

Abstract
Parallel spectroscopic and computational studies of iron(III) cysteine dioxygenase (CDO) and synthetic models are presented. The synthetic complexes utilize the ligand tris(4,5-diphenyl-1-methylimidazol-2-yl)phosphine (Ph2TIP), which mimics the facial three-histidine triad of CDO and other thiol dioxygenases. In addition to the previously reported [FeII(CysOEt)(Ph2TIP)]BPh4 (1; CysOEt is the ethyl ester of anionic l-cysteine), the formation and crystallographic characterization of [FeII(2-MTS)(Ph2TIP)]BPh4 (2) is reported, where the methyl 2-thiosalicylate anion (2-MTS) resembles the substrate of 3-mercaptopropionate dioxygenase (MDO). One-electron chemical oxidation of 1 and 2 yields ferric species that bind cyanide and azide anions, which have been used as spectroscopic probes of O2 binding in prior studies of FeIII-CDO. The six-coordinate FeIII-CN and FeIII-N3 adducts are examined with UV–vis absorption, electron paramagnetic resonance (EPR), and resonance Raman (rRaman) spectroscopies. In addition, UV–vis and rRaman studies of cysteine- and cyanide-bound FeIII-CDO are reported for both the wild-type (WT) enzyme and C93G variant, which lacks the Cys-Tyr cross-link that is present in the second coordination sphere of the WT active site. Density functional theory (DFT) and ab initio calculations are employed to provide geometric and electronic structure descriptions of the synthetic and enzymatic FeIII adducts. In particular, it is shown that the complete active space self-consistent field (CASSCF) method, in tandem with n-electron valence state second-order perturbation theory (NEVPT2), is capable of elucidating the structural basis of subtle shifts in EPR g values for low-spin FeIII species.
Graphical Abstract
INTRODUCTION
Thiol dioxygenases (TDOs) are a family of mononuclear nonheme iron dioxygenases that catalyze the O2-dependent oxidation of l-cysteine (Cys) or its derivatives to the corresponding sulfinic acids (RSO2H; Scheme 1).1 Two TDOs are known to operate in mammalian cells: namely, cysteine dioxygenase (CDO) and cysteamine dioxygenase (ADO). CDO regulates the intracellular concentration of exogenous Cys by performing the first step in its catabolism into small metabolites.2–4 ADO has been shown to catalyze a key step in the degradation of coenzyme A to (hypo)taurine,5 although a recent report argued that its primary function is to oxidize amino-terminal Cys residues of proteins in response to hypoxia.6 Bacterial CDOs have also been discovered.7,8 A third type of TDO, 3-mercaptopropionate (3-mpa) dioxygenase (MDO), has been isolated from certain soil bacteria.9–11 X-ray structures of CDOs from various organisms reveal a common iron coordination geometry consisting of three His residues in a facial orientation (the 3-His triad).8,12–14 The active sites of ADO15 and MDO8,11 also feature a 3-His triad, although a crystal structure of the former is currently lacking. The thiolate and amino donors of l-Cys bind directly to the FeII center in the enzyme–substrate complex, yielding a five-coordinate geometry primed for O2 binding.12,13 In contrast, a recent study suggested that only the thiolate moiety of 3-mpa binds to the Fe center of MDO,10 although bidentate coordination involving the carboxylate group has also been proposed by Jameson et al.16
Scheme 1.

Reactions Catalyzed by Thiol Dioxygenases
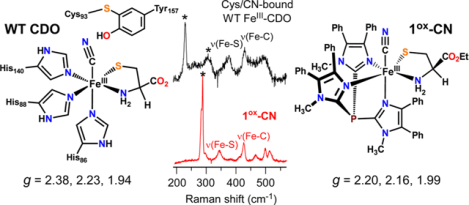
Even though the first-sphere coordination environment is conserved among all TDOs, these enzymes exhibit significant variability in the second (or outer) coordination sphere. Eukaryotic CDOs are distinguished by a post-translational cross-link between two conserved residues (Cys93 and Tyr157) located ~3.3 Å from the Fe center (Figure 1).13,17–19 This cross-link is generated during multiple turnovers of the newly formed enzyme,17–19 and although it is not required for catalysis, its presence causes a significant (at least 20-fold) increase in CDO activity.20 Alternatively, bacterial CDOs lack the Cys-Tyr cross-link yet maintain catalytic activities similar to those of eukaryotic CDOs.8 The catalytic function of the cross-link remains an open question, but recent studies suggest it plays a role in substrate binding/selectivity and discourages the binding of a water molecule to the Cys-bound FeII-CDO active site.13 CDOs also feature a conserved Arg residue (R60) that stabilizes the carboxylate group of bound l-Cys. MDOs lack this Arg residue, and its position in the active site is occupied instead by Gln. This change likely accounts for the ability of MDOs to oxidize all known TDO substrates (l-Cys, cysteamine, and 3-mpa),10,21 whereas CDOs exhibit a high degree of substrate specificity.22
Figure 1.

Crystallographic structure of the Cys-bound CDO active site derived from PDB entry 4IEV.12
The structural and functional effect of the protein environment on iron oxygenases can be probed by treating the enzyme with small molecules that bind to the iron center in a manner similar to that of O2. Surrogates of O2 and its one-electron-reduced derivative, superoxide, include nitrosyl, carbonyl, azido, and cyano ligands. Binding of these ligands to the active site gives rise to distinctive spectroscopic features that report on noncovalent interactions with second-sphere groups.23–27 Recently, the Liu group reported X-ray structures of cross-linked and non-cross-linked FeII-CDO bound to both NO and Cys.28 The Brunold and Pierce groups applied a similar strategy to FeIII-CDO by treating the oxidized enzyme with either azide29 or cyanide.30 These studies found that azide occupies a prebinding site in the enzyme but is unable to coordinate directly to the FeIII center of substrate-bound CDO. Alternatively, upon treatment with excess cyanide, the high-spin (S = 5/2) electron paramagnetic resonance (EPR) signal of substrate-bound FeIII-CDO is replaced by a low-spin (S = 1/2) signal, indicating the formation of a six-coordinate ternary complex.30 Significantly, the EPR g values of the cyano adduct were shown to be sensitive to changes in the second coordination sphere, such as the formation of the Cys-Tyr cross-link.
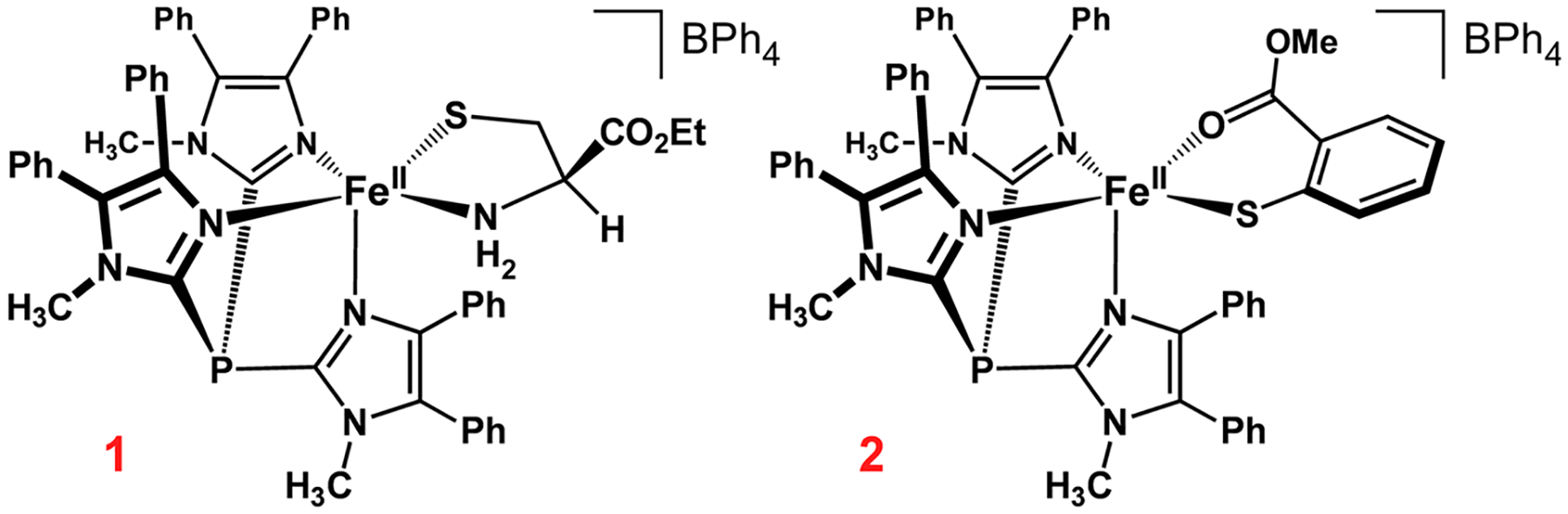
The interpretation of spectroscopic data collected for metalloenzymes is often aided by comparison to data obtained for synthetic complexes that replicate key aspects of the active site. In 2016, we reported the synthesis of a functional CDO model, [FeII(CysOEt)(Ph2TIP)]BPh4 (1; Scheme 2), where Ph2TIP is tris(4,5-diphenyl-1-methylimidazol-2-yl)phosphine and CysOEt is the ethyl ester of anionic l-Cys.31 Two factors make complex 1 the most structurally accurate model of the CDO active sites prepared to date: (i) the 2-TIPPh2 supporting ligand reproduces the neutral charge and all-imidazole coordination of the 3-His triad and (ii) the “substrate” ligand is not tethered to the supporting ligand, as in several other CDO mimics.32–35 Because complex 1 accurately reproduces the first coordination sphere of the enzyme, while lacking the corresponding active-site pocket, comparison of its spectroscopic features to those of the enzyme provides a unique opportunity for elucidating first- and second-sphere contributions to the geometric and electronic structures of CDO.
Scheme 2.
