

Abstract
Molecular decision‐makers of photoreceptor (PRC) membrane organization and gene regulation are critical to understanding sight and retinal degenerations that lead to blindness. Using Mfrprd6mice, which develop PRC degeneration, we uncovered that membrane‐type frizzled‐related protein (MFRP) participates in docosahexaenoic acid (DHA, 22:6) enrichment in a manner similar to adiponectin receptor 1 (AdipoR1). Untargeted imaging mass spectrometry demonstrates cell‐specific reduction of phospholipids containing 22:6 and very long‐chain polyunsaturated fatty acids (VLC‐PUFAs) in Adipor1−/−and Mfrprd6 retinas. Gene expression of pro‐inflammatory signaling pathways is increased and gene‐encoding proteins for PRC function decrease in both mutants. Thus, we propose that both proteins are necessary for retinal lipidome membrane organization, visual function, and to the understanding of the early pathology of retinal degenerative diseases.
Keywords: Adipor1, inflammatory signaling, matrix‐assisted laser desorption/ionization imaging mass spectrometry (MALDI IMS), retinal degenerations, RPE cell, VLC‐PUFAs
Abbreviations
- AdipoR1
Adiponectin Receptor 1
- AMD
age‐related macular degeneration
- C1QTNF5/CTRP5
C1q tumor necrosis factor‐related protein‐5
- DHB
dihydroxybenzoic acid
- ELOVL4
ELOngation of Very Long‐chain fatty acids‐4
- ELVs
elovanoids
- EPA
eicosapentaenoic acid
- ERG
electroretinograms
- JNK
c‐Jun kinases
- MALDI IMS
mass spectrometry‐based molecular imaging
- MALDI IMS
matrix‐assisted laser desorption/ionization imaging mass spectrometry
- MFRP
membrane‐type frizzled‐related protein
- NPD1
neuroprotectin D1
- OCT
optical coherence tomography
- PC
phosphatidylcholine
- PCA
Principal Component Analysis
- PE
phosphatidylethanolamine
- PRC
photoreceptor
- PS
phosphatidylserine
- RPE
retinal pigment epithelium
- SD‐OCT
spectral domain‐optical coherence tomography
- UOS
uncompensated oxidative stress
- VLC‐PUFAs
very long‐chain polyunsaturated fatty acids
- VLC‐SFAs
very long‐chain saturated fatty acids
- WT
wild‐type
1. INTRODUCTION
MFRP is a glycosylated transmembrane protein with an extracellular frizzled‐related cysteine‐rich domain1, 2 that recognizes frizzled Wnt receptors,2, 3 and participates in cell fate and development.1, 3 MFRP is expressed in the retinal pigment epithelium (RPE) and ciliary bodies,2 and gene mutations encoding this protein are associated with photoreceptor cell (PRC) degeneration, nanophthalmos, posterior microphthalmia, retinitis pigmentosa, foveoschisis, and optic disc drusen.4 The rd6 mice displays a mutated MFRP and also leads to retinal degeneration.2 Fundus analysis of these mice reveals discrete dots across the retina due to macrophage infiltration in the subretinal space5, 6, 7 as in flecked retinal diseases8 with similarities to human retinitis punctata albescens. 7, 9 Mfrprd6 has a 4 bp deletion in a splice donor sequence resulting in exon 4 being skipped, and in a truncated protein.2
Although MFRP mutations are linked to PRC degeneration, there is an incomplete understanding of MFRP protein function10, 11 and its potential association with ADIPOR1 (Adipor1) including a shared pathogenic mechanism due to phenotypic similarities of the mutants of these two proteins.10 Moreover, comparisons of Mfrprd6 mice with wild‐type (WT) littermates revealed the absence of ADIPOR1 in the RPE,10 but retention in the retina, which suggested that an ADIPOR1 deficiency in RPE may initiate retinal degeneration in rd6 mice.10
ADIPOR1 is expressed in the retina and RPE, and the mutant of the Adipor1 gene in these cells results in inability to take up and incorporate docosahexaenoic acid (DHA, 22:6,n‐3), loss of PRC, and retinal degeneration.10, 12 A single amino acid mutation of Adipor1 occurs in different forms of retinitis pigmentosa.13, 14 Polymorphisms of this receptor have been found in age‐related macular degeneration (AMD)15 as well as in other forms of retinal degenerations. 22:6 is an omega‐3 (n‐3) essential fatty acid highly enriched in and required for the biogenesis of PRC membranes16 and is supplied either directly by diet or synthesized from dietary linolenic acid in the liver (18:3,n‐3).17 22:6 supplied by the liver 18 is taken up from the choriocapillaris by the RPE and then delivered to the PRC inner segments.16, 19 Here, it is incorporated at the sn‐2 position of the glycerol backbone, especially in phosphatidylcholine (PC), which comprises more than 50% of PRC phospholipids20 mainly used for the biogenesis of PRC outer segments.21, 22 22:6‐containing phospholipids modulate lipid rafts,23 become associated with RHODOPSIN,24 and permit efficient conformational changes of RHODOPSIN and its associated G proteins as photons are absorbed.25, 26, 27 The inability of the retina to take up and incorporate 22:6 leads to homeostatic compromise and PRC death as in retinal degenerative diseases.12, 22, 28 PRC outer segment tips containing 22:6‐rich disks are phagocytized daily by the RPE, and the 22:6 is recycled back to PRC.16, 19 PRC also contain very long‐chain polyunsaturated fatty acids (VLC‐PUFAs; ≥ 28 carbons) formed by ELOVL4 (ELOngation of Very Long chain fatty acids‐4)29, 30 that elongates 26:6,n‐3 derived from 22:6 or 22:5,n‐3 (eicosapentaenoic acid, EPA).31, 32 Despite the low abundance of retinal 22:5 compared to 22:6, 22:5 is the favored substrate for VLC‐PUFA production.32 Generated by retro‐conversion from 22:6 in peroxisomes, 22:5 provides the 22:6 substrate for ELOVL4.32 ELOVL4 synthesizes VLC‐PUFAs in retina21, 33 and testes,34 and generates VLC‐saturated fatty acids (VLC‐SFAs) in skin and brain.35, 36 VLC‐PUFAs in PRC then become acyl chains of PC, incorporating at the sn‐1 position.37 Under uncompensated oxidative stress (UOS), 32C and 34C VLC‐PUFAs are released and converted to elovanoids (ELVs) through enzymatic lipoxygenation as a PRC protective response.37 Because UOS also triggers 22:6 release and conversion to neuroprotectin D1 (NPD1),28 22:6 availability is part of a dual protective signaling which sustains PRC integrity.28
To maintain fundamental PRC functions, membrane phospholipids provide an environment for phototransduction to proceed, and precursors of biologically active lipid mediators in the inner segment of PRC, as well as in the RPE.28, 37 Therefore, a tight balance on retinal lipidome homeostasis must be maintained because UOS affects highly unsaturated fatty acids, forming 22:6 protein adducts that contribute to retinal degeneration.38 Overall, a healthy retinal lipidome is essential for PRC function within a highly stressful environment of bright light, high‐oxygen demands, and an abundance of polyunsaturated fatty acids which are highly susceptible to oxidative damage.19, 28
Here, utilizing the Mfrprd6 mouse, we have characterized key events that lead to PRC degeneration by mass spectrometry imaging, LC‐MS/MS, physiology, and transcriptomic approaches. We have shown phenotypical, histological, and physiological similarities between Adipor1−/− and Mfrprd6 mouse retinas with comparable onset of PRC degeneration. Retinas and RPE in both mice presented a dramatic and selective deficit of 22:6,n‐3‐containing PC phospholipids, but not of arachidonic acid (20:4,n‐6)‐containing molecules, and an absence of VLC‐PUFAs in the retina. We compared gene expression profiles of Adipor1−/− and Mfrprd6 mice and found Casp1 and Pycard, part of the pyroptotic cell death pathway, upregulated in both mutants. Further analysis of target genes in enriched pathways indicated that the mutation of ADIPOR1 in the retina resulted in upregulation of genes belonging to JNK phosphorylation and activation mediated by the activated human TAK1 pathway, while the mutation of MFRP was linked to activation of the NOD‐like receptor signaling pathway. Overall, our results indicate that the inability to enrich the retina with 22:6 results in the absence of VLC‐PUFAs from the lipidomes of Adipor1−/− and Mfrprd6 mice, and, consequently, the inability to synthesize the neuroprotective elovanoids, which correlates with the activation of inflammatory signaling pathways and the onset of retinal degeneration.
2. MATERIALS AND METHODS
2.1. Animals
Throughout this study, C57BL/6J (wild‐type; WT, The Jackson Laboratory, stock # 000 664), B6.129P2‐Adipor1tm1Dgen/Mmnc (Adipor1−/−; MMRRC, stock # 011599‐UNC), and B6.C3Ga‐Mfrprd6/J (Mfrprd6; The Jackson Laboratory, stock # 003684) male and female mice were used. Mice were maintained in the LSU animal colony on a 12 h:12 h light cycle at ~ 30 lux (light onset was at 0600 h), and given water and food ad libitum. Adipor1−/− and Mfrprd6 were screened for Crb1 (rd8) mutation by PCR according to Mattapallil et al., 2012,39 and we confirmed that they do not contain an inherent Crb1 mutation; hence, the retinal phenotypes observed in these mice derive from the mutations of Adipor1 and Mfrp. Summary of the Mouse Universal Genotyping Array (MUGA) and MMRRC computational tools used to assess the genetic background of the Adipor1−/− mice can be found at: http://www.csbio.unc.edu/MMRRC/index.py?run=StatsTable.viewSample%26sample=3826%26mm=0. All experiments were conducted in accordance with the Association for Research of Vision and Ophthalmology (ARVO) statement for the use of animals in ophthalmic and vision research, and the protocols were approved by the Institutional Animal Care and Use Committee (IACUC) for the LSU Health Sciences Center.
