

Abstract
Many members of the AAA+ ATPase family function as hexamers that unfold their protein substrates. These AAA unfoldases include spastin, which plays a critical role in the architecture of eukaryotic cells by driving the remodeling and severing of microtubules, which are cytoskeletal polymers of tubulin subunits. Here, we demonstrate that a human spastin binds weakly to unmodified peptides from the C-terminal segment of human tubulin α1A/B. A peptide comprising alternating glutamate and tyrosine residues binds more tightly, which is consistent with the known importance of glutamylation for spastin microtubule severing activity. A cryo-EM structure of the spastin-peptide complex at 4.2 Å resolution revealed an asymmetric hexamer in which five spastin subunits adopt a helical, spiral staircase configuration that binds the peptide within the central pore, whereas the sixth subunit of the hexamer is displaced from the peptide/substrate, as if transitioning from one end of the helix to the other. This configuration differs from a recently published structure of spastin from Drosophila melanogaster, which forms a six-subunit spiral without a transitioning subunit. Our structure resembles other recently reported AAA unfoldases, including the meiotic clade relative Vps4, and supports a model in which spastin utilizes a hand-over-hand mechanism of tubulin translocation and microtubule remodeling.
Keywords: ATPases associated with diverse cellular activities (AAA), protein structure, peptide interaction, cryo-electron microscopy, structure-function, microtubule severing mechanism, molecular machine
Introduction
AAA+ ATPases constitute a large family of macromolecular machines that are classified into clades on the basis of sequence similarity (1). Many members function as hexamers that unfold or disassemble protein molecules or complexes (2), and are referred to as AAA unfoldases. Spastin belongs to the meiotic clade of AAA unfoldases that also includes katanin, which shares spastin's microtubule-severing activity, and Vps4, which performs an analogous activity against ESCRT-III filaments (3). These meiotic clade enzymes display a single MIT domain near the N terminus followed by a flexible linker and a single AAA cassette that assembles as a hexamer to perform the mechanical work of polymer remodeling and disassembly.
Mutations of spastin are the main cause of hereditary spastic paraplegia (4). Such disease-causing mutations reflect the importance of remodeling microtubules, which function in cellular architecture, transport, and motion. Microtubules comprise polymers of α- and β-tubulin heterodimers (5), which in humans are expressed from eight α and nine β isoform genes (6). The folded regions of tubulins, which create the microtubule filaments, are followed by flexible C-terminal tails that project from the microtubule surface and are important for the microtubule-binding and severing activity of spastin (7). Spastin binding is promoted by glutamylation of the tubulin C-terminal tails up to a threshold level, beyond which additional glutamylation reduces the microtubule-severing activity (8).
Molecular mechanisms of spastin and other microtubule-severing enzymes are debated (9), but an attractive possibility is that the spastin hexamer translocates from the C-terminal residues of tubulin into the folded domain to destabilize intersubunit contacts, thereby promoting subunit exchange or severing (10). Note that, depending upon the frame of reference, spastin translocation along the substrate polypeptide is equivalent to polypeptide translocation through the ring formed by the spastin hexamer. This model is consistent with recently determined structures of other AAA unfoldases with peptides that appear to mimic translocating substrates (11–22). These structures typically display 4–5 subunits in a helical configuration that binds the peptide within the central pore. These structures suggest that translocation along the substrate polypeptide proceeds by a hand-over-hand mechanism of subunits hydrolyzing ATP, disengaging from substrate, and transferring from one end of the helix to the other (23). Despite the emergence of the hand-over-hand model, the mechanism of the microtubule-severing enzymes is not yet settled (9). In part, this is due to recent structures of inactive mutants of Caenorhabditis elegans katanin (24) and Drosophila melanogaster spastin (25) that show all six subunits in a continuous helical configuration without a transitioning subunit.
To study how spastin engages and translocates along the tubulin polypeptide, we expressed a human spastin construct and demonstrated binding to tubulin C-terminal peptides. The cryo-EM structure showed that five subunits adopt the canonical helical configuration, whereas the sixth subunit is disengaged from the peptide as if transitioning from one end of the spastin helix to the other. These findings support a common mechanism for spastin and other AAA unfoldases in which ATP binding and hydrolysis drives a sequential hand-over-hand mechanism, with two amino acid residues being translocated for each ATP hydrolyzed.
Results and discussion
Expression of human spastin
Human spastin is expressed from a single gene as four different isoforms (26). The full-length (M1) protein comprises 616 amino acid residues, whereas use of an alternative initiator methionine produces the more abundant M87 isoforms (27, 28). Alternative splicing of both M1 and M87 spastin produces two other isoforms that differ by the presence or absence of exon 4, which encodes residues 197–228 and lies between the MIT domain and the ATPase cassette (26).
The M87 protein lacking exon 4 is expressed in mammalian cells (27, 29) and retains ATPase and microtubule-severing activity (30, 31). We used this construct (hereafter referred to as spastin) to avoid including a hydrophobic membrane spanning segment (residues 1–86) and exon 4, which promoted aggregation (not shown). Spastin was expressed in Escherichia coli as a GST4 fusion that, following cleavage by PreScission protease, started with three nonnative residues (GPH) prior to M87, and continued to the C terminus of the full-length protein. This protein construct was used in all of the binding and structure determination studies reported herein.
Spastin binds weakly to tubulin-derived peptides
We used fluorescence polarization spectroscopy to examine the binding of recombinant spastin to peptides derived from the C-terminal region of human tubulins α1A and α1B, which are identical in this region. Our choice of the nucleotide analog ADP·BeFx for these studies was guided by our experience with Vps4, which bound peptides in the presence of ADP·BeFx or ADP·AlFx, but not other nonhydrolyzable analogs such as ATPγS or AMPPNP (32).
