

Abstract
Heat shock proteins of 70 kDa (Hsp70s) are ubiquitous and highly conserved molecular chaperones. They play multiple essential roles in assisting with protein folding and maintaining protein homeostasis. Their chaperone activity has been proposed to require several rounds of binding to and release of polypeptide substrates at the substrate-binding domain (SBD) of Hsp70s. All available structures have revealed a single substrate-binding site in the SBD that binds a single segment of an extended polypeptide of 3–4 residues. However, this well-established single peptide-binding site alone has made it difficult to explain the efficient chaperone activity of Hsp70s. In this study, using purified proteins and site-directed mutagenesis, along with fluorescence polarization and luciferase-refolding assays, we report the unexpected discovery of a second peptide-binding site in Hsp70s. More importantly, the biochemical analyses suggested that this novel binding site, named here P2, is essential for Hsp70 chaperone activity. Furthermore, cross-linking and mutagenesis studies indicated that this second binding site is in the SBD adjacent to the first binding site. Taken together, our results suggest that these two essential binding sites of Hsp70s cooperate in protein folding.
Keywords: chaperone, chaperone DnaK (DnaK), heat shock protein (HSP), peptide interaction, substrate specificity, protein folding, heat shock protein 70 (Hsp70), P2 site, peptide-binding site, TRP2 peptide
Introduction
The 70-kDa heat shock proteins (Hsp70s)5 are one of the major classes of molecular chaperones that are most frequently stress-induced to combat protein denaturation caused by various environmental stresses, such as elevated temperature, radiation, and inflammation. Hsp70s are not only crucial for cell survival under various stress conditions, but also play multiple essential roles in almost every process in maintaining cellular proteostasis, including facilitating protein folding, assembly, translocation into organelles, degradation, and dismantling protein aggregates (1–11). Due to their importance in the fundamental process of maintaining protein homeostasis, Hsp70s are implicated in many human diseases, including cancers and neurodegenerative diseases (1, 12–19). Thus, they are potential key targets for designing novel and specific treatment and prevention strategies for these diseases.
In general, the chaperone activities of Hsp70s are facilitated by two classes of well-characterized co-chaperones: Hsp40s and the nucleotide-exchange factors (NEFs) (1, 2, 5, 6, 20, 21). Hsp40s are also called J-domain–containing proteins. The well-recognized role of Hsp40s is directly speeding up the rate of ATP hydrolysis by Hsp70s (21–27). At the same time, many Hsp40s have been shown to bind unfolded or partially folded polypeptides directly to prevent their aggregation. Thus, it was proposed that Hsp40s also serve as a substrate-scanning factor for Hsp70s and bring polypeptide substrates to Hsp70s. NEFs speed up the chaperone cycle of Hsp70s through promoting the exchange of ADP for ATP after ATP hydrolysis (1, 28, 29). It is well-established that Hsp70s, Hsp40s, and NEFs constitute the most essential chaperone machinery for protein folding across all kingdoms of life. Moreover, recent studies have supported an important role of Hsp90s in assisting the Hsp70-folding machinery (30, 31) and an emerging role of nucleotide-exchange inhibitor (NEI) in regulating Hsp70s in protein homeostasis (32, 33).
Consistent with their importance, Hsp70s are ubiquitous, abundant, and highly conserved (1, 5, 6, 34, 35). All Hsp70s contain two functional domains, a nucleotide-binding domain (NBD) at the N terminus and a substrate binding domain (SBD) at the C terminus (1, 3, 5, 13, 21, 36). Connecting these two domains is a short linker segment. Each domain carries an essential intrinsic activity: ATPase activity for the NBD and polypeptide substrate–binding activity for the SBD. The NBD captures the energy of ATP hydrolysis at the production of ADP to power chaperone activity. The SBD is the site where polypeptide substrates bind. In general, the SBD recognizes unfolded hydrophobic segments in extended conformations, which are normally sequestered inside natively folded proteins (37–43). Various biochemical and NMR studies have shown that in the ADP-bound or nucleotide-free state there is little contact between NBD and SBD; each domain acts independently as if isolated (17, 36, 42, 44–48). This state, like the isolated SBD, exhibits high affinity and slow kinetics for peptide substrates (44–46, 48). In contrast, ATP binding allosterically couples the two domains: the peptide substrate–binding affinity is reduced by 2–3 orders of magnitude (37, 49), and reciprocally, peptide substrate binding stimulates the ATPase rate (5, 37). This allosteric coupling ensures that energy from ATP hydrolysis is efficiently used to control peptide substrate binding and release. In conjunction with various biochemical, NMR, and FRET studies (1, 8, 44, 46, 47, 50–54), a number of recent crystal structures of intact Hsp70s in the ATP-bound state have provided revolutionary insights into this essential mechanism (8, 21, 52, 55–60).
Various X-ray crystallography and NMR studies have revealed the structural basis of each domain in binding their substrates. The crystal structure of the isolated NBD from the bovine Hsc70 with ADP bound is the first structure of NBD (61). Two large lobes form a deep pocket for binding the adenine nucleotides. The first crystal structure of the SBD was obtained with the isolated SBD from DnaK, a major Escherichia coli Hsp70, in complex with a short peptide substrate NR (NRLLLTG) (41). The SBD is composed of two subdomains: SBDβ and SBDα. SBDβ forms a single narrow peptide-binding pocket between two loops, whereas SBDα functions as a lid covering the peptide binding pocket on SBDβ. All available crystal and NMR structures report essentially the same conformation of the SBD (32, 41, 48, 55, 62–70). One of the most intriguing features of these structures is that Hsp70s mainly form contacts with 3–4 hydrophobic residues of the peptide substrates, such as the three middle leucine residues in the NR peptide. This feature is consistent with various biochemical characterizations on the peptide substrate–binding specificities of Hsp70s (39, 40, 42, 43). However, this well-established single peptide-binding site has made it difficult to explain the efficient chaperone activity of Hsp70s in protein folding and dismantling protein aggregates, considering the average size of proteins is several hundreds of amino acids long. Could there be additional peptide-binding sites on Hsp70s that account for their high efficiency? Interestingly, a very recent study using NMR unexpectedly showed that each monomer of Hsp40s has 1–3 transient binding sites for polypeptide substrates (71) in contrast to the long-speculated single binding site. It is possible that there is more than one binding site for polypeptide substrates for Hsp70s.
In this study, we have presented biochemical data including mutagenesis and peptide competition to support a second novel peptide-binding site (P2) in Hsp70s besides the aforementioned well-characterized classic peptide-binding site (P1). These two binding sites have different preference for polypeptide substrates. More importantly, this novel P2 peptide-binding site is crucial for Hsp70s' chaperone activity in protein folding while displaying different properties from the classic site. Our cross-linking results suggested that this novel P2 site is in the vicinity of the L5,6 region of the SBD. We propose that these two peptide substrate–binding sites cooperate to achieve the powerful chaperone activity of Hsp70s in protein folding. Thus, this surprising discovery of a novel second peptide-binding site on Hsp70s has opened a new direction for characterizing the basic mechanism of Hsp70s in assisting protein folding.
Results
Identification of a surprising new peptide substrate–binding site on E. coli Hsp70 DnaK
It is well-established that Hsp70s have a single peptide substrate–binding site on the SBDβ (1, 6, 8, 21, 32, 34, 41, 48, 55, 62–70, 72). This binding site is represented by the crystal structure of the isolated SBD of DnaK, a major Hsp70 in E. coli, in complex with the NR peptide (Fig. 1A) (41). The NR peptide (NRLLLTG) is a well-characterized peptide substrate for DnaK (41, 42). When simultaneously studying both Hsp70s and Hsp110s (73, 74), distant homologs of Hsp70s, to understand the chaperone activity difference between these two essential chaperones, we found that Hsp70s also bind to another type of peptide besides the NR peptide using a fluorescence polarization assay (75). This new type of peptide is represented by the TRP2 peptide (VYDFFVWLHYY), a peptide from TRP2 (tyrosinase-related protein 2), a tumor antigen. There is an important difference between these two types of peptides: NR is rich in aliphatic residues, whereas TRP2 is rich in aromatic residues. Binding of both the NR and TRP2 peptides to Hsp70s is consistent with the previous studies on peptide substrate specificities (40, 42, 43). Our previous studies have shown that, unlike Hsp70s, Hsp110s only showed strong binding to the TRP2 peptide and not to the NR peptide. Furthermore, our subsequent mutational studies suggested that the TRP2 peptide does not bind Hsp110s at the same binding site as the NR-binding site in the Hsp70s' structures. Because Hsp70s bind both the NR and TRP2 peptides, it is possible that there are two peptide-binding sites on Hsp70s with different properties. One is the classic Hsp70 substrate-binding site for peptides like NR, and the other one is shared with Hsp110s for binding to peptides like TRP2. To test this hypothesis, we carried out two tests.
Figure 1.
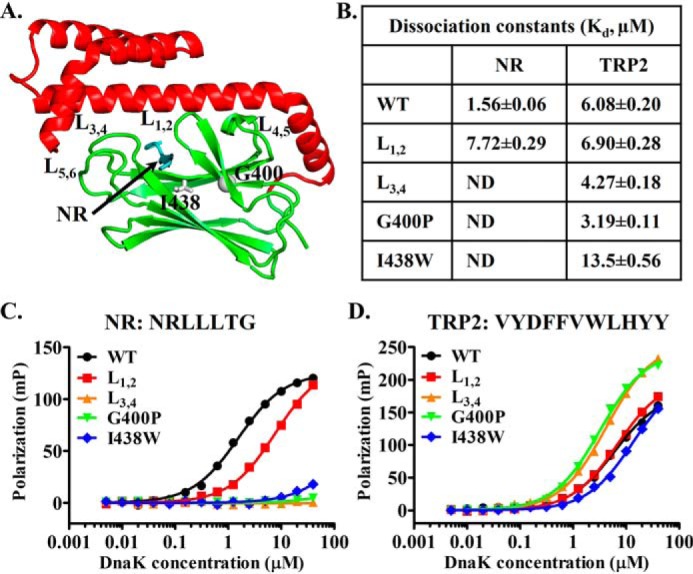
Mutations in the classic peptide substrate–binding site of DnaK have little impact on binding the TRP2 peptide. A, ribbon diagram of the isolated DnaK SBD structure (Protein Data Bank code 1DKX). Ile-438 and Gly-400 are highlighted in gray sticks and ball, respectively. The bound NR peptide is in cyan, and the peptide-binding loops and their supporting loops are labeled. B, the dissociation constants (Kd) of the NR and TRP2 peptides binding from C and D. ND, not detectable. C and D, the binding of DnaK proteins to the NR (C) and TRP2 peptides (D). Serial dilutions of DnaK proteins were incubated with peptides (10 nm), and fluorescence polarizations were recorded after binding reached equilibrium. The sequences of the peptides are labeled on the top of each plot.