2.2. Spectral domain‐optical coherence tomography (SD‐OCT)
Fundus images of 3‐month old and SD‐OCT images of WT, Adipor1−/−, and Mfrprd6 retinas of 1, 3, and 5 months of age were obtained (Heidelberg Spectralis HRA OCT system; Heidelberg Engineering, Heidelberg, Germany) as previously described,12 and viewed with ImageJ (http//imagej.nih.gov/ij). The thickness of the outer retina (PRC + RPE) was defined as the distance from the proximal edge of the outer plexiform layer nuclear layer to the tips of the photoreceptor outer segments; the inner retina thickness was defined as the distance from the distal edge of the inner nuclear layer to the proximal edge of the ganglion cell/nerve fiber layer.40 Three animals/genotype/time point were analyzed.
2.3. Electroretinograms
Conventional electroretinograms (ERGs) techniques were employed to determine general health and to follow physiological changes within the retinas of the experimental animals. Animals were dark adapted overnight (12‐16 hours), anesthetized, and pupils dilated with topical 1.0% atropine (Mydriacyl 1%, Alcon). Body temperature was maintained at 38°C throughout by the recording platform of the Espion ERG apparatus (Diagnosys, Lowell, MA). A drop of 1% methylcellulose placed on the cornea prevented corneal desiccation and provided improved electrical contact. Fiber optics contacted the corneas and delivered the light flashes generated by the Espion system. Scotopic ERG responses were elicited with short duration LED flashes delivered from the Espion ERG apparatus, with interstimulus intervals of 0.5‐2 minutes, depending on intensity. Flash intensities ranged from 0.0001 to 10 cd‐s/m2. Three to four responses were averaged for each step, depending on the stimulus intensity (the flash interval was adjusted to assure dark adaptation following the previous flash). ERG responses were filtered (low‐pass 0.125 Hz, high‐pass 300 Hz) and archived for later analysis. Intensity‐response amplitude data were displayed on log‐linear coordinates.
2.4. Matrix‐assisted laser desorption/ionization imaging mass spectrometry (MALDI IMS)
Whole eyes of 2‐month‐old mice were embedded in gelatin, frozen, cryosectioned (20 μm thickness), and then collected on alternating glass slides (for staining with hematoxylin and eosin) and coverslips (for MALDI IMS). Coverslips were attached to MALDI stainless steel plates using thermally conductive dual tape. Plates were then attached to a magnet (thermally conductive) glued to the bottom of a sublimation chamber. 2,5‐dihydroxybenzoic acid (DHB) (Fisher Scientific, Pittsburg, PA) formed the matrix for the positive ion mode. A Synapt G2‐Si (Waters, Milford, MA), equipped with a MALDI source that uses a solid‐state laser (355 nm) at a firing rate of 2000 Hz for positive ion mode data collection, was employed. HDImaing software (Waters, Milford, MA) was used to design the pattern of tissue scanning (30 μm spatial resolution for both horizontal and vertical movement) and data analysis. Each image spot consisted of a collection of 1 seconds data acquisition. Ions created by the MALDI source were further separated by ion‐mobility‐separation with helium gas in the TriWave region of the instrument, with an ion‐mobility‐separation wave velocity of 600 m/s and height of 40.0 V. Data processed with HDImaging was converted with an in‐house program, and BioMap software (Novartis) was used to generate images.
2.5. Phospholipids molecular species as regional markers
Prior to analysis, specific markers for retinal layers/cell types were determined. To ensure good separation of retina and RPE‐eyecup data, anterior segments were removed and retinas and RPE‐eyecups were isolated separately on nitrocellulose filters, frozen, cryosectioned, and analyzed by MALDI IMS. PRCs were highly enriched with m/z 756.6 PC(16:0/16:0), m/z 760.6 PC(16:0/18:1) labeled the inner retina, m/z 1046.8 PC(34:6/22:6) denoted an n‐3 derived VLC‐PUFA, and m/z 738 (not identified) demarked the uvea/RPE complex. The shorthand notation used for lipid species here follows those outlined by Liebisch.41 The PC lipids observed (positive ion mode) were identified to lipid species level by collision‐induced dissociation (CID) and are indicated by the number of acyl carbons (C) and the number of double bonds, for example PC(56:12). Lipids were identified to the fatty acyl level by CID and are indicated by the fatty acyl groups esterified to the sn‐1 and sn‐2 position, such as PC(34:6/22:6). To determine which phospholipids were most abundant between retinas, difference spectra were generated. Equal areas were selected from identical regions of retinal sections and total spectra obtained from 700 to 1100 m/z. One spectrum was then subtracted from a spectrum of another animal to produce a difference spectrum, which revealed molecular abundance within the two retinas. If the remaining peaks extended upward, there was more of that molecule within retina 1, while peaks extending downward indicated more within retina 2. Possible identification was then assigned to these PCs, and corresponding MALDI IMS retinal images obtained. WT and mutant retinas were compared in this manner. Since spectrum amplitudes differ among retinas, even if normalized, this method only revealed very large differences that were considered interesting; quantitation using this method is inaccurate.
2.6. Lipid extraction and mass spectrometry for analysis by XevoTQ‐S
Lipid extraction was performed similarly to our previous work.12 Briefly, each sample was homogenized in MeOH (3 mL) followed by addition of CHCl3 (6 mL) and the internal standard (PC 28:0). After sonication in a water bath, samples were centrifuged, and the supernatant was added with pH 3.5 H2O for phase separation. The bottom phase (organic phase) was dried down under N2 and re‐constituted in an AcN:MeOH:CHCl3 (90:5:5) solution. A Xevo TQ‐S equipped with Acquity UPLC BEH HILIC 1.7 µm 2.1 × 100 mm column was used with solvent A (acetonitrile:water, 1:1; 10 mM ammonium acetate pH 8.3) and solvent B (acetonitrile:water, 95:5; 10 mM ammonium acetate pH 8.3) as the mobile phase. Solvent B (100%) ran for the first 5 minutes isocratically was graduated to 20% solvent A for 8 minutes, and then ran at 65% of A for 0.5 minutes. It ran isocratically at 65% of A for 3 minutes, and then returned to 100% of B for 3.5 minutes for equilibration. The capillary voltage was 2.5 kV, desolvation temperature was set at 550°C, the desolvation gas flow rate was 800 L/h, cone gas was 150 L/h, and nebulizer pressure was 7.0 Bars with the source temperature at 120°C. Mutant and WT samples for males and females (n = 3 each) were treated and analyzed separately. There was no difference observed between the two sexes; therefore, we combined the data for each type of mouse for n = 6.
2.7. LC‐MS/MS data analysis
Raw mass spectrometry data were based on percent composition of relative molecular species per biological replicate. Determination of differentially occurring molecular species was carried out using the loading score for the top ten PC molecular species for principle component 1, Principal component analysis (PCA), hierarchical clustering, and visualization of significant overlapping PCs (Venn diagram). Visualization of selected 22:6‐containing and 20:4‐containing PCs was obtained using BioVinci software (v1.1.3, r20180606), © 2017 BioTuring Inc, San Diego, CA, USA.
2.8. RNA isolation and cDNA preparation
Retinas and RPE‐eyecups from 1‐month‐old WT, Adipor1−/−, and Mfrprd6animals (n = 6 each) were isolated. Mutant and WT samples for males and females (n = 3 each) were treated and analyzed separately. There was no difference observed between the two genders; therefore, we combined the data for each type of mouse for n = 6. Total RNA was extracted using RNeasy Plus Mini Kit (Qiagen, Germantown, MD) according to the manufacturer's instructions. One microgram of RNA was reverse transcribed using an iScript cDNA Synthesis Kit (Bio‐Rad, Hercules, CA). Diluted cDNA (100 ng, 1.25 µL) was used for the preamplification reaction by adding 1 µL of PreAmp Master Mix (Fluidigm, San Francisco, CA), 0.5 µL of a mix of all primers, and water to a final volume of 5 µL. The temperature profile was 95°C for 2 minutes followed by 12 cycles of amplification (95°C for 15 s, and 60°C for 4 min using the Bio‐Rad CFX96 thermocycler). Each preamplification reaction of cDNA was then subjected to Exonuclease I treatment, to remove unincorporated primers. The resulting preamplified and treated cDNA was then diluted 5 times in TE Buffer.