Three overlapping peptides spanning the C-terminal 50 residues of tubulin α1A/B were synthesized with a fluorescein at the N terminus. The two most N-terminal tubulin peptides bound spastin with a KD of ∼30 μm, whereas the C-terminal peptide bound even more weakly, with a KD in excess of 100 μm (Fig. 1). These data are consistent with the finding that the interaction of spastin with tubulin C-terminal residues is functionally important but inherently weak (31), and that posttranslational modifications enhance the microtubule-severing activity of spastin (8).
Figure 1.
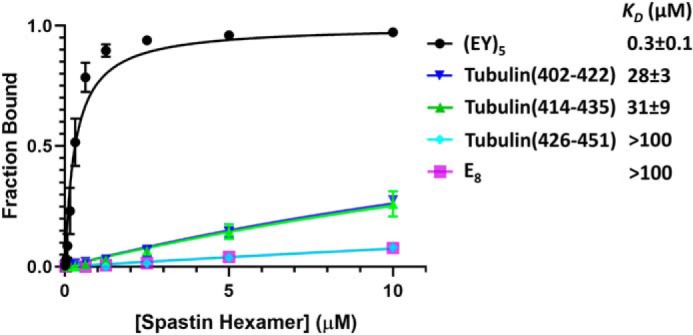
Binding of spastin to substrate peptides. Fluorescence polarization anisotropy isotherms for spastin binding to the indicated peptides. Error bars are shown for all data points, but are often smaller than the data point symbol. E8 is an 8-residue glutamylated peptide. (EY)5 is a 10-residue peptide comprised of alternating glutamate and tyrosine residues.
Enhanced binding of spastin to a glutamate-tyrosine peptide
The modest affinity seen for the tubulin peptides is notably weaker than the affinity demonstrated for an 8-residue peptide that binds Vps4 (11). Therefore, guided by the finding that tubulin glutamylation is important for microtubule recognition by spastin (8) and because Vps4 binds four dipeptide units, we tested the binding of E8, an 8-glutamylate peptide, and (EY)5, a 10-residue peptide comprised of alternating glutamate and tyrosine residues. E8 binding was weak, whereas (EY)5 bound with a KD of 0.3 ± 0.1 μm (Fig. 1), which is similar to that of an ESCRT-III–derived peptide binding to Vps4 (11). This relatively tight binding affinity may reflect the importance of an intermediate level of posttranslational glutamylation for optimal tubulin severing (8), and made (EY)5 an attractive target for determination of a spastin-peptide complex structure.
Cryo-EM structure of a spastin-peptide complex
Spastin was incubated with (EY)5 and Mg2+ ADP·BeFx. Cryo-EM and image analysis of frozen-hydrated specimens yielded a complex structure with an overall resolution of 4.2 Å, as indicated by the Fourier shell correlation (FSC) value of 0.143 for comparison of two independent reconstructions from separate groups of particles (33) (Fig. 2, Table S1, Figs. S1, S2 and S3, and Video S1). The assembly of the AAA cassettes is clearly defined. A cloud of additional density seen on the N-terminal side of the hexamer (not shown) presumably corresponds to the N-terminal linker residues and MIT domains. The indistinct appearance of this density indicates inherent flexibility of those regions.
Figure 2.

Cryo-EM structure of the spastin-peptide complex. A, top view of the cryo-EM density map, color-segmented for the six spastin subunits and the peptide. The contour shown is 6.8x the map root mean square deviation. B, side view of the density map with spastin shown as a transparent surface to reveal the peptide density (green) in the central pore. C, top view of the model. The large and small ATPase domains of subunit B are indicated with dashed lines. ATP (ADP·BeFx) and ADP (pink) are indicated at the subunit interfaces. D, side view of the model with subunit F removed to expose the binding groove and the peptide (green). E, sequence alignment of meiotic clade AAA+ ATPases of known structure. Human spastin secondary structures as defined by DSSP (49) are indicated above, and are numbered according to the scheme used for D. melanogaster spastin (25). Walker A, Walker B, pore loop 1 (PL1), pore loop 2 (PL2), and the arginine fingers are indicated.
Five of the spastin subunits (A–E) form a helix in which their tightly-packed subunit interfaces are stabilized by binding of ADP·BeFx in what appears to be an ATP-like state at the active sites of subunits A–D (Fig. 2C). As with other AAA+ ATPases, binding of nucleotide at these active sites is completed by coordination of finger arginine residues from the following subunit (Fig. 3A). Subunit E appears to bind ADP, which is consistent with the displacement of subunit F and its finger arginines (Fig. 3B). Subunit F does not continue the helix of subunits A–E, but rather is displaced from the helix axis and contacts subunits A and E at each end of the helix, primarily through interactions of small domains toward the periphery of the hexamer. Overall density for subunit F is relatively weak, and we were not able to model a coordinating nucleotide. In contrast to the tightly-packed interfaces that define the helical assembly of subunits A–E (Fig. 3A), the EF and FA interfaces are relatively open at the nucleotide-binding sites in the large domain, as if to allow for nucleotide exchange (Fig. 3, B and C).
Figure 3.
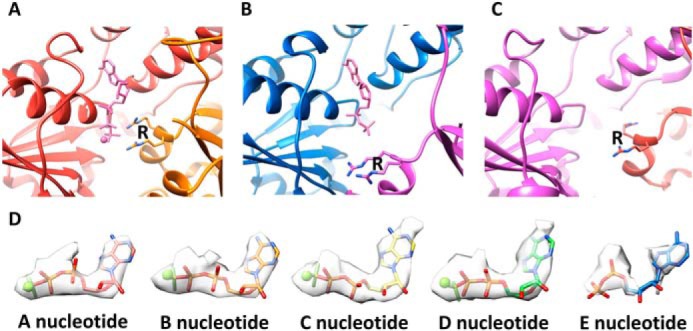
Binding poses of ATP and ADP. A, coordination of Mg2+ ADP−BeFx (ATP) at the active site of subunit A. Coordination of the β phosphate and BeFx is completed by the two arginine finger (R) residues of subunit B. The BC, CD, and DE interfaces are very similar. B, ADP at the subunit E active site, where the subunit F finger arginines are displaced ∼14 Å relative to the AB, BC, CD, and DE interfaces. C, the subunit F active site lacks sufficient density to determine whether or not nucleotide is bound. D, modeling of nucleotides in the density for nucleotide at the A–E active sites.