The first test was mutagenesis. The structure of the isolated DnaK SBD in complex with the NR peptide has revealed the structural basis of the NR binding to DnaK (41). The NR peptide binds in a narrow pocket formed between two loops, L1,2 and L3,4, the peptide-binding loops (Fig. 1A). Previously, we have made a mutation on each of these peptide-binding loops in DnaK: DnaK-L1,2 (replaced the original sequence of MGG with TAEDNQS) and DnaK-L3,4 (replaced the original sequence of TAEDNQS with MGG) (75). Consistent with the structure, mutating either of these peptide-binding loops has drastically reduced the binding affinity for the NR peptide (56, 75). Surprisingly, neither of these loop mutations showed appreciable reduction in binding the TRP2 peptide. In fact, the DnaK-L3,4 mutant protein showed even stronger binding toward the TRP2 peptide than the WT DnaK protein, whereas the binding for the NR peptide is almost completely abolished. We reproduced these results in this study (Fig. 1, B–D). These results support our hypothesis of a novel second binding site for peptide substrates like TRP2. To provide further support using mutagenesis, we took advantage of two mutations: G400P and I438W. Gly-400 is on L1,2. Our previous studies have shown that Gly-400 is crucial for opening L1,2 to bind the NR peptide, and the G400P mutation completely knocks out NR binding due to a failure to open L1,2, which leads to L1,2 covering the peptide-binding pocket (58). Consistent with our hypothesis, G400P showed little reduction in binding to the TRP2 peptide (Fig. 1, B and D), whereas the NR binding is completely abolished, as shown in our previous studies (58) (Fig. 1, B and C). In fact, the G400P mutant behaves like the L3,4 mutation with a slightly increased affinity. Ile-438 is on β4 of SBD (Fig. 1A) and sits at the bottom of the classic peptide-binding pocket. Mutating Ile-438 to tryptophan, a residue with a much larger hydrophobic side chain, is supposed to reduce the size of the classic peptide-binding pocket and therefore reduce NR binding and compromise chaperone activity. Consistent with this structural hypothesis, the NR binding to this I438W mutant DnaK is drastically compromised (Fig. 1, B and C), and the in vivo chaperone activity is almost completely abolished, although the expression level of this mutant protein is comparable with that of the WT control (Fig. S1, A and B). In contrast, the binding of the TRP2 peptide to the I438W mutant protein is comparable with that of the WT DnaK protein. Taken together, these mutational analyses indicated that the TRP2 peptide binds to a novel binding site in DnaK different from the classic binding site where the NR peptide binds.
The second test performed was a competition analysis between the NR and TRP2 peptides. If the NR and TRP2 peptides bind to the same peptide-binding pocket, they will compete with each other for binding to the DnaK protein. If they bind to different sites in DnaK, there will be little competition. We performed the aforementioned peptide-binding assays using fluorescence polarization in the presence of a large excess of the unlabeled NR peptide (200 and 800 μm). As expected, the binding of fluorescein-labeled NR, F-NR, is almost completely competed off by the large excess amount of the unlabeled NR (Fig. 2A). Consistent with our hypothesis, this large excess unlabeled NR peptide did not appreciably affect the binding of the fluorescently labeled TRP2 peptide (Fig. 2B). The poor solubility of the TRP2 peptide in solution prevented us from doing an analogous competition assay using the unlabeled TRP2 peptide.
Figure 2.
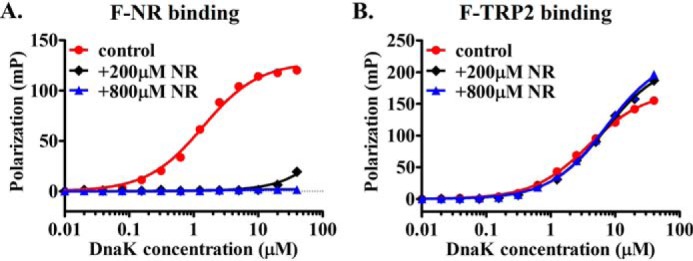
The TRP2 peptide does not compete with the NR peptide for binding to DnaK. A, the unlabeled NR peptide efficiently competed with F-NR for binding to DnaK protein. The concentrations (200 and 800 μm) of the unlabeled NR peptide are listed, and the concentration of the F-NR was 10 nm. The control is the F-NR binding in the absence of the unlabeled NR. B, the F-TRP2 peptide binding to DnaK was not appreciably competed by the presence of a large amount of the unlabeled NR peptide (200 and 800 μm). The F-TRP2 concentration was 10 nm. Peptide binding in the absence of the unlabeled NR is shown as a control.
In summary, there are two noncompeting peptide-binding sites on Hsp70 DnaK: 1) the classic peptide-binding site, as shown in the isolated SBD structure of DnaK for peptides like NR (rich in aliphatic residues), and 2) a second novel peptide-binding site for peptides like TRP2 (rich in aromatic residues). We named these two sites P1 and P2, respectively. Similar competition results were obtained for two eukaryotic Hsp70s: Ssa1 (76), a major yeast Hsp70, and BiP (77), a human Hsp70 in the ER (Fig. S2). Thus, these two noncompeting peptide-binding sites are most likely conserved in Hsp70s in general.
The novel peptide-binding site is on SBDβ and demonstrates properties different from those of the canonical binding site
We first dissected the domain location of this novel P2 peptide-binding site using truncations of DnaK. Hsp70s are highly conserved in domain organization. Each Hsp70, including DnaK, contains two functional domains: an NBD and an SBD (Fig. 3A). The SBD is further divided into two subdomains: SBDβ and SBDα. The classic peptide-binding site is in SBDβ. To explore the domain location of this novel second binding site, we made a number of truncations of DnaK containing only the NBD, SBD, SBDβ, and SBDα (Fig. 3B). Consistent with the previous structural and biochemical analysis, both the SBD and SBDβ bind the NR peptide with comparable affinities as the full-length DnaK protein, whereas the NBD and SBDα showed little binding to the NR peptide (Fig. 3, C and E). Like the NR peptide, the TRP2 peptide binds to both the SBD and SBDβ with similar affinities as the full-length DnaK protein (Fig. 3, D and E), although the absolute values of fluorescence polarization for the isolated SBD were lower than those of the full-length and isolated SBDβ. This difference in the absolute values could be due to the influence of SBDα on the binding environment of the P2 site. Importantly, the NBD and SBDα only showed background level of binding toward the TRP2 peptide. Thus, this novel P2 binding site is in the SBDβ, the same subdomain where the classic P1 peptide-binding site is located.
Figure 3.
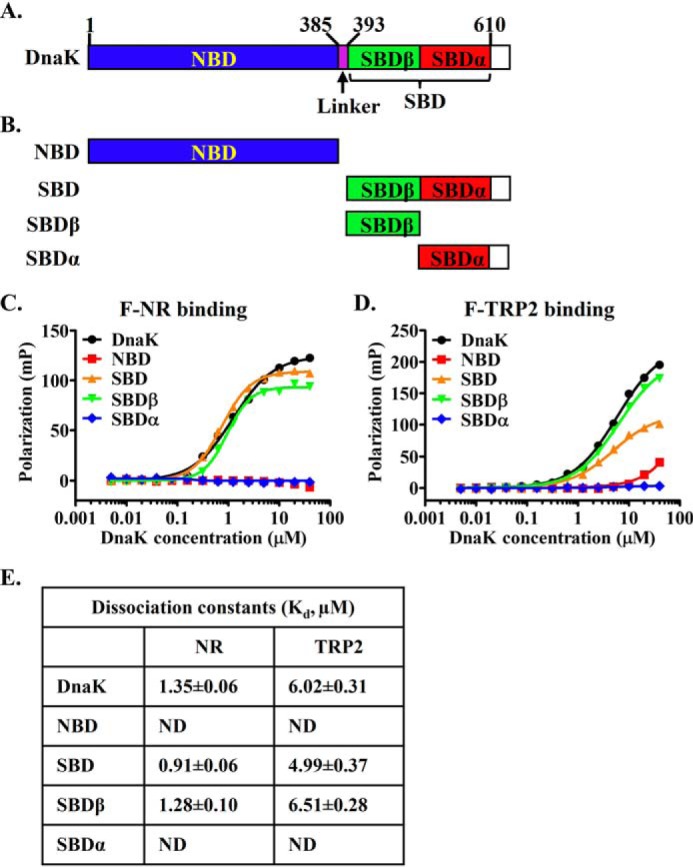
The novel second binding site for peptide substrate is on the SBDβ subdomain. A, schematic diagram of the domain organization of DnaK. Domain boundaries are labeled at the top. B, DnaK domain truncations. C and D, the NR (C) and TRP2 (D) peptide binding to the isolated domains of DnaK. E, the dissociation constants (Kd) of the NR and TRP2 peptides binding from C and D. ND, not detectable.
It is well-established that the NR peptide binding to DnaK is ATP-sensitive, and this binding stimulates the ATPase activity of DnaK (37, 49). These properties are the essential ATP-induced allosteric coupling in Hsp70s. We tested whether this novel P2 peptide-binding site shows these properties. Consistent with the published results, the NR binding is drastically reduced in the presence of ATP (more than 20-fold based on estimated Kd; Fig. 4A), whereas the binding of the TRP2 peptide in the presence of ATP is quite similar to that of in the absence of ATP with some small differences (Fig. 4A). The estimated Kd of the TRP2 peptide in the presence of ATP is only about 2-fold higher than that in the absence of ATP. For the NR peptide binding, this strong ATP-induced reduction in binding affinity is caused by the accelerated binding kinetics, especially the release rate (49). In contrast, in the absence of ATP, the binding kinetics for the NR peptide is much slower. As shown previously, the TRP2 peptide binds Hsp110 Sse1 with remarkably fast kinetics even in the absence of ATP (75). Here, we tested the binding kinetics of the TRP2 peptide to the DnaK protein. As shown in Fig. 4B and Fig. S3A, the binding kinetics of the TRP2 peptide to DnaK is at least several times faster than the binding kinetics of the NR peptide in the absence of ATP. Due to the fast kinetics and the limitation of our fluorescence polarization instrument, we were not able to determine accurate binding kinetics for the TRP2 peptide binding.
Figure 4.