Schematic Drawing of Complexes 1 and 2
In the present study, we have generated oxidized derivatives of 1 bound to azide and cyanide ligands and examined these FeIII species with an assortment of spectroscopic and computational methods. We also report the synthesis and characterization of a new TDO model, [FeII(2-MTS)-(Ph2TIP)]BPh4 (2; Scheme 2), where 2-MTS is the anion of methyl 2-thiosalicylate. The structure of 2 has similarities to the substrate-bound form of MDO, and analysis of the resulting FeIII-CN adduct illuminates the effect of thiolate ligand variations (alkyl vs aryl) on electronic and spectral features. Our approach has employed both EPR and resonance Raman (rRaman) spectroscopies, which are complementary and responsive to minor changes in structure. Herein, the resulting spectra are compared to those previously obtained for the analogous enzymatic species, and we also present newly collected rRaman spectra of substrate-bound FeIII-CDO treated with cyanide. In addition, we use the combined enzyme/model data to test the ability of density functional theory (DFT) and ab initio calculations to predict subtle shifts in spectroscopic parameters upon changes in the FeIII coordination environment. These studies demonstrate that the complete active space self-consistent field (CASSCF) method, when it is used in conjunction with n-electron valence state second-order perturbation theory (NEVPT2), provides very accurate g values for low-spin FeIII sites in both synthetic and protein environments.
EXPERIMENTAL AND COMPUTATIONAL METHODS
General Considerations.
All reagents and solvents were purchased from commercial sources and used as received without further purification unless stated otherwise. Dichloromethane (CH2Cl2) was purified and dried using a solvent purification system purchased from Vacuum Atmospheres. The synthetic FeII complexes (1 and 2) were prepared and stored under an inert atmosphere in a Vacuum Atmospheres Omni-Lab glovebox. Solvents were deoxygenated prior to use in the glovebox and stored over molecular sieves. The syntheses of [Fe(Ph2TIP)(MeCN)3](OTf)236 and complex 131 were reported previously.
Synthesis of [FeII(2-MTS)(Ph2TIP)]BPh4 (2).
Methyl 2-thiosalicylate (27.5 μL, 0.20 mmol) and NEt3 (30.7 μL, 0.22 mmol) were dissolved in CH2Cl2 and stirred for 30 min. Addition of [Fe(Ph2TIP)(MeCN)3](OTf)2 (0.242 g, 0.20 mmol) yielded a red-orange solution that was stirred for 1 h, followed by removal of solvent under vacuum. The resulting orange solid (i.e., 2-OTf) was dissolved in CH3OH (10 mL) and filtered. Combination with a methanolic solution of NaBPh4 (0.068 g, 0.20 mmol) caused immediate formation of an orange-yellow precipitate, which was isolated by filtration. This solid was taken up in a small amount of CH2Cl2 and layered with MeOH, which provided an orange-yellow solid that was collected after 1 day and dried in vacuo (yield 62%). Anal. Calcd for FeC80H66BN6O2PS (Mw = 1273.11 g mol−1): C, 75.47; H, 5.22; N, 6.60. Found: C, 74.10; H, 5.68; N, 6.80. UV–vis [λmax, nm (ε, M−1 cm−1) in CH2Cl2]: 380 (5100), 675 (80). FTIR (cm−1, CH2Cl2): ν 1651 [ν(C = O)], 1582, 1504, 1062, 1030. 1H NMR (400 MHz, CDCl3): δ 43.6 (s, 1H, 2-MTS), 36.8 (s, 1H, 2-MTS), 17.2 (s, 9H, −NCH3), 16.5 (s, 3H, −OCH3), 8.47 (s, 3H, Ph2TIP), 8.29 (s, 8H, BPh4) 7.65 (s, 8H, BPh4), 7.38 (s, 4H, BPh4), 6.73 (s, 6H, Ph2TIP), 5.83 (s, 3H, Ph2TIP), 4.85 (s, 6H, Ph2TIP), 3.10 (s, 6H, Ph2TIP), −0.52 (s, 1H, 2-MTS), −15.2 (br s, 6H, Ph2TIP), −17.1 (s, 1H, 2-MTS) ppm.
X-ray Crystallography.
Orange crystals of 2-OTf suitable for crystallographic analysis were grown by layering a concentrated CH2Cl2 solution with pentane. Details concerning the acquisition of crystallographic data are provided in the Supporting Information. The structure of 2-OTf contains two symmetrically independent units. In the second crystallographic unit, one of the phenyl substituents of the Ph2TIP ligand is rotationally disordered, as is the entire triflate counterion. In addition, two partially occupied positions containing CH2Cl2 molecules were positively localized within the layers, while the remaining solvent was accounted for using a solvent-mask procedure.
Crystallographic data for complex 2-OTf: C57H46F3FeN6O5PS2 • 0.5CH2Cl2, Mr = 1145.42 g mol−1, monoclinic, space group C2/c, a = 46.4436(4) Å, b = 16.71596(15) Å, c = 30.6899(2) Å, α = 90°, β = 89.9026(8)°, γ = 90 °, V = 23826.0(3) Å3, Z = 16, ρ = 1.282 g cm−3, μ = 3.902 mm−1, λ = 1.5418 Å, reflections collected 120390, independent reflections 23968 (Rint = 0.0324), 1518 parameters (127 restraints), R1 = 0.0503 and wR2 = 0.1477 for I ≥ 2σ(I), R1 = 0.0547 and wR2 = 0.1532 for all data. Crystallographic data (CIF) can be obtained from the Cambridge Crystallographic Data Centre using the deposition number 1945809.
Preparation of Synthetic FeIII–Cyano and –Azido Adducts.
Solutions of 1 (or 2) in CH2Cl2 were prepared in the glovebox and then cooled to either −78 or −70 °C. An anaerobic stock solution of [AcFc]BF4 (AcFc = acetylferrocenium) in CH2Cl2 was also generated. Injection of 1 equiv of [AcFc]BF4 into the cooled solution caused a color change indicative of 1ox or 2ox formation. Formation of the cyano adducts was achieved by subsequent addition of 10–50 equiv of CN− salt (countercation = K+, NEt4+, NBu4+) at low temperature. The 13CN and 13C15N adducts were generated by addition of isotopically labeled KCN salts that were solubilized in CH2Cl2 using 18-crown-6 ether. Formation of 1ox-N3 required addition of 5–50 equiv of N3− salt (countercation = K+, NBu4+) to 1ox at low temperature, followed by warming to room temperature. The 1ox-N3 adduct with 15N-labeled azide was prepared by addition of excess K15N14N2 solubilized in CH2Cl2 using 18-crown-6 ether.
Protein Purification.
Expression of the recombinant Mus musculus cdo gene was performed using Rosetta 2(DE3) E. coli cells (MilliporeSigma, Massachusetts) under control of the Lac operator in the pVP16 plasmid. The cdo gene was expressed as a fusion protein with an N-terminal 8×His and maltose binding protein (MBP) tags as previously described.14
The C93G CDO variant was created using site-directed mutagenesis (SDM) via PCR as previously described,37 with the exception that Phusion Master Mix with GC buffer (Thermo Scientific, Massachusetts) and E. coli 10G chemically competent cells (Lucigen, Wisconsin) were used instead. The primers used for SDM can be found in the Supporting Information. Cell growth and protein purification were performed as before,38 with some changes. Cells were grown in lysogeny broth (LB) containing chloramphenicol, ampicillin, and ferrous ammonium sulfate [(NH4)2Fe(SO4)2] (34 μg/mL, 100 μg/mL, and 110 μM, respectively). Gene overexpression was induced by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 80.5 μM, d-lactose to a final concentration of 8 mM, and Casamino acids to a final concentration of 0.20% (w/v). All buffers used had a pH of 8. Cell pellets were resuspended in 25 mM HEPES buffer with rLysozyme (MilliporeSigma, Massachusetts) and sonicated on ice for 30 min. The lysed solution was then centrifuged at 48400g and 4 °C for 75 min. Protein bound to the amylose column was eluted with buffer containing 25 mM HEPES, 200 mM NaCl, and 50 mM maltose. The N-terminal 8×His and MBP tags were then removed from CDO by incubating the solution with tobacco etch virus (TEV) protease overnight at room temperature. Tag-free CDO was isolated using subtractive immobilized metal affinity chromatography (IMAC) with a NiII nitrilotriacetic acid (NTA) agarose resin (GE Healthcare, Illinois).
C93G CDO samples for spectroscopy were reconstituted with FeII to increase the increase the fraction of Fe-bound CDO active sites. This was accomplished by adding a 2-fold excess of [(NH4)2Fe(SO4)2] to C93G CDO aerobically. After the protein and [(NH4)2Fe(SO4)2] solution was stirred for 30 min at 4 °C, a 3-fold excess of ammonium hexachloroiridate(IV) ((NH4)2[IrCl6]) was added and the solution was again stirred for 30 min at 4 °C. To remove any excess [(NH4)2Fe(SO4)2] and (NH4)2[IrCl6], a buffer exchange using 25 mM HEPES and 200 mM NaCl solutions, in conjunction with a 10 kDa centrifugal filter unit, was performed three times. The iron content of C93G CDO was then determined via a colorimetric assay using 2,4,6-tris(2-pyridyl)-s-triazine (TPTZ).39 A summary of this assay can be found in the Supporting Information. To generate Cys- and CN-bound C93G FeIII-CDO, a 10-fold molar excess of Cys over FeIII-bound variant protein was added to the sample. After 5 min, a 10-fold molar excess of KCN over C93G FeIII-CDO was then added. The sample was frozen after another 5 min incubation period.
WT CDO samples for spectroscopy were prepared similarly to C93G CDO with two modifications. Namely, iron reconstitution was performed anaerobically in a glovebox and samples were subsequently treated with Chelex 100 sodium form (Sigma-Aldrich, Missouri) for 30 min to remove any unbound iron in solution. Samples were not treated with (NH4)2[IrCl6]; thus, no buffer exchange was performed.
Spectroscopic Data.
UV–vis absorption spectra were collected with either an Agilent 8453 diode array spectrometer (synthetic samples) or Varian Cary 4 Bio spectrometer (protein samples). The former was equipped with a Unisoku Scientific Instruments (Osaka, Japan) cryostat for low-temperature measurements. 1H NMR spectra were measured on a Varian 400 MHz spectrometer. Elemental analyses were performed at Midwest Microlab, LLC, in Indianapolis, IN. Infrared (IR) spectra were measured with a Thermo Scientific Nicolet iS5 FTIR spectrophotometer. X-band EPR spectra were gathered using a Bruker EMX instrument equipped with an ER4112 SHQ resonator. EPR data were collected at liquid He temperatures (5–15 K) using a ColdEdge/Bruker RDK-408 Stinger recirculating cryocooler. The program EasySpin (version 5)40 was employed to conduct simulations of experimental EPR spectra.
