

Abstract
Increasing evidence highlights the central role of neurotoxic oligomers of the 42-residue-long β-amyloid (Aβ42) in Alzheimer's disease (AD). However, very limited information is available on the structural transition from oligomer to fibril, particularly for pathologically relevant amyloids. To the best of our knowledge, we present here the first site-specific structural characterization of Aβ42 misfolding, from toxic oligomeric assembly yielding a similar conformation to an AD-associated Aβ42 oligomer, into a fibril. Transmission EM (TEM) analysis revealed that a spherical amyloid assembly (SPA) of Aβ42 with a 15.6 ± 2.1-nm diameter forms in a ∼30-μm Aβ42 solution after a ∼10-h incubation at 4 °C, followed by a slow conversion into fibril at ∼180 h. Immunological analysis suggested that the SPA has a surface structure similar to that of amylospheroid (ASPD), a patient-derived toxic Aβ oligomer, which had a diameter of 10–15 nm in negative-stain TEM. Solid-state NMR analyses indicated that the SPA structure involves a β-loop-β motif, which significantly differed from the triple-β motif observed for the Aβ42 fibril. The comparison of the 13C chemical shifts of SPA with those of the fibril prepared in the above conditions and interstrand distance measurements suggested a large conformational change involving rearrangements of intermolecular β-sheet into in-register parallel β-sheet during the misfolding. A comparison of the SPA and ASPD 13C chemical shifts indicated that SPA is structurally similar to the ASPD relevant to AD. These observations provide insights into the architecture and key structural transitions of amyloid oligomers relevant for AD pathology.
Keywords: amyloid-beta (AB), amyloid, solid state NMR, protein misfolding, structural biology, oligomer, Alzheimer disease, neurodegeneration, protein aggregation, amylospheroid (ASPD), fibrillization, neurotoxic protein, spherical amyloid assembly (SPA), Abeta42
Introduction
Growing evidence suggests that subfibrillar diffusible aggregates and oligomers of β-amyloid (Aβ)3 play crucial roles in Alzheimer's disease (AD). The formation of neurotoxic Aβ fibrils, a primary component of senile plaque, has long been considered a trigger for neural degeneration in AD (1). Recent studies, however, suggested that some diffusible Aβ assemblies exhibit much higher cytotoxicity than Aβ fibrils (2–11). These studies provided support for a revised amyloid-cascade hypothesis, asserting that neural cell death in early AD may be caused by diffusible aggregates of Aβ rather than fibrils (10, 12, 13). In the past few decades, the potential pathological importance of such diffusible Aβ assemblies or oligomers has prompted an increasing number of studies that have identified misfolding intermediates of 40- or 42-residue Aβ (Aβ40 and Aβ42, respectively) (2–4, 14–20). Using immunological analysis, (3, 4, 6, 21–23) size-exclusion chromatography, transmission EM (TEM), and atomic force microscopy (24–26), these studies revealed various morphological and biological features of the diffusible Aβ assemblies. For example, cytotoxic Aβ-derived diffusible ligands (ADDLs) are small off-pathway globular oligomers of 5–6-nm size (2, 4, 15, 16). The size of AD brain-derived amylospheroid (ASPD) species (3) (12.5 ± 2.5-nm diameter by negative stain TEM) is larger than that of the ADDL ones. ASPD is believed to play an important role in AD pathologies and is considered a promising therapeutic target of AD (3, 14).
The atomic structures of amyloid intermediates pathogenically relevant for AD have attracted considerable attention, as the structural features of the Aβ oligomers would provide important understanding of the amyloid formation in AD, which may lead to plausible therapeutic interference against Aβ oligomers or fibrils. So far, only a handful of structural studies, based on solid-state NMR (SSNMR) approaches, have focused on Aβ oligomers with limited structural resolution (24, 27–29). For example, a fibrillar oligomer (23) of Aβ40, named β-sheet intermediate (Iβ), was reported to display a β-turn-β motif (29), which is commonly observed for Aβ40 fibril structures (30–33). A disk-shaped pentamer of Aβ42, identified by SSNMR and atomic force microscopy analyses, displayed a triple-β motif; the β1-turn-β2 motif of the pentamer was proposed to be stabilized by the hydrophobic contact between Phe19 and Leu34 (24). An SDS stable oligomer of Aβ42 showed a supramolecular structure with an antiparallel alignment (28). More recently, we have shown that the structure of ASPD reconstituted in vitro with synthetic Aβ42 at low temperature exhibits a β-loop-β motif (27). ASPD is a neurotoxic Aβ oligomer derived from human AD brain, with a positive correlation with AD (3, 14, 27, 34). TEM analysis revealed that brain-derived ASPD possesses a spherical morphology with a diameter of 10–15 nm. However, almost no structural information has been reported for other pathologically relevant Aβ oligomers.
Another important task in structural studies of amyloid intermediates associated with AD and other neurodegenerative diseases is the characterization of the misfolding pathway. The structural relationship between amyloid intermediate and fibril (the end product of misfolding) provides insight into the key structural transition that distinguishes the metastable and more toxic oligomer and the fibril species in the misfolding pathway. A few studies successfully characterized the stepwise structural transitions in the amyloid misfolding into fibril (24, 30). In particular, no previous studies provided atomic-level insight into the structural transition from Aβ oligomer to fibril for pathologically relevant amyloid species. The very limited amount of available samples has hindered the structural characterization of brain-derived (in or ex vivo) soluble Aβ oligomers (4, 35). Although Western blotting and immune analysis provided some insight into the size and structure of brain-derived oligomers (4, 35), the detailed site-specific structural analysis of these species is difficult. Thus, despite significant efforts, the structural evolution of Aβ and other amyloid proteins during the misfolding process associated with AD is still elusive.
In this work, we have studied the structural evolution of an amyloid intermediate by establishing a protocol to prepare an ASPD-like spherical assembly of Aβ42. Our group has recently succeeded in characterizing the site-resolved structure of ASPD, which was reconstituted in vitro in an F12 medium for the first time (27). However, in these conditions, Aβ42 tends to be metastable in the oligomer form; thus, a clear structural transition to the fibril morphology was not observed (3). To monitor the structural transition, in this work, we optimized the incubation conditions by replacing the F12 medium with a low-salt phosphate buffer at pH 7. Under these altered conditions, we could observe the formation of a structurally well-defined and stable Aβ oligomer, labeled “spherical assembly” (SPA), and characterize its subsequent transition to Aβ42 fibril by a combination of TEM, thioflavin T (ThT) fluorescence, and SSNMR analyses. The SSNMR structural analysis of the SPA and fibril samples showed drastic differences in the secondary structural profile and supramolecular structure, indicating that a significant conformational change and β-sheet rearrangements along the misfolding pathway may explain the stability of the Aβ42 oligomer in the misfolding process.
