

Abstract
Megalencephalic leukoencephalopathy (MLC) with subcortical cysts, also known as Van der Knaap disease (MIM #604004) is an autosomal recessive neurological disorder characterized by early onset macrocephaly, epilepsy, neurological deterioration with cerebellar ataxia and spasticity. An 8-month-old boy was admitted to our pediatric neurology clinic with macrocephaly. His brain magnetic resonance imaging (MRI) revealed bilateral, diffuse, symmetric structural white matter abnormalities, relatively sparing the cerebellum and bilateral subcortical temporal cysts. The diagnosis of Van der Knaap disease was suspected based on the clinical features and imaging findings and the genetic analysis revealed a novel homozygous c.768+2T>C mutation of the MLC1 gene. For determination of the novel splice-site mutation’s effect, cDNA amplification was performed. cDNA analysis showed that the splice-site c.768+2T>C mutation gave rise to exon 9 skipping.
Keywords: cDNA sequencing, Exon skipping, Leuko-encephalopathy, MLC1 gene, Splice-site mutation
Introduction
Megalencephalic leukoencephalopathy (MLC) with subcortical cysts (MIM #604004) is an autosomal recessive neurological disorder characterized by early onset macrocephaly, epilepsy, neurological deterioration with cerebellar ataxia and spasticity [1]. The majority of MLC cases (~75.0%) are associated with MLC1 gene mutations and in a few patients (~20.0%) is associated with HEPACAM gene mutations [2]. In this study, we present a novel homozygous c.768+2T>C (IVS9+2T>C) splice-site mutation in the MLC1 gene in a Turkish boy and a functional study showing exon 9 skipping.
Case Report
An 8-month-old boy was admitted to our pediatric neurology clinic with macrocephaly. He was born as the third child to consanguineous parents. His developmental milestones were normal; however, his parents noted that he had a large head. At admission, his head circumference was 52 cm (>97 percentile) and his neurological examination was unremarkable. His magnetic resonance imaging (MRI) scan revealed bilateral, diffuse, symmetric structural white matter abnormalities, relatively sparing the cerebellum and bilateral subcortical temporal cysts (Figure 1). Routine hematological, biochemical, and blood gas analyses were normal. Metabolic tests including tandem mass spectrometry (MS/MS), plasma amino acids, urine organic acids, lysosomal enzyme panel and very long chain fatty acids test were all normal. The diagnosis of Van der Knaap disease was suspected based on the clinical features and imaging findings and the genetic analysis revealed a novel homozygous c.768+2T>C mutation of the MLC1 gene. His Denver II Developmental screening test at 16 months revealed mild delay in gross and fine motor domains and he was referred to physical therapy and rehabilitation. He began walking at 18 months of age. He is now 4 years old. His head circumference is 56 cm (>97 percentile). He has difficulties in walking alone and his family reported that they noticed recently that his speech has become slurred. On neurological examination, he has increased tonus prominent in the lower extremities associated with other pyramidal signs including increased deep tendon reflexes, extensor plantar responses and ankle clonus. His Denver II Developmental screening test revealed delay in all developmental domains mainly in gross motor domain and language is the least affected domain.
Figure 1.
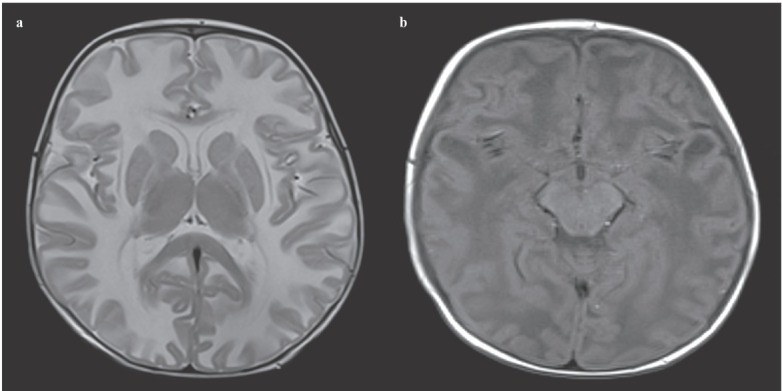
(a) Axial T2 weighted MRI shows bilateral, diffuse, symmetric hyperintense lesions in the cerebral white matter. (b) Axial T1 weighted MRI shows bilateral temporal cysts.
Molecular genetic studies
All experimental procedures were conducted in accordance with the recommendations of the Ethics Committee of Marmara University, İstanbul, Turkey, and informed written consent was obtained from parents. DNA was isolated from leucocytes. All coding exons and exon-intron boundaries of the MLC1 gene were sequenced using the MiSeq system (Illumina, Inc, San Diego, CA, USA). Total mRNA was isolated using the Qiagen RNeasy Mini Kit (Qiagen GmbH, Hilden, Germany) and reverse-transcribed using the QuantiTect Reverse Transcription Kit (Qiagen GmbH) following the manufacturer’s protocol. The cDNA amplification of the MLC1 gene was examined using the following primers: forward (5’-CAA CAT TCT GGA CGA AGT GCC AT-3’) and reverse (5’-CAG CCT TGC ACT GAC CTT GA-3’). All amplified products were purified directly and sequencing reaction was accomplished in an ABI PRISM® 3130xl genetic analyzer (Applied Biosystems, Foster City, CA, USA).