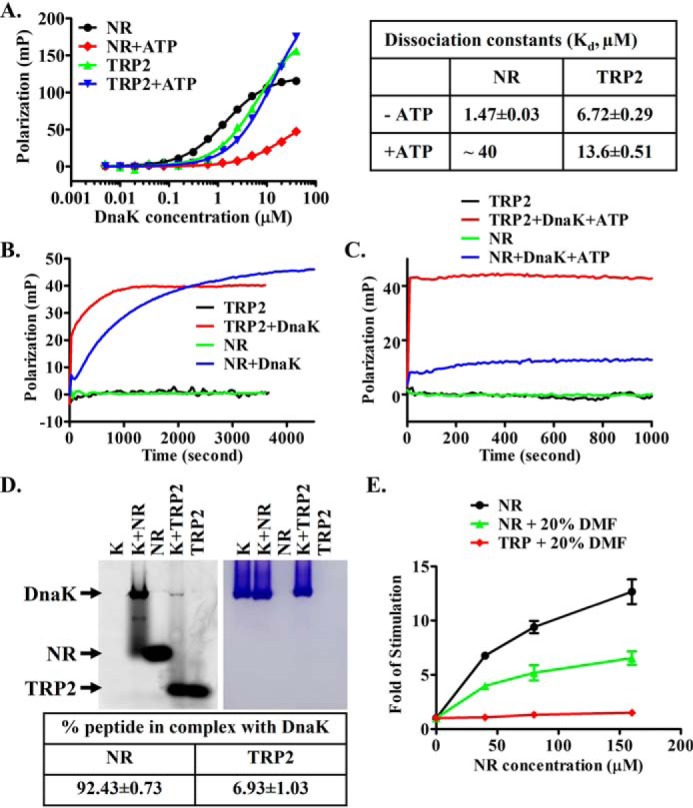
The novel second binding site has different properties from the classic peptide-binding site. A, the F-NR and F-TRP2 peptide binding to DnaK protein in the presence of and absence of ATP. The fitted and estimated dissociation constants are listed on the right. B and C, peptide binding kinetics in the absence (B) and presence (C) of ATP. The F-NR or F-TRP2 peptide was mixed with DnaK (2.5 μm) to initiate binding, and binding kinetics was recorded immediately as a function of time. D, native gel analysis on the stability of the DnaK-peptide complexes. The fluorescein-labeled peptides were visualized using a phosphorimaging system (left), and the DnaK proteins on the same gel were stained with Coomassie Blue (right). The positions of DnaK and the peptides are labeled. The percentage of each peptide forming a complex with DnaK was calculated after quantification of the bands containing the fluorescein-labeled peptides. The data in the table are averages from five independent repeats with freshly prepared samples. E, unlike the NR peptide, the TRP2 peptide showed little stimulation of the ATPase activity of DnaK. -Fold stimulation was calculated by setting the intrinsic ATPase activity (the ATPase activity in the absence of peptides) as 1. Error bars, S.E.
The fast binding kinetics and low affinity of the TRP2 peptide binding relative to the NR peptide binding suggests that the complex formation between DnaK and the TRP2 peptide may be less stable than that of the NR peptide. Importantly, our native gel analysis on the DnaK-peptide complexes provided support for this hypothesis (Fig. 4D). Unlike the fluorescence polarization assay, which is suitable for detecting both stable and transient interactions, strong complex bands are usually observed on native gels for stable interactions but not for transient interactions. At the concentrations of DnaK and peptides (20 and 10 μm, respectively) close to saturation of binding, the amount of complex formed between the TRP2 peptide and DnaK is much less than that of the NR peptide: almost all of the NR peptide formed a complex with DnaK, whereas only a very small amount of complex was formed between the TRP2 peptide and DnaK (Fig. 4D). Furthermore, in the presence of ATP, the binding kinetics of the TRP2 peptide to DnaK are even faster than the NR peptide binding to DnaK in the presence of ATP (Fig. 4C and Fig. S3B). These binding properties are similar to those of the TRP2 peptide binding to Hsp110s, suggesting a similar nature of binding of the TRP2 peptide to Hsp70s and Hsp110s.
Next, we tested the stimulation of the ATPase activity of DnaK. Consistent with the previous publications (78, 79), the NR peptide stimulated the ATPase activity of DnaK close to 15-fold in a well-established single-turnover ATPase assay (Fig. 4E). To achieve the concentrations of the TRP2 peptide high enough for the ATPase assay, 20% DMF was included in the reaction buffer. Although 20% DMF significantly influenced the fluorescence polarization assay for both the TRP2 and NR peptides (Fig. S4), the NR peptide showed robust stimulation on the ATPase activity of DnaK in the presence of 20% DMF (Fig. 4E). In contrast, no significant stimulation on the ATPase activity of DnaK was observed for the TRP2 peptide over the concentrations used in our assay (Fig. 4E). This is consistent with the peptide-binding kinetics in the presence of ATP (Fig. 4C and Fig. S3B). For the NR peptide, after the initial fast phase of binding, the polarization reading (indication of peptide binding) keeps on increasing over time although slow, indicating ATP hydrolysis stimulated by the NR binding, whereas for the TRP2 peptide, there was no such slow increase. Furthermore, peptide substrate showed little simulation on the ATPase activity of yeast Hsp110 Sse1 (80), further supporting the similarity of both Hsp70s and Hsp110s in interacting with the TRP2 peptide.
Taken together, the TRP2 peptide binding to DnaK has properties different from the NR peptide binding to DnaK, although both peptide-binding sites are located in the SBDβ. These differences between the two binding sites indicate their different roles in DnaK's chaperone activity.
The new P2 substrate-binding site is essential for DnaK's chaperone activity
To test the role of this novel P2 site in the chaperone activity of Hsp70s, we performed a luciferase-refolding assay in the presence of either NR or TRP2 peptide using the DnaK chaperone system (20). The DnaK chaperone system includes DnaK and its two essential co-chaperones DnaJ and GrpE, an Hsp40 and NEF, respectively. Consistent with the previous studies, the DnaK chaperone system refolds heat-denatured luciferase and leads to the recovery of the luciferase activity (Fig. S5A). In this assay, the heat-denatured luciferase is the substrate for DnaK. If this novel P2-binding site is essential for the chaperone activity, binding of TRP2 peptide at this site will prevent DnaK from binding to the heat-denatured luciferase and thus hinder the refolding of the luciferase. As expected, both peptides inhibit the refolding activity of DnaK (Fig. 5A and Fig. S5A), supporting the importance of both sites for the chaperone activity of DnaK. Surprisingly, the TRP2 peptide inhibits the refolding activity of DnaK even more effectively than NR, although the affinity of TRP2 is about 4-fold lower than that of NR (Fig. 1B). Thus, the P2 site is crucial for DnaK's chaperone activity and maybe even more important than the classic P1 binding site.
Figure 5.
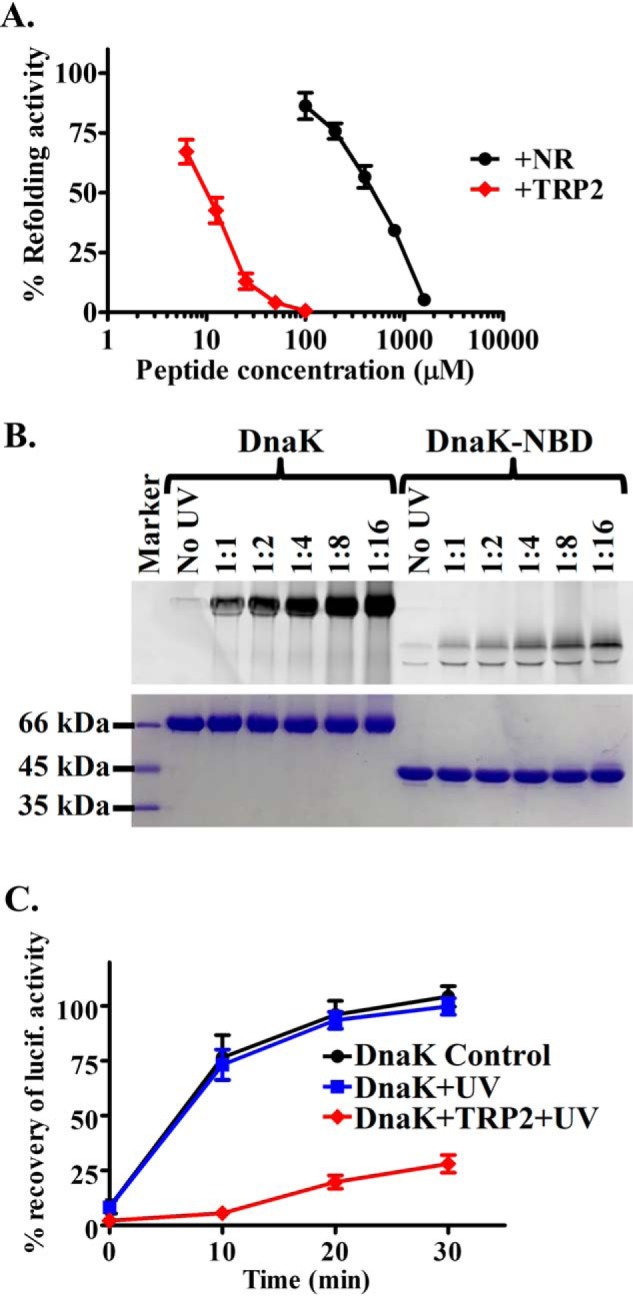
The new second peptide-binding site is essential for DnaK's chaperone activity. A, the TRP2 peptide inhibits the refolding activity of DnaK even more efficiently than the NR peptide. The refolding activities of DnaK were determined in the presence of the indicated concentrations of peptides. The refolding activity of DnaK in the absence of peptide was set as 100%. B, the TRP2-Bpa peptide was efficiently cross-linked to DnaK protein. DnaK protein was incubated with the TRP2-Bpa peptide at the indicated molar ratios and treated with UV light on ice. The isolated NBD of DnaK was used as a negative control. After SDS-PAGE, the cross-linked DnaK-peptide complex was visualized by fluorescence scan (top) and Coomassie Blue staining (bottom), respectively. C, the DnaK protein cross-linked with the TRP2-Bpa peptide has a compromised chaperone activity in refolding heat-denatured luciferase. The DnaK protein with only UV treatment was used as a control. The percentage of recovery of luciferase activity was calculated by setting the native luciferase activity as 100%. Error bars, S.E.
To further confirm that the TRP2 inhibition on the chaperone activity of the DnaK chaperone machinery is due to the binding of TRP2 to DnaK, we have designed a TRP2 peptide with p-benzoyl-l-phenylalanine (Bpa), an unnatural amino acid, replacing the tryptophan in the original TRP2 peptide. We named this new TRP2 peptide as TRP2-Bpa (SVYDFFVBpaLK) and labeled the N terminus with fluorescein for easy detection. Because Bpa and tryptophan share a similar structure, we do not expect that the Bpa replacement will have an appreciable effect on the properties of the TRP2 peptide. As expected, this TRP2-Bpa peptide binds DnaK with a similar affinity as the original TRP2 peptide (Kd = 5.59 ± 0.38 μm). More importantly, like the TRP2 peptide, unlabeled NR peptide did not show appreciable competition with this TRP2-Bpa peptide, and the DnaK L3,4 mutant protein binds to this TRP2-Bpa peptide similarly to the WT DnaK (Fig. S6). Upon treatment with UV light at 360 nm, a significant amount of this peptide was cross-linked to DnaK, whereas only a very small amount of cross-linking was observed for the NBD fragment of DnaK (Fig. 5B). This result is consistent with the observation that the TRP2 peptide binds the SBD of DnaK (Fig. 3D), supporting the specificity of this cross-linking. After cross-linking, we removed the free TRP2-Bpa peptide from the cross-linked complex containing DnaK and TRP2-Bpa using size-exclusion chromatography. We tested the refolding activity of this DnaK protein after cross-linking with the TRP2-Bpa peptide. Consistent with our hypothesis, the refolding activity of this cross-linked DnaK was drastically compromised, whereas UV treatment alone had little impact on the chaperone activity of DnaK (Fig. 5C). Taken together, this novel P2 binding site is essential for the chaperone activity of DnaK.
