

Abstract
A series of 2-alkyl/alkenyl pyridine C-region derivatives of 2-(3-fluoro-4-methylsulfonylaminophenyl)propanamides were investigated as hTRPV1 antagonists. Multiple compounds showed excellent and stereospecific TRPV1 antagonism with better potency than previous lead 2. Among them, compound 15f demonstrated a strong analgesic profile in a rat neuropathic pain model and blocked capsaicin-induced hypothermia in a dose-dependent manner. Docking analysis of (S)-15f with our hTRPV1 homology model provided insight into its specific binding mode.
Keywords: Vanilloid receptor 1, TRPV1 antagonist, Capsaicin, Resiniferatoxin, Molecular modeling
The transient receptor potential V1 (TRPV1) receptor is a non-selective cation channel with high Ca2+ permeability which functions as a molecular integrator of nociceptive stimuli predominantly expressed on sensory neurons.1 The receptor is activated by a diverse range of stimuli including protons,2 noxious heat,3 endogenous mediators4,5 and natural products such as capsaicin (CAP)6 and resiniferatoxin (RTX).7 Since the increase in intracellular Ca2+ upon TRPV1 activation causes excitation of the primary sensory neurons and the consequent central perception of pain, the blocking of TRPV1 provides a promising strategy for the development of novel analgesics, particularly for neuropathic pain.8 In consequence, an intense effort has been made to identify potent and selective TRPV1 antagonists, with numerous reviews summarizing their therapeutic development and documenting their clinical development.9–15
Recently, we reported that compound 2, the lead structure from series 1, showed highly potent antagonism to multiple TRPV1 activators including capsaicin, pH, heat (45 °C) and NADA (for example, Ki(CAP) = 0.2 nM, IC50(pH) = 6.3 nM) (Fig. 1). In addition, compound 2 demonstrated strong analgesic activity in the rat neuropathic pain model with almost no off-target effects and it blocked capsaicin-induced hypothermia, consistent with its in vitro mechanism of action.16 The modeling analysis of 2 using our hTRPV1 homology model suggested that its high potency could be attributed to a new hydrophobic interaction provided by the 4-methylpiperidine moiety in addition to that by the 6-trifluoromethyl group in the C-region. Consistent with the modeling, the SAR analysis with other 2-substituents in the pyridine C-region, 2-oxy17 and 2-thio18 derivatives, indicated that a hydrophobic interaction by the 2-substituents with the receptor was critical for its potent antagonism.
Figure 1.
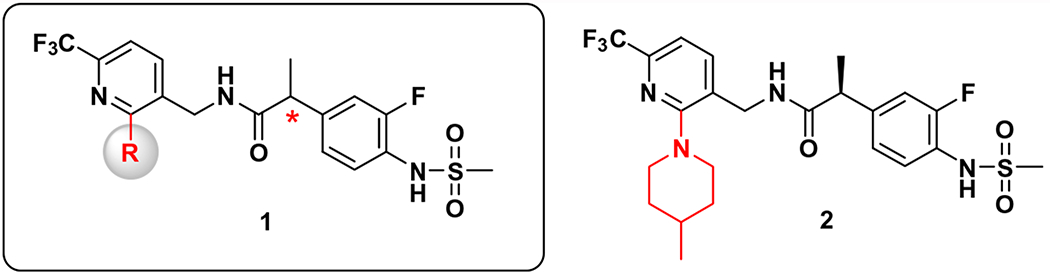
Structural series of TRPV1 antagonists (1) and lead antagonist (2).
In order to further optimize the 2-substituent in the N-(6-trifluoromethyl-pyridin-3-ylmethyl) C-region, here we have investigated the structure activity relationships of 2-alkyl type derivatives as hTRPV1 antagonists. With a selected potent antagonist in the series, we have further characterized its analgesic activity and its inhibition of capsaicin-induced hypothermia in animal models and we have performed molecular modeling with our hTRPV1 homology model.