Peptide binding in the spastin pore
Two conserved loops from each of the five A–E helical spastin subunits project into the central pore. These pore loop 1 and pore loop 2 residues form a double spiral staircase with helical steps of ∼6 Å translation and 60° rotation (Fig. 4A). Subunit E is slightly displaced from this symmetry, as was also noted for the previously reported structures of Vps4 (11) and YME1 (18). Pore loop residues from the five helical subunits bind the peptide, whereas the sixth subunit (subunit F) is displaced from contact with the peptide. The overall dimensions of the pore are defined by the distance from the helix axis to the closest pore loop 1 and pore loop 2 Cα atoms, which is 6 and 7 Å, respectively, and by the vertical distance between the top (A) and bottom (E) subunit pore loop 1 residues, which is 24 Å (Fig. 4A).
Figure 4.
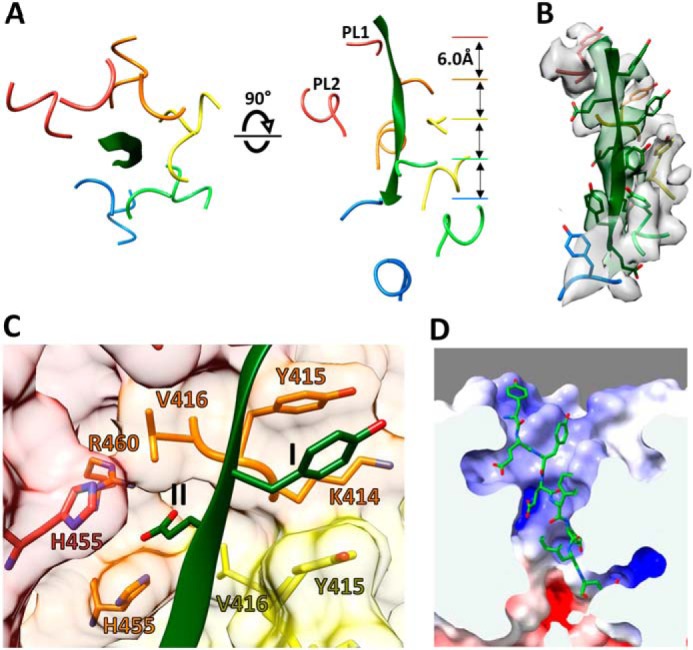
Binding of substrate peptide as an extended strand. A, top and side views of peptide (green) and residues from pore loop 1 (PL1; 414–416) and pore loop 2 (PL2; 455–460) of subunits A–E. The pore loops of these subunits form a double helix that binds the peptide. Pore loops of subunit F (not shown) are displaced away from the helix axis and the peptide. B, density for the bound peptide and pore loop 1 residues, contoured at 7.0 × σ. All side chains of the peptide are resolved except for the two terminal residues. C, close-up views of the class I and class II pockets that bind the alternating side chains of the peptide. This view shows pockets at the interface of subunits B and C (and His-455 of subunit A). The equivalent binding pockets at the AB, CD, and DE interfaces are very similar. D, electrostatic potential surface within the pore computed for subunits A–E. The scale was −10 (red) to 10 (blue) kb·T·ec−1. The calculation was performed at pH 7.2.
All 10 residues of the bound peptide were seen in the density map, although side chain density was not apparent for the first and last residues (Fig. 4B). As expected for an extended conformation at medium resolution, the peptide orientation is not apparent from the density. We have modeled the peptide in the N-to-C top-to-bottom direction of the side views in the figures because that orientation is consistent with the expected direction of substrate translocation by spastin and Vps4 (11, 12).
The (EY)5 peptide binds within the central pore in an extended (β) conformation, with alternating side chains projecting from either side of the strand. The alternating side chains are bound in two different types of pockets, termed class I and class II (23), which are formed by the pore loop residues of subunits A–E (Fig. 4C). The four class I-binding pockets are created by the pore loop 1 Tyr-415 residues of adjacent subunits and flanked by Lys-414 of the first subunit. The class II pockets are created by pore loop 1 Val-416 residues of adjacent subunits and are flanked by pore loop 2 His-455 residues of the first and preceding subunit and by Arg-460 of the first subunit. These pockets bind the peptide side chains, with residues 2, 4, 6, and 8 fitting into class I pockets and residues 3, 5, 7, and 9 into class II pockets. (Note that residue number 1 of the shorter Vps4-bound peptide (13) corresponds to residue 2 of (EY)5.) The density does not distinguish between peptide glutamate and tyrosine side chains, although the positioning of glutamates in class II pockets is consistent with the presence of His-455, which is conserved in spastins and katanins, and is the only potentially charged residue at the binding surface that differs between the microtubule-severing AAA unfoldases and their meiotic clade relative Vps4, where it is a serine residue. Consistent with binding of negatively charged residues of the substrate, the binding groove displays positive electrostatic potential (Fig. 4D).
