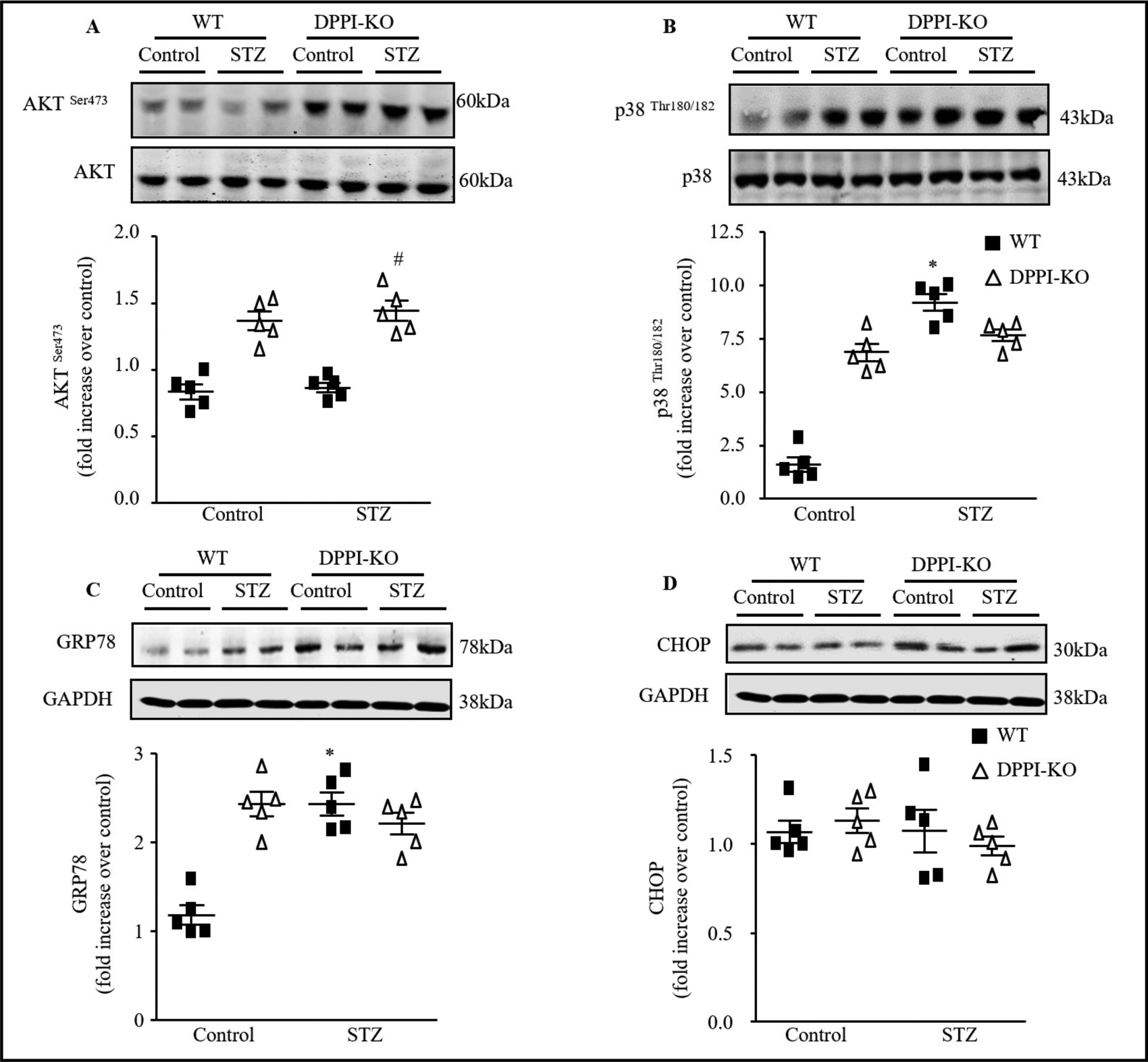
Signal transduction mechanisms involved in the induction of apoptosis by inflammatory serine protease. (A) Representative Immunoblots of LV lysates from control and diabetic WT or DPPI-KO animals. (A) Top, Immunoblots analysis of phosphor-AKTSer473, (B) phopsho-p38hr180/182, (C) GRP78, and (D) CHOP expression in the LV heart tissue. Botton, Quantification of the experiments represented as fold change compared to WT animals Data are expressed as means ± SEM (n=5).*=p< 0.05 vs control and #=p<0.05 vs STZ-treated WT. One-way ANOVA followed by the Tukey post hoc test was used to compare multiple groups.