The key intermediates of the C-region, 2-chloropyridine 3 and 2-bromopyridine 4, were prepared from the corresponding pyridone using POCl3 or PBr3, respectively, using the procedure in our previous report.16 The nitrile reduction of 3 and 4 followed by coupling with propanoic acid 719 provided the final compounds 8 and 9. The 2-methyl derivative 12 was synthesized starting from commercially available nicotinic acid 10 in conventional 4 steps (Scheme 1).
Scheme 1.
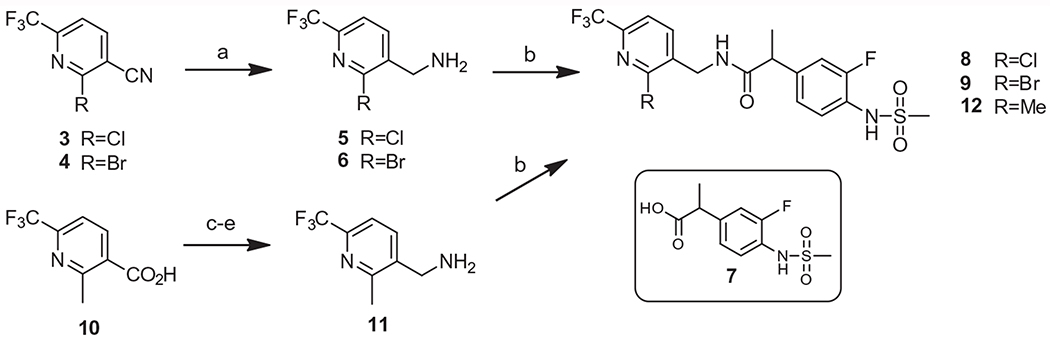
Synthesis of 2-halo and 2-methyl derivatives. Reagents and conditions: (a) BH3–SMe2 in THF, 80–90%; (b) Compound 7, EDC, HOBt, TEA, CH3CN, 70–80%; (c) LiAlH4, THF, 75%; (d) DPPA, NEt3, toluene, 72%; (e) PPh3, H2O, 85%.
A library of alkynyl, alkenyl and alkyl groups were reacted with 2-chloropyridine 3 to afford 2-substituted pyridines 13 using a palladium-catalyzed coupling reaction (Suzuki reaction for 13a and 13f–13n and Sonogashira reaction for 13b–13e). Nitriles 13 were either fully reduced to yield the corresponding primary amines 14a–14n with 2-alkyl substituents or chemoselectively reduced to give the amines 14o–14u with 2-alkenyl substituents. The amines of 14 were coupled with racemic or chiral propanoic acid 7 to provide the corresponding final compounds 15a–15u, (Scheme 2). Alternatively, the 2-alkenyl analogues (16a–e) were synthesized directly from the 2-chloro substituted compound 8 by Suzuki coupling (Scheme 3).