Moreover, to test whether this P2 site is essential for the chaperone activity of eukaryotic Hsp70s, we carried out a luciferase-refolding assay with the Ssa1 chaperone system, the major Hsp70 chaperone system in the yeast cytosol. This chaperone system includes Ssa1 and its two cochaperones Ydj1 and Sse1, an Hsp40 and NEF, respectively. As shown in Fig. S5B, the chaperone activity of this Ssa1 chaperone system is inhibited by both the TRP2 and NR peptides, supporting the importance of these two substrate-binding sites in the chaperone activity. Thus, this novel P2 site most likely is essential for the chaperone activity of Hsp70s in general.
The P2 binding site is in the vicinity of the loop L5,6 on SBD
To pinpoint the location of this novel P2 site on SBDβ, we took advantage of the aforementioned TRP2-Bpa peptide and a previously characterized DnaK SBD construct with an L3,4 deletion mutation, DnaK-SBD-L3,4 (56). Previous studies have shown that this L3,4 deletion mutation completely abolished the binding of DnaK to the NR peptide, whereas the binding to the TRP2 peptide is largely intact (75). Like the full-length WT DnaK protein shown in Fig. 5B, after treatment with UV light, a significant amount of the TRP2-Bpa peptide was cross-linked to this DnaK-SBD-L3,4 construct (Fig. 6A). We digested this cross-linked band with trypsin and did peptide fingerprinting using MS. Interestingly, the MS result suggested that the cross-linked position is Met-469 on the loop L5,6 of the SBDβ, suggesting that the loop L5,6 is in close proximity to this novel P2 peptide-binding site.
Figure 6.
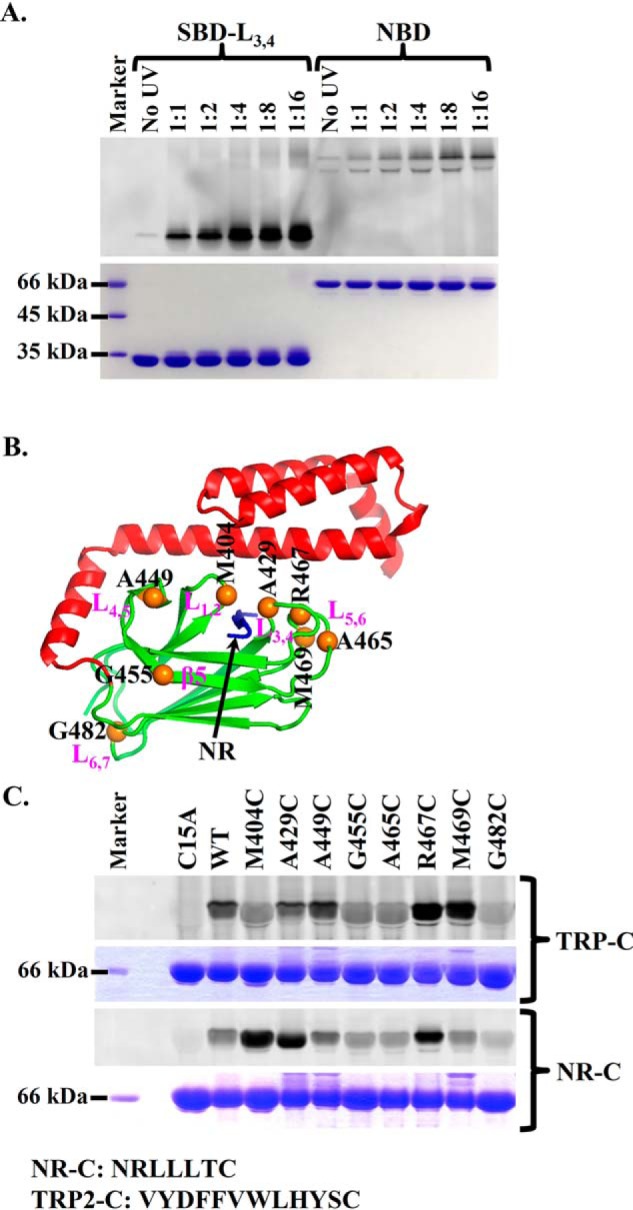
The novel second binding site is located in the vicinity of L5,6. A, the TRP2-Bpa peptide was efficiently cross-linked to the isolated DnaK SBD carrying the L3,4 mutation, SBD-L3,4. The isolated NBD of DnaK was used as a negative control. After SDS-PAGE, the cross-linked DnaK-peptide complexes were visualized by fluorescence scan (top) and Coomassie Blue staining (bottom), respectively. B, locations of the designed cysteine mutations in the structure of the isolated DnaK SBD (Protein Data Bank code 1DKX). The designed cysteine mutations were highlighted as orange balls. The bound NR peptide is in blue. C, the TRP2-C and NR-C peptides were cross-linked to the different cysteine mutations in DnaK. Purified DnaK proteins carrying the indicated cysteine mutations were incubated with the TRP2-C or NR-C peptide and then cross-linked through disulfide bonds using copper-phenanthroline. The cross-linked complexes were visualized by fluorescence scan (top) and Coomassie Blue staining (bottom), respectively, after separating on SDS-PAGE. The sequences of the NR-C and TRP2-C peptides are shown below the gel.
To test the above cross-linking results and further explore the P2 binding site, we designed derivatives of the NR and TRP2 peptides with a cysteine at the C terminus, NR-C (NRLLLTC) and TRP2-C (VYDFFVWLHYSC), respectively. At the same time, we mutated a number of residues in the SBDβ to cysteine: Met-404, Ala-429, Ala-449, Gly-455, Ala-465, Arg-467, Met-469, and Gly-482. All of these residues are on the surface of SBDβ (Fig. 6B). More importantly, none of these mutations affect either the in vivo chaperone activity of DnaK or the peptide-binding affinities for either NR-C or TRP2-C appreciably (Figs. S7 and S8), suggesting that none of these mutations has significant influence on either the structure or the activity of DnaK. Based on the published crystal structure (41), Met-404 and Ala-429 are located in the classic P1 binding site. Upon oxidation with Cu-phenanthroline, these residues are supposed to be cross-linked to the NR-C peptide through disulfide bonds. Consistent with this structural observation, a strong cross-linked band was observed for both proteins, whereas only weak background cross-linking was observed for the rest of the mutants except for R467C (Fig. 6C). For R467C, the cross-linking band was significantly above the background control (WT) but lower than the two peptide-binding loop mutants M404C and A429C. Arg-467 is on the loop L5,6, the supporting loop for L3,4, one of the peptide-binding loops for the canonical P1 peptide-binding site. The side chain of Arg-467 is close to the classic P1 site in the isolated SBD structure of DnaK (41). This may be the reason for the significant cross-linking with R467C. Although Arg-467 forms a salt bridge with Asp-540, a residue on SBDα, in the isolated SBD structure (41), mutating Arg-467 to cysteine has little influence on the in vivo chaperone activity of DnaK (Fig. S7), suggesting that the salt bridge formed between Arg-467 and Asp-540 is dispensable for the function of DnaK. DnaK has an endogenous cysteine in the NBD, Cys-15. The background cross-linking is most likely due to this Cys-15 because mutating Cys-15 to alanine almost completely abolished the background cross-linking. Interestingly, neither M404C nor A429C showed significant cross-linking to the TRP2-C peptide (Fig. 6C). This is consistent with the binding and mutagenesis data in Fig. 1. Excitingly, M469C formed a strong disulfide bond–linked band with TRP2-C, supporting the above MS analysis result on the TRP2-Bpa cross-linking. Moreover, the R467C mutant, another residue on L5,6, also formed a strong disulfide bond with the TRP2-C peptide. Taken together, this novel P2 peptide-binding site is most likely in the vicinity of the loop L5,6. Moreover, the specific cross-linking to the L5,6 region indicates that the TRP2 binding to the P2 site is specific.
To further confirm the involvement of L5,6 in binding the TRP2 peptide and the new P2 binding site, we have made a deletion mutation of L5,6, DnaK-L5,6 (Fig. 7A). Deleting L5,6, including Met-469, abolished the in vivo chaperone activity (Fig. 7B and Fig. S9A), although the expression level was similar to that of the WT DnaK (Fig. S9B). Interestingly, both the TRP2 and NR peptide binding to the DnaK-L5,6 mutant were compromised (Fig. 7, C and D), consistent with the lack of in vivo chaperone activity. The estimated Kd values were severalfold higher than those of the WT protein, although the binding curves for the L5,6 mutant had not reached a plateau. This defect in binding the TRP2 peptide is consistent with the involvement of L5,6 in this novel P2 binding site, as shown above. Moreover, supporting the binding defect, the cross-linking to the TRP2-Bpa peptide was also significantly reduced by this L5,6 mutation compared with the WT protein (Fig. 7E). The amount of TRP2-Bpa peptide crossed to the DnaK-L5,6 mutant was reduced to about half of that of the WT protein (50.5 ± 3.4% of the level of the WT protein averaged from six repeats). Because there was still significant binding to the TRP2 peptide for the DnaK-L5,6 mutant, L5,6 was only a part of this binding site. A similar defect for binding the NR peptide was observed for the DnaK-L1,2 mutant: L1,2 is a part of the classic P1 peptide-binding site, and mutating it compromises the NR peptide binding but does not abolish it. The reduced binding to the NR peptide for the DnaK-L5,6 mutant is because L5,6 is a supporting loop for the peptide-binding loop L3,4 of the classic P1 binding site (Fig. 1A). As a control, we deleted L4,5, another supporting loop for the classic P1 binding site (Figs. 1A and 7 (A and B)). This L4,5 mutation mainly affected the NR binding, whereas it left the TRP2 binding largely unaffected (Fig. 7, C and D), suggesting that this part of SBD is most likely not directly involved in the P2 binding site. Furthermore, consistent with the previous structural analysis (55–59), neither of these loops was involved in allosteric coupling when measured using tryptophan fluorescence spectra (Fig. S9C). In summary, L5,6 is most likely involved in the novel P2 binding site.
Figure 7.