Discussion
Megalencephalic leukoencephalopathy is a rare disorder, two phenotypes are observed in MLC. The classic phenotype of MLC is characterized by early-onset macrocephaly, mild motor developmental delay, seizures, ataxia, spasticity, and sometimes extrapyramidal findings; and generally mild mental regression. An improving phenotype of MLC has similar clinical features but these findings recover in time [1]. The diagnosis of MLC is established in individuals with typical clinical and brain MRI findings [3]. Description of homozygous mutations in the MLC1 or HEPACAM genes can prove the diagnosis of classic phenotype of MLC. Identification of a heterozygous HEPACAM pathogenic variant can establish the diagnosis of MLC with improving phenotype [4]. Our case was diagnosed on the basis of the classic phenotype’s clinical and radiological features including macrocephaly, mild motor developmental delay, bilateral, diffuse, symmetric structural white matter abnormalities, relatively sparing the cerebellum and bilateral subcortical temporal cysts.
In the molecular study, a novel homozygous splice-site variant (NM_015166: c.768+2T>C) in intron 9 of the MLC1 gene was identified (Figure 2). The present variant has not been previously reported in population studies (ESP, ExAC, 1000G, and gnomAD). His father and mother were heterozygous. In silico analysis using Human Splicing Finder (HSF) and Mutation Taster was supportive of pathogenicity. According to the HSF prediction, the splice-site c.768+2T>C mutation most probably affecting splicing via alteration of the wild type donor splice site.
Figure 2.
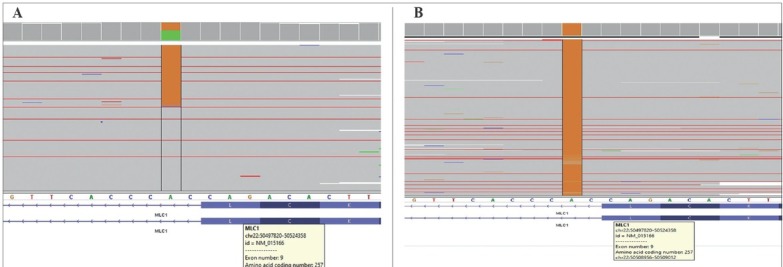
(A) Integrative Genomics Viewer (IGV) sequence data of the parents showed heterozygous c.768+ 2T>C mutation. (B) The IGV sequence data of the patient showed homozygous c.768+2T>C mutation.
For determination of the novel splice-site mutation’s effect, the exons 7-11 of MLC1 cDNA were amplified. Only a mutated single band (403 bp) appeared in the patient, but in his parents, a 460 bp wild-type and 403 bp mutated bands were detected. The Sanger sequencing of polymerase chain reaction (PCR) products showed that the 403 bp mutated allele matched the deletion of exon 9 in the MLC1 gene (Figure 3). In conclusion, cDNA analysis showed that the splice-site c.768+2T>C mutation gave rise to exon 9 skipping.
Figure 3.
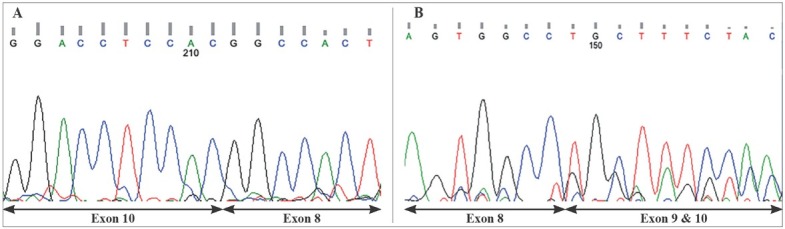
(A) Exon 9 was not found in cDNA (cDNA sequence of the patient, reverse strand). (B) The parents had two strands for the heterozygous mutation. Exons 9 and 10 were overlapping in cDNA (cDNA sequence of the parents, forward strand).
In summary, we report the novel homozygous splice-site mutation in the MLC1 gene causing a classic phenotype of MLC. Functional effect of the splice-site mutation was explained by cDNA analysis and was contributed to the literature.
Footnotes
Declaration of Interest. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Funding. (if applicable)
References
- 1.van der Knaap MS, Barth PG, Stroink H, van Nieuwenhuizen O, Arts WF, Hoogenraad F. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol. 1995;37(3):324–334. doi: 10.1002/ana.410370308. et al. [DOI] [PubMed] [Google Scholar]
- 2.Choi SA, Kim SY, Yoon J, Choi J, Park SS, Seong MW. A unique mutational spectrum of MLC1 in Korean patients with megalencephalic leukoencephaopalopathy with subcortical cysts: p.Ala275Asp founder mutation and maternal uniparental disomy of chromosome 22. Ann Lab Med. 2017;37(6):516–521. doi: 10.3343/alm.2017.37.6.516. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Voorn JP, Pouwels PJ, Hart AA, Serrarens J, Willemsen MA, Kremer HP. Childhood white matter disorders: Quantitative MR imaging and spectroscopy. Radiology. 2006;241(2):510–517. doi: 10.1148/radiol.2412051345. et al. [DOI] [PubMed] [Google Scholar]
- 4.Sugiura C, Shiota M, Maegaki Y, Yoshida K, Koeda T, Kitahara T. Late-onset neuropsychological symptoms in a Japanese patient with megalencephalic leuko-encephalopathy with subcortical cysts. Neuro-pediatrics. 2006;37(5):286–290. doi: 10.1055/s-2006-955930. et al. [DOI] [PubMed] [Google Scholar]