2.9. High‐throughput qPCR
The qPCR reaction mixture had a volume of 5 µL and contained 2.25 µL of diluted preamplified cDNA, 0.25 µL of DNA Binding Dye (Fluidigm), and 2.5 µL SsoFast EvaGreen Supermix with low ROX (Bio‐Rad). The primer reaction mixture had a final volume of 5 µL and contained 2.5 µL Assay Loading Reagent (Fluidigm) and 0.25 µL of a mix of all reverse and forward primers, corresponding to a final concentration of 500 nM in the reaction. The Biomark 96.96 IFC™ (Integrated Fluidic Circuit) was first primed with an oil solution in the Juno™ Controller (Fluidigm) to fill the fluidic circuit. In all, 96 sample reactions (5 µL each) were loaded into individual sample wells, and 96 forward and reverse primer mixtures were loaded into each assay wells (5 µL each). The IFC was then placed in the Juno™ Controller for automatic loading and mixing. After 90 minutes, the IFC was then transferred to the Biomark™ HD qPCR platform (Fluidigm). The cycling program consisted of Thermal Mix at 70°C for 40 minutes followed by 60°C for 30 seconds. Hot Start was 1 minutes at 95°C, followed by 30 cycles of denaturation at 96°C for 5 seconds, and annealing at 60°C for 20 seconds. Melting curves were collected between 60°C and 95°C with 1°C increments/3 seconds.
2.10. qPCR data processing and analysis
The Ct value of target genes was normalized to the housekeeping genes Actb, Gapdh, Tbp, and Tfrc. For hierarchical clustering, the gene expressions were calculated as number of transcripts with the housekeeping gene set as 10 000 transcripts. Mouse groups (columns) and genes (rows) were clustered using one minus Pearson correlation metric. For differential gene expression analysis, the fold‐change value was calculated using the comparative threshold cycle method (ΔΔCt) with the WT mouse sample as control. Calculated data were presented as box‐plots with averaged Ct from six animals as a data point. Student's t test analysis was used to compare the differences between mutants and WT. All significant genes that were upregulated or downregulated to the WT were plotted as Venn diagrams. To interpret the distinct and shared transcriptional modulation between Adipor1−/− and Mfrprd6 mice, we used ConsensusPathDB42 (http://cpdb.molgen.mpg.de/MCPDB) and uploaded the lists of genes that are increased or decreased in the mutant datasets (Supplemental Table 1). We picked the top five pathways (only four pathways found with the common downregulated genes) and plotted the percentage of genes within the total pathway population. All graphs were made using the Bio Vinci program (Bioturing Inc, San Diego, CA).
2.11. Statistics
The Kolmogorov‐Smirnov test was employed to determine the assumption of normality for the SD‐OCT measurements; an AR (1) as covariance structure was used for ERG measurements. A MANOVA test showed that differences existed among the retinal phosphatidylcholine (PC) data from the WT, Adipor1−/−, and Mfrprd6 animals; all P values < 0.0001 from Wilk's lambda, Pillai's Trace, Hotelling‐Lawley Trace, and Roy's Greatest Root. Tukey‐Kramer's multiple comparisons gave the final results (see Supplemental Table 2 for statistical details). *P ≤ .05, **P ≤ .01, ***P ≤ .001,****P ≤ .0001 throughout.
3. RESULTS
3.1. PRC degeneration in Mfrp and Adipor1 mutant mice
Fundus imaging of 3‐month‐old Mfrprd6 mice demonstrated a uniformly flecked retina similar to that found in the Adipor1−/− retina 12 and in human fundus albipunctatus. These white spots of uniform size were evenly distributed within the fundus and exhibited similar density in the Mfrprd6and Adipor1−/− retinas (Figure 1A). Optical coherence tomography (OCT) revealed a reduction in outer retinal thickness; by three months of age, about 40% of PRC nuclei had been lost, and at five months only about 40% remained (Figure 1B and C). Interestingly, the inner retina of the Mfrprd6 appeared thicker (Figure 1B and C). Overall, the Mfrprd6 phenotype closely resembles the Adipor1−/−.12
Figure 1.
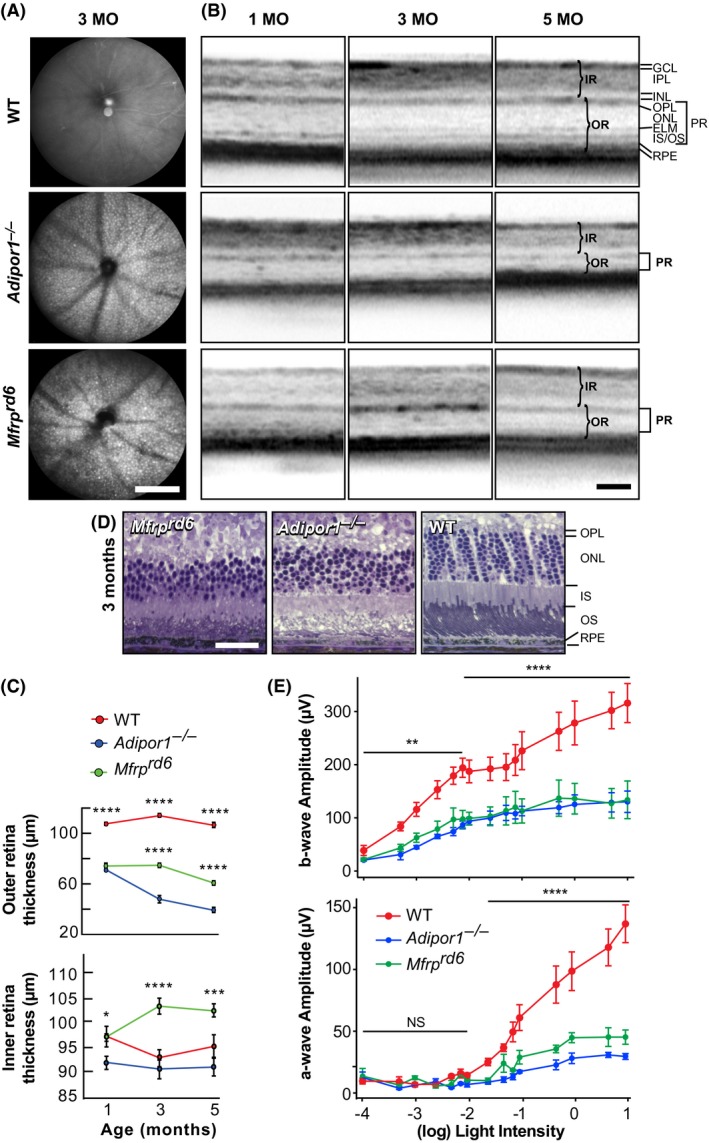
Progressive PRC degeneration is similar in Mfrprd6 and Adipor1 mutants. A, Fundus imaging revealed dense, evenly distributed white, punctate spots deep within 3‐month‐old mouse retinas, as observed in the Adipor1−/− mouse. Images are representative of four mice. B, Optical coherence tomography (OCT) for 1‐, 3‐, and 5‐month‐old Mfrprd6 and Adipor1−/− retinas demonstrated a gradual reduction in the photoreceptor layer thickness when compared to wild‐type (WT) mice. C, The outer retina (OR, photoreceptors + RPE) thickness in both mutants at 1 month of age is reduced compared to the WT retina; the Mfrprd6 decreases slightly by 5 months, while the Adipor1−/− OR continually decreases. The inner retinas (IR) remain unchanged for the WT and the Adipor1−/−; however, the Mfrprd6 IR is thicker at 3 and 5 months. Error bars represent SEM of three individuals. D, Histological sections illustrating cellular morphology and changes in thickness of the Mfrprd6 and Adipor1−/− photoreceptor layers, relative to WT mice, especially evident in the reduction of photoreceptors nuclei within the ONL. Each image is representative of three mice. E, a‐ and b‐wave electroretinographic (ERG) responses of 2‐month‐old animals, to increasing intensities of light (cd•s/m2). Mfrprd6 and Adipor1−/− retinal responses were reduced by approximately 75% and 50%, respectively, from those of WT mice. Generally, both mutant mice displayed similar reductions. Error bars represent SEM of three individuals. GCL, ganglion cell layer; IPL, Inner Plexiform Layer; INL, Inner Nuclear Layer; OPL, Outer Plexiform Layer; ONL; Outer Nuclear Layer; ELM, External Limiting Membrane; IS/OS, Inner/Outer Segments; RPE, Retinal Pigment Epithelium; IR, inner retina; OR, outer retina; PR, photoreceptors. Magnification bars: (A) 200 μm; (B) 100 μm; (D) 100 μm. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001
3.2. Retinal physiology is similarly impaired in Mfrp and Adipor1 mutants
ERGs were recorded from 2‐month‐old WT, Mfrprd6, and Adipor1−/− during the onset of PRC death when the PRC layer began to decrease (Figure 1B‐D). ERGs obtained from light flashes ranging in intensity from 0.0001 to 10 cd·s/m2 revealed a 50%‐75% decreased response in a‐waves, for both the Mfrprd6 and Adipor1−/− mice vs WT, indicating a reduction of PRC response. The b‐wave was reduced by about 50% throughout, displaying an inner retinal effect (largely bipolar cells). Interestingly, the dimmer light rod response in all mice plateaued through 0.005 cd·s/m2 to 0.075 cd·s/m2, followed by a similar rise in the brighter light cone response for all mice, indicating that reduced 22:6 affected both rods and cones (Figure 1E). Since there is no apparent loss of inner retinal cells in these mutations, this suggests that the reduced signal originated in functionally impaired PRC.
3.3. Perturbations in the retinal lipidome containing highly unsaturated molecular species of phospholipids precedes PRC death in the Mfrprd6 retina
22:6 is important for visual function and retinal homeostasis. However, this essential fatty acid must be obtained by diet, collected and packaged by the liver, and released into the circulation for targeted delivery to the RPE and PRC. Failure to achieve adequate delivery results in impaired photoreceptor function and eventual cell death.