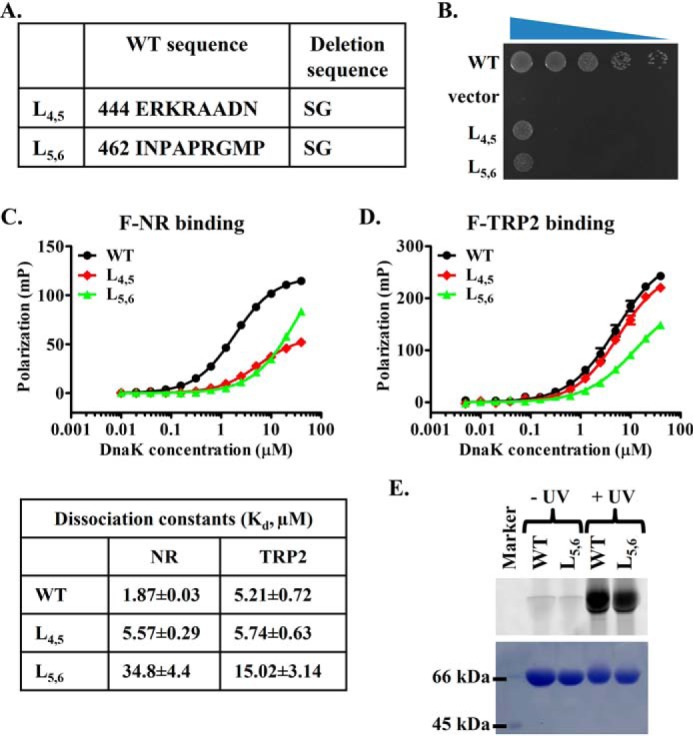
The L5,6 is involved in binding both the NR and TRP2 peptides. A, the loop deletion mutations in DnaK. B, growth test of the loop deletion DnaK mutations. Serial dilutions of fresh cultures carrying the loop deletion mutations were spotted on LB agar plates and grown for 1 overnight at 37 °C. WT DnaK and empty vector were used as positive and negative controls, respectively. C and D, the binding to the F-NR (C) and F-TRP2 (D) peptides of the DnaK loop mutations. The fitted Kd values for curves in both C and D are listed in the table in C. E, the TRP2-Bpa peptide cross-linked to the DnaK-L5,6 mutant is reduced compared with that of the WT DnaK. The DnaK proteins and the TRP2-Bpa peptide (at a 1:4 molar ratio) were cross-linked with UV on ice and separated on SDS-PAGE. Top, fluorescence scan; bottom, Coomassie Blue staining. The cross-linked bands were quantified, and the ratio of the TRP2-Bpa peptide cross-linked to the DnaK-L5,6 and the WT DnaK was calculated. The average ratio is 0.505 ± 0.034 from six independent repeats.
Discussion
In this study, we have provided biochemical evidence for the discovery of a novel second peptide substrate–binding site in Hsp70s and showed that this novel binding site is essential for the chaperone activity of Hsp70s. Moreover, we have demonstrated that this novel binding site has different properties from the classic first binding site. This discovery challenges the well-established notion of a single peptide substrate–binding site on Hsp70s, on which all of the current paradigms of Hsp70 chaperone activities are based. We believe that this surprising discovery of a novel second peptide-binding site on Hsp70s will open a new direction for characterizing the folding mechanism of Hsp70s and, at the same time, provide a new way to target Hsp70s for designing novel therapeutics to treat various diseases such as cancers.
A key question is what the functional role of the P2 site is in the chaperone activity of Hsp70s. The preference for different hydrophobic residues may provide an answer. For any given protein, there are both aliphatic and aromatic residues. Both types of residues are hydrophobic and contribute to the hydrophobic cores of the folded native proteins. It was surprising that previous screens on the peptide substrate properties of Hsp70s have discovered that Hsp70s mainly prefer aliphatic residues, and not so much aromatic residues, especially for DnaK (39, 40, 42, 43). In all of the available structures of Hsp70s in complex with peptide substrates, the peptide substrates are rich in aliphatic residues and contain no aromatic amino acids in the central binding pocket (32, 41, 48, 55, 62–68). This could partially be due to the fact that aromatic residues are more hydrophobic than the aliphatic residues, and it is difficult to work with peptides containing aromatic amino acids. It had been a puzzle as to why Hsp70s do not like aromatic residues as much as the aliphatic residues. This study provides an explanation. Here we have shown that Hsp70s have two binding sites and prefer both types of the hydrophobic residues, with the P2 site preferring aromatic residues. However, the binding to the P2 site is unstable, characterized by fast kinetics. This could explain why previous screens using either peptide libraries bound to nitrocellulose membrane or phage display did not reveal this P2 site because these screens require relatively stable binding. These screens are ideal for characterizing the classic P1 site binding to aliphatic residues with stable complex formation in the absence of ATP. Various structural analyses on isolated SBD in complex with peptide substrates have provided support (41, 48, 55, 62–69). With different preferences for hydrophobic residues, these two sites could work together in efficiently binding to polypeptide substrates through binding to different segments of the same polypeptide substrate by the same molecule of Hsp70.
We propose that both binding sites cooperate in chaperoning protein folding and homeostasis (Fig. 8). Due to the fast kinetics for both binding sites in the presence of ATP, binding polypeptide substrate most likely occurs in the ATP-bound state of Hsp70s. This is consistent with the previously proposed models. With these two binding sites, a single Hsp70 molecule may bind two separate segments of a polypeptide substrate simultaneously. As shown previously, binding to the P1 site stimulates ATP hydrolysis. With two separate segments of the polypeptide bound, bound ATP is hydrolyzed, and Hsp70 is now in the ADP-bound state. Our biochemical results showed that the kinetics for the P2 site binding are faster than the P1 site, and the peptide binding to the P2 site is far less stable than the P1 site. Thus, the binding to the P2 site may be dynamic even in the ADP-bound state. Upon nucleotide exchange, Hsp70 returns to the ATP-bound state, polypeptide substrate is released due to the fast kinetics for both sites, and the chaperone cycle restarts.
Figure 8.
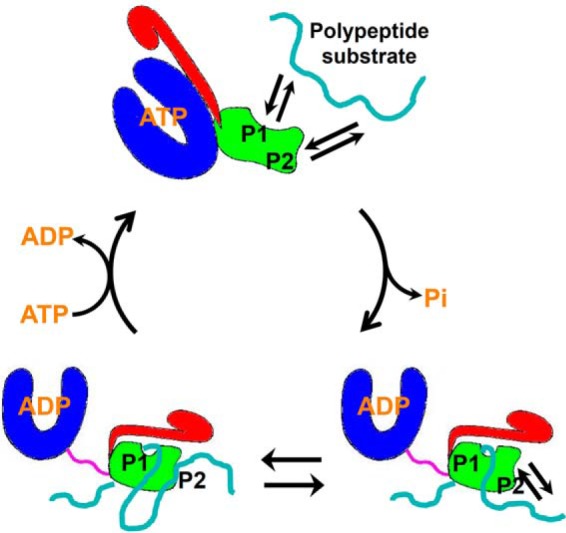
The model of the Hsp70 chaperone cycle with two essential peptide substrate–binding sites. The NBD, SBDβ, SBDα, and interdomain linker are colored in blue, green, red, and purple. The peptide substrates are highlighted in cyan. The two peptide substrate–binding sites are labeled as P1 and P2.
This hypothesis is consistent with a recent NMR study on the substrate interaction with Hsp40s, cochaperones for Hsp70s. This study surprisingly revealed that there are 1–3 binding sites on each monomer of Hsp40s for substrate binding, although all of these interactions with substrates are transient (71). Thus, as a functional dimer, Hsp40s have 2–6 binding sites for polypeptide substrates. More interestingly, the substrate preferences are different for the binding sites within the same Hsp40s. For ttHsp40, an Hsp40 from Thermus thermophiles, there are two binding sites: one site prefers aromatic residues, and the other prefers hydrophobic sequences containing no aromatic residues. These properties of ttHsp40 substrate binding are analogous to the two binding sites of Hsp70s reported in this study. Another interesting feature shared by the P2 site and the Hsp40 substrate interactions is the transient nature of substrate binding. The substrate binding to Hsp110s also shares this transient nature. This type of transient interaction may be important for chaperone activity in general. It is possible that both Hsp40s and Hsp110s deliver substrates to Hsp70s during efficient protein folding. The transient nature of substrate binding by Hsp40s, Hsp110s, and the P2 site of Hsp70s may be essential for efficient substrate transfer from Hsp40s and Hsp110s to Hsp70s. This could partially explain the efficient inhibition of chaperone activity by the TRP2 peptide in our refolding assay.
Our cross-linking and mutation analysis have provided support for the involvement of the L5,6 in this new P2 site. At the same time, L5,6 is also important for the classic P1 binding site through supporting the peptide-binding loop L3,4. L3,4 is part of the classic P1 binding site and crucial for binding the classic peptides like the NR peptide. Interestingly, the binding affinity for the TRP2 peptide is moderately increased for the L3,4 deletion mutation, suggesting that there may be some subtle involvement of this loop in the novel P2 site. Moreover, G400P, which is on L1,2, the other peptide-binding loop of the classic P1 site, has similar binding properties as the L3,4 deletion mutation. Thus, there seems to be some connection between the two peptide substrate–binding sites. It is possible that during ATP hydrolysis, the conformations and distance between these two sites may also change, and these changes could lead to an active remodeling of the polypeptide substrates. Consistent with this hypothesis, Hsp70s have been suggested to unfold polypeptide substrates, including two recent publications on Hsp70/Hsp40 unfolding a native protein P53 (81–83). More in-depth biochemical and structural analysis will help unravel the key question of the mechanistic role of the P2 site. Although our study suggests that L5,6 is involved in the P2 site, the P2 site must include regions beyond the L5,6 because deletion of L5,6 only partially compromises the binding of the TRP2 peptide. It is similar to the L1,2 situation for the P1 site: L1,2 is part of the P1 site, and deletion of L1,2 only partially compromises the binding of the NR peptide (Fig. 1C). Instead of a narrow cavity for the classic P1 binding site, the P2 site could be more like the Hsp40-substrate interaction with flat interfaces because both interactions are transient. Structural analysis using either NMR or crystallography will eventually reveal the structural basis of the P2 site. However, the poor solubility of the TRP2 peptide and transient nature of the TRP2 binding to the P2 site have presented challenges for such studies. This may be the same reason why the classic P1 peptide substrate–binding site for Hsp70s has been known for more than 2 decades, whereas we just started to reveal the P2 site. This study serves a starting point for understanding the unique nature and importance of this P2 site and molecular mechanism of substrate binding by Hsp70s.
It is interesting that both Hsp70s and Hsp110s bind the TRP2 peptide, and their binding properties share some common features such as fast kinetics and unstable complex formation. Because Hsp70s and Hsp110s are homologs, it is possible that the binding sites for the TRP2 peptide on these two classes of chaperones are located in an analogous position. Supporting this hypothesis, deletion of the L5,6 in Sse1, a yeast Hsp110, abolishes the in vivo chaperone activity, whereas deletion of either L1,2 or L4,5 has no detectable effect on the in vivo chaperone activity (data not shown), consistent with the involvement of the L5,6, but neither the L1,2 nor L4,5, in the P2 binding site of Hsp70s revealed by this study. Intriguingly, the sequence conservation in the L5,6 region is quite limited (Fig. S10), consistent with the overall low sequence conservation between Hsp70s and Hsp110s in the SBD region. Although the sequence conservation between Hsp70s and Hsp110s is low, this newly discovered P2 site may be functionally conserved between Hsp70s and Hsp110s. Consistent with this hypothesis, the sequence conservation of some of the newly discovered Hsp40 substrate-binding sites is also quite limited among Hsp40s.