DPPI deletion prevents diabetes-induced endoplasmic reticulum (ER) stress as well as the associated ER cell death pathway
Increasing evidence suggesting that ER stress and associated apoptotic cell death are involved in the development of DCM [37–40]. In the present study, immunoblot analysis revealed significant increases in GRP78 (Fig. 7C) but no changes has been found with CHOP (Fig. 7D) in diabetic mice; however, these changes were not seen in DPPI-KO-T1DM mice.
Discussion
As the incidence and prevalence of diabetes continued to rise, it is becoming increasingly important to understand its cardiovascular complications. This study establishes DPPI as a critical component of the early development of impaired cardiac function in mice with T1DM and illustrates the impact of global DPPI deletion on cardiac remodeling following T1DM induction.
The role of DPPI in the development of DCM in T1DM has never been examined prior to this study. To shed light on the importance of DPPI to cardiologic impairment, we demonstrated that DPPI-KO-T1DM mice experience drastic reductions in nearly all inflammatory markers. DPPI deletion causes a reversal of cardiac dysfunction induced by T1DM to near baseline levels, most likely by inhibiting p38 phosphorylation, which reduces ER stress and apoptosis.
We found that diabetes induction leads to heart dysfunction by triggering a spike in DPPI expression which was found to be predominantly expressed in inflammatory cells, especially neutrophils, mast cells, and cytotoxicT cells within myocardium. Out of the three cell types, neutrophils constituted the largest DPPI-positive population found from co-expressions of NIMP-R14 and DPPI together in LV heart. As a positive feedback of the inflammatory cascade, all the DPPI activated downstream serine proteases like cathepsin G, granzyme B, and chymase were found to have high activity levels in WT-T1DM hearts. As a possible consequence several major pro-inflammatory cytokines (such as TNF-α and IL6) were significantly increased by WT-T1DM mice compared to control group. The overall inflammation found to be an early event during cardiac remodeling orchestrated by DPPI. Since the blood glucose levels remained high in T1DM group even after DPPI deletion, it supports the previously published data [41, 42] that suggests that DPPI does not affect glucose metabolism but it prevents cardiac inflammation to prevent downstream damage to prevent DCM.
T1DM led to changes in cardiac structure and function. Echocardiographic findings were drastically abnormal (e.g., reduced LV EF and LV FS and decreased E/A ratio) in the diabetic mice compared to controls. The overall measurements in the T1DM mice were representative of decreased cardiac function and cardiac efficiency. Cardiac hypertrophy has been shown to be associated with characteristic alterations in gene expression, including up-regulation of ANP and BNP [43, 44], and recapitulation of fetal patterns for expression of myosin heavy chains [45–47]. In agreement with the later, message levels of ANP, BNP and β-MHC were significantly elevated in the T1DM cardiac tissue compared to its control counterpart. In addition, myocyte CSA displayed a significant increase in T1DM-WT mice compared to WT control. Both interstitial and perivascular deposition of collagen increased significantly in T1DM cardiac tissue compared to WT-control mice. DPPI-KO-T1DM group ameliorated the above mentioned structural and functional changes including collagen deposition.