Comparison with other meiotic clade AAA unfoldases
The human spastin-peptide complex structure overlaps closely with previously reported structures of other meiotic clade AAA unfoldases, including Vps4, katanin, and D. melanogaster spastin. As discussed below, the most notable difference between these structures is that katanin and Drosophila spastin have been reported in a continuous spiral conformation that has a different position for subunit F. In all of these structures, however, the A–E subunits are quite similar and overlap with root mean square deviation values of 1.7–3.2 Å (Table S2). The largest differences are seen at the termini and surface loops where poor density indicates conformational flexibility for one or more of the structures, whereas regions in the core of the structure are closely similar. This structural similarity is consistent with the sequence alignment of AAA cassettes (Fig. 2E), which shows 46–76% residue identity (Table S2).
The overlap with Vps4 is of particular interest because, like spastin, its subunit F contacts both subunits A and E at either end of the A–E helix, and it was a foundational structure for proposal of the hand-over-hand mechanism (11, 12). Rigid body superposition of Vps4 and spastin subunits A–E shows a root mean square deviation of 1.7 Å for 1,175 pairs of equivalent Cα atoms (Fig. 5A). This similarity extends to the arrangement of the pore loops of subunits A–E (Fig. 5B) and coordination of substrate side chains by the class I- and II-binding pockets. As with spastin, Vps4 subunit F is poorly ordered, and focused classification of four Vps4 datasets has led to the identification of 12 subunit F positions that together span a proposed transition path between the ends of the helix formed by subunits A–E (13). The position of subunit F resolved in the spastin reconstruction falls on the spectrum of the positions seen from focused classification of the Vps4 reconstructions (Fig. 5C).
Figure 5.
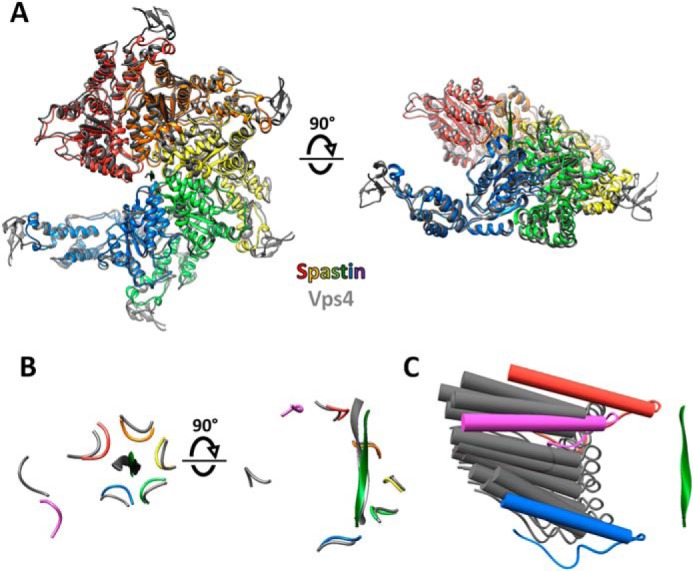
Close similarity between spastin and Vps4 and mechanistic implications. A, top and side views of spastin subunits A–E overlapped with Vps4 (12) using PyMol (Schrödinger, LLC). B, top and side views of spastin (colors) and Vps4 (gray) peptide and pore loop 1 residues after overlap on the large ATPase domains. Subunit F (magenta for spastin) is distant from the bound peptide. C, spastin subunit F (magenta) lies on the spectrum of subunit F positions seen from focused classification of multiple Vps4 structures (gray) (13). Pore loop 1 is shown with the preceding strand and following helix. Spastin subunits A (red) and E (blue) are indicated.
Implications for mechanism
The structural similarity implies that spastin uses the same hand-over-hand mechanism that has been proposed for substrate translocation by Vps4 and other AAA unfoldases (23), in this case to destabilize microtubules by translocating from the C-terminal tubulin tails toward the folded N-terminal domain. In this model (Fig. 6), ATP binding stabilizes the interfaces that give rise to the helical assembly of subunits A–E that is optimal for binding the substrate polypeptide. Hydrolysis and phosphate release at the subunit D active site weakens the interface with subunit E, which is then able to disengage from the substrate and transition to the more open position of subunit F. Exchange of ADP for ATP subsequently allows the subunit in position F to dock at the top of the helix of spastin subunits and bind the next substrate dipeptide.
Figure 6.

Proposed mechanism of substrate translocation. Spastin is colored white with the pore loop 1 Tyr-415 of all six subunits in color. Substrate (green) is modeled on the bound peptide, extended at either end by continuing the β-strand conformation, and shown with modeled glutamate side chains.
Consistent with the structure, the hand-over-hand model involves opening of the large domain interfaces between subunit F and the neighboring A and E subunits, while contacts are maintained by the small domains at the periphery of the hexamer. As a result of these small domain contacts, conformational transitions can be coordinated so that as subunit E moves from the bottom of the helical stack to the subunit F position, subunit F moves to the subunit A position. This enables four or five subunits to maintain contact with the peptide in the helical conformation as subunits ABCDEF transition to the BCDEFA states, respectively. Thus, important features of this mechanism are that each subunit maintains an identical grip on its substrate dipeptide as it translocates (with respect to the other subunits) through the A, B, C, D, and E states, and events at the opposite ends of the helix are coordinated by maintenance of contacts with the transitioning subunit.
Because tubulin sequences are much more varied than the (EY)5 peptide used in this study, the model implies that spastin binds different amino acid residues using equivalent class I- and II-binding pockets. This is presumably enabled by the highly solvated nature of the pore, which in the case of Vps4 can even accommodate a second substrate strand without distortion (13). We envision that the branched polypeptide created by polyglutamylation could be accommodated by spastin in an analogous manner. The mechanism also implies a translocation of two amino acids of tubulin for each molecule of ATP hydrolyzed. We expect that some deviations from this model could occur. For example, distortions or slippage might result from substrate residues such as proline that cannot adopt a canonical β-strand conformation, from glycine, which lacks a side chain and is therefore expected to bind only weakly, or upon encountering stable domains that are resistant to unfolding and translocation.