Unlike Hsp70s, previous studies suggested that Hsp110s lack the hallmark activity in directly facilitating protein folding except for one study (84–86). However, a potent chaperone activity in preventing protein aggregation was reported for Hsp110s besides functioning as the major NEFs for the cytosolic Hsp70s (28, 29, 87, 88). Because Hsp110s showed little binding to the NR peptide and mutating the analogous residues for the classic P1 binding site did not affect the chaperone function appreciably (75), most likely Hsp110s do not have a functional classic P1 peptide-binding site. Losing the functional P1-binding site could contribute to the loss of the folding activity for Hsp110s. Further evolvement of the P2 site in Hsp110s during evolution may have contributed to the potent chaperone activity in preventing protein aggregation, which is even more efficient than Hsp70s. This may also explain the low sequence conservation in the SBD between Hsp70s and Hsp110s including the L5,6 region (Fig. S10), which is involved in the P2 site.
Experimental procedures
Protein expression and purification
All of the full-length DnaK proteins used in this study were expressed and purified as described before (56, 78). Briefly, all of the DnaK constructs were cloned into the dnak expression plasmid pBB46 with a His6 tag at the extreme C terminus and expressed in the deletion strain BB205 (89) with 200 μm isopropyl 1-thio-β-d-galactopyranoside at 30 °C for about 6 h. After breaking open the cells, the DnaK proteins were first purified on a HisTrap column with 2× PBS buffer. After dialysis to reduce the salt concentration, the DnaK proteins were further purified on a HiTrap Q column using buffers containing 25 mm Hepes-KOH, pH 7.5, and 1 mm DTT.
The DnaK truncation constructs were cloned into the pSMT3 vector and expressed in BL21(DE3) as Smt3 fusion proteins with a His6 tag at the C terminus of Smt3. The induction was carried out at 30 °C to reduce the expression of endogenous DnaK. The Smt3 fusion proteins were purified on a HisTrap column. After the Smt3 tag was cleaved by the Ulp1 protease, all of the DnaK truncation proteins were put on a second HisTrap column to remove the Smt3 tag. The NBD construct was further purified on a HiTrap Q column, and all the SBD constructs were further purified on a Superdex 75 column.
The DnaJ, GrpE, and human BiP proteins were expressed in BL21(DE3) and purified as described previously (55, 58, 78). Briefly, these proteins were expressed as Smt3 fusion proteins using the pSMT3 vector. After a nickel column, the Smt3 was cleaved off using Ulp1 and then removed by passing through a second nickel column. HiTrap Q column was used to further purify GrpE and human BiP, and DnaJ was further purified on a Superdex 200 16/600 column (GE Healthcare Life Sciences). The Ssa1 protein was expressed in the yeast Pichia pastoris and purified as described before (75, 90). The Ssa1-expressing Pichia strain was a generous gift from Dr. Johannes Buchner. After purification, all of the proteins were concentrated to >10 mg/ml, aliquoted, flash-frozen in liquid nitrogen, and stored in a −80 °C freezer.
Site-directed mutagenesis and growth tests
Mutagenesis in DnaK and growth tests were carried out as described previously (59, 78, 89). Briefly, all of the DnaK mutations were cloned into the aforementioned dnak expression plasmid pBB46 using the QuikChange Lightning site-directed mutagenesis kit (Stratagene) with mutagenic primers. Incorporation of the mutations was confirmed by sequencing. After these mutants were transformed into the dnak deletion strain BB205, fresh transformants were dropped onto an LB agar plate containing 50 μg/ml ampicillin, 25 μg/ml kanamycin, 25 μg/ml chloramphenicol, and 20 μm isopropyl 1-thio-β-d-galactopyranoside. Growth tests were done with one overnight incubation at 37 °C with control growth done at 30 °C. All assays were performed more than three times with two or more different transformations.
Fluorescence polarization assay for peptide substrate binding
All of the peptides in this study were purchased from NeoScientific or Ontores (at >95% purity). Binding affinity assays were carried out as described previously with some modifications (55, 56, 75, 78). Briefly, serial dilutions of Hsp70 proteins were prepared in buffer A (25 mm Hepes-KOH, pH 7.5, 100 mm KCl, 10 mm Mg(OAc)2, 10% glycerol, and 1 mm DTT) and incubated with the indicated fluorescein-labeled peptide at a final concentration of 10 nm. After the binding reached equilibrium, fluorescence polarization measurements were carried out on Beacon 2000 (Invitrogen), and dissociation constants (Kd) were calculated using Prism (GraphPad Software). For the binding kinetics measurements, right after the Hsp70 proteins were mixed with either the F-NR or F-TRP2 peptide, fluorescence polarization measurements were collected over time on the Beacon 2000. The readings for the peptides only were used as the zero time point. All biochemical assays, including the polarization assay, were carried out at least three times with more than two different protein purifications. All of the reported data were the averages of more than three repeats.
Single-turnover ATPase assay
The assay was performed as described previously with some modifications (55, 78, 79, 91). Briefly, DnaK protein (20 μg) was incubated with 25 μCi of [α-32P]ATP (NEG503H250UC, 3000 Ci/mmol; PerkinElmer Life Sciences) in buffer A in the presence of 20 μm unlabeled ATP for 2 min on ice. Using a spin column, the DnaK-ATP complex was quickly isolated from the free ATP at 4 °C. The ATPase assay was started by mixing an equal volume of the DnaK-ATP complex with the indicated peptides and then incubating at 20 °C. For assays done in 20% DMF, the peptides were diluted in buffer A containing 40% DMF. At the indicated time points, aliquots of the ATPase reactions were taken and stopped by adding 1 m formic acid, 0.5 m LiCl, and 250 μm ATP. ATP and ADP were separated using polyethyleneimine-cellulose thin-layer chromatography plates (Sigma-Aldrich), and the amount of each radioactive nucleotide was quantified after being visualized with a Typhoon phosphorimaging system (GE Healthcare). The rates of ATP hydrolysis (kcat) were calculated by fitting the data to the first-order rate equation using nonlinear regression (GraphPad Prism).
Luciferase-refolding assay
We performed the refolding assay using the DnaK and Ssa1 chaperone systems as described previously with modifications (56). First, the purified firefly luciferase (purchased from Promega) was diluted to a final concentration of 0.1 μm using buffer B (25 mm Hepes-KOH, pH 7.5, 100 mm KCl, and 10 mm Mg(OAc)2, 2 mm DTT, and 3 mm ATP) and heat-denatured at 42 °C in the presence of a 10 μm concentration of either DnaK or Ssa1. The refolding reaction was started by mixing this denatured luciferase with a reaction mixture containing either the DnaK system (3 μm DnaK, 0.67 μm DnaJ, and 0.33 μm GrpE) or the Ssa1 system (3 μm Ssa1, 3 μm Ydj1, and 1 μm Sse1) in buffer B. At the indicated time point, 2 μl of the refolding reaction was mixed with 50 μl of the luciferase substrate, and the luciferase activity was determined using a Berthold LB9507 luminometer. For the peptide inhibition, the indicated concentrations of peptides were included in the refolding reaction. For the titration curves with different peptide concentrations, the refolding activities were determined after a 30-min incubation, when the refolding activity in the absence of any peptide reaches the maximum.
UV cross-linking with the TRP2-Bpa peptide
To cross-link the TRP2-Bpa peptide to the DnaK proteins, the DnaK proteins were diluted to 14 μm in buffer A, and the TRP2-Bpa peptide was added at the indicated molar ratio. UV cross-linking was carried out on ice using a VWR UV cross-linker for 30 min. The wavelength of the UV light is 365 nm, and the distance between the UV lamps and the samples was kept at about 1–2 cm. For the isolation of the cross-linked complex between DnaK and the TRP2-Bpa peptide, a 1:10 molar ratio of DnaK and the peptide was used for cross-linking with UV. Then the reaction was concentrated and loaded onto a Superdex 200 10/300 column to remove the free TRP2-Bpa peptide. The fractions containing the complex were pooled, concentrated, and stored in a −80 °C freezer.
Disulfide cross-linking with Cu-phenanthroline
The oxidation reactions to form disulfide bonds using Cu-phenanthroline were carried out as described previously with modifications (92, 93). Before the cross-linking, all of the DnaK proteins were fully reduced by incubating with 5 mm DTT in buffer A for 2 h on ice. After DTT was quickly removed by a spin column pre-equilibrated with buffer A, each DnaK protein was diluted to 0.5 mg/ml using buffer C (25 mm Hepes-KOH, pH 7.5, 100 mm KCl, 10 mm Mg(OAc)2, and 10% glycerol). The TRP2-C or NR-C peptide was added, and the cross-linking by oxidation was started by adding freshly prepared CuSO4 and 1,10-phenanthroline to final concentrations of 50 and 100 μm, respectively. To reduce background nonspecific cross-linking, the ratio of DnaK over TRP2-C was 2:1 and DnaK over NR-C was 10:1. This ratio difference between the TRP2-C and NR-C peptide used in the oxidation is consistent with the ∼20-fold difference in their binding affinities for DnaK. The reaction was allowed to proceed on ice for 1 h and stopped by adding EDTA to a final concentration of 5 mm. SDS-PAGE was used to separate the complex from the peptide.
Tryptophan fluorescence assay
The tryptophan fluorescence assay on the DnaK proteins was performed as described before (56, 78). Briefly, the DnaK proteins were diluted to 1 μm in buffer A in the absence or the presence of 2 mm ATP. After incubating at room temperature for 2 min, emission spectra were collected at room temperature from 315 to 400 nm with an excitation wavelength at 295 nm.
Author contributions
H. L., H. Z., E. B. S., Qingdai Liu, X. T., Y. Y., C. L., L. Z., and Qinglian Liu data curation; H. L. and Qinglian Liu validation; H. L., H. Z., E. B. S., Qingdai Liu, X. T., Y. Y., C. L., L. Z., and Qinglian Liu investigation; H. L. and Qinglian Liu visualization; H. L., H. Z., E. B. S., and Qinglian Liu methodology; H. L., Qingdai Liu, L. Z., and Qinglian Liu writing-review and editing; Qinglian Liu writing-original draft; H. L., H. Z., E. B. S., Qingdai Liu, X. T., Y. Y., C. L., L. Z., and Qinglian Liu formal analysis; L. Z. and Qinglian Liu conceptualization; Qinglian Liu resources; Qinglian Liu supervision; Qinglian Liu funding acquisition; Qinglian Liu project administration.
Supplementary Material
Acknowledgments
We thank Dr. Jiao Yang, Jiayue Su, Baoxiu Zhang, SungJi Jung, Chenxuan Zhou, Yan Zhu, and Dr. Vanessa Ledesma for technical support and discussion; Dr. John Hackett for discussion; and Dr. Cancan Sun, Ce Liang, and Crist Cuffee for critically reading the manuscript. The Massey Cancer Center Proteomics Resource is supported by NCI, National Institutes of Health, Cancer Center Support Grant 5O30CA16059.