MALDI IMS revealed relative molecular abundance (spectra from 400 to 1200 m/z) from scanned histological sections (Figure 2A). MALDI analysis by positive ion mode of Adipor1−/−, Mfrprd6, and WT retinas demonstrated that the same phospholipids were present in the three genotypes, although the abundance of specific species within the mutant retinas was different from those of the WT. Analysis of MALDI sections revealed reliable identifying markers for the retinal layers (Figure 2C‐G): m/z 738 Uvea and RPE, m/z 756 PC(32:0) photoreceptor layer, m/z 760 PC(34:1) inner retina. m/z 900 PC(22:6/22:6), an n‐3 phospholipid, labeled the PRCs of the WT, but was absent in both mutants; m/z 832 PC(20:4/18:0), an n‐6 phospholipid, was present in all three animals, but was absent (greatly reduced) in the PRCs of the WT, indicating an n‐3 phospholipid preference in WT PRCs. m/z 1046 PC(34:6/22:6), an n‐3 VLC‐PUFA‐containing molecule, was prevalent at the outer edge of the PRC layer in the WT, but was absent in both mutants, indicating that the lack of 22:6 impaired the synthesis of the VLC‐PUFA phospholipids.
Figure 2.
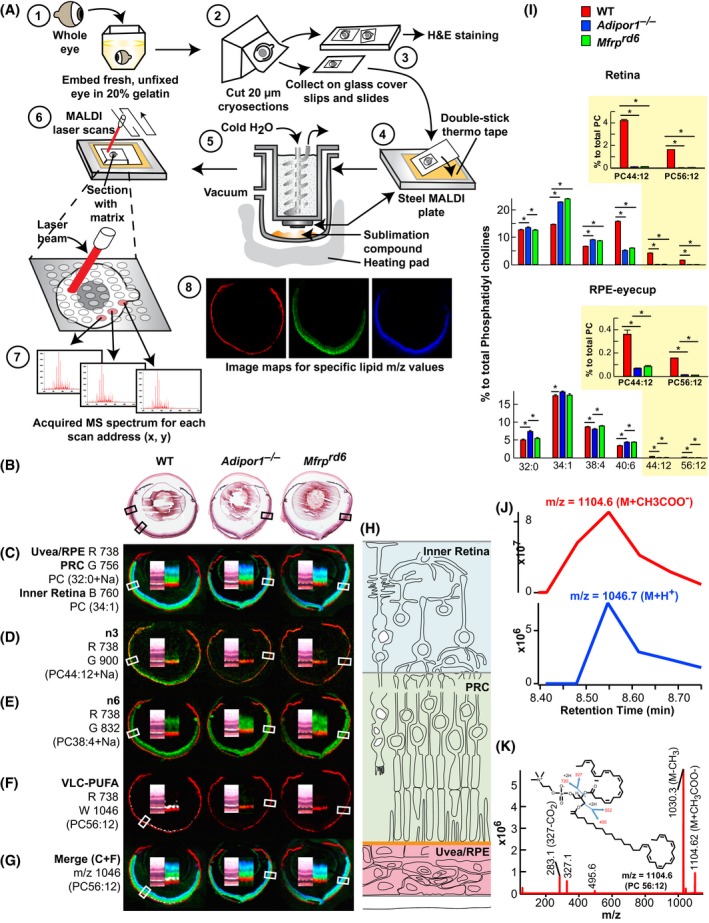
MALDI IMS reveals loss of PC containing 22:6 and VLC‐PUFAs in the outer nuclear layer of Mfrprd6 and Adipor1−/− retinas. A, Whole eyes embedded in gelatin (1), frozen, cryosectioned, 20 μm (2), collected on alternating glass slides (for H&E) and coverslips (for MALDI) (3). Coverslips attached to MALDI plates (4) placed within sublimation chamber,43 matrix (2,5‐dihydroxybenzoic acid, DHB) applied for positive ion mode analysis (5) MALDI Synapt G2‐Si MS. Sections rastered by laser, 355 nm, 2000 Hz (6). Scanning control (30 μm, horizontal and vertical movement) and analysis. Image spot consisted of a collection of one second of data acquisition (7). Image processing (8). B, H&E sections of adjacent MALDI section for WT, Adipor1−/−, and the Mfrprd6 mice. C, Lipid markers for uvea/RPE (m/z 738, red, R), PRC (m/z 756, green, G), and inner retina (m/z 760, blue, B). D, n‐3 containing PC(22:6/22:6), m/z 900 (green) is present in WT but absent in Adipor1−/− and Mfrprd6 retinas. E, n‐6‐containing phospholipids compensate D. Here, PC(18:0/20:4 n‐6, m/z 832, green) is reduced in WT PRC, but still abundant within the inner retina while present within PRC of Adipor1−/− and Mfrprd6. F, 22:6 is elongated to produce VLC‐PUFAs. WT retina contains 34:6 within PC(34:6/22:6); 34:6 is indicated by the white label, while the Adipor1−/− and the Mfrprd6 mice show no 34:6. G, The merge of C and F illustrates the location of 34:6 just inside the uvea/RPE marker (red) at the outer region of PRC (green) in WT, but no label within mutant retinas (absence of 22:6). H, Diagram on retinal location of C color markers. Double inset within each retina image is an enlargement of MALDI label and the corresponding area from H&E in B, indicated by boxes. N = 3 for each condition. I, LC‐MS/MS quantitative PC molecular species distributions in bar graphs for retina (top) and RPE (bottom). 22:6‐containing PCs, which are abundant in PRC outer segments, are reduced in Adipor1−/− and Mfrprd6 retinas and RPE, especially PC 44:12 and PC 56:12 (yellow insets). Student's t test (*P < .05). (J) Retention time of negative ion mode of PC 56:12 (M + CH3COO) at m/z = 1104.6 (top) matches with positive ion mode (M + H+) at m/z = 1047.7, confirming that m/z = 1046.7 measured in MALDI is from PC 56:12 (M + H+). (K) Full fragmentation spectrum of PC 56:12 (M + CH3COO‐) measured in negative mode shows composition of PC 56:12 (34:6/22:6)
LC‐MS/MS‐based retinal lipidomic analysis displays the quantitative PC molecular species distributions in bar graphs for the retina (top) and the RPE (bottom) (Figure 2I). Clearly, 22:6‐containing PCs are reduced in both Adipor1−/− and Mfrprd6 retinas and RPE, especially PC(44:12) and PC(56:12), which are abundant in PRC outer segments (Figure 2I, insets). The composition of the mass numbers used for retinal layer specification was determined by full fragmentation in negative ion mode (Figure 2K and Supplemental Figure 1). Matching of the peaks in positive and negative ion modes confirmed the identity of the molecules selected for retina specification (Figure 2J). In our study, m/z 1104.6 (negative ion) and 1046.7 (positive ion) was identified as the m/z of VLC‐PUFAs (Figure 2F, J and K) that was not detectable in Adipor1−/− and Mfrprd6. These spectra indicate that a dysfunctional MFRP results in a remarkable altered retinal lipidome, and, based on the importance of 22:6 in retinal maintenance and function, suggests that reduction of 22:6 contributes to eventual PRC degeneration in the Mfrprd6 mouse.
To further define differences in the retinal lipidome of the WT, Mfrprd6, and Adipor1−/−, retinal sections were obtained and imaged by MALDI IMS. Total spectra from m/z 700 to m/z 1200 were collected from small retina regions for comparison (Figure 3). A difference spectrum was then constructed by subtracting the Adipor1−/− or Mfrprd6 profile from the WT profile (Figure 3) A and B. The resulting plots, while not quantitative, emphasized lipid abundance, with the prevalent WT lipids (pointing up) and Mfrprd6, and Adipor1−/− lipids indicated as downward peaks. 22:6‐containing PCs (m/z 828.61 PC(16:0/22:6) and m/z 856.64 PC(18:0/22:6)) were more prevalent in the WT retina, whereas the 20:4‐containing PCs (m/z 804.6 PC(16:0/ 20:4) and m/z 832.64 PC(18:0/20:4)) were enhanced in mutant retinas. Analysis of VLC‐PUFAs spectra (from m/z 1000 to m/z 1200) revealed them to be almost non‐existent in both mutant retinas. Supplemental Table 3 depicts the more abundant phospholipid species revealed by the differential spectra of the WT, the Mfrprd6, and the Adipor1−/− retinas, emphasizing the lack of VLC‐PUFA species in the mutants. To further highlight the retinal lipidome differences between Adipor1−/− and Mfrprd6, we generated differential spectrum of the two mutants (Figure 3C). The relative amount of 22:6‐containig PCs, although severely depleted in Mfrprd6, was still detectable compared to Adipor1−/−. Supplemental Table 3 depicts the distribution of the more abundant phospholipid species revealed by the differential spectra of the Mfrprd6 and the Adipor1−/− retinas. Overall, this comparison suggests that the 22:6‐containing molecules are reduced in the Mfrprd6 and Adipor1−/− retinas, whereas the 20:4‐containing species are either retained or enhanced, indicating that products of n‐3 fatty acid synthesis are reduced in the Mfrprd6, while the n‐6 fatty acid pathway may be conserved.
Figure 3.