The resonance Raman data were measured using 647.1 nm excitation from a Coherent I-302C Kr+ laser. The laser power at the sample was between 30 and 50 mW for the synthetic samples and ~20 mW for the protein samples. The synthetic samples had concentrations of 5–10 mM on the basis of the initial quantity of precursor complex (1 or 2). The preparation of protein samples for rRaman analysis is described in further detail above. Light scattered from frozen samples in liquid N2 was collected using a 135° backscattering arrangement and dispersed by an Acton Research triple monochromator featuring a 1200 grooves/mm grating. The scattered light was detected and analyzed with a Princeton Instruments Spec X:100BR deep depletion, back-thinned CCD camera. Baselines of the rRaman spectra were corrected using piecewise functions in the program IGOR to remove broad background contributions. Peak positions were calibrated against the 984 cm−1 peak of a K2SO4 standard or ice peak at 228 cm−1 for protein samples.
Computational Methods.
All calculations were performed with the ORCA software package (version 3.0 or 4.0) developed by Dr. F. Neese (MPI for Chemical Energy Conversion).41 Computational models of 1ox-CN, 2ox-CN, and 1ox-N3 were generated via density functional theory (DFT) calculations that employed Becke’s three-parameter hybrid functional for exchange along with the Lee–Yang–Parr correlation functional (B3LYP).42,43 The geometry optimizations used Ahlrichs’ valence triple-ζ basis set combined with polarization functions on main-group and transition-metal elements (TZVP).44–46 The Ph2TIP supporting ligand was modified by replacing the 5-phenyl substituents with hydrogen atoms. Computational times were reduced by applying the resolution of identity and chain of sphere (RIJCOSX) approximation47 and the TZV/J auxiliary basis set.48 Only real vibrational frequencies were computed for the optimized structures, indicating that they correspond to local energy minima. Active-site models of CDO in its cross-linked (α) and non-cross-linked (β) forms were obtained from previous QM/MM calculations reported by Li et al.30 Atomic coordinates of all computational models are provided in Tables S4–S10. For each FeIII-CN adduct, the composition of the singly occupied molecular orbital (SOMO) was determined through a natural orbital calculation on the basis of the DFT/B3LYP results. Computational results were visualized with the ChemCraft program.
Two different approaches were employed to compute the g tensor of 1ox-CN. The first method determined spin–orbit coupling contributions by solving the coupled-perturbed self-consistent field (CP-SCF) equations.49–52 These calculations employed the B3LYP functional, RIJCOSX approximation, and TZVP basis set. The second approach utilized the complete active space self-consistent field (CASSCF) based method along with n-electron valence state second-order perturbation theory (NEVPT2).53,54 These calculations were based on unrestricted natural orbitals derived from single-point DFT/B3LYP calculations. The CASSCF/NEVPT2 calculations used either a split valence basis set with polarization (SVP with auxiliaries) or the TZVP basis set described above. The impact of active-space size and basis set size on computed g values was analyzed for 1ox-CN, and the results are provided in Table S2. These results suggested that the most appropriate active space is CAS(11,13), which consists of the five Fe 3d orbitals, a second Fe d shell, and three CysOEt-based orbitals. The CAS(11,13) approach was then applied to the other FeCN adducts. Ten doublet roots were calculated in each case.
RESULTS AND DISCUSSION
Synthesis and X-ray Structure of a Substrate-Bound MDO Model.
Following our earlier procedure for the synthesis of complex 1,31 [FeII(2-MTS)(Ph2TIP)]OTf (2-OTf) was prepared by treating the [FeII(Ph2TIP)(MeCN)3](OTf)2 precursor with methyl 2-thiosalicylate in the presence of NEt3. Counteranion metathesis with NaBPh4 in MeOH provided [FeII(2-MTS)(Ph2TIP)]BPh4 (2) as a precipitate that was collected and dried to give a yellow powder. Yellow-orange crystals of 2-OTf for X-ray diffraction experiments were grown by layering a concentrated CH2Cl2 solution with pentane. The unit cell of 2-OTf consists of two symmetrically independent FeII complexes with similar five-coordinate geometries. The three imidazole donors of Ph2TIP are arranged facially with an average Fe–NTIP distance of 2.15 Å (Figure 2). These bond distances reflect the high-spin (S = 2) state of the FeII center, which is also apparent in the paramagnetically shifted peaks between −20 and +50 ppm observed by 1H NMR spectroscopy (Figure S1). The overall geometry is best described as distorted trigonal bipyramidal (average τ value 0.57)55 with the thiolate and carbonyl donors of 2-MTS occupying equatorial and axial positions, respectively. Metric parameters for complexes 1 and 2-OTf are compared in Table S1. The two complexes exhibit nearly identical Fe–S bond distances (~2.32 Å) despite the change from alkanethiolate (1) to arenethiolate (2-OTf). The distance between the Fe center and O atom donor of 2-MTS is 2.13 Å, which is considerably shorter (by 0.11 Å) than the Fe–Namino distance measured for the CysOEt ligand of 1. These results highlight the stability of the six-membered chelate formed by the bidendate S,O-coordination of 2-MTS.
Figure 2.

Thermal ellipsoid plot (50% probability) obtained from the X-ray crystal structure of 2-OTf. The triflate counteranion, noncoordinating solvent molecules, and all hydrogen atoms are omitted for clarity.
Because of the structural similarities between 2-MTS and 3-mpa, complex 2 serves as an active-site mimic of substrate-bound FeII-MDO. Recently, Jameson and co-workers employed docking methods to discern the position of 3-mpa within the MDO active site.16 In the resulting model (Figure S2), the thiolate and carboxylate groups of 3-mpa coordinate trans to His91 and His142, respectively. This bidentate conformation is favored by the formation of a salt bridge between the substrate carboxylate and Arg168, which places one of the O atoms in proximity to the Fe center. In contrast, Pierce has suggested that the carboxylate of 3-mpa participates in a hydrogen bond with a conserved Tyr residue, resulting in thiolate-only coordination.10 Although the X-ray structure of 2-OTf resembles the docking model of Jameson, the absence of outer-sphere residues makes it impossible to draw firm conclusions from our synthetic efforts. Thus, future structural and spectroscopic studies of MDOs are required to determine the mode of substrate binding.
Formation and Spectroscopic Features of Ferric Cyano and Azido Species.
Analysis with cyclic voltammetry (CV) revealed that complex 1 undergoes irreversible oxidation at +65 mV (vs Fc+/0) in CH2Cl2 at room temperature. On the basis this result, we reckoned that it would be possible to generate the ferric derivative by chemical oxidation with acetylferrocenium (AcFc+; E1/2 = +270 mV).56 Indeed, treatment of 1 with 1 equiv of [AcFc]BF4 in CH2Cl2 at −78 °C generates a new chromophore (1ox) with intense absorption bands at 445 and 595 nm (Figure 3a). The oxidized species is stable at reduced temperatures but decays upon warming to room temperature, consistent with the irreversibility observed by CV (Figure S3). Oxidation of 2 under similar conditions (T = −70 °C) yields a reddish-brown species (2ox) with a similar pattern of UV–vis features, albeit red-shifted in comparison to 1ox. Like 1ox, the absorption features of 2ox decay upon warming to room temperature to give a featureless spectrum (Figure S3). The 1ox and 2ox absorption bands likely derive from thiolate to FeIII charge transfer (CT) transitions. In support of this conclusion, X-band EPR spectra of both species (Figure S4) feature an intense derivative feature at g = 4.3 and a much weaker peak near g = 9.4. Such spectra are typical of mononuclear high-spin FeIII complexes with rhombic symmetry (E/D ≈ 0.33).
Figure 3.

UV–vis and EPR spectra of oxidized derivatives of complexes 1 and 2. Absorption spectra were measured in CH2Cl2 at either −78 °C (a) or −70 °C (b, c), with the exception of the 1ox-N3 spectrum (20 °C). Samples of 1ox and 2ox were generated by treatment of 1 and 2, respectively, with 1 equiv of [AcFc]BF4. Further addition of 50 equiv of (a) [NBu4]CN, (b) [NEt4]CN, or (c) [NBu4]N3 gave rise to the cyano and azido adducts (formation of the latter required warming to 20 °C). Initial concentration of Fe complex: (a) 0.52 mM, (b) 0.59 mM, and (c) 0.45 mM. Insets: X-band EPR spectra of (a) 1ox-CN, (b) 2ox-CN, and (c) 1ox-N3 measured in frozen CH2Cl2 solutions at the indicated temperature. The microwave power was 2 mW, and the frequency (ν) is indicated. The features designated by an asterisk (*) arise from a copper impurity in the sample cavity.
Addition of excess cyanide anion to solutions of 1ox at low temperature results in the formation of a new teal species (1ox-CN). Complete conversion of 1ox → 1ox-CN requires at least 10 equiv of cyanide anion, suggesting that the affinity is rather low. The same procedure was used to generate 2ox-CN. As shown in Figure 3, these cyanide-treated species are characterized by a strong S → FeIII charge transfer (CT) band at 645 (1ox-CN) or 720 nm (2ox-CN), in addition to a second CT feature at shorter wavelength (λmax ≈ 440 nm). The energies and intensities of these UV–vis features resemble those reported for the thiolate-ligated nonheme FeIII-CN adduct of superoxide reductase (SOR).23 Unlike their 1ox/2ox precursors, the cyanide-treated species are stable at room temperature.
As expected, cyanide coordination causes the FeIII complexes to adopt a low-spin configuration. The EPR spectrum of 1ox-CN in frozen CH2Cl2 exhibits a nearly axial S = 1/2 signal (Figure 3a; inset) that was simulated with g values of 2.204, 2.161, and 1.988. No high-spin S = 5/2 features were observed even at 10 K, indicating full conversion of 1ox to 1ox-CN. The spectrum of 2ox-CN (Figure 3b; inset) displays slightly greater rhombicity with g values of 2.231, 2.161, and 1.964. Table 1 compares these g values to those measured by Pierce et al. for Cys-bound FeIII-CDO treated with cyanide.30 It is clear that the EPR signals of the CDO adducts are significantly more rhombic and anisotropic than those exhibited by the synthetic analogues. The structural factors behind these disparities will be explored in Computational Studies.