Comparisons with the continuous spiral structures of katanin and spastin
Although we favor the hand-over-hand model, the extent to which spastin utilizes the same mechanism as other AAA unfoldases is debated, in part because recent structures of D. melanogaster spastin (25) and C. elegans katanin (24) both display continuous six-member spiral architectures. These spirals are essentially identical to our spastin and Vps4 closed ring structures over subunits A–E (Table S2), whereas the sixth subunit of the spiral structures continues the helix of subunits A–E rather than adopting a transitioning position that connects the E and A subunits (Fig. 7). The similarity of the closed ring and continuous spiral structures over subunits A–E is emphasized by the finding that a polyglutamate peptide binds within the D. melanogaster spastin spiral in the same conformation as peptides bind to the Vps4 and human spastin ring structures (25). Interestingly, inspection of the deposited katanin map shows a previously unrecognized peptide in the substrate-binding groove of that structure also (Fig. S4). The close relationship between the continuous spiral and ring configurations is further reinforced by the observation that, in addition to the spiral configuration, the katanin structure determination resolved a second configuration that is essentially identical to our spastin and Vps4 structures (24). Moreover, a reconstruction of Vps4EQ, which has an active site mutation (34), displayed the same spiral configuration as katanin and D. melanogaster spastin.
Figure 7.

Comparison of ring and spiral configurations of katanin. A, top and side views of the katanin ring structure (PDB 5WCB). The human spastin structure described herein, and Vps4 (12) are essentially identical. B, schematic representation of the side view of the ring structure. C, top and side views of the katanin spiral structure (PDB 5WC0) structure (24). The D. melanogaster spastin structure (25) is essentially identical. D, schematic representation of the side view of the spiral structures. The ring and spiral configurations are essentially identical to each other over the red-blue subunits, and vary only in the position of the magenta subunit.
We note that the continuous spiral structures of D. melanogaster spastin (25), katanin (24), and Vps4EQ (34) were all determined using proteins that contain an Glu to Gln mutation of the Walker B glutamate. This widely used mutation allows ATP binding but does not allow ATP hydrolysis. Thus, the spiral configuration may be promoted by biochemical conditions that stabilize the additional ATP-bound interface (with concomitant loss of a subunit FA contact) to the point that the continuous spiral configuration outcompetes the alternative ring configuration in which smaller interactions with both subunits A and E are maintained as F moves from the bottom to the top of the AAA helix.
For these reasons, we believe that the spiral structures are off-pathway conformations that are slightly distorted from the authentic translocating conformation. The main distinction being that in the translocating conformation subunit F maintains contacts with both subunits A and E. We propose that multiple AAA unfoldases, including members of the meiotic clade discussed herein, translocate their substrates using the hand-over-hand mechanism described in the legend to Fig. 6. In this manner, tubulin subunits can be translocated through the spastin pore, starting from their flexible and exposed C-terminal residues and progressing into their folded microtubule-forming N-terminal domains, thereby facilitating microtubule remodeling.
Experimental procedures
Peptides
The unlabeled (EY)5 peptide and the tubulin peptides labeled with an N-terminal fluorescein at greater than 98% purity were purchased from GenScript. The labeled (EY)5 peptide with an N-terminal fluorescein was synthesized by the University of Utah Health Sciences Center Core Facility in DNA/Peptide Synthesis at 98% purity. The labeled E8 peptide was synthesized on a Gyros Peptide Technologies Prelude X automated peptide synthesizer using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) solid-phase peptide synthesis methods and purified by C18 RP-HPLC to >95% purity. In all cases, the fluorescein was covalently linked to the N-terminal amine and the C terminus was capped as a carboxyamide. The N terminus of the unlabeled (EY)5 peptide was capped by acetylation. All peptide identities were all confirmed by MS.
Protein expression and purification
The spastin expression plasmid has been submitted to the Addgene plasmid repository (plasmid ID 128794). Spastin was expressed in E. coli BL21(DE3)RIL from a pGEX-based vector with a cleavable N-terminal GST tag. Expression cultures were grown in ZY autoinduction media at 37 °C to A600 = 0.5 and then at 19 °C for 16 h (35). Cells were harvested by centrifugation and stored at −80 °C. Cell pellets were thawed and resuspended in lysis buffer (50 mm Tris/HCl, pH 8.0, 350 mm NaCl, 5 mm MgCl2, and 1 mm DTT) supplemented with protease inhibitors (phenylmethylsulfonyl fluoride, Biosynth; leupeptin, Roche; pepstatin A and aprotinin, GoldBio), DNase I (0.01 mg/ml), and lysozyme (1 mg/ml). After incubation on ice for 30 min, cells were re-frozen in liquid N2 and thawed again. The cell lysate was clarified by centrifugation, and the resulting supernatant was filtered using a 0.45-μm filter and loaded onto a GSH column that was pre-equilibrated with lysis buffer. Following a wash with 150 ml of lysis buffer, GST-tagged spastin was eluted with lysis buffer containing 10 mm reduced GSH. The GST tag was cleaved by incubation with 1 mg of precision protease during dialysis against 25 mm Tris/HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, and 1 mm DTT overnight at 4 °C. The sample was then bound to a 5-ml HiTrap SP ion exchange column equilibrated with buffer A (25 mm Tris/HCl, pH 8.0, 150 mm NaCl, 5 mm MgCl2, and 1 mm DTT) and was eluted with a gradient of 0–50% buffer B (25 mm Tris/HCl, pH 8.0, 1 m NaCl, 5 mm MgCl2, 1 mm DTT) over a 500-ml volume. Fractions containing spastin were pooled, concentrated to 5 ml, and further purified by gel filtration into 20 mm HEPES/NaOH, pH 8.0, 150 mm NaCl, 5 mm MgCl2, and 1 mm DTT using a 120-ml Superdex 200 column (GE Healthcare Life Sciences). Spastin protein identity was confirmed by MS (calculated mass = 54,708.8 Da, experimental mass = 54,708.3 Da).