Scheme 2.
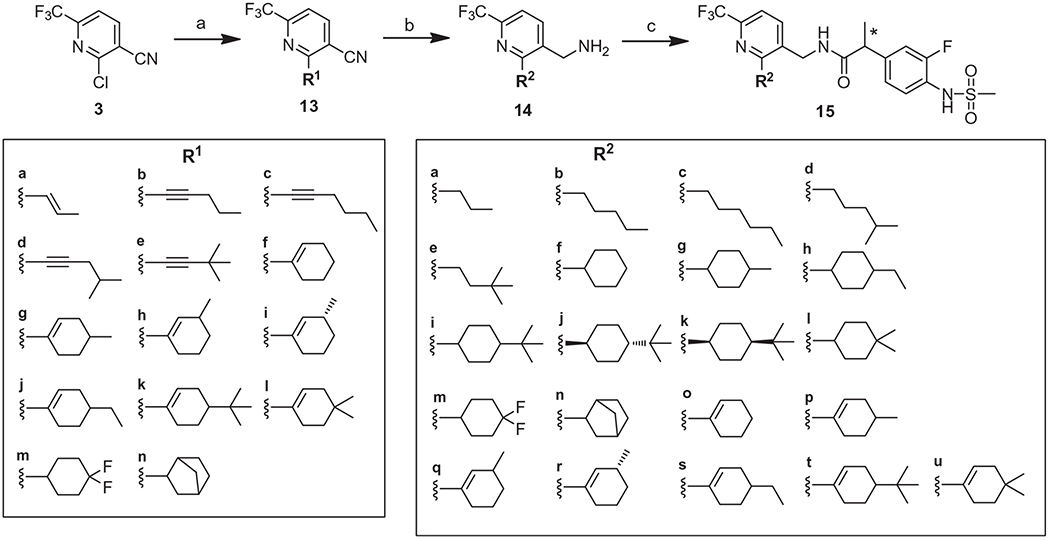
Synthesis of 2-alkyl, 2-cycloakyl, and 2-cycloalkenyl derivatives. Reagents and conditions: (a) 13a: CH3CH═CH–B(OH)2, Pd(PPh3)4, Na2CO3, toluene/1,4-dioxane, reflux, overnight, 63%, 13b–13e: Pd(PPh3)4, CuI, NEt3 or DIPEA, toluene or NMP, 90 °C, overnight, 50–75%, 13f–13n: R-B(OH)2, Pd(PPh3)4, Na2CO3, toluene/EtOH, 45–80%; (b) 14a–14n: H2, Pd-C, c-HCl, MeOH, RT, overnight, 40–92%, 14o–14u: Raney Ni, NH3 in MeOH, 50–75%; (c) Compound 7, EDC, HOBt, TEA (or DIEA), CH3CN (or DMF), 70–90%.
Scheme 3.
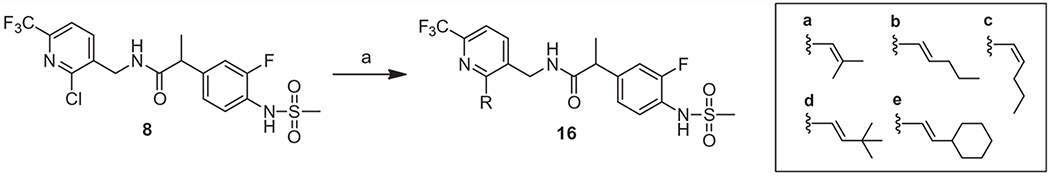
Synthesis of 2-alkenyl derivatives. Reagents and conditions: (a) R-B(OH)2, Pd(PPh3)4, Na2CO3, toluene/EtOH, 100 °C, 1 h, microwave, 62–72%.
The synthesized TRPV1 ligands were evaluated in vitro for antagonism as measured by inhibition of activation by capsaicin (100 nM). The assays were conducted using a fluorometric imaging plate reader (FLIPR) with human TRPV1 heterologously expressed in Chinese hamster ovary (CHO) cells.16 The results are summarized in Tables 1 and 2, together with the potencies of the previously reported racemate of 2 (Ki(CAP) = 0.3 nM) as a reference.16
Table 1.