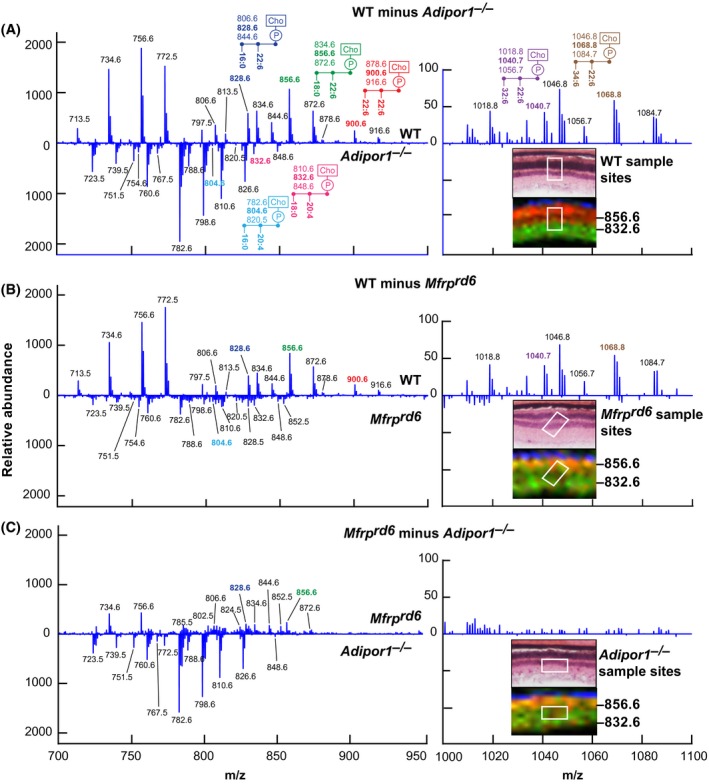
Differential MALDI spectra reveal compensatory PCs generated in Adipor1−/− and the Mfrprd6 retina. Difference spectra show relative abundances detected by MALDI IMS. A, Molecules more abundant in WT retina vs Adipor1−/− retina are presented in the upper part of the graph, while molecules more abundant in the Adipor1−/− retina are displayed at the bottom. 22:6‐ and/or VLC‐PUFA‐containing PCs are much more abundant in WT while 20:4‐containing PCs are increased in the mutant retina. The regions from which the spectra are extracted are shown in the insets H and E (top) and MALDI (bottom), showing PC40:6 (m/z = 856.6, red) and PC38:4 (m/z = 832.6, green). The identification of the mass numbers is shown in Supplemental Table 3. B, The difference spectra of Mfrprd6 to WT. (C) The difference spectra of Adipor1−/− to Mfrprd6. Inset images show that in both mutants, the PRC layers contain both PCs 40:6 and 38:4 (yellow) while WT has clear separation of the two PCs species between PRC layer and inner retina layer
3.4. VLC‐PUFA‐containing phosphatidylcholines are negligible in Mfrprd6 mouse eyes
MALDI IMS suggests that abundance of 22:6‐containing PCs in the Mfrprd6 is drastically decreased (Figures 2F and 3B). Inability to take up and incorporate 22:6 in retina and RPE was discovered in the Adipor1−/− mouse.12 Similarly, in the Mfrprd6, there is almost complete loss of PC‐containing fatty acids generated through the n‐3 fatty acid synthesis pathway, especially the VLC‐PUFAs (Figures 2F,4 and 5). Since the Mfrprd6 may have either lost the ability to take up 22:6 and/or synthesize PCs containing molecules elongated from 22:6 as in the Adipor1 mutant, we investigated the overall retinal lipidome of 22:6‐containing phospholipids in Mfrprd6 retinas and RPE‐eyecup. PC, phosphatidylethanolamine (PE), and phosphatidylserine (PS) molecular species in retinas and RPE‐eyecups contain either 20:4 or 22:6, except PE(22:1/22:0) and PS(22:1/22:0) (Figures 4 and 5). Overall, retinal PCs show that 20:4‐containing species are more abundant in the mutants, whereas 22:6‐containing species are highly reduced (Figure 4). In PEs, 20:4‐containing species are higher in the mutants, but this phospholipid with a single 22:6 (eg, PE[16:0/22:6] and PE[18:0/22:6]) is not different from WT. However, PE(22:6/22:6) and PE(24:6/22:6) are greatly reduced in the mutants (Figure 4). At the same time, PS shows a large increase in single‐22:6‐containing species (PS[16:0/22:6] and PS[18:0/22:6]), while PS(22:6/22:6) and PS(24:6/22:6) are greatly reduced as in the PEs (Figure 4). In the RPE‐eyecup, single 22:6 containing species (16:0/22:6, 18:0/22:6) for both PC and PE are more abundant in the mutants (Figure 5), but the 22:6/22:6 and 24:6/22:6 species are greatly reduced as they are in retinas. The 22:6‐containing PSs in the RPE‐eyecups were not detectable.
Figure 4.
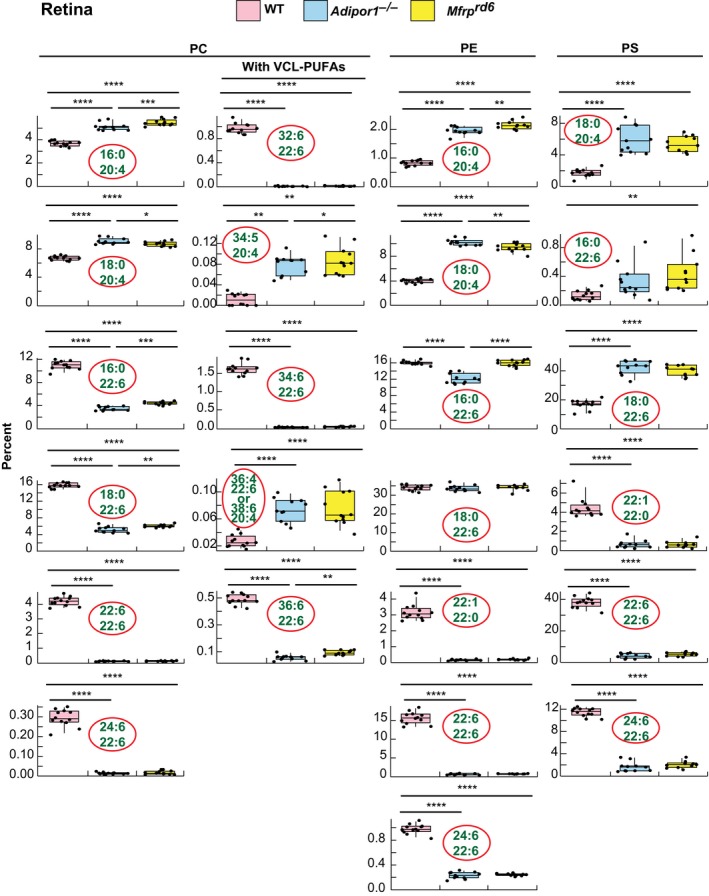
Selected phospholipid molecular species of WT, Adipor1−/−, and Mfrprd6 mouse retinas. 22:6‐ or 20:4‐containing PC, PE and PS (except for PE(22:1/22:0) and PS(22:1/22:0)) are shown in the box‐plots. 20:4‐containing PC, PE, and PS are abundant in the mutant retinas. In both mutants, there is a great reduction in all the phospholipids containing two 22:6 molecules (PC, PE, or PS(22:6/22:6)) and PCs composed of 22:6 and VLC‐PUFAs (PC(22:6/32:6), PC(22:6/34:6), and PC(22:6/36:6)). On the other hand, single 22:6‐containing PEs, such as PE(18:0/22:6), do not show differences among the groups; however, PE(22:6/24:6) shows greatly reduced amounts in both mutant retinas compared to WT. Interestingly, non‐22:6 or −20:4 containing PE(20:0/22:1) also shows great reduction in the mutants. For PS species, single 22:6‐containing molecular species show higher abundances in the mutants while PS(22:6/24:6) and PS(22:0/22:1) show significant reduction in mutants
Figure 5.