This work was supported by National Institutes of Health Grants R01GM098592 and R21AI140006 (to Q. L.), a Blick Scholar Award from VCU (to Q. L.), and American Heart Association Grant 17GRNT33660506 (to Q. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S10.
- Hsp70
- heat shock protein of 70 kDa
- NEF
- nucleotide-exchange factor
- NEI
- nucleotide-exchange inhibitor
- NBD
- nucleotide-binding domain
- SBD
- substrate-binding domain
- DMF
- N,N-dimethylformamide
- Bpa
- p-benzoyl-l-phenylalanine.
References
- 1. Mayer M. P., and Bukau B. (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62, 670–684 10.1007/s00018-004-4464-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hartl F. U., and Hayer-Hartl M. (2009) Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16, 574–581 10.1038/nsmb.1591 [DOI] [PubMed] [Google Scholar]
- 3. Bukau B., Weissman J., and Horwich A. (2006) Molecular chaperones and protein quality control. Cell 125, 443–451 10.1016/j.cell.2006.04.014 [DOI] [PubMed] [Google Scholar]
- 4. Bukau B., Deuerling E., Pfund C., and Craig E. A. (2000) Getting newly synthesized proteins into shape. Cell 101, 119–122 10.1016/S0092-8674(00)80806-5 [DOI] [PubMed] [Google Scholar]
- 5. Bukau B., and Horwich A. L. (1998) The Hsp70 and Hsp60 chaperone machines. Cell 92, 351–366 10.1016/S0092-8674(00)80928-9 [DOI] [PubMed] [Google Scholar]
- 6. Young J. C. (2010) Mechanisms of the Hsp70 chaperone system. Biochem. Cell Biol. 88, 291–300 10.1139/O09-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu Q., and Craig E. A. (2016) Molecular biology: mature proteins braced by a chaperone. Nature 539, 361–362 10.1038/nature20470 [DOI] [PubMed] [Google Scholar]
- 8. Mayer M. P., and Gierasch L. M. (2019) Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 294, 2085–2097 10.1074/jbc.REV118.002810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hendershot L. M. (2004) The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 71, 289–297 [PubMed] [Google Scholar]
- 10. Fernández-Fernández M. R., Gragera M., Ochoa-Ibarrola L., Quintana-Gallardo L., and Valpuesta J. M. (2017) Hsp70: a master regulator in protein degradation. FEBS Lett. 591, 2648–2660 10.1002/1873-3468.12751 [DOI] [PubMed] [Google Scholar]
- 11. Fernandez-Fernandez M. R., and Valpuesta J. M. (2018) Hsp70 chaperone: a master player in protein homeostasis. F1000Res 7, 10.12688/f1000research.15528.1 10.12688/f1000research.15528.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jolly C., and Morimoto R. I. (2000) Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 92, 1564–1572 10.1093/jnci/92.19.1564 [DOI] [PubMed] [Google Scholar]
- 13. Evans C. G., Chang L., and Gestwicki J. E. (2010) Heat shock protein 70 (hsp70) as an emerging drug target. J. Med. Chem. 53, 4585–4602 10.1021/jm100054f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muchowski P. J., and Wacker J. L. (2005) Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 6, 11–22 10.1038/nrn1587 [DOI] [PubMed] [Google Scholar]
- 15. Morimoto R. I. (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22, 1427–1438 10.1101/gad.1657108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Balch W. E., Morimoto R. I., Dillin A., and Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 10.1126/science.1141448 [DOI] [PubMed] [Google Scholar]
- 17. Zuiderweg E. R., Bertelsen E. B., Rousaki A., Mayer M. P., Gestwicki J. E., and Ahmad A. (2013) Allostery in the Hsp70 chaperone proteins. Top. Curr. Chem. 328, 99–153 10.1007/128_2012_323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen B., Retzlaff M., Roos T., and Frydman J. (2011) Cellular strategies of protein quality control. Cold Spring Harb. Perspect. Biol. 3, a004374 10.1101/cshperspect.a004374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brodsky J. L., and Chiosis G. (2006) Hsp70 molecular chaperones: emerging roles in human disease and identification of small molecule modulators. Curr. Topics Med. Chem. 6, 1215–1225 10.2174/156802606777811997 [DOI] [PubMed] [Google Scholar]
- 20. Schröder H., Langer T., Hartl F. U., and Bukau B. (1993) DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 12, 4137–4144 10.1002/j.1460-2075.1993.tb06097.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu Q., Liang C., and Zhou L. (2019) Structural and functional analysis of the Hsp70/Hsp40 chaperone system. Protein Sci. 10.1002/pro.3725 10.1002/pro.3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Craig E. A., Huang P., Aron R., and Andrew A. (2006) The diverse roles of J-proteins, the obligate Hsp70 co-chaperone. Rev. Physiol. Biochem. Pharmacol. 156, 1–21 10.1007/s10254-005-0001-0 [DOI] [PubMed] [Google Scholar]
- 23. Fan C. Y., Lee S., and Cyr D. M. (2003) Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones 8, 309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J., Qian X., and Sha B. (2009) Heat shock protein 40: structural studies and their functional implications. Protein Pept. Lett. 16, 606–612 10.2174/092986609788490159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kampinga H. H., Andreasson C., Barducci A., Cheetham M. E., Cyr D., Emanuelsson C., Genevaux P., Gestwicki J. E., Goloubinoff P., Huerta-Cepas J., Kirstein J., Liberek K., Mayer M. P., Nagata K., Nillegoda N. B., et al. (2019) Function, evolution, and structure of J-domain proteins. Cell Stress Chaperones 24, 7–15 10.1007/s12192-018-0948-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alderson T. R., Kim J. H., and Markley J. L. (2016) Dynamical structures of Hsp70 and Hsp70-Hsp40 complexes. Structure 24, 1014–1030 10.1016/j.str.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kampinga H. H., and Craig E. A. (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592 10.1038/nrm2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hendrickson W. A., and Liu Q. (2008) Exchange we can believe in. Structure 16, 1153–1155 10.1016/j.str.2008.07.002 [DOI] [PubMed] [Google Scholar]
- 29. Bracher A., and Verghese J. (2015) The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2, 10 10.3389/fmolb.2015.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morán Luengo T., Kityk R., Mayer M. P., and Rüdiger S. G. D. (2018) Hsp90 breaks the deadlock of the Hsp70 chaperone system. Mol. Cell 70, 545–552.e9 10.1016/j.molcel.2018.03.028 [DOI] [PubMed] [Google Scholar]
- 31. Genest O., Hoskins J. R., Camberg J. L., Doyle S. M., and Wickner S. (2011) Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling. Proc. Natl. Acad. Sci. U.S.A. 108, 8206–8211 10.1073/pnas.1104703108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan Y., Rato C., Rohland L., Preissler S., and Ron D. (2019) MANF antagonizes nucleotide exchange by the endoplasmic reticulum chaperone BiP. Nat. Commun. 10, 541 10.1038/s41467-019-08450-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li Z., Hartl F. U., and Bracher A. (2013) Structure and function of Hip, an attenuator of the Hsp70 chaperone cycle. Nat. Struct. Mol. Biol. 20, 929–935 10.1038/nsmb.2608 [DOI] [PubMed] [Google Scholar]
- 34. Zuiderweg E. R., Hightower L. E., and Gestwicki J. E. (2017) The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones 22, 173–189 10.1007/s12192-017-0776-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hartl F. U., Bracher A., and Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- 36. Mayer M. P. (2010) Gymnastics of molecular chaperones. Mol. Cell 39, 321–331 10.1016/j.molcel.2010.07.012 [DOI] [PubMed] [Google Scholar]
- 37. Flynn G. C., Chappell T. G., and Rothman J. E. (1989) Peptide binding and release by proteins implicated as catalysts of protein assembly. Science 245, 385–390 10.1126/science.2756425 [DOI] [PubMed] [Google Scholar]
- 38. Flynn G. C., Pohl J., Flocco M. T., and Rothman J. E. (1991) Peptide-binding specificity of the molecular chaperone BiP. Nature 353, 726–730 10.1038/353726a0 [DOI] [PubMed] [Google Scholar]
- 39. Rüdiger S., Buchberger A., and Bukau B. (1997) Interaction of Hsp70 chaperones with substrates. Nat. Struct. Biol. 4, 342–349 10.1038/nsb0597-342 [DOI] [PubMed] [Google Scholar]
- 40. Rüdiger S., Germeroth L., Schneider-Mergener J., and Bukau B. (1997) Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 16, 1501–1507 10.1093/emboj/16.7.1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu X., Zhao X., Burkholder W. F., Gragerov A., Ogata C. M., Gottesman M. E., and Hendrickson W. A. (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272, 1606–1614 10.1126/science.272.5268.1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gragerov A., Zeng L., Zhao X., Burkholder W., and Gottesman M. E. (1994) Specificity of DnaK-peptide binding. J. Mol. Biol. 235, 848–854 10.1006/jmbi.1994.1043 [DOI] [PubMed] [Google Scholar]
- 43. Blond-Elguindi S., Cwirla S. E., Dower W. J., Lipshutz R. J., Sprang S. R., Sambrook J. F., and Gething M. J. (1993) Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell 75, 717–728 10.1016/0092-8674(93)90492-9 [DOI] [PubMed] [Google Scholar]
- 44. Buchberger A., Theyssen H., Schröder H., McCarty J. S., Virgallita G., Milkereit P., Reinstein J., and Bukau B. (1995) Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J. Biol. Chem. 270, 16903–16910 10.1074/jbc.270.28.16903 [DOI] [PubMed] [Google Scholar]
- 45. Bertelsen E. B., Chang L., Gestwicki J. E., and Zuiderweg E. R. (2009) Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. U.S.A. 106, 8471–8476 10.1073/pnas.0903503106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Swain J. F., Dinler G., Sivendran R., Montgomery D. L., Stotz M., and Gierasch L. M. (2007) Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell 26, 27–39 10.1016/j.molcel.2007.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Swain J. F., and Gierasch L. M. (2006) The changing landscape of protein allostery. Curr. Opin. Struct. Biol. 16, 102–108 10.1016/j.sbi.2006.01.003 [DOI] [PubMed] [Google Scholar]
- 48. Chang Y. W., Sun Y. J., Wang C., and Hsiao C. D. (2008) Crystal structures of the 70-kDa heat shock proteins in domain disjoining conformation. J. Biol. Chem. 283, 15502–15511 10.1074/jbc.M708992200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schmid D., Baici A., Gehring H., and Christen P. (1994) Kinetics of molecular chaperone action. Science 263, 971–973 10.1126/science.8310296 [DOI] [PubMed] [Google Scholar]
- 50. Mapa K., Sikor M., Kudryavtsev V., Waegemann K., Kalinin S., Seidel C. A., Neupert W., Lamb D. C., and Mokranjac D. (2010) The conformational dynamics of the mitochondrial Hsp70 chaperone. Mol. Cell 38, 89–100 10.1016/j.molcel.2010.03.010 [DOI] [PubMed] [Google Scholar]