We also found that T1DM increased intracellular stress and damage. When we measured protein levels of ER stress markers GRP78. The CHOP expression level may change at the later time point. Also, we performed TUNEL staining in order to assess the degree of apoptotic cell death following ER stress and CHOP activation. We found a significantly higher number of TUNEL positive cells in WT-T1DM-LV heart compared to WT-control. Subsequently pro-apoptotic proteins like Bax and cleaved caspase-3 were significantly increased while anti-apoptotic Bcl-2 was significantly reduced in WT-T1DM heart. All of these detrimental changes in terms of apoptosis and ER stress reverted to normal levels in DPPI-KO-T1DM mice compared to WT-T1DM group.
The induction of myocyte death has been proposed to be a principal factor leading to the tissue fibrosis [26]. Our study reinforces this concept through the finding of both decreased apoptosis and collagen deposition in DPPI-KO-T1DM mice compared to WT-T1DM mice. Some studies have proposed a direct effect of DPPI on fibrosis through TGF-β and stimulation of fibroblast differentiation prompting ECM synthesis [48]. In this study, we show that DPPI deletion decreases the expression of fibronectin, collagen, TGF-β, CTGF and MMP-9 in cardiac tissue, hence prevents fibrosis. Inhibition in TGF-β signaling leads to deactivation of p38 signaling through TAK1 mediated pathway [49]. This can inhibit caspase-mediated apoptosis [50, 51]. In addition, GRP78 mediated ER stress can activate p38 signaling that can execute caspase mediated apoptosis mediated DNA fragmentation and cell death [52–54].
Based on the findings from our study, it appears likely that DPPI deletion in diabetic heart inhibits inflammatory and fibrotic development through attenuation of pro-inflammatory cytokines and TGF-β signaling. This downregulates TGF-β/TAK1 mediated P38/JNK pathway or GRP78 mediated CHOP activation. Both in turn prevent cell death that leads to cardiac fibrosis during development of DCM in diabetic mice. Our study is unique in the sense that it first describes how DPPI-KO halts the acute stages of inflammation, which is necessary to the development of DCM. It also suggests that early stage in DCM is very important and can be clinically targeted for drug development since DPPI mediated inflammatory cascade remains maximally expressed at this point that causes myocyte death and cardiac dysfunction in long run. Our data suggests that modulation of the expression of DPPI or its downstream serine proteases could eventually evolve into a therapy given to individuals long before they develop the irreversible symptoms of overt heart failure. This has been described in many inflammatory models, but this is the first time it has been documented in a T1DM model of DCM [55, 56].
Conclusion
In conclusion, DPPI likely plays an important role in the early stages of DCM. T1DM is a lifelong, chronic disease, and it is well known that the initial stages of DCM in humans are characterized by asymptomatic abnormalities in cardiac function. Our findings indicate that DPPI is a potential pharmacological target that may be useful in ameliorating the inflammatory cascade responsible for DCM. Further studies focused on identifying the specific cells responsible and key mechanisms will be integral to developing specific targets for therapy. Together, these studies may be used to engineer preventative medications, vaccines, or direct cures for DCM.
Acknowledgements
This work was supported by grants from the National Institutes of Health (HL111278). DPPI-KO mice were a generous gift from Dr. Pham of Washington University School of Medicine, St. Louis, MO.
Footnotes
Disclosure Statement
The authors declare no conflict of interests.