Results and discussion
Formation of spherical assembly and conversion into Aβ42 fibril
First, we examined the TEM images and ThT fluorescence data of an aqueous Aβ42 solution to examine the morphological and kinetic features of Aβ aggregates and to establish the optimum conditions for isolating SPA. Fig. 1 shows the incubation time (t) dependence of the (a) ThT fluorescence intensity and (b) TEM images for an Aβ42 solution (29 μm) in 10 mm phosphate buffer (pH 7.5) at 4 °C (see details under “Materials and methods”). In the TEM images of the sample (Fig. 1b), the spherical morphology of SPA emerged at t = 12–79 h. The ThT fluorescence is a sensitive indicator of the formation of amyloid fibrils or amyloid-like extended β-sheet structures (36). The results show that Aβ42 did not form ThT-positive amyloid assemblies until t reached ∼80 h. The experiments are reproducible (Figs. S1 (a and b) and S2). The TEM results from a separate test involving incubation of 32 μm Aβ42 (Fig. S1, a and b) show that SPA was formed at t = 7–50 h and was still the dominant species after 50 h of incubation. These results indicate that Aβ42 was misfolded into SPA at an early stage of the incubation (7–12 h) and that SPA remained stable at t ∼50 h. Therefore, we selected 12–14 h as a suitable incubation period for harvesting SPA samples for the SSNMR experiments. The spherical morphologies of SPA species in the TEM images are similar to those observed for the ASPD ones (27). Neither SPA nor ASPD could be detected by ThT fluorescence. Despite the similarities between the two morphologies, the diameters of the SPA species (in the range of 11–21 nm and with an average value of 15.6 ± 2.1 nm) are slightly larger than those of the ASPD ones (10–15 nm). As discussed below, although our immunological analysis using ASPD-specific antibodies highlighted some similarities between ASPD and SPA, the reactivity of the antibodies to SPA was ∼40% of that to ASPD. Thus, SPA and ASPD exhibit a considerably different behavior while retaining conformational similarity.
Figure 1.
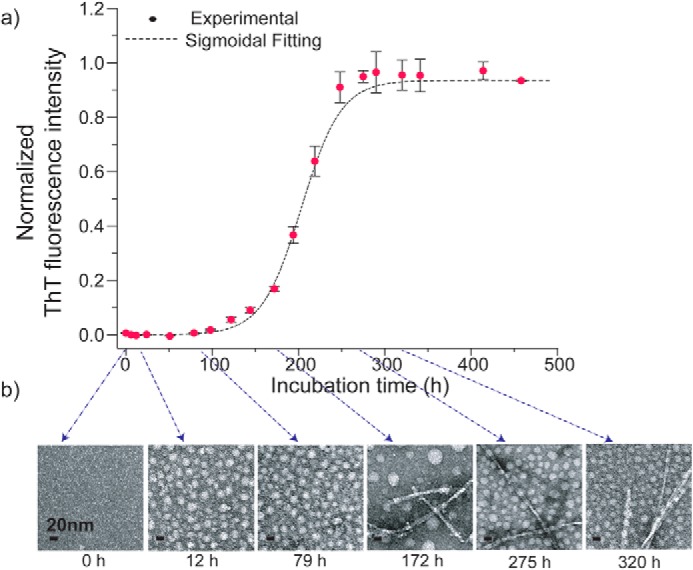
ThT fluorescence (a) and TEM (b) images monitored at different incubation times (t) for 29 μm Aβ42 in 10 mm phosphate buffer (pH 7.5, 0.02% (w/v) NaN3) at 4 °C. The TEM images show the morphological changes in the Aβ42 aggregates at t = 0–320 h. Error bars, S.D.
In the incubation conditions used here, SPA was reproducibly observed after 12–14 h of incubation. The established protocol was employed to prepare the SPA samples for the SSNMR-based structural analysis. Unlike previous results for ASPD (3), the present TEM results showed that mature fibrils of Aβ42 were successfully formed at a later stage of the incubation (t ∼180 h) in the optimized conditions (Fig. 1b). After 1 month (720 h) of incubation, we observed matured fibrils as a dominant species (∼70% in weight) coexisting with minor amounts of SPA, as confirmed by the TEM (Fig. S1, c and d) and SSNMR (Fig. S9) measurements. These results indicate that SPA, the kinetically favored amyloid intermediate, was converted to the energetically favored fibril along the Aβ42 misfolding pathway at a later stage of the incubation; then the two species reached a quasi-equilibrium state where they coexisted as a mixture (see the images at 275 and 320 h in Fig. 1b). It was also shown that the fibril (Fig. S1c) and SPA (Fig. S1d) species could be separately collected from the bottom pellet and supernatant of the Aβ solution, respectively, after centrifuging the solution at 8,000 × g for 45 min, as shown in Fig. S1d. Interestingly, immediately before the conversion into fibril at ∼175 h, spherical species with noticeably larger diameter (30–40 nm) were consistently observed. Unlike the SPA, these species disappeared rapidly as the fibril became the dominant species. Thus, we speculate that these species may be transient amyloid intermediates that trigger the conversion into the fibril, although the characterization of this intermediate is beyond the scope of the present work. In this work, we focus on the SPA that forms at an early stage of amyloid assembly, before the fibril formation.
As discussed below, the structure of the Aβ42 fibril formed under the incubation conditions may differ from the S-shaped triple-β-sheet structure reported for Aβ42 fibrils (triple-β fibril) prepared at room temperature (RT), (37) denoted as RT-fibril. Thus, the present study also reports the isolation of a new form of Aβ42 fibril, denoted as low-temperature fibril (LT-fibril). Whereas the structural characterization of the LT-fibril species will be the subject of future investigations, in the present study, we mainly focus on the structural analysis of SPA based on SSNMR measurements.
SSNMR analysis of SPA and fibril samples
For the SSNMR structural analysis, we harvested SPA samples prepared with 13C- and 15N-labeled Aβ42 at an incubation time of 12 h and subsequently lyophilized these samples after rapid freezing in liquid nitrogen (see “Materials and methods”). The effect of lyophilization of SPA was confirmed by TEM. The TEM images of SPA samples without and with lyophilization showed nearly identical images (see Fig. S3). Fig. 2 (a and b) shows the 2D 13C-13C correlation SSNMR spectra of the lyophilized SPA for Aβ42 samples in which uniformly 13C- and 15N-labeled amino acids were incorporated at several selected residues in two different labeling schemes (see the legend to Fig. 2; further SSNMR data for additional samples are shown in Fig. S5). The data were obtained with a dipolar-assisted rotational resonance (DARR) recoupling sequence (38) using a mixing time of 50 ms. We were able to uniquely assign the signals for the 13C sites (color-coded lines) of all isotope-labeled residues. In most cases, each chemically bonded 13C-13C pair produced a single set of cross-peaks, suggesting that SPA is an oligomer made of Aβ molecules with a relatively homogeneous structure. The line widths of 13C-13C cross-peaks (3–4 ppm) are reasonably narrow, compared with those of lyophilized amyloid fibrils (30). Although the line widths are subject to mild broadening due to conformational heterogeneity in the lyophilized state (29, 31, 33), relatively narrow lines were still observed for structured regions. The cross-peak intensities are generally suppressed in regions with dynamic or structural heterogeneities. For the SPA samples, the residues near the N terminus, namely Ala2 (Fig. 2a), Phe4, Val12 (Fig. S5a), and His13 (Fig. S5b), showed weak cross-peaks, suggesting that the structure of SPA in the N-terminal region is dynamic or heterogeneous, which is consistent with the SSNMR results for the Aβ42 fibril. The C-terminal residues of SPA, such as Val39 and Ile41 (Fig. 2a), showed strong cross-peaks, indicating that the C terminus of SPA has a rigid conformation. Besides the N-terminal region, the weak cross-peaks for the Ala21 and Val24 residues in the 2D 13C-13C SSNMR spectrum (Fig. 2b) revealed the dynamic nature of these residues. As discussed later, this finding is consistent with the conclusion based on the secondary structure analysis, that these residues are located in the non-β segment.
Figure 2.

2D 13C SSNMR chemical shift correlation spectra of SPA (a and b) and LT-fibril (c) samples. The samples were uniformly labeled with 13C and 15N at Ala2, Gly9, Phe20, Val39, and Ile41 (a) and Phe20, Ala21, Val24, Gly25, and Leu34 residues (b and c). The overall experiment time was 24–72 h. The fibril sample used in c was hydrated, whereas the SPA samples in a and b were lyophilized. The assignments are indicated by color-coded dotted lines for Ala (orange), Val (green), Ile/Leu (cyan), and Phe (red).