Table 1.
Comparison of EPR g Values Measured for (l-Cys/CN)-Bound FeIII-CDO and Related Model Complexes
| species | g1 | g2 | g3 | ref |
|---|---|---|---|---|
| WT (l-Cys/CN)-bound FeIII-CDOa | 1.937 | 2.234 | 2.379 | 30 |
| C93A (l-Cys/CN)-bound FeIII-CDOb+ | 1.951 | 2.208 | 2.343 | 30 |
| 1ox-CN | 1.988 | 2.161 | 2.204 | this work |
| 2ox-CN | 1.964 | 2.161 | 2.231 | this work |
WT enzyme with Cys93-Y157 cross-link.
The C93A CDO variant lacks the Cys93-Y157 cross-link
The ability of 1ox to bind azide was also examined. Treatment of 1ox with 50 equiv of [NBu4]N3 at −80 °C results in only modest spectral changes; however, the reaction at 20 °C generates a dark brown chromophore (1ox-N3) that displays intense absorption features in the 400–600 nm region (Figure 3c). Comparison to synthetic precedents suggests that these features arise from N3− → FeIII CT transitions,57 although S → FeIII CT bands probably also contribute to the absorption envelope. The EPR spectrum of 1ox-N3 consists of a rhombic S = 5/2 signal (Figure 3c, inset) similar to that measured for 1ox, indicating the complex remains high spin upon azide coordination.
The properties of the ferric-cyano and -azido adducts were further probed with rRaman spectroscopy. Excitation into the S → FeIII CT band using 647.1 nm laser light provided the spectra of 1ox-CN and 2ox-CN shown in Figures 4 and 5, respectively. These spectra display a series of features in the 300–550 cm−1 region arising from vibrations of the NC–Fe–SR unit. Peak assignments were made by comparing spectra of samples prepared with different cyanide isotopes (i.e., 12C14N, 13C14N, and 13C15N). The frequency of the peak at 341 cm−1 in the 1ox-CN spectrum is characteristic of ν(Fe–S) modes in nonheme iron enzymes and complexes with thiolate coordination.23,58–65 This peak downshifts slightly (1 cm−1) upon 13CN labeling due to kinematic coupling between the Fe–S and Fe–C stretching motions. The peak at 464 cm−1 in the natural abundance (NA, 12C14N) spectrum exhibits a similarly small isotope shift, likely corresponding to the Fe–N stretching mode of the −NH2 donor of CysOEt.
Figure 4.

Resonance Raman spectra of 1ox-CN collected using 647.1 nm laser excitation (~40 mW at the sample). The samples were prepared by treating 1ox with (a) natural abundance KCN (50 equiv) in CD2Cl2, (b) K13CN (50 equiv) in CD2Cl2, and (d) K13C15N (10 equiv) in CH2Cl2. The KCN salts were solubilized using 18-crown-6 ether. The difference spectrum (a–b) generated by digital subtraction is provided in (c). The 13C and 13C15N isotope shifts relative to the NA spectrum are indicated in cm−1.
Figure 5.

Resonance Raman spectra of 2ox-CN collected at 77 K using 647.1 nm laser excitation (30 mW at the sample). The samples were prepared at −78 °C by treating 2ox with (a) natural-abundance KCN (10 equiv) or (b) K13CN (10 equiv) in CH2Cl2. The KCN salts were solubilized using 18-crown-6 ether. The 13C isotope shifts relative to the NA spectrum are indicated in cm−1. Solvent-derived peaks are indicated with an asterisk (*).
Much larger 13C isotope shifts are displayed by the three peaks at 426, 499, and 513 cm−1 in the NA spectrum. These features arise from ν(Fe–CN) stretching and δ(Fe–C–N) bending modes that often exhibit a substantial degree of mixing. On the basis of prior studies of FeIII–CN adducts in proteins, it is known that modes with predominant ν(Fe–CN) character are distinguished by large shifts upon isotopic substitution of both the carbon and nitrogen atoms of the cyano ligand, whereas the frequencies of δ(Fe–C–N) modes are predominantly sensitive to isotopic substitution at the carbon atom.23,25,66 As shown in Figure 4, the peak at 426 cm−1 in the NA spectrum decreases by 15 cm−1 upon 13C15N labeling, which is 4 cm−1 greater than the change caused by 13C labeling alone. In contrast, the 13C15N shifts of the peaks at 499 and 513 cm−1 are nearly identical with the 13C shifts. Thus, the peak at 426 cm−1 is assigned to the ν(Fe–CN) stretching mode, while those at 499 and 513 cm−1 are attributed to modes with predominately δ(Fe–C–N) bending character. Clay et al. also observed δ(Fe–C–N) bending modes around 500 cm−1 in the spectrum of CN-treated FeIII–SOR.23 The observation of two δ(Fe–C–N)-based peaks for 1ox-CN may indicate the presence of two conformations in frozen solution. An alternative explanation is that the FeCN geometry is nearly linear, since the frequencies of the two bending modes merge as the bond angle approaches 180°. The latter scenario is supported by DFT calculations (vide infra), which predict a Fe–C–N angle of 176° for 1ox-CN.
The rRaman spectrum of 2ox-CN exhibits peaks due to ν(Fe–S) and ν(Fe–CN) stretching modes at 347 and 403 cm−1, respectively (Figure 5). The feature at 452 cm−1 with a small 13C isotope shift of −2 cm−1 is tentatively assigned to a mode primarily involving stretching of the Fe–O(carbonyl) bond. Unlike 1ox-CN, the spectrum of 2ox-CN displays a single δ(Fe–C–N) peak at 495 cm−1, although the higher frequency mode at 539 cm−1 likely has significant δ(Fe–C–N) and/or ν(Fe–CN) character.
Raman studies of 1ox-N3 employed 488.1 nm laser excitation into the overlapping N3− → FeIII and S → FeIII CT bands. The resulting spectrum (Figure S5) exhibits an intense peak at 377 cm−1 that is assigned to the ν(Fe–S) stretching mode on the basis of its frequency. This peak downshifts by 2 cm−1 when the sample is prepared with 15N14N2-labeled azide, indicative of weak coupling to the ν(Fe–N) mode of the azido ligand. For reasons that are not clear, the stretching and bending modes of the FeIII-N3 unit are not observed directly. Nevertheless, the clear shift in the ν(Fe–S) frequency upon isotopic labeling of N3− provides direct evidence of azide coordination to the FeIII center. The formation of 1ox-N3 is notable because spectroscopic and crystallographic studies13 have shown that azide does not form an inner-sphere complex with Cys-bound FeIII-CDO. Our results thus support the proposal that the inability of Cys-bound FeIIICDO to form an FeIII-N3 adduct is due to a hydroxide ion occupying the sixth coordination site rather than an intrinsic lack of azide affinity by the FeIII site.29
Resonance Raman Studies of CN/Cys-Bound FeIII-CDO.
Parallel studies of Cys- and CN-treated FeIII-CDO were conducted to discern the effect of the protein environment on the geometric and electronic structures of the FeIII center. Wild-type (WT) Cys/CN-bound FeIII-CDO exhibits a bluish color due to the presence of a S → FeIII CT band at λmax = 667 nm (Figure S6). This peak is red-shifted by ~700 cm−1 relative to the same feature displayed by high-spin Cys-bound FeIII-CDO. A red shift of similar magnitude was observed upon CN binding to SOR.23 We also prepared rRaman samples for the Cys/CN adduct of the C93G CDO variant, which lacks the C93–Y157 cross-link (vide supra). In this case, the S → FeIII CT band appears at 650 nm, indicating that loss of the cross-link modulates the Fe–S bonding interaction.
rRaman spectra of the enzyme samples were obtained by 647.1 nm laser excitation into the S → FeIII CT band (Figure 6). In addition to the ice lattice peaks (marked by asterisks), the spectrum of WT Cys/CN-bound FeIII-CDO exhibits two strong peaks at 377 and 429 cm−1 that are assigned to the ν(Fe–S) and ν(Fe–CN) stretching modes, respectively, on the basis of a comparison to the 1ox-CN results. The CDO spectrum also contains a weaker band at 495 cm−1 that is attributed to a δ(Fe–C–N) bending mode. In the rRaman spectrum of the C93G CDO variant, the ν(Fe–S)- and ν(Fe–CN)-based peaks appear at slightly lower frequencies, which proves that the cross-link perturbs both the Cys and CN ligands. These peaks are sensitive to 13CN substitution with isotope shifts similar to those observed for 1ox-CN and 2ox-CN.
Figure 6.

Resonance Raman spectra of Cys/CN adducts of CDO collected at 77 K using 647.1 nm laser excitation (20–25 mW at the sample): (upper spectrum) WT FeIII-CDO treated with Cys and natural abundance KCN; (middle spectrum) C93G FeIII-CDO treated with Cys and natural-abundance KCN; (lower spectrum) C93G FeIII-CDO treated with Cys and K13C14N. Peaks labeled with an asterisk arise from the lattice modes of ice. Frequencies and 13C isotope shifts (in cm−1) are provided for select peaks.
The ν(Fe–S) frequency of 377 cm−1 observed for the Cys/CN adduct of low-spin WT CDO is significantly higher than the value of 340 cm−1 measured for high-spin Cys-bound FeIII-CDO.59 This trend is opposite to that found for the synthetic complexes, as the ν(Fe–S) frequency of low-spin 1ox-CN is lower (by 36 cm−1) than that of high-spin 1ox-N3. Interestingly, Johnson and co-workers reported that the ν(Fe–S) frequency of SOR is largely unchanged upon CN binding.23 These findings suggest that the ν(Fe–S) frequency is not a reliable indicator of spin state or Fe–S bond distance, and this issue will be addressed in greater detail in the Conclusions.
COMPUTATIONAL STUDIES
Geometry-Optimized Synthetic and Enzyme Models.