References
- 1.Pappachan JM, Varughese GI, Sriraman R, Arunagirinathan G: Diabetic cardiomyopathy: Pathophysiology, diagnostic evaluation and management. World J Diabetes 2013;4:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingelfinger JR, Jarcho JA: Increase in the Incidence of Diabetes and Its Implications. N Engl J Med 2017;376:1473–1474. [DOI] [PubMed] [Google Scholar]
- 3.Stokes A, Preston SH: Deaths Attributable to Diabetes in the United States: Comparison of Data Sources and Estimation Approaches. PLoS One 2017;12:e0170219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A: New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 1972;30:595–602. [DOI] [PubMed] [Google Scholar]
- 5.Sarkar A, Shukla SK, Alqatawni A, Kumar A, Addya S, Tsygankov AY, Rafiq K: The Role of Allograft Inflammatory Factor-1 in the Effects of Experimental Diabetes on B Cell Functions in the Heart. Front Cardiovasc Med 2018;5:126. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Rafiq K, Kolpakov MA, Abdelfettah M, Streblow DN, Hassid A, Dell’Italia LJ, Sabri A: Role of protein-tyrosine phosphatase SHP2 in focal adhesion kinase down-regulation during neutrophil cathepsin G-induced cardiomyocytes anoikis. J Biol Chem 2006;281:19781–19792. [DOI] [PubMed] [Google Scholar]
- 7.Rafiq K, Hanscom M, Valerie K, Steinberg SF, Sabri A: Novel mode for neutrophil protease cathepsin G-mediated signaling: membrane shedding of epidermal growth factor is required for cardiomyocyte anoikis. Circ Res 2008;102:32–41. [DOI] [PubMed] [Google Scholar]
- 8.DeLeon-Pennell KY, Meschiari CA, Jung M, Lindsey ML: Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog Mol Biol Transl Sci 2017;147:75–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan D, Takawale A, Lee J, Kassiri Z: Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korkmaz B, Horwitz MS, Jenne DE, Gauthier F: Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 2010;62:726–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Craciun I, Fenner AM, Kerns RJ: N-Arylacyl O-sulfonated aminoglycosides as novel inhibitors of human neutrophil elastase, cathepsin G and proteinase 3. Glycobiology 2016;26:701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer-Hoffert U, Wiedow O: Neutrophil serine proteases: mediators of innate immune responses. Curr Opin Hematol 2011;18:19–24. [DOI] [PubMed] [Google Scholar]
- 13.Pechous RD: With Friends Like These: The Complex Role of Neutrophils in the Progression of Severe Pneumonia. Front Cell Infect Microbiol 2017;7:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mortaz E, Alipoor SD, Adcock IM, Mumby S, Koenderman L: Update on Neutrophil Function in Severe Inflammation. Front Immunol 2018;9:2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caughey GH: Mast cell proteases as pharmacological targets. Eur J Pharmacol 2016;778:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turk D, Janjic V, Stern I, Podobnik M, Lamba D, Dahl SW, Lauritzen C, Pedersen J, Turk V, Turk B: Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J 2001;20:6570–6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molgaard A, Arnau J, Lauritzen C, Larsen S, Petersen G, Pedersen J: The crystal structure of human dipeptidyl peptidase I (cathepsin C) in complex with the inhibitor Gly-Phe-CHN2. Biochem J 2007;401:645650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pham CT, Ley TJ: Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc Natl Acad Sci U S A 1999;96:8627–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W, Yan M, Liu Y, McLeish KR, Coleman WG, Jr., Rodgers GP: Olfactomedin 4 inhibits cathepsin C-mediated protease activities, thereby modulating neutrophil killing of Staphylococcus aureus and Escherichia coli in mice. J Immunol 2012;189:2460–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adkison AM, Raptis SZ, Kelley DG, Pham CT: Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest 2002;109:363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolters PJ, Pham CT, Muilenburg DJ, Ley TJ, Caughey GH: Dipeptidyl peptidase I is essential for activation of mast cell chymases, but not tryptases, in mice. J Biol Chem 2001;276:18551–18556. [DOI] [PubMed] [Google Scholar]