Next, we compared the SSNMR data for LT-fibril harvested at 746 h (31 days; Fig. 2c) with the corresponding spectrum of SPA in the same labeling scheme (Fig. 2b). The spectrum of LT-fibril shows a completely different spectral profile from that of SPA; for example, the strong cross-peaks for Ala21 and Val24 indicate the lack of dynamics for the residues. The narrower line widths in Fig. 2c compared with those in Fig. 2b are attributed partly to the fact that the fibril sample of Fig. 2c was hydrated, whereas the sample in Fig. 2b was lyophilized. For comparison, we confirmed that the line widths of a lyophilized control Aβ42 fibril sample (prepared at a room temperature) decreased from 2–3 ppm to 1.5–2 ppm upon hydration (see Fig. S6). Moreover, the 13Cα and 13Cβ chemical shift positions for Phe20 and Val24 show large differences between SPA and LT-fibril. These results indicate different loop or turn locations in the LT-fibril and SPA samples. Thus, in addition to the morphological change, SPA undergoes a large conformational change at the atomic level upon misfolding into fibril. This suggests that SPA is a structurally distinct intermediate from the fibril. As discussed later, the LT-fibril sample also showed 13C chemical shifts that are markedly different from those of the RT Aβ42 fibril. As only one set of cross-peaks was observed for a directly bonded 13C-13C pair, it appears that SPA misfolded into LT-fibril with a unique conformation. Whereas the structural details of the LT-fibril species will be investigated in future work, the SSNMR analysis of SPA provides an effective tool to characterize the structural evolution of Aβ42 from amyloid intermediate to fibril.
Secondary structure of SPA
Based on the SSNMR analysis of six SPA samples with different labeling schemes (Fig. 2 (a and b) and Fig. S5), we completed signal assignments for 29 residues, which cover 83% of the structurally ordered residues 12–42 (Table S2). Then the secondary structure of SPA was analyzed using the chemical shift data. The secondary chemical shifts for 13CO, 13Cα, and 13Cβ (39, 40) are shown in Fig. 3 (a–c), whereas the torsion angles and secondary structures predicted by the TALOS-N software (41) are displayed in Fig. 3d. Both analyses indicate the presence of two β-strand regions (β1 (His13–Ala21) and β2 (Ala30–Ile41)) that are connected with a turn/loop region at Glu22–Gly29. Interestingly, the two β-sheet regions of SPA are considerably different from the triple β-sheet regions reported for Aβ42 fibrils (β1 (Val12–Val18), β2 (Val24–Gly33), and β3 (Val36–Ile40)) (37). These results are consistent with the marked conformational change from SPA to LT-fibril suggested by the data in Fig. 2. Instead, the above regions are similar to those previously reported for Aβ40 fibrils (30, 33, 42) and Aβ42 ASPD (27) structure models (Fig. 3d; see also Fig. S7 (a, b, and d)). We also found that the 13C chemical shifts of SPA are similar to those of ASPD, except for the Phe20 and Val24 residues, as discussed below. Overall, our analysis indicates that the SPA is a structurally ordered amyloid oligomer exhibiting a β-turn-β (or β-loop-β) motif.
Figure 3.
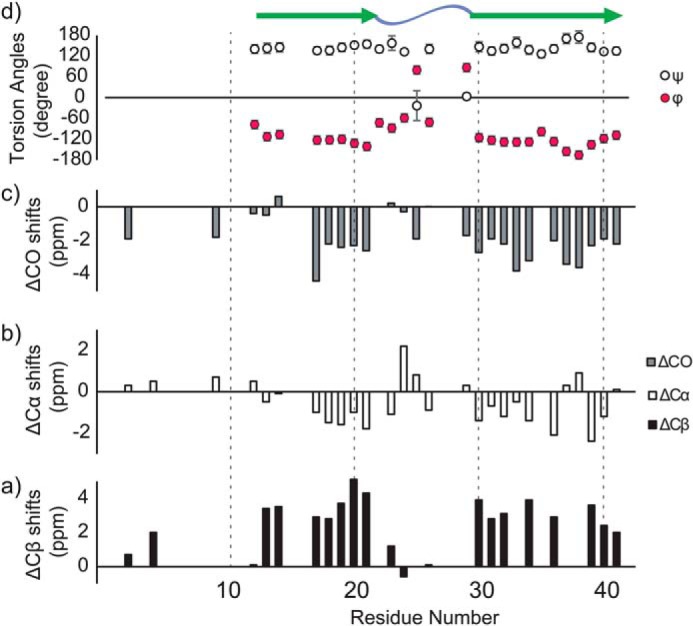
13C chemical shift and secondary structural profile of SPA. a–c, secondary 13C chemical shifts obtained by SSNMR for 13Cβ (a), 13Cα (b), and 13CO (c) in the SPA form of Aβ42 (40). d, dihedral angles (ϕ, ψ) obtained by the TALOS-N software according to the 13C chemical shift analysis (Table S2) of Aβ42 in the SPA form (41). As shown in the top region of d, the secondary structure analysis performed by the TALOS software identified two β-strand regions (green arrows) separated by loop/turn regions (blue wave) at residues 22–29.
Probing the supramolecular structure of SPA
Although SPA adopts an extended β-sheet structure, which is commonly observed for many Aβ fibrils (18, 27, 30, 31, 33), our data show that this species is not detected by ThT fluorescence like ASPD. To identify unique differences in the tertiary structure of the SPA and fibril samples, we examined intermolecular contacts that would characterize the tertiary arrangement of Aβ in the SPA form. Because only a few structural studies of Aβ oligomers and fibrils suggest the occurrence of antiparallel β-sheet motifs, (28, 43, 44), we performed a SSNMR analysis based on the hypothesis that SPA forms a parallel β-sheet structure. Three Aβ42 SPA samples prepared for SSNMR experiments were selectively labeled at 13CO of Ala30, Leu34, or Val39 residues, to perform interstrand 13CO-13CO distance measurements at each site. As previously described (45), the 13C dephasing curves (Fig. 4, a–c) measured by constant-time finite-pulse radiofrequency-driven recoupling (fpRFDR-CT) SSNMR experiments were compared with simulated dephasing curves for varied distances, which were calculated with the SpinEvolution software. (46) The experimental protocols were described previously (37). The interstrand distances for Ala30, Leu34, and Val39 in SPA were determined to be 6.1 ± 0.2, 5.9 ± 0.2, and 6.0 ± 0.1 Å, respectively. Interestingly, these distances consistently deviated from the 4.7–5.0 Å values that were reported for interstrand distances of the Aβ40 and Aβ42 fibrils in in-register parallel β-sheet arrangements (Fig. 4d) (31, 37).
Figure 4.