The geometric and electronic structures of our synthetic FeIII-CN and FeIII-N3 species were further examined with DFT calculations employing the B3LYP functional. The computational models generated via geometry optimization are shown in Figure 7, and key metric parameters are summarized in Table 2 for the Fe-CN adducts. The Fe–S bond distances of 2.18 and 2.20 Å computed for 1ox-CN and 2ox-CN, respectively, are significantly shorter than those measured for their FeII precursors, reflecting the increase in Fe oxidation state and the change from high-spin to low-spin state. In both cases, the Fe-CN units are nearly linear with Fe–C–N bond angles of 176°. The aryl ring of the 2-MTS ligand in 2ox-CN is tilted by nearly 40° out of the plane created by the S–Fe–O chelate ring in order to minimize steric interactions between 2-MTS and the nearby Ph ring of Ph2TIP. In the geometry-optimized model of 1ox-N3, the Fe-N3 unit adopts a highly bent orientation with a Fe–N–N bond angle of 136°. The azido ligand points away from the CysOEt chelate to avoid steric interactions with the adjacent phenyl rings (Figure 7). The high-spin nature of the FeIII center in 1ox-N3 results in a lengthy Fe–S bond distance of 2.32 Å.
Figure 7.

DFT geometry-optimized structures of 1ox-CN, 2ox-CN, and 1ox-N3.
Table 2.
DFT-Computed Bond Lengths (Å) and Angles (deg) for 1ox-CN and 2ox-CN Compared to Those for QM/MM-Optimized Cys/CN-Bound FeIII-CDO Active Sites
DFT calculations of 1ox-CN and 2ox-CN predict ν(Fe–S) stretching frequencies of 378 and 365 cm−1, respectively, in reasonable agreement with the experimental data.67 The calculations somewhat overestimate the frequencies of the ν(Fe–CN) modes, providing values of 457 and 472 cm−1 for 1ox-CN and 2ox-CN, respectively. Analysis of the computed Δ13C isotope shifts reveals that several modes in the 420–480 cm−1 region contain partial δ(Fe–C–N) character, making it difficult to determine the precise frequencies of these modes. However, the presence of multiple isotopically active modes with ν(Fe–CN) and δ(Fe–C–N) character is consistent with the rRaman data measured for the synthetic FeIII-CN adducts.
To better understand the role of outer-sphere residues in modulating the coordination geometry of the Fe center, it is worthwhile to compare the DFT-generated structures of our synthetic CDO mimics to analogous computational models of the FeIII-CDO active site. Recently, the Pierce and Brunold groups used a quantum mechanical/molecular mechanics (QM/MM) approach to generate whole-protein models of FeIII-CDO bound to l-Cys substrate and CN.30 In these calculations, the QM region was treated with the same DFT/B3LYP methodology as employed here for the synthetic complexes. In the QM/MM-optimized protein structures, the carboxylate group of the l-Cys substrate forms a salt bridge to a conserved Arg60 residue, as illustrated in Figure 1. Due to this interaction, the Fe–S bond distance of 2.28 Å in the CN/Cys-bound FeIII-CDO active site is significantly longer than the value of 2.18 Å computed for 1ox-CN (Table 2). In addition, the protein active site features shorter Fe–NHis distances in comparison to the Fe–NTIP distances in 1ox-CN and 2ox-CN, which contributes to the longer Fe–S and Fe–C(N) bonds in the active-site models (Table 2).
The QM/MM studies also examined the structural effects of the Cys-Tyr cross-link on the Cys/CN FeIII-CDO adducts. Here, we adopt the labeling scheme of Pierce et al. and refer to the cross-linked and non-cross-linked enzymes as α-CDO and β-CDO, respectively.27 In the absence of the cross-link, the Fe–C–N unit is nearly linear with an angle of 176° (nearly identical to the values computed for 1ox-CN and 2ox-CN). However, α-CDO exhibits a more bent Fe–C–N unit with an angle of 169°. In addition, by shifting the position of the Tyr157 residue, the cross-link disrupts the network of hydrogen bonds between the side chain of Arg60, the phenol of Tyr157, and the substrate Cys carboxylate (Figure 1), which causes the Fe–S bond to lengthen by 0.02 Å. Although seemingly minor, this perturbation has a measurable effect on the electronic and spectroscopic properties of the enzyme active site, as described below.
Electronic Structure and Bonding Interactions.
The DFT/B3LYP-computed molecular orbital (MO) diagram of 1ox-CN shown in Figure 8a provides a useful starting point for discussing the electronic structures of these thiolate-ligated FeIII-CN adducts, both synthetic and enzymatic. As expected for a distorted-octahedral complex, the five 3d electrons of the low-spin FeIII center occupy the t2g set of orbitals (dxy, dxz, dyz), while the eg orbitals (, ) are unoccupied (see Figure 8c for the molecular coordinate system). The singly occupied Fe 3dxy-based MO is destabilized relative to the doubly occupied Fe 3dxz- and 3dyz-based MOs by a strong π-antibonding interaction with the sulfur 3py orbital. Following the convention established by Griffith and Taylor for low-spin FeIII systems,68,69 the energy difference between the 3dxy orbital and 3dxz/3dyz pair is referred to as the tetragonal (Δ) splitting, while the gap between the 3dxz/3dyz orbitals is the rhombic splitting (V; Figure 8b). The Δ and V splittings can be estimated experimentally through analysis of EPR g values, as discussed in the next section.
Figure 8.

(a) MO diagram derived from a spin-unrestricted DFT calculation (TZVP/B3LYP) of 1ox-CN. MOs are labeled according to their principal contributor, and energies correspond to the spin-down (β) orbitals. (b) Schematic depiction of Δ and V splittings of the t2g set of Fe 3d orbitals. (c) DFT-generated isosurface plot of the singly occupied natural MO of 1ox-CN (phenyl rings of TIP ligand omitted for clarity).
The covalent nature of the Fe–S bond in 1ox-CN is reflected in the composition of the singly occupied MO (SOMO), as determined through a natural orbital calculation based upon the DFT/B3LYP results. The SOMO of 1ox-CN shown in Figure 8c contains 75% Fe 3d and 20% S 3py character. Because of the Fe–S π-bonding interaction, the unpaired electron is partially delocalized onto the thiolate ligand, as indicated by the Mulliken spin population of 0.18 for the S atom of 1ox-CN. The Fe–S bond of 2ox-CN is similarly covalent, although the thiolate-based π-donor MO is partially delocalized over the aromatic ring.70 The longer Fe–S bonds of the enzymatic models reduce the S character of the SOMOs to 14% and 11% in β-CDO and α-CDO, respectively, indicative of markedly less covalent π-interactions.
Computational Analysis of EPR g Values.
For low-spin d5 complexes in six-coordinate environments, the anisotropy of the g tensor is mainly due to spin–orbit coupling (SOC) among the three states arising from the parent 2T2g ground state in Oh symmetry. However, the near-degeneracy of the 2T2g-derived states poses a challenge for the calculation of magnetic and EPR properties. While coupled-perturbed self-consistent field (CP-SCF) equations are often employed to account for SOC effects in DFT calculations, previous studies (by us and others) have found that this method often fails to reproduce the g values of low-spin nonheme FeIII systems.30,71,72 Indeed, as shown in Table 3, DFT/CP-SCF calculations for 1ox-CN greatly underestimate the g3 value and incorrectly predict a g1 value greater than 2.0, regardless of the functional used. To overcome such deficiencies, we recently developed an alternative approach that utilizes time-dependent DFT to calculate the Δ and V splittings (Figure 8b) of the t2g orbitals.72 These values are then entered into equations developed by Taylor to arrive at the computed g values. While this strategy proved effective in predicting the g values of various states of iron-type nitrile hydratase, it fails to explicitly treat metal–ligand covalency and also requires prior calibration with structurally characterized models to determine the SOC constant (λ).
Table 3.
Comparison of Computed and Experimental g Values and Ligand-Field Splittings for Relevant FeCN Adductsa
| model | method | g1 | g2 | g3 | rms devc | Δ (cm−1) | V (cm−1) |
|---|---|---|---|---|---|---|---|
| 1ox-CN | exptl | 1.988 | 2.161 | 2.204 | 4731d | 1094d | |
| DFT/PBE0/CP-SCF | 2.026 | 2.127 | 2.145 | 0.045 | |||
| DFT/B3LYP/CP-SCF | 2.020 | 2.110 | 2.131 | 0.055 | |||
| CASSCF/NEVPT2 | 1.946 | 2.183 | 2.234 | 0.032 | 4321 | 512 | |
| 2ox-CN | exptl | 1.964 | 2.161 | 2.231 | 4063d | 1292d | |
| CASSCF/NEVPT2 | 1.953 | 2.191 | 2.256 | 0.023 | 3886 | 431 | |
| α-CDO | exptlb | 1.937 | 2.235 | 2.379 | 2776d | 1171d | |
| CASSCF/NEVPT2 | 1.914 | 2.258 | 2.425 | 0.032 | 2833 | 1056 | |
| β-CDO | exptlb | 1.951 | 2.207 | 2.344 | 3126d | 1416d | |
| CASSCF/NEVPT2 | 1.930 | 2.228 | 2.350 | 0.017 | 3366 | 1013 | |
| Hybrid-1 | CASSCF/NEVPT2 | 1.931 | 2.206 | 2.389 | nae | 3280 | 1422 |
| Hybrid-2 | CASSCF/NEVPT2 | 1.971 | 2.283 | 2.352 | nae | 2849 | 785 |
The CASSCF/NEVPT2 calculations employed a CAS(11,13) active space and TZVP basis set.
rms dev = root-mean-square deviation between experimental and computed g values.
The experimental Δ and V values were calculated using Taylor’s equations for low-spin FeIII complexes in the absence of iron–ligand covalency, assuming a SOC parameter (λ) of 400 cm−1. See ref 68 for more details.
na = not applicable.