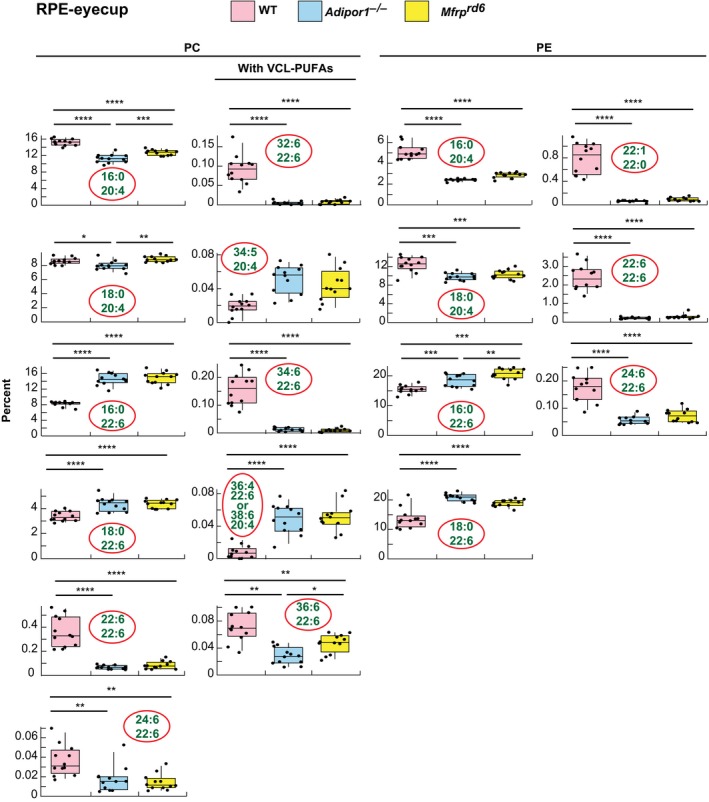
Box‐plots of selected 22:6‐ or 20:4‐containing PCs and PEs of WT, Adipor1−/−, and Mfrprd6 mouse RPE‐eyecups. Most of the single 22:6‐containing PC species in the RPE‐eyecups show higher abundance in the mutants, unlike in the retinas. PC(22:6/22:6) or PC(22:6 combined with a VLC‐PUFA) show great reduction in the mutants. 20:4‐containing PE is reduced in the mutants, which is also an opposite observation from the retinas. Single 22:6‐containing PEs are also increased in mutants, but PE (22:6/22:6) shows a great reduction in mutants, similar to the retina. Non‐22:6 or 20:4 containing PE(20:0/22:1) also show great reduction in the mutants
Principal component analysis (PCA) of WT and mutant retinas and RPE‐eyecups show good separation, particularly with retina (Figure 6A). Hierarchical clustering reveals two major clusters in retina (Figure 6B) and RPE‐eyecup (Figure 6C), with red and blue indicating increased and decreased abundance, respectively. A Venn diagram (Figure 6D) depicts the overlap of significant differentially occurring PCs across strains and tissues. The numbers in each oval show the number of PC species statistically different vs WT.
Figure 6.
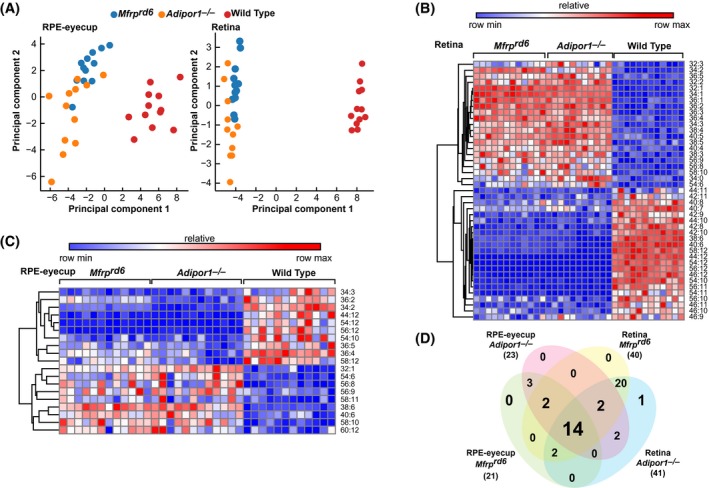
PCs containing VLC‐PUFAs in Adipor1−/− and Mfrprd6 eyes are greatly reduced. A, Principal component analysis of targeted PCs illustrates tissue‐specific lipid profiles in mutants and WT mice (n = 12). B, C, Hierarchical clustering of significantly differentially occurring PC species (ANOVA one‐way) in Adipor1−/− and Mfrprd6 retinas and RPE‐eyecups compared to WT (P ≤ .05, absolute value fold‐change ≥ 1.5) suggests compensatory PC dynamics. D, Venn diagram illustrates overlap of differentially occurring PCs across strains and tissues
3.5. Phosphatidylcholines containing LC‐PUFAs with 4‐7 double bonds are enhanced in Mfrprd6 and Adipor1−/− mice
While there is a reduction of VLC‐PUFAs in the mutants, even within PCs containing fewer than 10 double bonds, additional LC‐MS/MS analyses of retinas and RPE‐eyecups revealed increases in some 4‐7 double bond, 50‐58C PC molecular species (Supplemental Figure 3). These increases, which may be compensatory, were observed in retinas and RPE‐eyecups of both Mfrp and Adipor1 mutants. However, since PRCs were eventually lost, compensatory replacement with alternate PC species was only a temporary fix, signifying a potential transient mechanism to sustain the retinal lipidome until adequate 22:6 could be achieved.
3.6. VLC‐PUFAs abundance and Elovl4 expression decline in Mfrprd6 retinas
Functional Mfrp gene yields a retinal lipidome rich in PC species (Figure 6) required for Elovl4 expression. We found reduced Elovl4 expression in the Mfrprd6 RPE‐eyecups and retina (Figure 7C, Supplemental Figure 6A), which further explains the decreased abundance of VLC‐PUFAs that we found.
Figure 7.
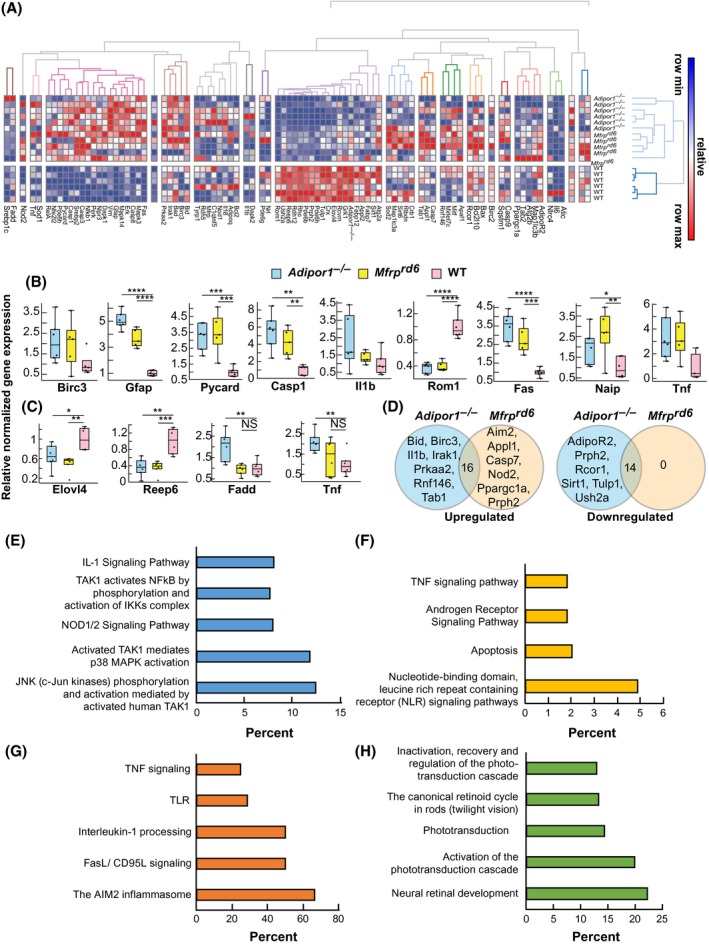
The expression of inflammatory markers is increased and visual system markers decreased in Adipor1−/− and Mfrprd6. A, Heat map illustrating the hierarchical clustering of 88 target genes for the retinas of Adipor1−/− (n = 6), Mfrprd6 (n = 5) and WT mice (n = 6). Color key represents row scaling of normalized fold‐change (FC) expression values from RT‐qPCR. B, Box‐plots representation of selected genes for the retinas with their FC below − 2 or above + 2, relative to the WT. Boxes denote median values with upper and lower quartiles, and whiskers, minimum and maximum outliers. C, Box‐plots representation of selected genes for the RPE‐eyecup samples with their FC below − 2 or above + 2, relative to WT. D, Venn diagrams of differentially expressed genes within Adipor1−/− and Mfrprd6 datasets that are upregulated or downregulated in a statistically significant manner in the retina compared to the WT. The number in the intersection represents the differentially expressed genes that are common between the two datasets (Supplemental Table 1). Top pathways resulting from the upregulation of genes in Adipor1−/− retina (E) or Mfrprd6 retina (F) compared to the WT, or common upregulated (G) or downregulated (H) genes in both mutant retinas. Student's t test was used. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, NS = not significant. Error bars represent standard deviation. When there was no statistical difference in the mutant datasets compared to the WT, statistic bars were not added
3.7. Genes signatures of pro‐inflammatory signaling pathways are increased and genes encoding proteins for PRC function are decreased in AdipoR1−/− and Mfrprd6
Mutation of Mfrp or Adipor1 affects differentially signaling pathways leading to PRC death, as revealed by RT‐qPCR analysis of the expression of 88 candidate genes in retina and RPE‐eyecups. Target genes were selected for their function in the visual system and their role in inflammatory and immune system pathways. We found a higher hierarchical clustering of the selected genes for retina (Figure 7A) compared to the RPE‐eyecup (Supplemental Figure 4), likely because the RPE‐eyecup is less sensitive to the Mfrp or Adipor1 mutation, as opposed to PRCs, which subsequently degenerates. Box‐plots show the expression range of individual genes with the most significant fold‐change in retina (Figure 7B) and RPE‐eyecup (Figure 7C) across the three different genotypes (Mfrprd6, Adipor1−/−, and WT). Genes with higher fold‐change differences in retina were linked to inflammatory pathways: Birc3, Pycard, Casp1, Il1b, Fas, Naip, and Tnf. Interestingly, genes that are part of the pyroptosis cell death pathway, such as Casp1 and Pycard, were upregulated more than 4‐ and 3‐fold, respectively, in mutants. Next, we followed the expression of genes associated with visual function, and linked to retinopathies when dysregulated. ROM1 is part of the protein complex containing PRPH2 that contributes to PRC disk morphogenesis. We found a 3‐fold downregulation of Rom1 in both mutants (Figure 7B), and although attenuated, the mutant retinas also displayed a downward regulation of Prph2 expression (Supplemental Figure 5A). Gfap, a marker of reactive gliosis in the Müller glia cells, was upregulated 5‐fold in Adipor1−/− and more than 3‐fold in Mfrprd6 (Figure 7B). In the RPE‐eyecup, there were only a few genes that displayed more than a 2‐fold‐change between mutants and WT. Among these genes, Elovl4 (1.5‐ to 2‐fold) and Reep6 (3‐fold) were decreased in mutants, while genes associated with inflammatory pathways, Fadd and Tnf, were increased 2‐fold in Adipor1−/− RPE‐eyecups only (Figure 7C). We investigated the classical signal transduction pathway via Adiponectin receptors, ADIPOR1 and ADIPOR2, which modulates different biological events such as steroidogenesis, glucose uptake, cell survival, fatty acid oxidation, vasodilatation, and cytoprotection.44 Although most of the candidate genes did not have their FC ≤ 2 or > 2 (except for Adipor1 in the Adipor1−/− dataset), they displayed differences compared to the WT. Adipor1 expression was decreased by 1.5‐fold in the retina and 1.4‐fold in the RPE‐eyecup of Mfrprd6 (Supplemental Figure 5). Adipor2 was reduced in Adipor1−/− retina and RPE‐eyecup, possibly as a compensatory way to respond to Adipor1 deletion (Supplemental Figure 5). Interestingly, Mfrp expression was not significantly affected in either the retina or RPE‐eyecup of Adipor1−/−, suggesting a feedback loop for transcriptional regulation of Adipor1 by MFRP (Supplemental Figure 7). Overall, a downregulation of genes was linked to visual function in the retina of both mutants (Supplemental Figure 6A), implying that the dysregulation of ADIPOR1 and MFRP has direct impact on the physiology of the PRC and their survival.