In vitro hTRPV1 antagonistic activities for acyclic 2-alkyl and 2-alkenyl derivatives
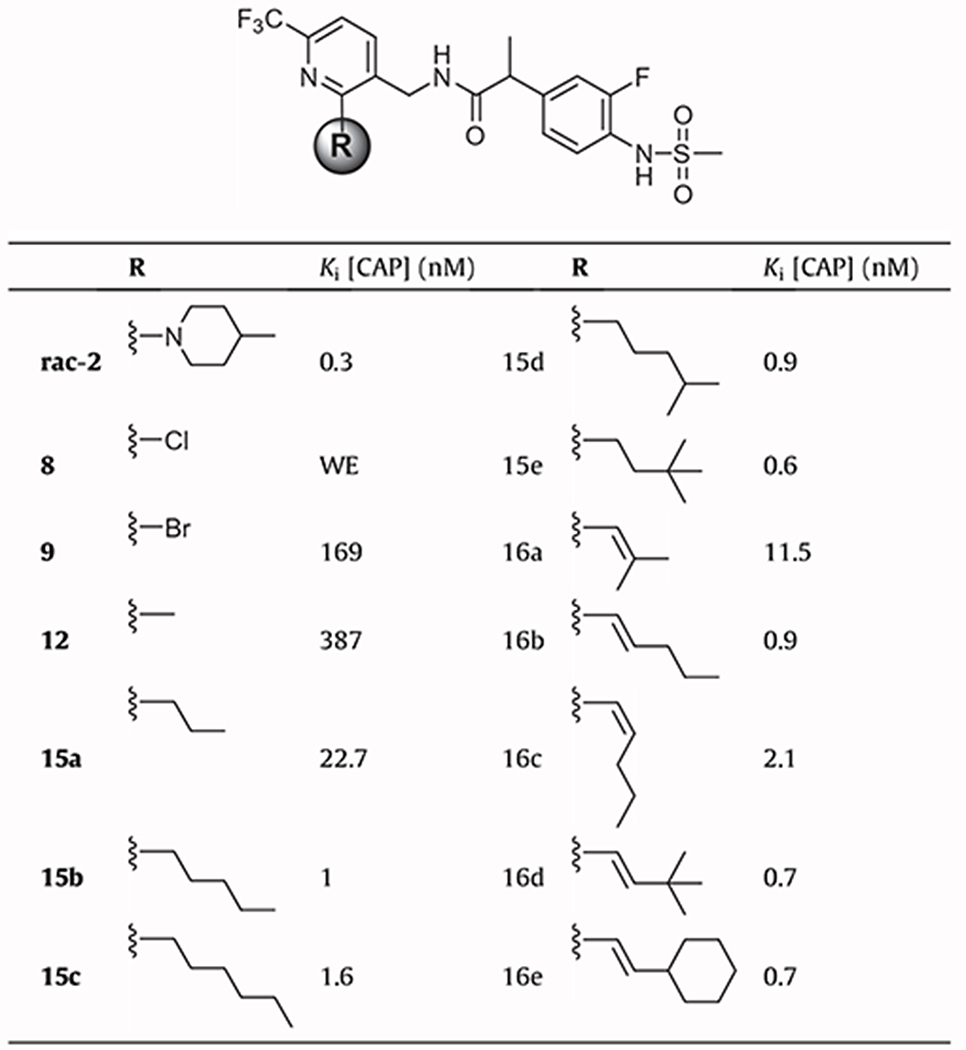 |
Table 2.
In vitro hTRPV1 antagonistic activities for 2-cyclohexyl derivatives
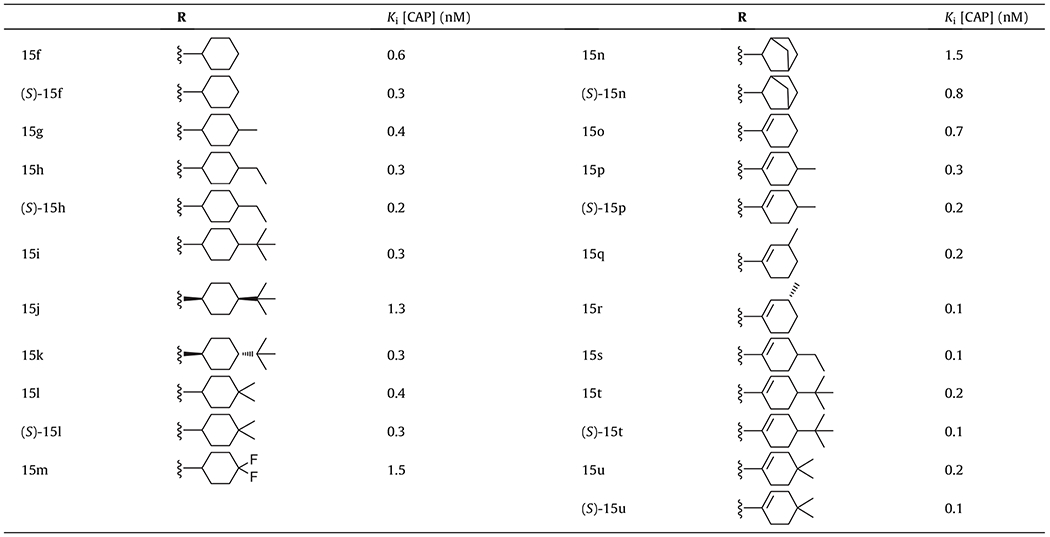 |
First, we examined the 2-chloro (8) and 2-bromo (9) derivatives which were the synthetic precursors for the synthesis of the 2-alkenyl derivatives. They were found to be weak antagonists, probably due to low lipophilicity or insufficient size to make hydrophobic interactions with the receptor.