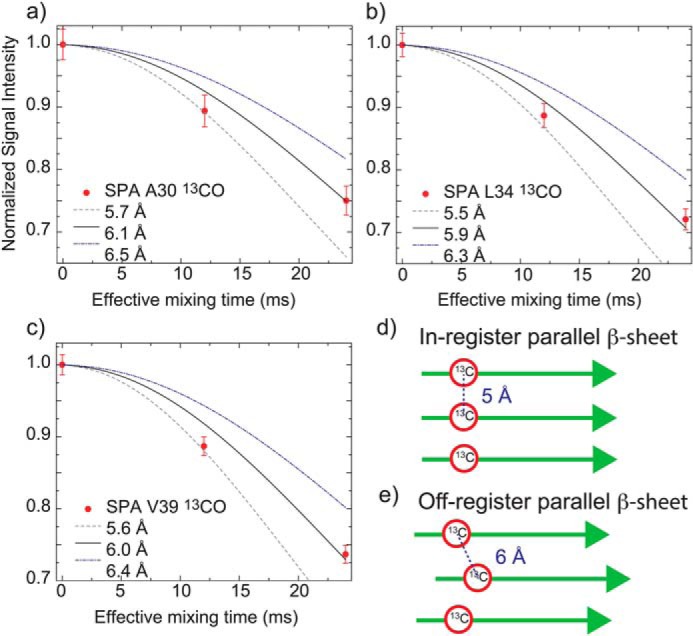
Signal dephasing curves obtained by fpRFDR-CT experiments (45) for the determination of interstrand 13CO–13CO distances in Aβ42 SPA samples with 13CO labeled at Ala30 (a), Leu34 (b), and Val39 (c). The interstrand distances at Ala30, Leu34, and Val39 residues were found to be 6.1 ± 0.2 Å (χ2min = 1.62), 5.9 ± 0.2 Å (χ2min = 2.16), and 6.0 ± 0.1 Å (χ2min = 6.34), respectively. χ2min analysis was employed to fit the experimental data to the simulated dephasing curves (see “Materials and methods”). According to the Δχ2 analysis (confidence level p = 90%, degree of freedom = 1, Δχ2 = 2.71), the errors in the 13CO–13CO interstrand distances for Ala30, Leu34, and Val39 are within 0.2, 0.2, and 0.1 Å, respectively. d and e, arrangements of in-register (d) and off-register (e) parallel β-sheets with corresponding interstrand 13CO–13CO distances. Error bars, S.D.
These results indicate that SPA is most likely to have an off-registered parallel β-sheet structure (Fig. 4e), rather than an in-register arrangement found in fibrils. It is the first time that such an off-registered parallel β-sheet structure was indicated for any amyloid species. Although other arrangement may be possible, the deviation from the typical fibril structural motifs for Aβ is consistent with the fact that SPA was not detected by ThT fluorescence despite its extended β-sheet structure.
We also performed DARR experiments with a mixing time of 200 ms, but no long-range contacts were identified due to the limited sensitivity. The 13C-15N distances, measured by frequency-selective rotational-echo double-resonance (47), also excluded the formation of a salt bridge between Asp23 and Lys28 (data not shown). Thus, the SPA structure is characterized by a fold distinct from those of both Aβ40 and Aβ42 fibrils.
Structural and toxicity comparison of SPA and ASPD
Here, we present a semiquantitative comparison of the structure of SPA and ASPD by immunological assays and NMR-based chemical shift analysis. First, we compared the surface structures of SPA and ASPD using an ASPD-specific polyclonal antibody purified from rabbit (rpASD1) (3). The rpASD1 antibody selectively binds both ex vivo and in vitro ASPD (158–669 kDa) with high affinity (Kd = 0.005 nm) (3), whereas it does not cross-react with other forms of Aβ42, such as monomer, fibrils, and other Aβ oligomers, including fibrillar oligomers (A11 or ThT-detectable) and ADDLs (2, 3). Dot-blot assays for SPA and ASPD samples were serially diluted from 24 μm to the desired concentrations, as shown in Fig. 5. Duplicated dilution series of the samples were loaded on the dots (Fig. 5, a and b) with rpASD1 (a) and 82E1 (b) antibodies; the latter is a conformation-independent antibody targeting the linear epitope Aβ(1–16) in all types of Aβ and was used for the quantification of the total Aβ amount. The average intensities representing the reactivity of the rpASD1 (c) and 82E1 (d) antibodies toward SPA and ASPD were plotted for different sample concentrations (Fig. 5, c and d). The emission intensities of the SPA and ASPD samples in the 82E1 assay (Fig. 5, b and d) were detected and showed a linear increase from 0.0625 to 1.0 pmol/dot, with a slight variation for partial dissociation into monomers, the inefficiency of the membranes in retaining SPA, and/or other unknown reasons. Although the reactivity of rpASD1 toward SPA was lower than that toward ASPD, SPA was clearly recognized by rpASD1. This is a strong indication that SPA has a similar surface structure to ASPD, unlike ADDLs or fibrillar oligomers. Despite their size difference, the similarity between SPA and ASPD is as high as 38 ± 6%, as estimated by the reactivity of rpASD1 normalized by that of 82E1 (see “Materials and methods”).
Figure 5.

a and b, dot-blot assays for different concentrations of SPA and ASPD using rpASD1 anti-ASPD antibody (a) and 82E1 antibody (b), the latter of which targets Aβ(1–16). Serially diluted samples were loaded on the dots in duplicate (a and b), and the average intensities of the dots were plotted in c and d. The rpASD1 assays (a and c) were used for the detection of ASPD species, whereas the 82E1 assays (b and d) were performed as a control for the quantification of Aβ species. Error bars, S.D.
Next, we compared the 13C SSNMR shifts of the SPA and ASPD species. Fig. 6 shows a comparison of the secondary 13Cα (red circle) and 13Cβ (black diamond) chemical shifts of the SPA sample with those of the ASPD species (a) (27), Aβ42 fibril (b), and Aβ40 fibril (c), where the secondary shifts denote deviations from the corresponding shift for a model peptide in a random coil conformation. As the secondary shifts sensitively reflect the secondary structure of a particular residue, similar secondary shifts indicate similar confirmations. The root mean square deviations (RMSDs) between the 13C chemical shifts of SPA and ASPD were calculated to be RMSDCα = 1.23 ppm (n = 17) and RMSDCβ = 1.40 ppm (n = 13), where n is the number of pairs of carbon atoms used in each calculation. In contrast, the RMSDs between the 13C chemical shifts of SPA and Aβ42 fibrils were larger; RMSDCα = 2.03 ppm (n = 27) and RMSDCβ = 2.01 ppm (n = 21). The deviations between SPA and Aβ40 fibrils were similarly larger: RMSDCα = 1.98 ppm (n = 23) and RMSDCβ = 1.86 ppm (n = 17). The smaller RMSD values reflect a higher structural similarity between SPA and ASPD. Most of the data points in the ASPD versus SPA plot (a) are distributed along the y = x (blue solid) line, within the y = x ± 1 region (cyan dashed lines), indicating conformational similarity between the two species. Except for the N-terminal residues (positions 1–12) and Ile41, the only large difference in Δ13C chemical shifts was observed for the Phe20 and Val24 residues, for which both |Δ13Cα| and |Δ13Cβ| (Fig. 6, green arrows in a) exceeded 1 ppm (∼4 ppm for Val24), indicating some conformational difference. In contrast, large deviations were observed in b and c, denoting structural differences between SPA and Aβ42 or Aβ40 fibrils. The SSNMR-based structural comparison between the SPA and ASPD forms of Aβ42 thus reveals a good consistency between their conformations, except for the Phe20 and Val24 residues at or near the non-β-sheet region. Such differences may be attributed to the different solvents (i.e. phosphate buffer and F12 medium) used for incubating the two species. In combination with the SSNMR results discussed above, it can be concluded that SPA shares similar surface and conformational structure properties with ASPD.
Figure 6.

Comparison of 13C secondary shifts (ΔCS) of SPA with those of ASPD (a), Aβ42 fibril (b), and Aβ40 fibril (quiescent) (c) samples. The secondary chemical shifts of the ASPD, Aβ42 fibril, and Aβ40 fibril species (Δ13Cα shown in red and Δ13Cβ in black) are plotted on the vertical axis against those of SPA on the horizontal axis, along with the y = x (blue solid line) and y = x ± 1 (cyan dashed lines) curves. Marked differences between SPA and ASPD were found in both ΔCα and ΔCβ shifts of Phe20 and Val24 (green arrows), indicating different conformations of the two species.