In Adipor1−/− and Mfrprd6 retina datasets, seven and six distinct genes, respectively, were upregulated (Figure 7D). To highlight the particularity of each mutant and to understand if they follow a specific degenerative pathway, we looked at the over‐representation of candidate genes in enriched pathway‐based sets (Figure 7E‐H). The most highly represented biological activities enriched in the mutants compared to the WT, mainly involved response to inflammation and cellular processes for programmed cell death. The highest score of over‐representation of candidate genes upregulated in Adipor1−/− dataset belonged to JNK (c‐Jun kinases) phosphorylation and activation mediated by the activated human TAK1 pathway (Figure 7E), while upregulated candidates in the Mfrprd6 dataset belonged mainly to the NOD‐like receptor signaling pathway (Figure 7F). The common upregulated candidate genes (Supplemental Table 1) in Adipor1−/− and Mfrprd6 datasets are mainly represented within the AIM2 inflammasome pathway (Figure 7G).
Regarding the downregulated candidates, we found six unique upregulated genes in the Adipor1−/− dataset, while there were none in the Mfrprd6 dataset (Figure 7D). The list of these genes did not hit any enriched pathway‐based sets, most likely because of the reduced list size. Common candidate genes downregulated in mutant datasets had the highest over‐representation score for the neural retinal development pathway (Figure 7H); these candidate genes have a function in the development and function of PRC; thus, ADIPOR1 and MFRP are essential for the maintenance of PRC homeostasis by acting directly or indirectly on effectors for cellular integrity.
4. DISCUSSION
A central question regarding onset and progression of retinal degenerative diseases is how acquisition of key molecules in the retinal lipidome takes place, and how they sustain transcriptomic programs for retinal function. To address this question, this study used sensitive and specific mass spectrometry‐based molecular imaging (MALDI IMS) for mapping the untargeted in situ spatial distribution of molecular species of phospholipids, complemented with LC‐MS/MS quantification and detailed characterization, with OCT imaging and analysis of electroretinograms (ERG) to assess retina structure and function. Additionally, transcriptomic approaches have allowed us to define gene signatures involved in PRC degeneration in the Mfrprd6 mouse, and to establish analogies and differences with Adipor1−/− mouse, particularly preceding retinal degeneration. These findings have revealed novel necessary molecular events for PRC function.
Mfrprd6 displays characteristics similar to those expressed in human retinitis punctata albescens.7, 9 Subretinal flecks also appear across the retina in this mouse, corresponding to accumulation of macrophages within the subretinal space, and this phenotypic characteristic is also evident in the Adipor1−/− retinas. While a detailed timeline has not been established for these mutants, histology demonstrated the accumulation of autofluorescence of undigested outer segments within the RPE, and OCT analysis revealed a progressive decline in the outer retina thickness as a result of PRC loss.12 Outer retinas (the PRC layer) of both mutants show a reduction from the normal to about 40% at one month with progressive loss up to 5 months, the Adipor1−/− declining more rapidly. Moreover, the ERG declined by P25, indicating slow loss of function,12 with both mutant ERGs reduced to about 50% at 2 months. Differences in lipids among 3‐month mutants and WT are described, but it is not known whether these differences are unchanged from birth in the mutants or are a result of an ongoing restructuring and compensation. An earlier study demonstrated that key retinal proteins were depressed and that rhodopsin was depleted prior to PRC death by P21 in the Mfrprd6 mice.10 Changes associated with the RPE cells in the Mfrprd6 mice have also been noted.9, 45 Developing Mfrprd6 mice show reduced RPE microvilli, altered PRC inner segments, and disorganized outer segments, as well as impaired phagocytosis of PRC apical outer segments.45 Conversely, in RPE cell cultures, an increased number of RPE microvilli occur in the Mfrp mutants.9 Alterations in microvilli formation, density, and/or distribution would affect outer segment shedding, disrupt the process of membrane disposal by the RPE, and upset the retinal lipidome and PRC homeostasis.
MFRP has been localized in the RPE apical membrane and along the entire length of the RPE microvilli, as well as in PRC inner segments. MFRP is expressed as a dicistronic transcript with the C1q tumor necrosis factor‐related protein‐5 (C1QTNF5/CTRP5) gene,2, 46, 47 which is associated with cell adhesion and basement membranes48 in the RPE and ciliary body,49 and the MFRP level is inversely proportional to CTRP5 expression.50 β‐actin is increased in MFRP‐deficient cells and in cells overexpressing CTRP5, and MFRP affects actin polymerization, RPE cell adhesion, and microvilli morphology.50 CTRP5 is also localized in the RPE microvilli,49 and CTRP5 molecules form multimers in adjacent RPE cells with their collagen domains binding to collagen receptors in the membranes and their globular heads interacting to enhance RPE cell‐cell adhesion.51 This has suggested a potential interaction with actin filaments52 at this level. An interaction here could be diminished since the Mfrprd6 mice have reduced/distorted RPE microvilli,9, 45 which could reduce interdigitation with outer segments and impede 22:6 trafficking.
22:6 is reduced in both retina and RPE. The RPE regulates 22:6 uptake from the choriocapillaris, but their altered microvilli in the Mfrp rd6 mice suggest that redistribution of the 22:6 from the RPE to photoreceptors may be impeded or prevented. Eventually, with no or little 22:6 to form the neuroprotective compounds, the presence/accumulation of unmitigated stress would trigger an inflammatory response. Thus, arachidonic acid (20:4) is noticeably increased in both mutants, while it is notably less in the healthy WT mice, especially as compared with the abundance of 22:6 in the WT.
Healthy RPE cells are necessary for PRC integrity, recycling 22:6 from phagosomes back to the PRCs for synthesis of new disk membranes.16, 45 Thus, RPE cells regulate 22:6 trafficking to the PRC, and impediments to this process limits the availability of 22:6 for elongation to VLC‐PUFAs and for the synthesis of NPD1 and the ELV neuroprotectants, diminishing the ability to counter unmitigated oxidative stress and retinal homeostatic upset.28 The availability of this dual protective mechanism in which stress triggers 22:6 release and its conversion to NPD1, as well as the VLC‐PUFAs and their conversion to ELV,28 is important to sustain PRCs. Therefore, we monitored 22:6 accumulation and elongation to VLC‐PUFAs in retina by observing the distribution of n‐3 PC(44:12), containing two 22:6 molecules, and n‐6 PC(38:4), containing (18:0/20:4). In those same retinas, MALDI imaging showed that PC(56:12), containing the n‐3 VLC‐PUFAs 34:6 and 22:6, was localized within the outer PRC layer in WT retinas, but was absent in both mutant retinas. Conversely, the n‐6 PC(38:4) was abundant within the PRC. Also, VLC‐PUFAs were undetectable in the MALDI spectra of the Adipor1−/− and rd6 mice. Our data show that depressed PRC 22:6 n‐3 in both mutants was accompanied by a compensatory response where less prevalent n‐6 molecules were increased in the photoreceptor layer. Absence of 22:6 and the VLC‐PUFAs, therefore, results in an increased n‐6/n‐3 ratio in the Adipor1−/− and rd6 retinas, as similarly noted in AMD donor retinas.53 This creates a 20:4 n‐6‐enriched phospholipid environment which likely results in a proinflammatory condition, which contributes to the onset of PRC dysfunction. Since the n‐3 VLC‐PUFAs are released from their PC molecules upon unmitigated stress, and converted to protective elovanoids (ELVs),37 absence of this pathway favors PRC degeneration.