To investigate the SAR for 2-alkyl/alkenyl derivatives of the pyridine C-region, we began by examining the effect of linear alkyl chains on the antagonism of capsaicin response. Antagonistic activity increased with the number of carbons in the chain up to 5 carbons as seen with 12, 15a, 15b. However, as seen with 15c, the antagonism decreased after that, indicating that the number of carbons in this series which appeared to be optimal for antagonism was five. A similar pattern was observed in our previous SAR analysis with the series of the corresponding linear 2-alkyloxy17 and 2-alkylthio18 derivatives, for which once again the 2-butyloxy and 2-butylthio derivatives, corresponding to 5 carbons, were optimal with low lipophilicity and potent antagonism. The branched derivatives 15d and 15e showed better antagonism than the corresponding straight derivative 15c by ca. 2-fold, suggesting that the hydrophobic pocket for the 2-alkyl chain could be better filled by the branched group.
The SAR of 2-alkenyl derivatives was also investigated. The isobutenyl derivative 16a exhibited moderate antagonistic activity intermediate between the activities of 15a (3 carbons) and 15b (5 carbons). Whereas the trans-pentenyl derivative 16b showed potency comparable to the corresponding saturated derivative 15b, the cis-pentenyl derivative 16c was 2-fold less potent than 15b. Compounds 16d and 16e exhibited similar, potent antagonism comparable to the corresponding saturated derivative 15e.
Next, we sought to evaluate the SAR of 2-cyclohexyl derivatives because previous reports indicated that the 2-cyclohexyl analogues in a series of 2-oxy and 2-thio derivatives provided potent antagonism (Table 2).17,18 The cyclohexyl derivative 15f showed potent antagonism with Ki(CAP) = 0.6 nM, which was a 2.5-fold improvement over that of the corresponding linear derivative 15c. A further 2-fold improvement in potency, with Ki(CAP) = 0.3 nM, was obtained with (S)-15f, the eutomer of 15f.
We next explored 4-alkyl cyclohexyl derivatives based on the previous SAR results from the series of 2-piperidinyl, 2-cyclohexyloxy and 2-cyclohexylthio derivatives,16–18 which indicated that alkyl substituents at the 4 position further improved potency. The 4-methyl cyclohexyl derivative 15g showed better antagonism than the parent 15f and the 4-ethyl derivative 15h exhibited further enhanced potency with a 2-fold increase compared to 15f. The 4-t-butyl cyclohexyl derivative 15i was found to be as potent as the 4-ethyl derivative 15h and its trans isomer 15k was found to be the preferred geometric isomer. The 4,4-dimethyl cyclohexyl derivative 15l also exhibited potent antagonism comparable to the 4-monoalkyl derivatives. However, the 4,4-difluoro cyclohexyl derivative displayed reduced potency, presumably due to its polarity. The bicyclic derivative 15n was 2.5-fold less potent than the corresponding monocycle 15f. We conclude from this SAR analysis of our series of cyclohexyl derivatives that 4-alkyl groups further improved the potency, providing better hydrophobic interaction with the receptor as found in 15h, 15i and 15l.