Our preliminary toxicity assay on rat hippocampal cells confirmed cytotoxicity of SPA samples both without and with lyophilization at a concentration of 3–6 μm (p < 0.05; see Fig. S4). The cytotoxicity of SPA was lower than that of ASPD, which required lower agonist concentration (0.4–1.3 μm) to cause cytotoxicity. The toxicity of the SPA was approximately the same as or slightly higher than that of fibril samples. The cytotoxicity of lyophilized SPA showed a relatively large variation among different trials compared with that of SPA without lyophilization, fibril, and ASPD for an unknown reason. The present data confirmed that the SPA is a toxic oligomeric species of Aβ42. The observed toxicity levels indicated that ASPD was more toxic than SPA, confirming that SPA and ASPD are likely to be different species.
Structural transition from SPA to Aβ42 fibril
Finally, we examined the structural transition from SPA to fibril along the misfolding pathway of Aβ42 in low-salt and low-temperature conditions. Fig. 7a shows a comparison of the 2D 13C-13C SSNMR spectra of SPA (Fig. 7a, blue spectrum) and of the fibrils harvested after 1 month of low-temperature incubation (LT-fibrils, orange spectrum in Fig. 7 (a and b)). Fig. S8 shows a comparison of the secondary shifts. The completely different profiles of the SPA and Aβ42 fibril (both LT and RT) species show that SPA is structurally and morphologically distinct from the fibril species. Based on this finding, we hypothesize that SPA is an off-pathway oligomer in the fibril formation process. In contrast, a fibrillar oligomer of Aβ40 (Iβ) (29) was previously reported to be the on-pathway intermediate for the formation of the Aβ40 fibril (30), based on the fact that the SSNMR chemical shifts of the two species were almost identical.
Figure 7.

Comparison of 2D 13C-13C correlation SSNMR spectra of the LT fibril (orange) with the corresponding spectra of Aβ42 SPA (blue) (a) and RT-fibrils collected after 1 week of incubation (green) (b). All samples were uniformly labeled with 13C and 15N at Phe20, Ala21, Val24, Gly25, and Leu34 residues. The overall experimental time was 24–72 h.
SSNMR analysis reveals kinetic features of the Aβ42 misfolding process
Extended incubation of SPA at low temperature can lead to the formation of the LT-fibril Aβ42 polymorph. Fig. 7b compares the 2D 13C-13C spectrum of the LT-fibril (orange spectrum, which is identical to that in a) and RT-fibril (green spectrum) polymorphs. Again, the two Aβ42 fibril polymorphs are found to be conformationally different, according to the quite distinct 13C chemical shifts in their SSNMR spectra (Fig. 7b). This indicates that the LT-fibril species represents a new type of Aβ42 fibril. Thus, SPA shows a completely different structural profile from both the LT fibril and the triple-β fibril.
Fig. 8 shows a proposed mechanism for the Aβ42 misfolding into the three distinct Aβ42 assemblies discussed in this work. RT conditions lead to the rapid formation of the kinetically favored fibril in the S-shaped triple β-sheet structure (colored in green in Fig. 8). The latter was found to be the dominant species even in the unseeded incubation at RT (37). Low-temperature incubation conditions are likely to lead to a separate misfolding pathway, which produces SPA (Fig. 8, blue) and then LT-fibril (orange). As mentioned above, the SPA species, with a unique off-registered parallel β-sheet, is likely to have a structural profile very different from the LT-fibril one, indicating that SPA may be an off-pathway intermediate species in the misfolding process into fibrils at 4 °C. Our data suggest that SPA has to undergo a large structural transition for the formation of LT-fibril. Our data also confirm that SPA has a structure similar to that of ASPD (purple). These results may explain the considerable stability of the pathologically relevant Aβ42 oligomeric species such as ASPD due to the expected large energy barrier in the transition toward the fibril. Also, the study provides insight into a possible therapeutic strategy that stabilizing in-register parallel β-sheet may shift the equilibrium toward less toxic amyloid fibril from the oligomers for Aβ42. Although further structural studies are needed to examine the atomic details of the structural conversion, this study highlighted unique site-specific structural features characterizing the conversion of Aβ42 from the unique oligomer SPA into fibril.
Figure 8.
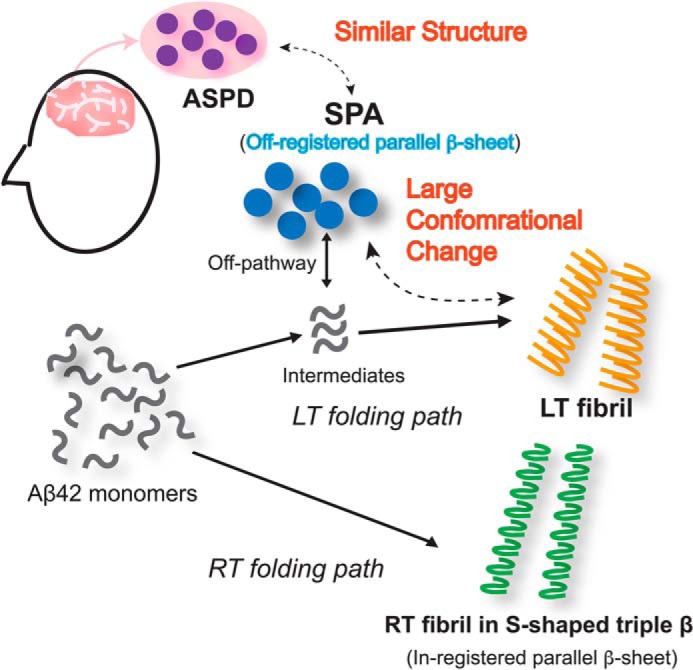
Proposed Aβ42 folding pathways at RT and LT (4 °C). The solid arrows at the top indicate the misfolding path at LT, whereas the solid arrow at the bottom marks the RT path. The SPA species is considered to be an off-pathway intermediate. The dotted arrows indicate structural relationships.
Conclusions
In conclusion, a new oligomeric Aβ42 species with a diameter of ∼16 nm, termed SPA, was obtained under low-salt incubation conditions. This study presents the first structural characterization at atomic level of the conversion to fibril of an Aβ42 amyloid oligomer conformationally similar to the Aβ oligomer associated with AD. At variance with previous studies (24), the present work showed that in the course of the misfolding process into fibril, Aβ42 can form a highly ordered and metastable oligomer with a unique β-sheet profile, which is recognized by ASPD-specific conformational antibodies and may thus be used as a therapeutic target. The structural similarity of SPA and ASPD may make SPA an excellent model for the structural characterization of ASPD, which is more pathologically relevant to the development of AD. The study also demonstrates that SPA is likely to involve a unique off-register parallel β-sheet alignment, which has never been observed for any Aβ fibrils or oligomers reported previously (24, 28, 29, 37, 48, 49). The unique structural motif and the large conformational change from SPA to fibril indicate a large energy barrier in fibrillization, which may explain the stability of the Aβ42 oligomer in the misfolding path. Last, our study indicates important clues for designing therapeutic agents for AD. First, the study indicates that parallel β-sheet is a common motif that characterizes both fibrils and oligomers. This may explain why β-sheet breakers often reduce amyloid toxicity, as they should break down both fibrils and oligomers. Second, although fibrils and oligomers share parallel β-sheet structures in common, our data show that each has a different β-sheet motif. SPA is likely to have an off-registered parallel β-sheet structure, whereas fibrils have an in-registered one. Based on this structural difference, we think it might be possible to develop reagents that stabilize preferentially in-parallel β-sheet structure and increase the amount of less toxic fibril by shifting the equilibrium from toxic oligomers to fibrils. Dissection of atomic level difference in the future would contribute to developing such a fibril stabilizer for AD therapy.