Neese and co-workers have demonstrated that complete active space self-consistent field (CASSCF) methods provide reliable g values for transition-metal complexes, particularly when dynamic correlation is incorporated via n-electron valence state second-order perturbation theory (NEVPT2).53 We therefore applied the CASSCF/NEVPT2 approach to each of the synthetic and enzymatic FeIII-CN models described above. Initial studies gauged the impact of active-space size on the calculated g values for 1ox-CN, starting with the minimal active space consisting of five active electrons in the five Fe 3d orbitals (i.e., CAS(5,5)). As shown in Table S2, the g values derived from the CAS(5,5) calculation (g1,2,3 = 1.932, 2.272, and 2.334) are significantly more anisotropic than the experimental values. However, improved results were obtained with the CAS(11,8) active space, which includes three occupied CysOEt-based orbitals. Further addition of a second Fe d shell (i.e., CAS(11,13)) yielded g values in excellent agreement with experiment, as evidenced by the root-mean-square deviation of just 0.023. Expansion of the basis set (from SVP to TZVP) caused only slight changes in the computed g values (Table S2), consistent with the basis-set stability of CASSCF/NEVPT2 calculations noted by Neese.53 Analysis of the g tensor revealed that the g1, g2, and g3 axes are aligned along the Fe–NH2, Fe–CCN, and Fe–S bonds, respectively, as indicated in Figure 8c. Following Taylor’s analysis, the average anisotropy of g2 and g3 (i.e., (g2 + g3)/2 − 2.0023) is dictated by Δ, while the rhombicity (V) accounts for the difference between g2 and g3.
Encouraged by our success with 1ox-CN, we performed CAS(11,13)/NEVPT2 calculations for the remaining FeIII-CN adducts using the TZVP basis set. In each case, the computed and experimental g values are in good agreement, as indicated by root-mean-square deviations of around 0.02 (Table 3). Most notably, the computational results reproduce the increased anisotropy of the enzymatic FeIII-CN adducts relative to the synthetic models, as well as the subtle differences between α-CDO and β-CDO. Through analysis of the ligand-field transition energies provided by the CASSCF/NEVPT2 calculations, we can extract the Δ and V splittings of the t2g orbitals for each model (Table 3). The computed Δ values of 1ox-CN and 2ox-CN are 4300 and 3900 cm−1, respectively, reflecting the considerable destabilization of the Fe 3dxy-based MO due to π-antibonding interactions with the thiolate donor (vide supra). In comparison, the Δ values of α-CDO and β-CDO are smaller by roughly 1000 cm−1, which increases the g tensor anisotropy by enhancing SOC-induced mixing among the 2T2g-derived states. The decrease in Δ is largely a consequence of the elongated Fe–S bonds in α-CDO and β-CDO relative to the synthetic complexes (Table 2). The computed V splittings are also greater for the enzymatic structures, although the experimentally determined rhombicities are roughly similar for both the synthetic and biological FeIII-CN adducts.
The CASSCF/NEVPT2 calculations help to illuminate the structural origins of the different g values exhibited by α-CDO and β-CDO. As described above, the presence of the Cys-Tyr cross-link lengthens the Fe–S bond and induces a decrease in the Fe–C–N bond angle. To determine whether these perturbations are responsible for the greater g anisotropy of α-CDO, we generated two hybrid structures. Hybrid-1 is identical with β-CDO apart from the Fe-CN unit, which adopts the bent orientation of α-CDO (Fe–C–N angle of 169°). In the Hybrid-2 structure, the Fe-CN unit is nearly linear as in β-CDO (Fe–C–N angle of 176°) but the Fe–SCys and Fe–NCys bond distances correspond to those found in α-CDO. As shown in Table 3, the Δ value is primarily determined by the length of the Fe–S bond, while the Fe–C–N bond angle has the greatest effect on the rhombicity parameter (V). Comparison of the computed g values indicates that the larger g3 value of α-CDO—in comparison to β-CDO—is mainly a consequence of the bend in the FeCN unit due to the Cys-Tyr cross-link. The g2 value is most sensitive to the Fe–S bond distance, while the effect of structural variations on g1 is less clear. These results support the conclusion that the observed shifts in g values between α-CDO and β-CDO arise from cross-linked-induced perturbations of both the CN and Cys ligands.
CONCLUSIONS
A variety of synthetic, spectroscopic, and computational methods have been used to elucidate the geometric and electronic structures of thiolate-ligated FeIII complexes of relevance to the active sites of thiol dioxygenases. These efforts included the preparation and structural characterization of [FeII(2-MTS)(Ph2TIP)]BPh4 (2), which serves as the first synthetic model of substrate-bound FeII-MDO (Figure 2). One-electron chemical oxidation of 2 and the previously reported 1 yielded ferric derivatives capable of binding small anions (namely, cyanide and azide) that are commonly used as spectroscopic probes of biological and synthetic Fe sites involved in O2 activation. The formation of six-coordinate FeIII-CN and FeIII-N3 adducts (1ox-CN, 2ox-CN, and 1ox-N3) was confirmed by examination with UV–vis absorption, EPR, and rRaman spectroscopies. DFT calculations determined that the unpaired electron of the FeIII-CN complexes occupies the Fe 3dxy-based MO that carries significant S character due to the covalent nature of the Fe–S π-bond. In addition, the g values derived from CASSCF/NEVPT2 calculations for 1ox-CN and 2ox-CN are in excellent agreement with the experimental data. The observed and calculated g tensors were rationalized on the basis of Δ and V splittings among the Fe 3d t2g orbitals.
Importantly, our studies of the synthetic CDO/MDO models facilitated the interpretation of data collected in parallel studies of CDO. We expanded upon the prior EPR studies of Pierce and co-workers by collecting rRaman spectra of Cys- and CN-bound FeIII-CDO. The assignment of ν(Fe–S), ν(Fe–CN), and δ(Fe–C–N) features in the rRaman spectra was aided by close similarities between the enzymatic and model spectra. The effect of the Cys-Tyr cross-link was examined by comparing rRaman spectra of Cys- and CN-treated WT CDO (cross-linked; α-CDO) and C93G CDO (non-cross-linked; β-CDO). Small shifts in the ν(Fe–S) and ν(Fe–CN) frequencies indicate that both the CN and Cys ligands are perturbed by formation of the cross-link. This finding is consistent with prior QM/MM calculations which provided the active site models that we used in our ab initio computational studies of CDO FeIII-CN adducts. To the best of our knowledge, this is the first time that the CASSCF/NEVPT2 approach has been applied to a metalloenzyme active site. The computed g values for α-CDO and β-CDO exhibit remarkably good agreement with experiment. Moreover, by analyzing the effect of structural variations on the computed g values and ligand-field splittings, we determined that the greater g anisotropy of α-CDO—in comparison to β-CDO—is the result of both the elongation of the Fe–S bond and the tilting of the Fe-CN unit caused by the Cys-Tyr cross-link.
It is somewhat curious that loss of the Cys-Tyr cross-link is accompanied by a slight decrease (9 cm−1) in the ν(Fe–S) frequency of Cys- and CN-bound FeIII-CDO (Figure 6), even though the computational results indicate that the Fe–S bond is shorter in the non-cross-linked active site. This finding would seem to contradict Badger’s rule, which states that bond length is inversely proportional to vibrational frequency.73,74 We noted a similar discrepancy in our studies of the synthetic adducts: the ν(Fe–S) frequency of 1ox-CN (341 cm−1) is lower than that of 1ox-N3 (377 cm−1), even though DFT calculations provide Fe–S bond distances of 2.18 and 2.32 Å, respectively.
To explore this matter further, we sought to compare the spin states, ν(Fe–S) frequencies, and computed Fe–S bond distances of biological and synthetic nonheme FeIII centers with mixed S/N coordination reported in the literature. From the data compiled in Table 4, and the corresponding plots in Figure S7, it is clear that the ν(Fe–S) modes of these nonheme iron sites do not behave according to Badger’s rule. To determine whether the lack of a clear relationship could be due to errors in the calculated Fe–S bonds, we performed geometry optimizations of 1ox-CN and 2ox-CN using a range of DFT functionals (nonhybrid, meta-GGA, and hybrid). As shown in Table S3, the absolute and relative Fe–S bond distances are largely independent of functional, suggesting that differences in computational methods cannot entirely account for the observed breakdown in Badger’s rule. Another likely explanation is that the Fe–S stretching motion is often strongly coupled to δ(S–C–C) and δ(C–C–N) motions, and thus ν(Fe–S) character is distributed across several modes. Because of this, the ν(Fe–S) frequency is not a dependable marker of either spin state or Fe–S bond distance in nonheme iron systems. In contrast, our results suggest that EPR studies of low-spin FeIII sites can provide more reliable structural insights when they are conducted in tandem with DFT and CASSCF/NEVPT2 calculations.
Table 4.
Spin States, Fe–S Bond Distances, and Fe–S Stretching Frequencies of Nonheme FeIII Centers with Mixed S/N Ligation
| spin | r(Fe–S), Åa | ν(Fe–S), cm−1 | ref | |
|---|---|---|---|---|
| Synthetic | ||||
| 1ox-CN | 1/2 | 2.18 | 341 | this work |
| 2ox-CN | 1/2 | 2.20 | 347 | this work |
| 1ox-N3 | 5/2 | 2.32 | 377 | this work |
| [FeIII(cyclam-PrS)(OOH)]+ | 5/2 | 2.36 | 352 | 61 |
| Biological | ||||
| Cys-bound WT FeIII-CDO | 5/2 | 2.27 | 340 | 59 |
| Cys/CN-bound FeIII-CDO (WT) | 1/2 | 2.28 | 377 | 30 |
| Cys/CN-bound FeIII-CDO (C93G) | 1/2 | 2.26 | 368 | 30 |
| FeIII-SOR | 5/2 | 2.36b | 323 | 58 |
| CN-bound FeIII-SOR | 1/2 | 2.28 | 323 | 23 |
| nitrile hydratase (low pH form) | 1/2 | 2.21c | 373 | 65 |
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for financial support from the National Institutes of Health (GM126522 to A.T.F. and GM117120 to T.C.B.). Initial funding for this project was provided by the National Science Foundation (CHE-1056845 to A.T.F.). Improvements to the X-band EPR instrument at Marquette University were funded through a grant from the National Science Foundation (CHE-1532168).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.9b02432.