Finally, we examined 2-cyclohexenyl derivatives as rigid analogues of the corresponding cyclohexyl derivatives. Generally, the cyclohexenyl derivatives exhibited better potency compared to the corresponding cyclohexyl derivatives. The cyclohexenyl derivative 15o showed similar potency compared to the parent cyclohexyl derivative 15f. Impressively, the introduction of alkyl groups on the cyclohexene such as 4-methyl (15p), 3-methyl (15q and 15r), 4-ethyl (15s), 4-t-butyl (15t), and 4,4-dimethyl (15u) led to extremely potent antagonists with a range of Ki(CAP) = 0.1–0.3 nM. As eutomers, the (S)-isomers of 15h, 15l, 15n, 15p, 15t and 15u were synthesized independently and found to be ca. 2-fold more potent than the corresponding racemates as expected.
Overall SAR analysis indicated that the antagonism in a series of 2-alkyl/alkenyl pyridine C-regions showed the following order of potency: cycloalkenyl > cycloalkyl > branched alkyl > linear alkyl. 4-Alkyl groups on the ring improved the potency further.
Next, the in vitro activity of compound 15f, taken as a representative antagonist in this series, was investigated by using other TRPV1 activators (Table 3). We found that compound 15f also showed excellent antagonism toward activators other than capsaicin such as pH, heat (45 °C) and N-arachidonoyl dopamine (NADA), with potencies in the low nanomolar range.
Table 3.
In vitro antagonism of 15f for various activators of human TRPV1
| Activators, parameter | 15f |
|---|---|
| CAP, Ki (nM) | 0.6 |
| pH, IC50 (nM) | 43.4 |
| heat 45 °C, IC50 (nM) | 14.1 |
| NADA, Ki (nM) | 0.2 |
Consistent with its in vitro mechanism of action as an hTRPVl antagonist, in vivo 15f also blocked response to capsaicin (Table 4). Compound 15f was administered orally at doses of 0.1 and 0.3 mg/kg 15 min before intraperitoneal injection of 3 mg/kg capsaicin, following the procedure described previously.16 These doses of 15f inhibited the hypothermic response to capsaicin, assayed 30 min after capsaicin injection, by 24% and 78%, respectively. We further evaluated the analgesic activity of compound 15f administered orally in the rat Bennett model20 for neuropathic pain (Table 4). We found that the 15f showed dose-dependent anti-allodynic activity with ED50 = 1.6 mg/kg po (max 60% at 10 mg/kg). It was thus more potent than pregabalin (ED50 = 42.2 mg/kg, max 69% at 100 mg/kg) and showed approximately comparable maximal effect.
Table 4.
Inhibition of capsaicin-induced hypothermia and analgesic activity of compound 15f on CCI-induced cold allodynia after oral administration in mouse. Data, n = 10, mean ± SEM, *p <0.05 versus vehicle. MPE, maximal possible effect
| 0.1 mg/kg | 0.3 mg/kg | 1 mg/kg | 10 mg/kg | |
|---|---|---|---|---|
| CAP-induced hypothermia | 24% inh. | 78% inh. | ||
| Bennett model | 25 MPE | 53 MPE | 60 MPE |
Using our human TRPV1 (hTRPV1) model16 built based on our rat TRPV1 (rTRPV1) model,21 we performed a flexible docking study of (S)-15f. As illustrated in Figure 2, (S)-15f displayed a binding mode generally similar to that of 2.16 The sulfonylaminobenzyl group (A-region) occupied the deep bottom hole and was involved in the hydrophobic interactions with Val508, Tyr511, Ile564, Tyr565, and Ile569. A fluorine atom of the A-region participated in hydrogen bonding with Ser512 and the NH of the sulfonamide group made hydrogen bonds with Tyr565. The amide group (B region) made a hydrogen bond with Tyr511 and also contributed to the appropriate positioning of the C-region for its hydrophobic interactions. In addition, the 3-trifluoromethyl group (C-region) extended toward Thr550 in the hydrophobic area. The pyridine ring (C-region) made hydrophobic interactions with Tyr554 and the adjacent monomer Phe587. Furthermore, the cyclohexyl group in the C-region made an additional hydrophobic interaction with Leu515.
Figure 2.