Materials and methods
Preparation of SPA and LT-fibrils of Aβ42
Monomeric Aβ42 was prepared as described previously (37). Aβ42 monomers were first dissolved in anhydrous DMSO at 2.0 mm for 30 min at room temperature. The solution was then diluted to 60 μm with 10 mm phosphate buffer (with 0.02% (w/v) NaN3 at pH 7.5, filtered through a 0.2-μm syringe filter) at 4 °C. The 60 μm Aβ solution was filtered through a 50-kDa molecular mass cut-off filter to remove preformed aggregates and resulted in a final Aβ concentration ranging from 29 to 55 μm, as evaluated by a UV-visible spectrophotometer at 280 nm. The SPA sample formed under 400 rpm circular agitation at 4 °C was collected at an incubation time between 12 and 14 h, by quick freezing of the incubated Aβ solution in liquid nitrogen and subsequent lyophilization, for the SSNMR and immunological experiments. For harvesting the LT-fibril species and separating them from the stable oligomers, the above incubation was extended to 1 month, and then the fibril pellet was collected after centrifuging at 8,000 × g for 45 min. The supernatant and pellets were lyophilized separately for the SSNMR analysis.
SSNMR analysis
All SSNMR experiments were performed on a Bruker Avance III spectrometer equipped with a 400-MHz Oxford wide-bore magnet, a home-built 2.5-mm triple-resonance magic-angle spinning (MAS) probe, and a Varian MAS/variable-temperature controller. Lyophilized SPA samples of 8–30 mg, which included phosphate salts, were packed into 2.5-mm zirconia rotors (see Table S1). The lyophilized fibril samples were rehydrated as described previously (37). The MAS frequency was set to 20,000 ± 5 Hz with an air temperature of −10 °C, unless otherwise mentioned. The natural abundance signal of adamantane at 38.48 ppm was used as a secondary reference for 13C chemical shifts referenced to neat tetramethylsilane. 2D 13C/13C correlation SSNMR experiments were carried out with 50-ms DARR mixing. The indirect dimension of the SPA samples was obtained from a total of 160–180 t1 complex points, with a t1 increment of 38.1 μs, except for the sample in Fig. S5 (e and f), for which a total of 112 t1 complex points, with a t1 increment of 50 μs, were used. The indirect dimension of the LT-fibril samples was obtained from a total of 260 t1 complex points, with a t1 increment of 50 μs. A two-pulse phase-modulation 1H decoupling sequence at 90 kHz was employed during the indirect detection and acquisition periods. The adiabatic cross-polarization transfer was performed with a contact time of 1.0–1.5 ms. All 2D SSNMR data were processed with the NMRPipe software (50); the data were apodized with a Lorenz-to-Gauss window function with an inverse exponential narrowing of 30 Hz and a Gaussian broadening of 120 Hz in both the t1 and t2 domains. The fpRFDR-CT data in Fig. 4 were analyzed as described previously (37) by minimizing χ2 = Σk = 0n(Sk,exp − Sk,sim)2/σk2, where Sk,exp and Sk,sim denote signal intensities at the kth data point of the experimental dephasing curve and of the corresponding simulated curve, respectively, whereas σk denotes the S.D. at the kth point. The 13CO–13CO distance was determined by minimizing the χ2 value.
TEM analysis
The TEM data were collected in the following manner unless otherwise mentioned. A 10-μl aliquot of Aβ solution, sampled at an incubation time of 12 h or 1 month (supernatant), was loaded onto a 300-mesh copper formvar/carbon grid for 1.5 min, followed by blotting away the excess solution using filter paper. The sample attached to the grid was negatively stained with a 10-μl solution of 5% (w/v) uranyl acetate for 1.5 min, followed by blotting away and air-drying the excess staining solution. The grids were stored in a desiccating chamber before data collection. TEM measurements were carried out with a JEOL 1220 instrument operated at 80 kV and a magnification of ×120,000. The diameter of SPA was measured by “eye-ball” fitting of the diameters of 115 SPA species in Fig. 1b and two other TEM images obtained with three separate grids.
Dot-blot assay methods
The details of the method used for the dot-blot assays are described in a previous study by Hoshi and co-workers (3). The protocols are briefly summarized below. Serial dilutions of both SPA and ASPD samples were performed by using an ice-cold 1× PBS solution to final concentrations of 2.0, 1.0, 0.5, 0.25, 0.125, 0.0625, and 0.03125 μm. A 2-μl aliquot of each sample was loaded on the dots in duplicate, on a 0.22-μm nitrocellulose membrane (PROTRAN 0.2 μm; Schleicher & Schuell), to ensure that the loaded sample amount on each dot was 4.0, 2.0, 1.0, 0.5, 0.25, 0.125, and 0.0625 pmol, respectively. The membrane blocked with 5% milk in 1× PBS was incubated with a blocking solution containing a primary antibody and then with a blocking solution containing a horseradish peroxidase–conjugated secondary antibody. The membrane was washed three times with 1× PBS before and after application to the secondary antibody. The membrane was then reacted with the reagent containing the horseradish peroxidase substrate, and the emission intensity per dot was detected by chemiluminescence. Two types of primary antibodies were used: rpASD1 is the anti-ASPD antibody, whereas 82E1 is a commercially available antibody that recognizes the N-terminal part of Aβ. The analysis of the similarity between SPA and ASPD was performed as follows. First, the reactivity detected by rpASD1 was normalized by that detected by 82E1 for each of the SPA and ASPD samples, with Aβ amounts of 2.0, 1.0, and 0.5 pmol/dot, for which a linear relationship between reactivity and amount of Aβ was observed in Fig. 5 (c and d). Then the ratios of the normalized reactivities toward SPA and ASPD were obtained for each Aβ amount. The average ratio, weighted by the uncertainty, yielded a similarity parameter of 38 ± 6%, which shows that the SPA species is recognized by rpASD1 to a ∼40% degree with respect to the corresponding recognition of ASPD by rpASD1.
SPA toxicity assay
The Animal Care and Experimentation Committees of the Foundation for Biomedical Research and Innovation at Kobe and TAO Health Life Pharma Co. Ltd. approved animal experiments. Primary rat hippocampal cultures at 3.1 × 104 cells/cm2 (96-well plates) were prepared from embryonic day 17 pups and cultured in Neurobasal medium containing B27 supplement, 2.5 μm l-glutamine, and astrocyte condition medium, as described previously by Hoshi and co-workers (51) At 22 days in vitro, the cultures were stimulated with SPA, fibril, or ASPD for 44 h, and then the cell viability levels or the cell death levels were measured using Cell Counting kit-8 (catalog no. CK04, Dojindo Molecular Technologies) and calcein-AM (catalog no. C396, Dojindo Molecular Technologies) or using propidium iodide (catalog no. P378, Dojindo Molecular Technologies) and the Cell Death Detection ELISAplus kit (catalog no. 11774425001, Roche Applied Science), respectively, according to the manufacturer's instructions. The cell staining area by calcein or the stained nuclear count by propidium iodide was quantified using a Yokogawa CQ1 system.