1H NMR spectra of 2, EPR spectra of 1ox and 2ox, rRaman spectra of 1ox-N3, UV–vis spectra of Cys/CN adducts of FeIII-CDO, plots of Fe–S bond distances versus stretching frequencies, metric parameters determined by X-ray crystallography, CASSCF/NEVTP2 computed g values for 1ox-CN, and coordinates for computational structures (PDF)
Accession Codes
CCDC 1945809 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).Stipanuk MH; Simmons CR; Andrew Karplus P; Dominy JE Jr. Thiol dioxygenases: unique families of cupin proteins. Amino Acids 2011, 41, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Stipanuk MH; Londono M; Hirschberger LL; Hickey C; Thiel DJ; Wang L Evidence for expression of a single distinct form of mammalian cysteine dioxygenase. Amino Acids 2004, 26, 99–106. [DOI] [PubMed] [Google Scholar]
- (3).Stipanuk MH Sulfur amino acid metabolism: Pathways for production and removal of homocysteine and cysteine. Annu. Rev. Nutr 2004, 24, 539–577. [DOI] [PubMed] [Google Scholar]
- (4).Chai SC; Jerkins AA; Banik JJ; Shalev I; Pinkham JL; Uden PC; Maroney MJ Heterologous expression, purification, and characterization of recombinant rat cysteine dioxygenase. J. Biol. Chem 2005, 280, 9865–9869. [DOI] [PubMed] [Google Scholar]
- (5).Dominy JE Jr.; Simmons CR; Hirschberger LL; Hwang J; Coloso RM; Stipanuk MH Discovery and Characterization of a Second Mammalian Thiol Dioxygenase, Cysteamine Dioxygenase. J. Biol. Chem 2007, 282, 25189–25198. [DOI] [PubMed] [Google Scholar]
- (6).Masson N; Keeley TP; Giuntoli B; White MD; Puerta ML; Perata P; Hopkinson RJ; Flashman E; Licausi F; Ratcliffe PJ Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science 2019, 365, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dominy JE Jr.; Simmons CR; Karplus PA; Gehring AM; Stipanuk MH Identification and characterization of bacterial cysteine dioxygenases: a new route of cysteine degradation for eubacteria. J. Bacteriol 2006, 188, 5561–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Driggers CM; Hartman SJ; Karplus PA Structures of Arg-and Gln-type bacterial cysteine dioxygenase homologs. Protein Sci. 2015, 24, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bruland N; Wuebbeler JH; Steinbuechel A 3-Mercaptopropionate dioxygenase, a cysteine dioxygenase homolog, catalyzes the initial step of 3-mercaptopropionate catabolism in the 3,3-thiodipropionic acid-degrading bacterium Variovorax paradoxus. J. Biol. Chem 2009, 284, 660–672. [DOI] [PubMed] [Google Scholar]
- (10).Pierce BS; Subedi BP; Sardar S; Crowell JK The “Gln-Type” Thiol Dioxygenase from Azotobacter vinelandii Is a 3-Mercaptopropionic Acid Dioxygenase. Biochemistry 2015, 54, 7477–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tchesnokov EP; Fellner M; Siakkou E; Kleffmann T; Martin LW; Aloi S; Lamont IL; Wilbanks SM; Jameson GNL The Cysteine Dioxygenase Homologue from Pseudomonas aeruginosa Is a 3-Mercaptopropionate Dioxygenase. J. Biol. Chem 2015, 290, 24424–24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Driggers CM; Cooley RB; Sankaran B; Hirschberger LL; Stipanuk MH; Karplus PA Cysteine Dioxygenase Structures from pH 4 to 9: Consistent Cys-Persulfenate Formation at Intermediate pH and a Cys-Bound Enzyme at Higher pH. J. Mol. Biol 2013, 425, 3121–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Driggers CM; Kean KM; Hirschberger LL; Cooley RB; Stipanuk MH; Karplus PA Structure-Based Insights into the Role of the Cys-Tyr Crosslink and Inhibitor Recognition by Mammalian Cysteine Dioxygenase. J. Mol. Biol 2016, 428, 3999–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McCoy JG; Bailey LJ; Bitto E; Bingman CA; Aceti DJ; Fox BG; Phillips GN, Jr Structure and mechanism of mouse cysteine dioxygenase. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 3084–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang Y; Griffith WP; Li J; Koto T; Wherritt DJ; Fritz E; Liu A Cofactor Biogenesis in Cysteamine Dioxygenase: C-F Bond Cleavage with Genetically Incorporated Unnatural Tyrosine. Angew. Chem., Int. Ed 2018, 57, 8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Aloi S; Davies CG; Karplus PA; Wilbanks SM; Jameson GNL Substrate specificity in thiol dioxygenases. Biochemistry 2019, 58, 2398–2407. [DOI] [PubMed] [Google Scholar]
- (17).Dominy JE Jr.; Hwang J; Guo S; Hirschberger LL; Zhang S; Stipanuk MH Synthesis of Amino Acid Cofactor in Cysteine Dioxygenase Is Regulated by Substrate and Represents a Novel Post-translational Regulation of Activity. J. Biol. Chem 2008, 283, 12188–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang Y; Griffith WP; Li J; Koto T; Wherritt DJ; Fritz E; Liu A Cofactor Biogenesis in Cysteamine Dioxygenase: C-F Bond Cleavage with Genetically Incorporated Unnatural Tyrosine. Angew. Chem., Int. Ed 2018, 57, 8149–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li J; Griffith WP; Davis I; Shin I; Wang J; Li F; Wang Y; Wherritt DJ; Liu A Cleavage of a carbon-fluorine bond by an engineered cysteine dioxygenase. Nat. Chem. Biol 2018, 14, 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ye S; Wu X. a.; Wei L; Tang D; Sun P; Bartlam M; Rao Z An Insight into the Mechanism of Human Cysteine Dioxygenase. Key Roles of the Thioether-Bonded Tyrosine-Cysteine Cofactor. J. Biol. Chem 2007, 282, 3391–3402. [DOI] [PubMed] [Google Scholar]
- (21).Crowell JK; Sardar S; Hossain MS; Foss FW Jr.; Pierce BS Non-chemical proton-dependent steps prior to O2-activation limit Azotobacter vinelandii 3-mercaptopropionic acid dioxygenase (MDO) catalysis. Arch. Biochem. Biophys 2016, 604, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li W; Pierce BS Steady-state substrate specificity and O2-coupling efficiency of mouse cysteine dioxygenase. Arch. Biochem. Biophys 2015, 565, 49–56. [DOI] [PubMed] [Google Scholar]
- (23).Clay MD; Yang T-C; Jenney FE Jr.; Kung IY; Cosper CA; Krishnan R; Kurtz DM Jr.; Adams MWW; Hoffman BM; Johnson MK Geometries and Electronic Structures of Cyanide Adducts of the Non-Heme Iron Active Site of Superoxide Reductases: Vibrational and ENDOR Studies. Biochemistry 2006, 45, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Clay MD; Cosper CA; Jenney FE Jr.; Adams MWW; Johnson MK Nitric oxide binding at the mononuclear active site of reduced pyrococcus furiosus superoxide reductase. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3796–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Simianu MC; Kincaid JR Resonance Raman Spectroscopic Detection of Both Linear and Bent Fe-CN Fragments for the Cyanide Adducts of Cytochrome P-450 Camphor and Its Substrate-Bound Forms. Relevance to the “Charge Relay” Mechanism. J. Am. Chem. Soc 1995, 117, 4628–36. [Google Scholar]
- (26).Jackson TA; Karapetian A; Miller A-F; Brunold TC Probing the Geometric and Electronic Structures of the Low-Temperature Azide Adduct and the Product-Inhibited Form of Oxidized Manganese Superoxide Dismutase. Biochemistry 2005, 44, 1504–1520. [DOI] [PubMed] [Google Scholar]
- (27).Pierce BS; Gardner JD; Bailey LJ; Brunold TC; Fox BG Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: Electronic structure of the active site and mechanistic implications. Biochemistry 2007, 46, 8569–8578. [DOI] [PubMed] [Google Scholar]
- (28).Li J; Koto T; Davis I; Liu A Probing the Cys-Tyr Cofactor Biogenesis in Cysteine Dioxygenase by the Genetic Incorporation of Fluorotyrosine. Biochemistry 2019, 58, 2218–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Blaesi EJ; Fox BG; Brunold TC Spectroscopic and Computational Investigation of Iron(III) Cysteine Dioxygenase: Implications for the Nature of the Putative Superoxo-Fe(III) Intermediate. Biochemistry 2014, 53, 5759–5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li W; Blaesi EJ; Pecore MD; Crowell JK; Pierce BS Second-sphere interactions between the C93-Y157 crosslink and the substrate-bound Fe site influence the O2 coupling efficiency in mouse cysteine dioxygenase. Biochemistry 2013, 52, 9104–9119. [DOI] [PubMed] [Google Scholar]
- (31).Fischer AA; Stracey N; Lindeman SV; Brunold TC; Fiedler AT Synthesis, x-ray structures, electronic properties, and O2/NO reactivities of thiol dioxygenase active-site models. Inorg. Chem 2016, 55, 11839–11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).McQuilken AC; Jiang Y; Siegler MA; Goldberg DP Addition of Dioxygen to an N4S(thiolate) Iron(II) Cysteine Dioxygenase Model Gives a Structurally Characterized Sulfinato-Iron(II) Complex. J. Am. Chem. Soc 2012, 134, 8758–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).McQuilken AC; Ha Y; Sutherlin KD; Siegler MA; Hodgson KO; Hedman B; Solomon EI; Jameson GNL; Goldberg DP Preparation of Non-heme {FeNO}7 Models of Cysteine Dioxygenase: Sulfur versus Nitrogen Ligation and Photorelease of Nitric Oxide. J. Am. Chem. Soc 2013, 135, 14024–14027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).McQuilken AC; Goldberg DP Sulfur oxygenation in biomimetic non-heme iron-thiolate complexes. Dalton Trans 2012, 41, 10883–10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Blakely MN; Dedushko MA; Poon PCY; Villar-Acevedo G; Kovacs JA Formation of a Reactive, Alkyl Thiolate-Ligated FeIII-Superoxo Intermediate Derived from Dioxygen. J. Am. Chem. Soc 2019, 141, 1867–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bittner MM; Baus JS; Lindeman SV; Fiedler AT Synthesis and Structural Characterization of Iron(II) Complexes with Tris(imidazolyl)phosphane Ligands: A Platform for Modeling the 3-Histidine Facial Triad of Nonheme Iron Dioxygenases. Eur. J. Inorg. Chem 2012, 2012, 1848–1856. [Google Scholar]