- 22.Mallen-St Clair J, Pham CT, Villalta SA, Caughey GH, Wolters PJ: Mast cell dipeptidyl peptidase I mediates survival from sepsis. J Clin Invest 2004;113:628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourbeau J, Johnson M: New and controversial therapies for chronic obstructive pulmonary disease. Proc Am Thorac Soc 2009;6:553–554. [DOI] [PubMed] [Google Scholar]
- 24.Seeley EJ, Sutherland RE, Kim SS, Wolters PJ: Systemic mast cell degranulation increases mortality during polymicrobial septic peritonitis in mice. J Leukoc Biol 2011;90:591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi GP: Role of cathepsin C in elastase-induced mouse abdominal aortic aneurysms. Future Cardiol 2007;3:591–593. [DOI] [PubMed] [Google Scholar]
- 26.Raptis SZ, Shapiro SD, Simmons PM, Cheng AM, Pham CT: Serine protease cathepsin G regulates adhesiondependent neutrophil effector functions by modulating integrin clustering. Immunity 2005;22:679–691. [DOI] [PubMed] [Google Scholar]
- 27.Deeds MC, Anderson JM, Armstrong AS, Gastineau DA, Hiddinga HJ, Jahangir A, Eberhardt NL, Kudva YC: Single dose streptozotocin-induced diabetes: considerations for study design in islet transplantation models. Lab Anim 2011;45:131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiMeglio LA, Evans-Molina C, Oram RA: Type 1 diabetes. Lancet 2018;391:2449–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao S, Ho D, Vatner DE, Vatner SF: Echocardiography in Mice. Curr Protoc Mouse Biol 2011;1:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin S, Le Lay J, Everett LJ, Gupta R, Rafiq K, Kaestner KH: CREB mediates the insulinotropic and antiapoptotic effects of GLP-1 signaling in adult mouse beta-cells. Mol Metab 2014;3:803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ares-Carrasco S, Picatoste B, Benito-Martin A, Zubiri I, Sanz AB, Sanchez-Nino MD, Ortiz A, Egido J, Tunon J, Lorenzo O: Myocardial fibrosis and apoptosis, but not inflammation, are present in long-term experimental diabetes. Am J Physiol Heart Circ Physiol 2009;297:H2109–2119. [DOI] [PubMed] [Google Scholar]
- 32.Xiao Q, Zhao XY, Jiang RC, Chen XH, Zhu X, Chen KF, Chen SY, Zhang XL, Qin Y, Liu YH, Luo JD: Increased expression of Sonic hedgehog restores diabetic endothelial progenitor cells and improves cardiac repair after acute myocardial infarction in diabetic mice. Int J Mol Med 2019;44:1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmes JW, Borg TK, Covell JW: Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng 2005;7:223–253. [DOI] [PubMed] [Google Scholar]
- 34.Bing R, Dweck MR: Myocardial fibrosis: why image, how to image and clinical implications. Heart 2019;105:1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang C, Wang K, Li Q, Bai J, Zhang H : Influence of the distribution of fibrosis within an area of myocardial infarction on wave propagation in ventricular tissue. Sci Rep 2019;9:14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fomovsky GM, Rouillard AD, Holmes JW: Regional mechanics determine collagen fiber structure in healing myocardial infarcts. J Mol Cell Cardiol 2012;52:1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tao S, Chen L, Song J, Zhu N, Song X, Shi R, Ge G, Zhang Y: Tanshinone IIA ameliorates diabetic cardiomyopathy by inhibiting Grp78 and CHOP expression in STZ-induced diabetes rats. Exp Ther Med 2019;18:729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun S, Yang S, An N, Wang G, Xu Q, Liu J, Mao Y: Astragalus polysaccharides inhibits cardiomyocyte apoptosis during diabetic cardiomyopathy via the endoplasmic reticulum stress pathway. J Ethnopharmacol 2019;238:111857. [DOI] [PubMed] [Google Scholar]
- 39.Hou H, Zhang Q, Dong H, Ge Z: Matrine improves diabetic cardiomyopathy through TGF-beta-induced protein kinase RNA-like endoplasmic reticulum kinase signaling pathway. J Cell Biochem 2019;120:1357313582. [DOI] [PubMed] [Google Scholar]