Author contributions
Y. X., I. M., and Y. I. data curation; Y. X., I. M., M. I., T. S., M. H., and Y. I. investigation; Y. X., I. M., M. H., and Y. I. methodology; Y. X., I. M., M. H., and Y. I. writing-original draft; Y. X., I. M., M. I., T. S., M. H., and Y. I. writing-review and editing; M. H. and Y. I. funding acquisition; M. H. and Y. I. project administration; Y. I. conceptualization; Y. I. supervision.
Supplementary Material
Acknowledgment
We thank the Biomaterials Analysis Division, Tokyo Institute of Technology, for technical assistance with TEM.
The initial part of this work was supported primarily by National Institutes of Health Grants U01 GM098033 and R01 GM098033 (to Y. I.). This work was also supported by JST-Mirai, Japan, Program Grant JPMJMI17A2 (to Y. I.) and Japan Society for the Promotion of Science (SSPS) KAKENHI Grant 17H04055 (to M. H.). The instruments employed in this work were supported in part by JSPS KAKENHI Grant JP15K21772 (to Y. I.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S8.
- Aβ
- β-amyloid
- AD
- Alzheimer's disease
- TEM
- transmission EM
- ADDL
- Aβ-derived diffusible ligand
- ASPD
- amylospheroid
- SSNMR
- solid-state NMR
- Iβ
- β-sheet intermediate
- SPA
- spherical amyloid assembly
- ThT
- thioflavin T
- RT
- room temperature
- LT
- low temperature
- 2D
- two-dimensional
- DARR
- dipolar-assisted rotational resonance
- fpRFDR-CT
- constant-time finite-pulse radiofrequency-driven recoupling
- RMSD
- root mean square deviation
- MAS
- magic-angle spinning.
References
- 1. Selkoe D. J. (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 10.1152/physrev.2001.81.2.741 [DOI] [PubMed] [Google Scholar]
- 2. Gong Y., Chang L., Viola K. L., Lacor P. N., Lambert M. P., Finch C. E., Krafft G. A., and Klein W. L. (2003) Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422 10.1073/pnas.1834302100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Noguchi A., Matsumura S., Dezawa M., Tada M., Yanazawa M., Ito A., Akioka M., Kikuchi S., Sato M., Ideno S., Noda M., Fukunari A., Muramatsu S., Itokazu Y., Sato K., et al. (2009) Isolation and characterization of patient-derived, toxic, high mass amyloid β-protein (Aβ) assembly from Alzheimer disease brains. J. Biol. Chem. 284, 32895–32905 10.1074/jbc.M109.000208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., and Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 10.1038/nature04533 [DOI] [PubMed] [Google Scholar]
- 5. Lasagna-Reeves C. A., Glabe C. G., and Kayed R. (2011) Amyloid-β annular protofibrils evade fibrillar fate in Alzheimer disease brain. J. Biol. Chem. 286, 22122–22130 10.1074/jbc.M111.236257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barghorn S., Nimmrich V., Striebinger A., Krantz C., Keller P., Janson B., Bahr M., Schmidt M., Bitner R. S., Harlan J., Barlow E., Ebert U., and Hillen H. (2005) Globular amyloid β-peptide1–42 oligomer: a homogenous and stable neuropathological protein in Alzheimer's disease. J. Neurochem. 95, 834–847 10.1111/j.1471-4159.2005.03407.x [DOI] [PubMed] [Google Scholar]
- 7. Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A., Regan C. M., Walsh D. M., Sabatini B. L., and Selkoe D. J. (2008) Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amar F., Sherman M. A., Rush T., Larson M., Boyle G., Chang L., Götz J., Buisson A., and Lesné S. E. (2017) The amyloid-β oligomer A β*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci. Signal. 10, eaal2021 10.1126/scisignal.aal2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Opazo P., Viana da Silva S., Carta M., Breillat C., Coultrap S. J., Grillo-Bosch D., Sainlos M., Coussen F., Bayer K. U., Mulle C., and Choquet D. (2018) CaMKII metaplasticity drives Aβ oligomer-mediated synaptotoxicity. Cell Rep. 23, 3137–3145 10.1016/j.celrep.2018.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cline E. N., Bicca M. A., Viola K. L., and Klein W. L. (2018) The amyloid-β oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis. 64, S567–S610 10.3233/JAD-179941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kotler S. A., Walsh P., Brender J. R., and Ramamoorthy A. (2014) Differences between amyloid-β aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer's disease. Chem. Soc. Rev. 43, 6692–6700 10.1039/C3CS60431D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Selkoe D. J. (2004) Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat. Cell Biol. 6, 1054–1061 10.1038/ncb1104-1054 [DOI] [PubMed] [Google Scholar]
- 13. Klein W. L. (2013) Synaptotoxic amyloid-β oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer's disease? J. Alzheimers Dis. 33, S49–S65 10.3233/JAD-2012-129039 [DOI] [PubMed] [Google Scholar]
- 14. Matsumura S., Shinoda K., Yamada M., Yokojima S., Inoue M., Ohnishi T., Shimada T., Kikuchi K., Masui D., Hashimoto S., Sato M., Ito A., Akioka M., Takagi S., Nakamura Y., et al. (2011) Two distinct amyloid β-protein (Aβ) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology, and toxicity analyses. J. Biol. Chem. 286, 11555–11562 10.1074/jbc.M110.181313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hepler R. W., Grimm K. M., Nahas D. D., Breese R., Dodson E. C., Acton P., Keller P. M., Yeager M., Wang H., Shughrue P., Kinney G., and Joyce J. G. (2006) Solution state characterization of amyloid β-derived diffusible ligands. Biochemistry 45, 15157–15167 10.1021/bi061850f [DOI] [PubMed] [Google Scholar]
- 16. Yoshihara T., Takiguchi S., Kyuno A., Tanaka K., Kuba S., Hashiguchi S., Ito Y., Hashimoto T., Iwatsubo T., Tsuyama S., Nakashima T., and Sugimura K. (2008) Immunoreactivity of phage library-derived human single-chain antibodies to amyloid β conformers in vitro. J. Biochem. 143, 475–486 10.1093/jb/mvm239 [DOI] [PubMed] [Google Scholar]
- 17. Williamson M. P., Suzuki Y., Bourne N. T., and Asakura T. (2006) Binding of amyloid β-peptide to ganglioside micelles is dependent on histidine-13. Biochem. J. 397, 483–490 10.1042/BJ20060293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu L., Edalji R., Harlan J. E., Holzman T. F., Lopez A. P., Labkovsky B., Pereda L., Labokovsky B., Hillen H., Barghorn S., Ebert U., Richardson P. L., Miesbauer L., Solomon L., Bartley D., et al. (2009) Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry 48, 1870–1877 10.1021/bi802046n [DOI] [PubMed] [Google Scholar]
- 19. Lopez del Amo J. M., Fink U., Dasari M., Grelle G., Wanker E. E., Bieschke J., and Reif B. (2012) Structural properties of EGCG-induced, nontoxic Alzheimer's disease Aβ oligomers. J. Mol. Biol. 421, 517–524 10.1016/j.jmb.2012.01.013 [DOI] [PubMed] [Google Scholar]