- (37).Blaesi EJ; Fox BG; Brunold TC Spectroscopic and computational Investigation of the H155A variant of cysteine dioxygenase: Geometric and electronic consequences of a third-sphere amino acid substitution. Biochemistry 2015, 54, 2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Blaesi EJ; Gardner JD; Fox BG; Brunold TC Spectroscopic and computational characterization of the NO adduct of substrate-bound Fe(II) cysteine dioxygenase: Insights into the mechanism of O2 activation. Biochemistry 2013, 52, 6040–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Fischer DS; Price DC Simple serum iron method using the new sensitive chromogen tripyridyl-s-triazine. Clin. Chem 1964, 10, 21–31. [PubMed] [Google Scholar]
- (40).Stoll S; Schweiger A EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson 2006, 178, 42–55. [DOI] [PubMed] [Google Scholar]
- (41).Neese F ORCA - An Ab Initio, DFT and Semiempirical Electronic Structure Package, versions 3.0 and 4.0; Max Planck Institute for Chemical Energy Conversion: Muelheim, Germany, 2017. [Google Scholar]
- (42).Becke AD Density-Functional Thermochemistry. 3. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (43).Lee CT; Yang WT; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B: Condens. Matter Mater. Phys 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (44).Schafer A; Huber C; Ahlrichs R Fully Optimized Contracted Gaussian-Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys 1994, 100, 5829–5835. [Google Scholar]
- (45).Schafer A; Horn H; Ahlrichs R Fully Optimized Contracted Gaussian-Basis Sets for Atoms Li to Kr. J. Chem. Phys 1992, 97, 2571–2577. [Google Scholar]
- (46).Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (47).Neese F; Wennmohs F; Hansen A; Becker U Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange. Chem. Phys 2009, 356, 98–109. [Google Scholar]
- (48).Eichkorn K; Treutler O; Oehm H; Haeser M; Ahlrichs R Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett 1995, 240, 283–290. [Google Scholar]
- (49).Neese F Quantum chemical calculations of spectroscopic properties of metalloproteins and model compounds: EPR and Moessbauer properties. Curr. Opin. Chem. Biol 2003, 7, 125–135. [DOI] [PubMed] [Google Scholar]
- (50).Neese F Prediction of electron paramagnetic resonance g values using coupled perturbed Hartree-Fock and Kohn-Sham theory. J. Chem. Phys 2001, 115, 11080–11096. [Google Scholar]
- (51).Sinnecker S; Neese F; Noodleman L; Lubitz W Calculating the electron paramagnetic resonance parameters of exchange coupled transition metal complexes using broken symmetry density functional theory: Application to a Mn(III)/Mn(IV) model compound. J. Am. Chem. Soc 2004, 126, 2613–2622. [DOI] [PubMed] [Google Scholar]
- (52).Neese F Metal and ligand hyperfine couplings in transition metal complexes: the effect of spin-orbit coupling as studied by coupled perturbed Kohn-Sham theory. J. Chem. Phys 2003, 118, 3939–3948. [Google Scholar]
- (53).Singh SK; Atanasov M; Neese F Challenges in Multireference Perturbation Theory for the Calculations of the g-Tensor of First-Row Transition-Metal Complexes. J. Chem. Theory Comput 2018, 14, 4662–4677. [DOI] [PubMed] [Google Scholar]
- (54).Singh SK; Eng J; Atanasov M; Neese F Covalency and chemical bonding in transition metal complexes: An ab initio based ligand field perspective. Coord. Chem. Rev 2017, 344, 2–25. [Google Scholar]
- (55).Addison AW; Rao TN; Reedijk J; Vanrijn J; Verschoor GC Synthesis, Structure, and Spectroscopic Properties of Copper-(II) Compounds Containing Nitrogen Sulfur Donor Ligands. J. Chem. Soc., Dalton Trans 1984, 1349–1356. [Google Scholar]
- (56).Connelly NG; Geiger WE Chemical Redox Agents for Organometallic Chemistry. Chem. Rev 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- (57).Gutman CT; Guzei IA; Brunold TC Structural, Spectroscopic, and Computational Characterization of the Azide Adduct of FeIII(2,6-diacetylpyridinebis(semioxamazide)), a Functional Analogue of Iron Superoxide Dismutase. Inorg. Chem 2013, 52, 8909–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Clay MD; Jenney FE Jr.; Noh HJ; Hagedoorn PL; Adams MWW; Johnson MK Resonance Raman Characterization of the Mononuclear Iron Active-Site Vibrations and Putative Electron Transport Pathways in Pyrococcus furiosus Superoxide Reductase. Biochemistry 2002, 41, 9833–9841. [DOI] [PubMed] [Google Scholar]
- (59).Gardner JD; Pierce BS; Fox BG; Brunold TC Spectroscopic and Computational Characterization of Substrate-Bound Mouse Cysteine Dioxygenase: Nature of the Ferrous and Ferric Cysteine Adducts and Mechanistic Implications. Biochemistry 2010, 49, 6033–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Fiedler AT; Brunold TC Combined Spectroscopic/Computational Study of Binuclear Fe(I)-Fe(I) Complexes: Implications for the Fully-Reduced Active-Site Cluster of Fe-Only Hydrogenases. Inorg. Chem 2005, 44, 1794–1809. [DOI] [PubMed] [Google Scholar]
- (61).Kitagawa T; Dey A; Lugo-Mas P; Benedict JB; Kaminsky W; Solomon E; Kovacs JA A Functional Model for the Cysteinate-Ligated Non-Heme Iron Enzyme Superoxide Reductase (SOR). J. Am. Chem. Soc 2006, 128, 14448–14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Tremey E; Bonnot F; Moreau Y; Berthomieu C; Desbois A; Favaudon V; Blondin G; Houee-Levin C; Niviere V Hydrogen bonding to the cysteine ligand of superoxide reductase: acid-base control of the reaction intermediates. JBIC, J. Biol. Inorg. Chem 2013, 18, 815–830. [DOI] [PubMed] [Google Scholar]
- (63).Siiman O; Carey PR Resonance Raman spectra of some ferric and cupric thiolate complexes. J. Inorg. Biochem 1980, 12, 353–62. [Google Scholar]
- (64).Clay MD; Emerson JP; Coulter ED; Kurtz DM; Johnson MK Spectroscopic characterization of the [Fe(His)4-(Cys)] site in 2Fe-superoxide reductase from Desulfovibrio vulgaris. JBIC, J. Biol. Inorg. Chem 2003, 8, 671–682. [DOI] [PubMed] [Google Scholar]
- (65).Brennan BA; Cummings JG; Chase DB; Turner IM Jr.; Nelson MJ Resonance Raman spectroscopy of nitrile hydratase, a novel iron-sulfur enzyme. Biochemistry 1996, 35, 10068–10077. [DOI] [PubMed] [Google Scholar]
- (66).Hirota S; Ogura T; Shinzawa-Itoh K; Yoshikawa S; Kitagawa T Observation of Multiple CN-Isotope-Sensitive Raman Bands for CN Adducts of Hemoglobin, Myoglobin, and Cytochrome c Oxidase: Evidence for Vibrational Coupling between the Fe-C-N Bending and Porphyrin In-Plane Modes. J. Phys. Chem 1996, 100, 15274–15279. [Google Scholar]
- (67).For both species, analysis of the computed 32S/34S isotope shifts revealed that several modes between 350 and 400 cm−1 contain partial ν(Fe–S) character. The frequencies of these modes were weighted according to the magnitude of their sulfur isotope shifts to obtain the “true” computed ν(Fe–S) frequency.
- (68).Taylor CPS The EPR of Low Spin Heme Complexes. Biochim. Biophys. Acta, Protein Struct 1977, 491, 137–149. [DOI] [PubMed] [Google Scholar]
- (69).Griffith JS Theory of electron resonance in ferrihemoglobin azide. Nature (London U. K.) 1957, 180, 30–1. [DOI] [PubMed] [Google Scholar]
- (70).Fiedler AT; Halfen HL; Halfen JA; Brunold TC Synthesis, Structure Determination, and Spectroscopic/Computational Characterization of a Series of Fe(II)-Thiolate Model Complexes: Implications for Fe-S Bonding in Superoxide Reductases. J. Am. Chem. Soc 2005, 127, 1675–1689. [DOI] [PubMed] [Google Scholar]
- (71).Light KM; Yamanaka Y; Odaka M; Solomon EI Spectroscopic and computational studies of nitrile hydratase: insights into geometric and electronic structure and the mechanism of amide synthesis. Chem. Sci 2015, 6, 6280–6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Stein N; Gumataotao N; Hajnas N; Wu R; Lankathilaka KPW; Bornscheuer UT; Liu D; Fiedler AT; Holz RC; Bennett B Multiple States of Nitrile Hydratase from Rhodococcus equi TG328–2: Structural and Mechanistic Insights from Electron Paramagnetic Resonance and Density Functional Theory Studies. Biochemistry 2017, 56, 3068–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Badger RM A relation between internuclear distances and bond force constants. J. Chem. Phys 1934, 2, 128–31. [Google Scholar]
- (74).The mathematical form of Badger’s rule is re = (C/ν2/3) + d, where re is the equilibrium bond distance, ν is the vibrational frequency, and C and d are empirically determined constants for a given combination of atoms.
- (75).Clay MD; Jenney FE Jr.; Hagedoorn PL; George GN; Adams MWW; Johnson MK Spectroscopic Studies of Pyrococcus furiosus Superoxide Reductase: Implications for Active-Site Structures and the Catalytic Mechanism. J. Am. Chem. Soc 2002, 124, 788–805. [DOI] [PubMed] [Google Scholar]
- (76).Scarrow RC; Brennan BA; Cummings JG; Jin H; Duong DJ; Kindt JT; Nelson MJ X-ray spectroscopy of nitrile hydratase at pH 7 and 9. Biochemistry 1996, 35, 10078–10088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.