Fluorescence polarization binding assay
Labeled peptides (1 nm) were incubated at room temperature with spastin (0–60 μm subunit concentration) in binding buffer (20 mm HEPES/NaOH, pH 8.0, 150 mm NaCl, 1 mm ADP·BeFx, 5 mm MgCl2, and 1 mm DTT) in a total volume of 60 μl. After attaining equilibrium, parallel and perpendicular fluorescence intensities were measured on a Biotek Synergy Neo HTS microplate reader using an excitation wavelength of 485 nm and an emission wavelength of 528 nm. The fluorescence polarization was plotted against the spastin hexamer concentration and the dissociation constants were estimated by global fitting of the equation FP = [spastin hexamer]/(KD + [spastin hexamer]) to data points from four independent experiments, where FP is the normalized fluorescence polarization or “fraction bound,” using GraphPad Prism 8.
Sample preparation and EM data collection
Purified spastin (final concentration 10 μm) was incubated on ice for 1 h with unlabeled peptide (EY)5 (final concentration 20 μm) in the presence of 1 mm ADP−BeFx. The protein-peptide complex was then cross-linked using glutaraldehyde (final concentration 0.01%), followed by concentration and purification by gel-filtration using a Superose 6 Increase column (GE Healthcare Life Sciences). An aliquot (3.5 μl) of the purified cross-linked spastin sample (diluted to 1 μm hexamer concentration) was applied to a glow-discharged (25 mA, 25 s) Quantifoil 1.2/1.3 holey carbon 400-mesh copper grid, and plunge frozen in liquid ethane using a Vitrobot Mark III set to 4 °C, 100% relative humidity, −1 mm offset, and 8-s blotting time.
Electron cryomicrographs were collected at the University of Virginia Molecular Electron Microscopy Core using a Titan Krios electron microscope (ThermoFisher) operating at 300 kV. A total of 1,200 movies were recorded at defocus values from −1.5 μm to −2 μm using a Falcon 3EC detector operating in counting mode, with two movies per hole. Each exposure lasted 90.77 s, which was collected as a 49-frame movie using a total electron dose of 57 e/Å2, and a pixel size of pixel size 1.056 Å.
Single particle image analysis
Unless indicated, image processing was performed using the RELION 3.0 software suite (36). Beam-induced movement was corrected over all movie frames using Relion_MotionCorr (37). CTF parameters were determined on nondose-weighted sums using GCTF.v.1.06 (38). Images with poor defocus, astigmatism, resolution, and CtfFOM were excluded from further analysis, yielding a total of 1,173 images for subsequent image processing. Laplacian picking of micrographs generated initial particle images that were subjected to 2D classification. Three distinct classes were used as a template to autopick the entire data set. A total of 1,021,617 particles were extracted using a box size of 256 pixels. After multiple rounds of full CTF-corrected 2D classification, 438,336 particles were used to generate an initial model for 3D classification. One class, containing 129,402 particles, was used for autorefinement, which indicated a resolution of 4.3 Å (FSC = 0.143) as calculated from independent ½-map reconstructions (33). Subsequent rounds of per particle CTF refinement and motion correction followed by a round of 3D classification reduced the particle count to 119,984. A final round of autorefinement and post processing using a full-structure soft mask (hexamer of ATPase cassettes) yielded a map with an average “gold-standard” resolution of 4.2 Å using a B-factor sharpening factor of −147 Å2. Local resolution was estimated using an automated procedure in RELION (39). Half-map and map-model FSC validation was performed using PHENIX.mtriage version 1.17 (40)
Model building, refinement, and validation
Model building and refinement followed the same approach we used for previously reported Vps4 structures (12). Briefly, six copies of a spastin crystal structure (PDB code 5Z6Q) (41) were docked into the 4.2-Å map as rigid bodies using UCSF Chimera (42). The missing features of the model, including pore loop 1 and pore loop 2 of subunits A–E, and the peptide, were built using Coot (43).
Subunits A to E (excluding residues 541–543 and 555–596 of subunit E) and the substrate were subjected to real-space refinement using Phenix (40). (Subunit F and residues 541–543 and 555–596 of subunit E were not refined beyond rigid body docking due to the low local resolution.) Secondary structure restraints were applied during refinement. NCS restraints were applied to spastin subunits A–E, except for residues 468–480 of subunit A, which appeared to adopt a distinct conformation. For subunits A, B, C, and D, the distance between the beryllium and the O3B of ADP was restrained to 1.6 Å, and the distance between Mg2+ and F1 of BeF3 was restrained to 2.0 Å. Structure validation used Molprobity (44), EMRinger (45), and 3D FSC (46).
Electrostatic potential fields were calculated using PDB 2PQR (47) at pH 7.2 using the AMBER forcefield and APBS (48), and texture-mapped to the molecular surface using Chimera (42). Molecular graphics figures and video were generated using Chimera.
The sharpened (B-factor −147 Å2) (EMD-20327) and unsharpened (EMD-20805) maps were deposited at the EMDB. Coordinate models were deposited to the PDB: a refined model comprising subunits subjected to atomic refinement (PDB 6PEK) and a comprehensive model that also includes subunit F and other segments that were fitted as rigid bodies (PDB 6PEN).