- 20. Sahoo B. R., Genjo T., Bekier M., Cox S. J., Stoddard A. K., Ivanova M., Yasuhara K., Fierke C. A., Wang Y. Z., and Ramamoorthy A. (2018) Alzheimer's amyloid-β intermediates generated using polymer-nanodiscs. Chem. Commun. 54, 12883–12886 10.1039/C8CC07921H [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang T., O'Malley T. T., Kanmert D., Jerecic J., Zieske L. R., Zetterberg H., Hyman B. T., Walsh D. M., and Selkoe D. J. (2015) A highly sensitive novel immunoassay specifically detects low levels of soluble Aβ oligomers in human cerebrospinal fluid. Alzheimers Res. Ther. 7, 14 10.1186/s13195-015-0100-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bieschke J., Herbst M., Wiglenda T., Friedrich R. P., Boeddrich A., Schiele F., Kleckers D., Lopez del Amo J. M., Grüning B. A., Wang Q., Schmidt M. R., Lurz R., Anwyl R., Schnoegl S., Fändrich M., et al. (2011) Small-molecule conversion of toxic oligomers to nontoxic β-sheet–rich amyloid fibrils. Nat. Chem. Biol. 8, 93–101 10.1038/nchembio.719 [DOI] [PubMed] [Google Scholar]
- 23. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 10.1074/jbc.R800016200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ahmed M., Davis J., Aucoin D., Sato T., Ahuja S., Aimoto S., Elliott J. I., Van Nostrand W. E., and Smith S. O. (2010) Structural conversion of neurotoxic amyloid-β(1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 17, 561–567 10.1038/nsmb.1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Freir D. B., Fedriani R., Scully D., Smith I. M., Selkoe D. J., Walsh D. M., and Regan C. M. (2011) Aβ oligomers inhibit synapse remodelling necessary for memory consolidation. Neurobiol. Aging 32, 2211–2218 10.1016/j.neurobiolaging.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fu Z., Aucoin D., Davis J., Van Nostrand W. E., and Smith S. O. (2015) Mechanism of nucleated conformational conversion of Aβ42. Biochemistry 54, 4197–4207 10.1021/acs.biochem.5b00467 [DOI] [PubMed] [Google Scholar]
- 27. Parthasarathy S., Inoue M., Xiao Y., Matsumura Y., Nabeshima Y., Hoshi M., and Ishii Y. (2015) Structural insight into an Alzheimer's brain-derived spherical assembly of amyloid β by solid-state NMR. J. Am. Chem. Soc. 137, 6480–6483 10.1021/jacs.5b03373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang D., Zimmerman M. I., Martin P. K., Nix A. J., Rosenberry T. L., and Paravastu A. K. (2015) Antiparallel β-sheet structure within the C-terminal region of 42-residue Alzheimer's amyloid-β peptides when they form 150-kDa oligomers. J. Mol. Biol. 427, 2319–2328 10.1016/j.jmb.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chimon S., Shaibat M. A., Jones C. R., Calero D. C., Aizezi B., and Ishii Y. (2007) Evidence of fibril-like β-sheet structures in neurotoxic amyloid intermediate for Alzheimer's β-amyloid. Nat. Struct. Mol. Biol. 14, 1157–1164 10.1038/nsmb1345 [DOI] [PubMed] [Google Scholar]
- 30. Petkova A. T., Ishii Y., Balbach J. J., Antzutkin O. N., Leapman R. D., Delaglio F., and Tycko R. (2002) A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747 10.1073/pnas.262663499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petkova A. T., Yau W. M., and Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry 45, 498–512 10.1021/bi051952q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bertini I., Gonnelli L., Luchinat C., Mao J., and Nesi A. (2011) A new structural model of Aβ(40) fibrils. J. Am. Chem. Soc. 133, 16013–16022 10.1021/ja2035859 [DOI] [PubMed] [Google Scholar]
- 33. Paravastu A. K., Leapman R. D., Yau W.-M., and Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 10.1073/pnas.0806270105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roychaudhuri R., Yang M., Hoshi M. M., and Teplow D. B. (2009) Amyloid β-protein assembly and Alzheimer disease. J. Biol. Chem. 284, 4749–4753 10.1074/jbc.R800036200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Benilova I., Karran E., and De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 10.1038/nn.3028 [DOI] [PubMed] [Google Scholar]
- 36. LeVine H. (1995) Thioflavine T interaction with amyloid β-sheet structures. Amyloid 2, 1–6 10.3109/13506129509031881 [DOI] [Google Scholar]
- 37. Xiao Y., Ma B., McElheny D., Parthasarathy S., Long F., Hoshi M., Nussinov R., and Ishii Y. (2015) Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nat. Struct. Mol. Biol. 22, 499–505 10.1038/nsmb.2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takegoshi K., Nakamura S., and Terao T. (2003) C-13-H-1 dipolar-driven C-13-C-13 recoupling without C-13 rf irradiation in nuclear magnetic resonance of rotating solids. J. Chem. Phys. 118, 2325–2341 10.1063/1.1534105 [DOI] [Google Scholar]
- 39. Wishart D. S., Sykes B. D., and Richards F. M. (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222, 311–333 10.1016/0022-2836(91)90214-Q [DOI] [PubMed] [Google Scholar]
- 40. Wishart D. S., Bigam C. G., Holm A., Hodges R. S., and Sykes B. D. (1995) 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 5, 67–81 10.1007/BF00227471 [DOI] [PubMed] [Google Scholar]
- 41. Shen Y., and Bax A. (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 10.1007/s10858-013-9741-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tycko R. (2006) Molecular structure of amyloid fibrils: insights from solid-state NMR. Q. Rev. Biophys. 39, 1–55 10.1017/S0033583506004173 [DOI] [PubMed] [Google Scholar]
- 43. Qiang W., Yau W.-M., Luo Y., Mattson M. P., and Tycko R. (2012) Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 4443–4448 10.1073/pnas.1111305109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gu L., Liu C., Stroud J. C., Ngo S., Jiang L., and Guo Z. (2014) Antiparallel triple-strand architecture for prefibrillar Aβ42 oligomers. J. Biol. Chem. 289, 27300–27313 10.1074/jbc.M114.569004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishii Y., Balbach J. J., and Tycko R. (2001) Measurement of dipole-coupled lineshapes in a many-spin system by constant-time two-dimensional solid state NMR with high-speed magic-angle spinning. Chem. Phys. 266, 231–236 10.1016/S0301-0104(01)00250-6 [DOI] [Google Scholar]
- 46. Veshtort M., and Griffin R. G. (2006) SPINEVOLUTION: a powerful tool for the simulation of solid and liquid state NMR experiments. J. Magn. Reson. 178, 248–282 10.1016/j.jmr.2005.07.018 [DOI] [PubMed] [Google Scholar]
- 47. Jaroniec C. P., Tounge B. A., Herzfeld J., and Griffin R. G. (2001) Frequency selective heteronuclear dipolar recoupling in rotating solids: accurate C-13-N-15 distance measurements in uniformly C-13,N-15-labeled peptides. J. Am. Chem. Soc. 123, 3507–3519 10.1021/ja003266e [DOI] [PubMed] [Google Scholar]
- 48. Tycko R. (2014) Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci. 23, 1528–1539 10.1002/pro.2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chimon S., and Ishii Y. (2005) Capturing intermediate structures of Alzheimer's β-amyloid, Aβ(1–40), by solid-state NMR spectroscopy. J. Am. Chem. Soc. 127, 13472–13473 10.1021/ja054039l [DOI] [PubMed] [Google Scholar]
- 50. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) Nmrpipe: a multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR 6, 277–293 10.1007/bf00197809 [DOI] [PubMed] [Google Scholar]
- 51. Ohnishi T., Yanazawa M., Sasahara T., Kitamura Y., Hiroaki H., Fukazawa Y., Kii I., Nishiyama T., Kakita A., Takeda H., Takeuchi A., Arai Y., Ito A., Komura H., Hirao H., et al. (2015) Na,K-ATPase α3 is a death target of Alzheimer patient amyloid-β assembly. Proc. Natl. Acad. Sci. U.S.A. 112, E4465–E4474 10.1073/pnas.1421182112 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.