- 51. Rist W., Graf C., Bukau B., and Mayer M. P. (2006) Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J. Biol. Chem. 281, 16493–16501 10.1074/jbc.M600847200 [DOI] [PubMed] [Google Scholar]
- 52. Zhuravleva A., Clerico E. M., and Gierasch L. M. (2012) An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 151, 1296–1307 10.1016/j.cell.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schlecht R., Erbse A. H., Bukau B., and Mayer M. P. (2011) Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat. Struct. Mol. Biol. 18, 345–351 10.1038/nsmb.2006 [DOI] [PubMed] [Google Scholar]
- 54. Marcinowski M., Höller M., Feige M. J., Baerend D., Lamb D. C., and Buchner J. (2011) Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat. Struct. Mol. Biol. 18, 150–158 10.1038/nsmb.1970 [DOI] [PubMed] [Google Scholar]
- 55. Yang J., Nune M., Zong Y., Zhou L., and Liu Q. (2015) Close and allosteric opening of the polypeptide-binding site in a human Hsp70 chaperone BiP. Structure 23, 2191–2203 10.1016/j.str.2015.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Qi R., Sarbeng E. B., Liu Q., Le K. Q., Xu X., Xu H., Yang J., Wong J. L., Vorvis C., Hendrickson W. A., Zhou L., and Liu Q. (2013) Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat. Struct. Mol. Biol. 20, 900–907 10.1038/nsmb.2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kityk R., Kopp J., Sinning I., and Mayer M. P. (2012) Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol. Cell 48, 863–874 10.1016/j.molcel.2012.09.023 [DOI] [PubMed] [Google Scholar]
- 58. Yang J., Zong Y., Su J., Li H., Zhu H., Columbus L., Zhou L., and Liu Q. (2017) Conformation transitions of the polypeptide-binding pocket support an active substrate release from Hsp70s. Nat. Commun. 8, 1201 10.1038/s41467-017-01310-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu Q., and Hendrickson W. A. (2007) Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell 131, 106–120 10.1016/j.cell.2007.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gumiero A., Conz C., Gesé G. V., Zhang Y., Weyer F. A., Lapouge K., Kappes J., von Plehwe U., Schermann G., Fitzke E., Wölfle T., Fischer T., Rospert S., and Sinning I. (2016) Interaction of the cotranslational Hsp70 Ssb with ribosomal proteins and rRNA depends on its lid domain. Nat. Commun. 7, 13563 10.1038/ncomms13563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Flaherty K. M., DeLuca-Flaherty C., and McKay D. B. (1990) Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature 346, 623–628 10.1038/346623a0 [DOI] [PubMed] [Google Scholar]
- 62. Cupp-Vickery J. R., Peterson J. C., Ta D. T., and Vickery L. E. (2004) Crystal structure of the molecular chaperone HscA substrate binding domain complexed with the IscU recognition peptide ELPPVKIHC. J. Mol. Biol. 342, 1265–1278 10.1016/j.jmb.2004.07.025 [DOI] [PubMed] [Google Scholar]
- 63. Jiang J., Prasad K., Lafer E. M., and Sousa R. (2005) Structural basis of interdomain communication in the Hsc70 chaperone. Mol. Cell 20, 513–524 10.1016/j.molcel.2005.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Morshauser R. C., Hu W., Wang H., Pang Y., Flynn G. C., and Zuiderweg E. R. (1999) High-resolution solution structure of the 18 kDa substrate-binding domain of the mammalian chaperone protein Hsc70. J. Mol. Biol. 289, 1387–1403 10.1006/jmbi.1999.2776 [DOI] [PubMed] [Google Scholar]
- 65. Pellecchia M., Montgomery D. L., Stevens S. Y., Vander Kooi C. W., Feng H. P., Gierasch L. M., and Zuiderweg E. R. (2000) Structural insights into substrate binding by the molecular chaperone DnaK. Nat. Struct. Biol. 7, 298–303 10.1038/74062 [DOI] [PubMed] [Google Scholar]
- 66. Stevens S. Y., Cai S., Pellecchia M., and Zuiderweg E. R. (2003) The solution structure of the bacterial HSP70 chaperone protein domain DnaK(393–507) in complex with the peptide NRLLLTG. Protein Sci. 12, 2588–2596 10.1110/ps.03269103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang H., Kurochkin A. V., Pang Y., Hu W., Flynn G. C., and Zuiderweg E. R. (1998) NMR solution structure of the 21 kDa chaperone protein DnaK substrate binding domain: a preview of chaperone-protein interaction. Biochemistry 37, 7929–7940 10.1021/bi9800855 [DOI] [PubMed] [Google Scholar]
- 68. Liebscher M., and Roujeinikova A. (2009) Allosteric coupling between the lid and interdomain linker in DnaK revealed by inhibitor binding studies. J. Bacteriol. 191, 1456–1462 10.1128/JB.01131-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang P., Leu J. I., Murphy M. E., George D. L., and Marmorstein R. (2014) Crystal structure of the stress-inducible human heat shock protein 70 substrate-binding domain in complex with peptide substrate. PLoS One 9, e103518 10.1371/journal.pone.0103518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Clerico E. M., Tilitsky J. M., Meng W., and Gierasch L. M. (2015) How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J. Mol. Biol. 427, 1575–1588 10.1016/j.jmb.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jiang Y., Rossi P., and Kalodimos C. G. (2019) Structural basis for client recognition and activity of Hsp40 chaperones. Science 365, 1313–1319 10.1126/science.aax1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Balchin D., Hayer-Hartl M., and Hartl F. U. (2016) In vivo aspects of protein folding and quality control. Science 353, aac4354 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- 73. Shaner L., and Morano K. A. (2007) All in the family: atypical Hsp70 chaperones are conserved modulators of Hsp70 activity. Cell Stress Chaperones 12, 1–8 10.1379/CSC-245R.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Easton D. P., Kaneko Y., and Subjeck J. R. (2000) The hsp110 and Grp1 70 stress proteins: newly recognized relatives of the Hsp70s. Cell Stress Chaperones 5, 276–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xu X., Sarbeng E. B., Vorvis C., Kumar D. P., Zhou L., and Liu Q. (2012) Unique peptide substrate binding properties of 110-kDa heat-shock protein (Hsp110) determine its distinct chaperone activity. J. Biol. Chem. 287, 5661–5672 10.1074/jbc.M111.275057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lindquist S., and Craig E. A. (1988) The heat-shock proteins. Annu. Rev. Genet. 22, 631–677 10.1146/annurev.ge.22.120188.003215 [DOI] [PubMed] [Google Scholar]
- 77. Pobre K. F. R., Poet G. J., and Hendershot L. M. (2019) The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: getting by with a little help from ERdj friends. J. Biol. Chem. 294, 2098–2108 10.1074/jbc.REV118.002804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kumar D. P., Vorvis C., Sarbeng E. B., Cabra Ledesma V. C., Willis J. E., and Liu Q. (2011) The four hydrophobic residues on the Hsp70 inter-domain linker have two distinct roles. J. Mol. Biol. 411, 1099–1113 10.1016/j.jmb.2011.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Davis J. E., Voisine C., and Craig E. A. (1999) Intragenic suppressors of Hsp70 mutants: interplay between the ATPase- and peptide-binding domains. Proc. Natl. Acad. Sci. U.S.A. 96, 9269–9276 10.1073/pnas.96.16.9269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Goeckeler J. L., Petruso A. P., Aguirre J., Clement C. C., Chiosis G., and Brodsky J. L. (2008) The yeast Hsp110, Sse1p, exhibits high-affinity peptide binding. FEBS Lett. 582, 2393–2396 10.1016/j.febslet.2008.05.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Boysen M., Kityk R., and Mayer M. P. (2019) Hsp70- and Hsp90-mediated regulation of the conformation of p53 DNA binding domain and p53 cancer variants. Mol. Cell 74, 831–843.e4 10.1016/j.molcel.2019.03.032 [DOI] [PubMed] [Google Scholar]
- 82. Dahiya V., Agam G., Lawatscheck J., Rutz D. A., Lamb D. C., and Buchner J. (2019) Coordinated conformational processing of the tumor suppressor protein p53 by the Hsp70 and Hsp90 chaperone machineries. Mol. Cell 74, 816–830.e7 10.1016/j.molcel.2019.03.026 [DOI] [PubMed] [Google Scholar]
- 83. Sharma S. K., De los Rios P., Christen P., Lustig A., and Goloubinoff P. (2010) The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat. Chem. Biol. 6, 914–920 10.1038/nchembio.455 [DOI] [PubMed] [Google Scholar]
- 84. Oh H. J., Chen X., and Subjeck J. R. (1997) Hsp110 protects heat-denatured proteins and confers cellular thermoresistance. J. Biol. Chem. 272, 31636–31640 10.1074/jbc.272.50.31636 [DOI] [PubMed] [Google Scholar]
- 85. Goeckeler J. L., Stephens A., Lee P., Caplan A. J., and Brodsky J. L. (2002) Overexpression of yeast Hsp110 homolog Sse1p suppresses ydj1–151 thermosensitivity and restores Hsp90-dependent activity. Mol. Biol. Cell 13, 2760–2770 10.1091/mbc.02-04-0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mattoo R. U., Sharma S. K., Priya S., Finka A., and Goloubinoff P. (2013) Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J. Biol. Chem. 288, 21399–21411 10.1074/jbc.M113.479253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Raviol H., Sadlish H., Rodriguez F., Mayer M. P., and Bukau B. (2006) Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 25, 2510–2518 10.1038/sj.emboj.7601139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dragovic Z., Broadley S. A., Shomura Y., Bracher A., and Hartl F. U. (2006) Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 25, 2519–2528 10.1038/sj.emboj.7601138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Burkholder W. F., Zhao X., Zhu X., Hendrickson W. A., Gragerov A., and Gottesman M. E. (1996) Mutations in the C-terminal fragment of DnaK affecting peptide binding. Proc. Natl. Acad. Sci. U.S.A. 93, 10632–10637 10.1073/pnas.93.20.10632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wegele H., Haslbeck M., and Buchner J. (2003) Recombinant expression and purification of Ssa1p (Hsp70) from Saccharomyces cerevisiae using Pichia pastoris. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 786, 109–115 10.1016/S1570-0232(02)00724-9 [DOI] [PubMed] [Google Scholar]
- 91. Liu Q., Krzewska J., Liberek K., and Craig E. A. (2001) Mitochondrial Hsp70 Ssc1: role in protein folding. J. Biol. Chem. 276, 6112–6118 10.1074/jbc.M009519200 [DOI] [PubMed] [Google Scholar]
- 92. Sarbeng E. B., Liu Q., Tian X., Yang J., Li H., Wong J. L., Zhou L., and Liu Q. (2015) A functional DnaK dimer is essential for the efficient interaction with Hsp40 heat shock protein. J. Biol. Chem. 290, 8849–8862 10.1074/jbc.M114.596288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu Q., Li H., Yang Y., Tian X., Su J., Zhou L., and Liu Q. (2017) A disulfide-bonded DnaK dimer is maintained in an ATP-bound state. Cell Stress Chaperones 22, 201–212 10.1007/s12192-016-0752-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.