Author contributions
H. H., H. L. S., J. M., and N. M. formal analysis; H. H., H. L. S., J. M., and C. P. H. validation; H. H., H. L. S., J. M., N. M., M. D. P., and M. Y. investigation; H. H. and H. L. S. visualization; H. H., H. L. S., J. M., N. M., M. D. P., M. Y., and W. I. S. writing-review and editing; W. I. S. and C. P. H. conceptualization; W. I. S. and C. P. H. resources; W. I. S. and C. P. H. supervision; W. I. S. and C. P. H. funding acquisition; W. I. S. and C. P. H. project administration; C. P. H. writing-original draft.
Supplementary Material
Acknowledgments
We thank James Fulcher and Michael Kay for the labeled E8 peptide. The (EY)5 peptide synthesis, sequencing of constructs, and validation of proteins was performed at the peptide synthesis, DNA sequencing, and MS core facilities at the University of Utah, which are supported by National Institutes of Health, NCI Grant P30CA042014. Mass spectrometry equipment in the Core was obtained through an National Institutes of Health Shared Instrumentation Grant S10 OD018210. Data processing and image reconstruction used the University of Utah Center for High Performance Computing.
This work was supported by National Institutes of Health Grants R01 GM112080 (to W. I. S. and C. P. H.), R01 GM128507 (to M. Y.), and U24 GM116790 (to M. Y.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4, Tables S1 and S2, and Video S1.
The electron microscopy data reported in this paper have been submitted to the Electron Microscopy data bank under accession nos. EMD20805 and EMD20327.
The atomic coordinates and structure factors (codes 6PEK and 6PEN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GST
- glutathione S-transferase
- AAA/AAA+
- ATPases associated with diverse cellular activities
- cryo-EM
- electron cryomicroscopy
- FSC
- Fourier shell correlation
- PDB
- Protein Data Bank
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate.
References
- 1. Erzberger J. P., and Berger J. M. (2006) Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93–114 10.1146/annurev.biophys.35.040405.101933 [DOI] [PubMed] [Google Scholar]
- 2. Nyquist K., and Martin A. (2014) Marching to the beat of the ring: polypeptide translocation by AAA+ proteases. Trends Biochem. Sci. 39, 53–60 10.1016/j.tibs.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Monroe N., and Hill C. P. (2016) Meiotic Clade AAA ATPases: protein polymer disassembly machines. J. Mol. Biol. 428, 1897–1911 10.1016/j.jmb.2015.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Plaud C., Joshi V., Kajevu N., Pous C., Curmi P. A., and Burgo A. (2018) Functional differences of short and long isoforms of spastin harboring missense mutation. Dis. Model Mech. 11, pii: dmm033704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nogales E., and Wang H. W. (2006) Structural mechanisms underlying nucleotide-dependent self-assembly of tubulin and its relatives. Curr. Opin. Struct. Biol. 16, 221–229 10.1016/j.sbi.2006.03.005 [DOI] [PubMed] [Google Scholar]
- 6. Roll-Mecak A. (2019) How cells exploit tubulin diversity to build functional cellular microtubule mosaics. Curr. Opin. Cell Biol. 56, 102–108 10.1016/j.ceb.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roll-Mecak A., and Vale R. D. (2005) The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr. Biol. 15, 650–655 10.1016/j.cub.2005.02.029 [DOI] [PubMed] [Google Scholar]
- 8. Valenstein M. L., and Roll-Mecak A. (2016) Graded control of microtubule severing by tubulin glutamylation. Cell 164, 911–921 10.1016/j.cell.2016.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McNally F. J., and Roll-Mecak A. (2018) Microtubule-severing enzymes: from cellular functions to molecular mechanism. J. Cell Biol. 217, 4057–4069 10.1083/jcb.201612104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vemu A., Szczesna E., Zehr E. A., Spector J. O., Grigorieff N., Deaconescu A. M., and Roll-Mecak A. (2018) Severing enzymes amplify microtubule arrays through lattice GTP-tubulin incorporation. Science 361, eaau1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Monroe N., Han H., Shen P. S., Sundquist W. I., and Hill C. P. (2017) Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. eLife 6, e24487 10.7554/eLife.24487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han H., Monroe N., Sundquist W. I., Shen P. S., and Hill C. P. (2017) The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. eLife 6, e31324 10.7554/eLife.31324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han H., Fulcher J. M., Dandey V. P., Iwasa J. H., Sundquist W. I., Kay M. S., Shen P. S., and Hill C. P. (2019) Structure of Vps4 with circular peptides and implications for translocation of two polypeptide chains by AAA+ ATPases. eLife 8, e44071 10.7554/eLife.44071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ripstein Z. A., Huang R., Augustyniak R., Kay L. E., and Rubinstein J. L. (2017) Structure of a AAA+ unfoldase in the process of unfolding substrate. eLife 6, e25754 10.7554/eLife.25754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gates S. N., Yokom A. L., Lin J., Jackrel M. E., Rizo A. N., Kendsersky N. M., Buell C. E., Sweeny E. A., Mack K. L., Chuang E., Torrente M. P., Su M., Shorter J., and Southworth D. R. (2017) Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273–279 10.1126/science.aan1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deville C., Carroni M., Franke K. B., Topf M., Bukau B., Mogk A., and Saibil H. R. (2017) Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. Sci. Adv. 3, e1701726 10.1126/sciadv.1701726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu H., Lupoli T. J., Kovach A., Meng X., Zhao G., Nathan C. F., and Li H. (2018) ATP hydrolysis-coupled peptide translocation mechanism of Mycobacterium tuberculosis ClpB. Proc. Natl. Acad. Sci. U.S.A. 115, E9560–E9569 10.1073/pnas.1810648115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Puchades C., Rampello A. J., Shin M., Giuliano C. J., Wiseman R. L., Glynn S. E., and Lander G. C. (2017) Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358, eaao0464 10.1126/science.aao0464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de la Peña A. H., Goodall E. A., Gates S. N., Lander G. C., and Martin A. (2018) Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 362, eaav0725 10.1126/science.aav0725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dong Y., Zhang S., Wu Z., Li X., Wang W. L., Zhu Y., Stoilova-McPhie S., Lu Y., Finley D., and Mao Y. (2019) Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 565, 49–55 10.1038/s41586-018-0736-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alfieri C., Chang L., and Barford D. (2018) Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature 559, 274–278 10.1038/s41586-018-0281-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. White K. I., Zhao M., Choi U. B., Pfuetzner R. A., and Brunger A. T. (2018) Structural principles of SNARE complex recognition by the AAA+ protein NSF. eLife 7, e38888 10.7554/eLife.38888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han H., and Hill C. P. (2019) Structure and mechanism of the ESCRT pathway AAA+ ATPase Vps4. Biochem. Soc. Trans. 