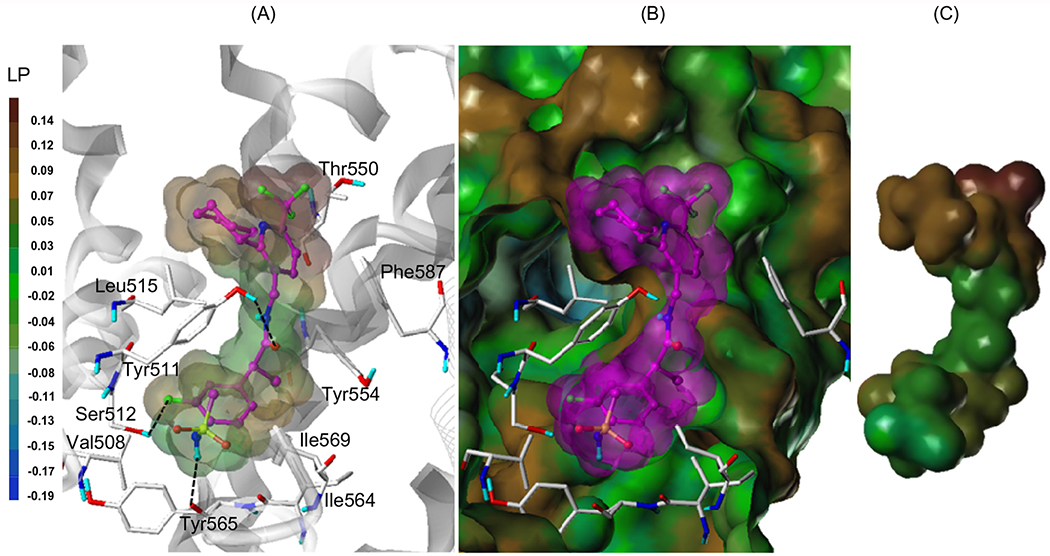
Flexible docking result of (S)-15f in the hTRPV1 model. (A) Predicted binding mode of the (S)-15f. The key interacting residues are marked and displayed as capped-stick with carbon atoms in white. The helices are colored by gray and the helices of the neighboring monomer are displayed in line ribbon. (S)-15f is depicted as a ball-and-stick with carbon atoms in magenta and the van der Waals surface of the ligand is presented with its lipophilic potential property. Hydrogen bonds are shown as black dashed lines and non-polar hydrogens are undisplayed for clarity. (B) Surface of hTRPV1 and the docked ligand. The fast connolly surface of hTRPV1 was generated by MOLCAD and colored by the lipophilic potential property. For clarity, the surface of hTRPV1 is Z-clipped and that of the ligand is in its carbon color. (C) Van dar Waals surface of the ligand colored by its lipophilic potential property.
In summary, we investigated the structure activity relationships of 2-alkyl/alkenyl pyridine C-region derivatives of the previous lead 2 as hTRPV1 antagonists. Multiple compounds in the series, particularly cyclohexenyl derivatives, showed excellent and stereospecific TRPV1 antagonism with better potencies than reference 2. Compound 15f was selected for further study and was shown to antagonize capsaicin-induced hypothermia in a dose-dependent manner, consistent with its action in vivo being through TRPV1, and it demonstrated strong analgesic activity in a rat neuropathic pain model. Docking analysis of (S)-15f with our hTRPV1 homology model indicated that (S)-15f showed a binding mode similar to that previously reported16 for compound 2.
Acknowledgments
This research was supported by Research Grants from Grunenthal, Germany, Grants from the National Research Foundation of Korea (NRF) (R11-2007-107-02001-0), Grants from the National Leading Research Lab (NLRL) program (2011-0028885), Republic of Korea and in part by the Intramural Research Program of NIH, Center for Cancer Research, NCI, USA (Project Z1A BC 005270).