- 40.Younce CW, Burmeister MA, Ayala JE: Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis via inhibition of endoplasmic reticulum stress and activation of SERCA2a. Am J Physiol Cell Physiol 2013;304:C508–518. [DOI] [PubMed] [Google Scholar]
- 41.Akk AM, Simmons PM, Chan HW, Agapov E, Holtzman MJ, Grayson MH, Pham CT: Dipeptidyl peptidase I-dependent neutrophil recruitment modulates the inflammatory response to Sendai virus infection. J Immunol 2008;180:3535–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pagano MB, Bartoli MA, Ennis TL, Mao D, Simmons PM, Thompson RW, Pham CT: Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc Natl Acad Sci U S A 2007;104:2855–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holditch SJ, Schreiber CA, Nini R, Tonne JM, Peng KW, Geurts A, Jacob HJ, Burnett JC, Cataliotti A, Ikeda Y: B-Type Natriuretic Peptide Deletion Leads to Progressive Hypertension, Associated Organ Damage, and Reduced Survival: Novel Model for Human Hypertension. Hypertension 2015;66:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, Lan Y, Wang Y, Nie M, Lu Y, Zhao E: Telmisartan suppresses cardiac hypertrophy by inhibiting cardiomyocyte apoptosis via the NFAT/ANP/BNP signaling pathway. Mol Med Rep 2017;15:2574–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alamo L, Ware JS, Pinto A, Gillilan RE, Seidman JG, Seidman CE, Padron R: Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 2017;6:pii:e24634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.England J, Loughna S: Heavy and light roles: myosin in the morphogenesis of the heart. Cell Mol Life Sci 2013;70:1221–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stelzer JE, Brickson SL, Locher MR, Moss RL: Role of myosin heavy chain composition in the stretch activation response of rat myocardium. J Physiol 2007;579:161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson TJ, Nannuru KC, Singh RK: Cathepsin G-mediated activation of pro-matrix metalloproteinase 9 at the tumor-bone interface promotes transforming growth factor-beta signaling and bone destruction. Mol Cancer Res 2009;7:1224–1233. [DOI] [PubMed] [Google Scholar]
- 49.Choi ME, Ding Y, Kim SI: TGF-beta signaling via TAK1 pathway: role in kidney fibrosis. Semin Nephrol 2012;32:244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma FY, Tesch GH, Ozols E, Xie M, Schneider MD, Nikolic-Paterson DJ: TGF-beta1-activated kinase-1 regulates inflammation and fibrosis in the obstructed kidney. Am J Physiol Renal Physiol 2011;300:F1410–1421. [DOI] [PubMed] [Google Scholar]
- 51.Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, Miyai K, Akira S, Brenner DA, Seki E: Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci U S A 2010;107:844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mishra R, Karande AA: Endoplasmic reticulum stress-mediated activation of p38 MAPK, Caspase-2 and Caspase-8 leads to abrin-induced apoptosis. PLoS One 2014;9:e92586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tatsuta T, Hosono M, Miura Y, Sugawara S, Kariya Y, Hakomori S, Nitta K: Involvement of ER stress in apoptosis induced by sialic acid-binding lectin (leczyme) from bullfrog eggs. Int J Oncol 2013;43:17991808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zerbini LF, de Vasconcellos JF, Czibere A, Wang Y, Paccez JD, Gu X, Zhou JR, Libermann TA: JunD-mediated repression of GADD45alpha and gamma regulates escape from cell death in prostate cancer. Cell Cycle 2011;10:2583–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan H, Zhou HF, Akk A, Hu Y, Springer LE, Ennis TL, Pham CTN: Neutrophil Proteases Promote Experimental Abdominal Aortic Aneurysm via Extracellular Trap Release and Plasmacytoid Dendritic Cell Activation. Arterioscler Thromb Vasc Biol 2016;36:1660–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pham CT: Neutrophil serine proteases fine-tune the inflammatory response. Int J Biochem Cell Biol 2008;40:1317–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]