47, 37–45 10.1042/BST20180260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zehr E., Szyk A., Piszczek G., Szczesna E., Zuo X., and Roll-Mecak A. (2017) Katanin spiral and ring structures shed light on power stroke for microtubule severing. Nat. Struct. Mol. Biol. 24, 717–725 10.1038/nsmb.3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sandate C. R., Szyk A., Zehr E. A., Lander G. C., and Roll-Mecak A. (2019) An allosteric network in spastin couples multiple activities required for microtubule severing. Nat. Struct. Mol. Biol. 26, 671–678 10.1038/s41594-019-0257-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lumb J. H., Connell J. W., Allison R., and Reid E. (2012) The AAA ATPase spastin links microtubule severing to membrane modelling. Biochim. Biophys. Acta 1823, 192–197 10.1016/j.bbamcr.2011.08.010 [DOI] [PubMed] [Google Scholar]
- 27. Claudiani P., Riano E., Errico A., Andolfi G., and Rugarli E. I. (2005) Spastin subcellular localization is regulated through usage of different translation start sites and active export from the nucleus. Exp. Cell Res. 309, 358–369 10.1016/j.yexcr.2005.06.009 [DOI] [PubMed] [Google Scholar]
- 28. Mancuso G., and Rugarli E. I. (2008) A cryptic promoter in the first exon of the SPG4 gene directs the synthesis of the 60-kDa spastin isoform. BMC Biol. 6, 31 10.1186/1741-7007-6-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Svenson I. K., Ashley-Koch A. E., Gaskell P. C., Riney T. J., Cumming W. J., Kingston H. M., Hogan E. L., Boustany R. M., Vance J. M., Nance M. A., Pericak-Vance M. A., and Marchuk D. A. (2001) Identification and expression analysis of spastin gene mutations in hereditary spastic paraplegia. Am. J. Hum. Genet. 68, 1077–1085 10.1086/320111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Errico A., Claudiani P., D'Addio M., and Rugarli E. I. (2004) Spastin interacts with the centrosomal protein NA14, and is enriched in the spindle pole, the midbody and the distal axon. Hum. Mol. Genet. 13, 2121–2132 10.1093/hmg/ddh223 [DOI] [PubMed] [Google Scholar]
- 31. White S. R., Evans K. J., Lary J., Cole J. L., and Lauring B. (2007) Recognition of C-terminal amino acids in tubulin by pore loops in Spastin is important for microtubule severing. J. Cell Biol. 176, 995–1005 10.1083/jcb.200610072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han H., Monroe N., Votteler J., Shakya B., Sundquist W. I., and Hill C. P. (2015) Binding of substrates to the central pore of the Vps4 ATPase is autoinhibited by the microtubule interacting and trafficking (MIT) domain and activated by MIT interacting motifs (MIMs). J. Biol. Chem. 290, 13490–13499 10.1074/jbc.M115.642355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Henderson R., Sali A., Baker M., Carragher B., Devkota B., Downing K., Egelman E., Feng Z., Frank J., Grigorieff N., Jiang W., Ludtke S., Medalia O., Penczek P., Rosenthal P., et al. (2012) Outcome of the First Electron Microscopy Validation Task Force Meeting. Structure 20, 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Su M., Guo E. Z., Ding X., Li Y., Tarrasch J. T., Brooks C. L. 3rd, Xu Z., and Skiniotis G. (2017) Mechanism of Vps4 hexamer function revealed by cryo-EM. Sci. Adv. 3, e1700325 10.1126/sciadv.1700325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Exp. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 36. Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J., Lindahl E., and Scheres S. H. (2018) New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 10.7554/eLife.42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., and Agard D. A. (2017) MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang K. (2016) Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 10.1016/j.jsb.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenthal P. B., and Henderson R. (2003) Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 10.1016/j.jmb.2003.07.013 [DOI] [PubMed] [Google Scholar]
- 40. Afonine P. V., Poon B. K., Read R. J., Sobolev O. V., Terwilliger T. C., Urzhumtsev A., and Adams P. D. (2018) Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 10.1107/S2059798318006551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fan X., Lin Z., Fan G., Lu J., Hou Y., Habai G., Sun L., Yu P., Shen Y., Wen M., and Wang C. (2018) The AAA protein spastin possesses two levels of basal ATPase activity. FEBS Lett. 592, 1625–1633 10.1002/1873-3468.13075 [DOI] [PubMed] [Google Scholar]
- 42. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comp. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 43. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B. 3rd, Snoeyink J., Adams P. D., Lovell S. C., Richardson J. S., and Richardson D. C. (2018) MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 10.1002/pro.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barad B. A., Echols N., Wang R. Y., Cheng Y., DiMaio F., Adams P. D., and Fraser J. S. (2015) EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 10.1038/nmeth.3541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tan Y. Z., Baldwin P. R., Davis J. H., Williamson J. R., Potter C. S., Carragher B., and Lyumkis D. (2017) Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 10.1038/nmeth.4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 10.1093/nar/gkh381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.