References and notes
- 1.Szallasi A; Blumberg PM Pharmacol. Rev 1999, 51, 159. [PubMed] [Google Scholar]
- 2.Tominaga M; Caterina MJ; Malmberg AB; Rosen TA; Gilbert H; Skinner K; Raumann BE; Basbaum AI; Julius D Neuron 1998, 21, 531. [DOI] [PubMed] [Google Scholar]
- 3.Caterina MJ; Schumacher MA; Tominaga M; Rosen TA; Levine JD; Julius D Nature 1997, 389, 816. [DOI] [PubMed] [Google Scholar]
- 4.Zygmunt PM; Petersson J; Andersson DA; Chuang H-H; Sorgard M; Di Marzo V; Julius D; Hogestatt ED Nature 1999, 400, 452. [DOI] [PubMed] [Google Scholar]
- 5.Hwang SW; Cho H; Kwak J; Lee SY; Kang CJ; Jung J; Cho S; Min KH; Suh YG; Kim D; Oh U Proc. Natl. Acad. Sci. U.S.A 2000, 97, 6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walpole CSJ; Wrigglesworth R Capsaicin in the Study of Pain; Academic Press: San Diego, CA, 1993; p 63. [Google Scholar]
- 7.Appendino G; Szallasi A Life Sci. 1997, 60, 681. [DOI] [PubMed] [Google Scholar]
- 8.Szallasi A; Cruz F; Geppetti P Trends Mol. Med 2006, 12, 545. [DOI] [PubMed] [Google Scholar]
- 9.Kym PR; Kort ME; Hutchins CW Biochem. Pharmacol 2009, 78, 211. [DOI] [PubMed] [Google Scholar]
- 10.Wong GY; Gavva NR Brain Res. Rev 2009, 60, 267. [DOI] [PubMed] [Google Scholar]
- 11.Gunthorpe MJ; Chizh BA Drug Discovery Today 2009, 14, 56. [DOI] [PubMed] [Google Scholar]
- 12.Lazar J; Gharat L; Khairathkar-Joshi N; Blumberg PM; Szallasi A Expert Opin. Drug Disc 2009, 4, 159. [DOI] [PubMed] [Google Scholar]
- 13.Voight EA; Kort ME Expert Opin. Ther. Pat 2010, 20, 1. [DOI] [PubMed] [Google Scholar]
- 14.Szolcsányi J; Sándor Z Trend Pharmacol. Sci 2012, 33, 646. [DOI] [PubMed] [Google Scholar]
- 15.Szallasi A; Sheta M Expert Opin. Investig. Drug 2012, 21, 1351. [DOI] [PubMed] [Google Scholar]
- 16.Kim MS; Ryu H; Kang DW; Cho S-H; Seo S; Park YS; Kim M-Y; Kwak EJ; Kim YS; Bhondwe RS; Kim HS; Park S-G; Son K; Choi S; DeAndrea-Lazarus I; Pearce LV; Blumberg PM; Frank R; Bahrenberg G; Stockhausen H; Kögel BY; Schiene K; Christoph T; Lee J J. Med. Chem 2012, 55, 8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorat SA; Kang DW; Ryu H; Kim MS; Kim HS; Ann J; Ha T-H; Kim SE; Son K; Choi S; Blumberg PM; Frank R; Bahrenberg G; Schiene K; Christoph T; Lee J Eur. J. Med. Chem 2013, 64, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ha T-H; Ryu H; Kim S-E; Kim HS; Ann J; Tran P-T; Hoang V-H; Son K; Cui M; Choi S; Blumberg PM; Frank R; Bahrenberg G; Schiene K; Christoph T; Frormann S; Lee J Bioorg. Med. Chem 2013, 21, 6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryu H; Jin M-K; Kang S-U; Kim SY; Kang DW; Lee J; Pearce LV; Pavlyukovets VA; Morgan MA; Tran R; Toth A; Lundberg DJ; Blumberg PM J. Med. Chem 2008, 51, 57–67. [DOI] [PubMed] [Google Scholar]
- 20.Bennett GJ; Xie Y-K Pain 1988, 33, 87. [DOI] [PubMed] [Google Scholar]
- 21.Lee JH; Lee Y; Ryu H; Kang DW; Lee J; Lazar J; Pearce LV; Pavlyukovets VA; Blumberg PM; Choi S J. Comput. Aided Mol. Des 2011, 25, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]