We suggest that the absence of 22:6, and, therefore, the lack of VLC‐PUFAs, in the Mrprd6 and Adipor1−/− mice leads to transcriptomic modifications, which then trigger an increase in pro‐inflammatory genes and a decrease of genes that are critical for visual system function. Transcriptome analysis provides insight into the relationship between Mfrp and Adipor1. Our gene signature shows that the JNK phosphorylation and activation mediated by the activated TAK1 pathway are initiated in Adipor1−/− retina. The induction of this pathway could be due to the relative abundance of arachidonoyl groups in phospholipids in Adipor1−/− retina that provides substrate for COX2 activation by c‐Jun kinases to foster the formation of pro‐inflammatory effectors that ultimately contribute to triggering cell death pathways.54 On the other hand, the favored pathway for Mfrprd6 is NOD‐like receptor signaling. Free radicals and lipid peroxidation products in the retina activate this pathway. This is facilitated because the recycling by the RPE is altered in Mfrprd6, enabling accumulation of these harmful molecules that, in turn, could establish a stress‐prone environment for PRC. As a consequence, the activation of the NOD‐like receptor signaling pathway takes place. It is probable that although there is a distinct induction of inflammatory pathways in Adipor1−/− and Mfrprd6, there is eventually a convergence of pathways and, in this case, the predicted AIM2 inflammasome signaling, which would then activate apoptotic and pyroptotic signals following the activation of caspase‐8 and −1.55
Taken together, our study demonstrates that the Mfrp mutation resembles the Adipor1 mutation on multiple levels, with some transcriptome differences: (i) phenotypically, the deletion of these proteins leads to flecked retina and slow PRC death onset; (ii) functionally, the mutations express the same degree of ERG attenuation; (iii) both mutants clearly demonstrate the inability to take up and incorporate 22:6 in the RPE and PRC, which results in remarkable changes in the retinal lipidome; and (iv) although the gene signature shows that both mutants are activated by signaling pathways involving proinflammatory cytokines, we found that each of these pathways have distinct features. Moreover, our results highlight the critical role of MFRP and ADIPOR1 in preserving PRC integrity through regulation of enrichment and distribution of 22:6 which, importantly, affects the synthesis of VLC‐PUFAs that provide the precursors for the neuroprotective ELVs. Hence, the maintenance of a balanced 22:6 retinal lipidome relies on MFRP and ADIPOR1, acting to ensure proper acquisition and distribution of key lipids from the RPE to the PRC, these events are necessary to sustain function in early stages of AMD and of other retinal degenerative deceases.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
NGB conceived the study; BJ conducted the lipidomic analysis (LC‐MS/MS and MALDI); histology was performed by WCG; MAK and KD performed the gene analysis; MAK conducted the OCT analysis; BM acquired the fundus images and performed ERG; NGB, BJ, MAK, and WCG designed experiments and analyzed the data; ZF performed the statistical analysis; WCG, MAK, and NGB wrote the paper with input from all other authors. All authors have read and approved the manuscript.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health (NIH) grants R01 EY005121 (National Eye Institute) and P30 GM103340 (National Institute of General Medical Sciences) to NGB, the Eye, Ear, Nose & Throat Foundation of New Orleans, and, in part, by the Research to Prevent Blindness, New York, NY. We thank Thang Pham for technical assistance with the transcriptomic analysis, and Jessica Heap for her help with the lipids Venn diagram figure.
Kautzmann M‐AI, Gordon WC, Jun B, et al. Membrane‐type frizzled‐related protein regulates lipidome and transcription for photoreceptor function. FASEB BioAdvances. 2020;34:912–929. 10.1096/fj.201902359R
REFERENCES
- 1. Katoh M. Molecular cloning and characterization of MFRP, a novel gene encoding a membrane‐type Frizzled‐related protein. Biochem Biophys Res Commun. 2001;282:116‐123. [DOI] [PubMed] [Google Scholar]
- 2. Kameya S, Hawes NL, Chang B, Heckenlively JR, Naggert JK, Nishina PM. Mfrp, a gene encoding a frizzled related protein, is mutated in the mouse retinal degeneration 6. Hum Mol Genet. 2002;11:1879‐1886. [DOI] [PubMed] [Google Scholar]
- 3. Van Raay TJ, Vetter ML. Wnt/frizzled signaling during vertebrate retinal development. Dev Neurosci. 2004;26:352‐358. [DOI] [PubMed] [Google Scholar]
- 4. Ayala‐Ramirez R, Graue‐Wiechers F, Robredo V, Amato‐Almanza M, Horta‐Diez I, Zenteno JC. A new autosomal recessive syndrome consisting of posterior microphthalmos, retinitis pigmentosa, foveoschisis, and optic disc drusen is caused by a MFRP gene mutation. Mol Vis. 2006;12:1483‐1489. [PubMed] [Google Scholar]
- 5. Fogerty J, Besharse JC. Subretinal infiltration of monocyte derived cells and complement misregulation in mice with AMD‐like pathology. Adv Exp Med Biol. 2014;801:355‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collery RF, Volberding PJ, Bostrom JR, Link BA, Besharse JC. Loss of zebrafish Mfrp causes nanophthalmia, hyperopia, and accumulation of subretinal macrophages. Invest Ophthalmol Vis Sci. 2016;57:6805‐6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hawes NL, Chang B, Hageman GS, et al. Retinal degeneration 6 (rd6): a new mouse model for human retinitis punctata albescens . Invest Ophthalmol Vis Sci. 2000;41:3149‐3157. [PubMed] [Google Scholar]
- 8. Krill AE.Flecked retina diseases In: Krill AE,Archer DB, eds. Hereditary Retinal and Choroidal Dystrophies. Vol II New York: Harper & Row; 1977: 739‐824. [Google Scholar]
- 9. Fogerty J, Besharse JC. 174delG mutation in mouse MFRP causes photoreceptor degeneration and RPE atrophy. Invest Ophthalmol Vis Sci. 2011;52:7256‐7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sluch VM, Banks A, Li H, et al. ADIPOR1 is essential for vision and its RPE expression is lost in the Mfrp rd6 mouse. Sci Rep. 2018;8:14339–14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Velez G, Tsang SH, Tsai Y‐T, et al. Gene therapy restores Mfrp and corrects axial eye length. Sci Rep. 2017;7:16151–16159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rice DS, Calandria JM, Gordon WC, et al. Adiponectin receptor 1 conserves docosahexaenoic acid and promotes photoreceptor cell survival. Nat Commun. 2015;6:6228–6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang J, Wang C, Shen Y, et al. A mutation in ADIPOR1 causes nonsyndromic autosomal dominant retinitis pigmentosa. Hum Genet. 2016;135:1375‐1387. [DOI] [PubMed] [Google Scholar]
- 14. Xu M, Eblimit A, Wang J, et al. ADIPOR1 is mutated in syndromic retinitis pigmentosa. Hum Mutat. 2016;37:246‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaarniranta K, Paananen J, Nevalainen T, et al. Adiponectin receptor 1 gene (ADIPOR1) variant is associated with advanced age‐related macular degeneration in Finnish population. Neurosci Lett. 2012;513:233‐237. [DOI] [PubMed] [Google Scholar]
- 16. Rodriguez de Turco EB, Gordon WC, Bazan NG. Rapid and selective uptake, metabolism, and cellular distribution of docosahexaenoic acid among rod and cone photoreceptor cells in the frog retina. J Neurosci. 1991;11:3667‐3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Metherel AH, Irfan M, Chouinard‐Watkins R, Trépanier M‐O, Stark KD, Bazinet RP. DHA cycling halves the DHA supplementation needed to maintain blood and tissue concentrations via higher synthesis from ALA in long‐evans rats. J Nutr. 2019;149:586‐595. [DOI] [PubMed] [Google Scholar]
- 18. Scott BL, Bazan NG. Membrane docosahexaenoate is supplied to the developing brain and retina by the liver. PNAS. 1989;86:2903‐2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bazan NG, Molina MF, Gordon WC. Docosahexaenoic acid signalolipidomics in nutrition: significance in aging, neuroinflammation, macular degeneration, Alzheimer’s, and other neurodegenerative diseases. Annu Rev Nutr. 2011;31:321‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate retina. Prog Lipid Res. 1983;22:79‐131. [DOI] [PubMed] [Google Scholar]
- 21. Aveldaño MI. A novel group of very long chain polyenoic fatty acids in dipolyunsaturated phosphatidylcholines from vertebrate retina. J Biol Chem. 1987;262:1172‐1179. [PubMed] [Google Scholar]
- 22. Bazan NG. Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci. 2006;29:263‐271. [DOI] [PubMed] [Google Scholar]
- 23. Wassall SR, Leng X, Canner SW, et al. Docosahexaenoic acid regulates the formation of lipid rafts: a unified view from experiment and simulation. Biochim Biophys Acta Biomembr. 2018;1860:1985‐1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soubias O, Teague WE, Gawrisch K. Evidence for specificity in lipid‐rhodopsin interactions. J Biol Chem. 2006;281:33233‐33241. [DOI] [PubMed] [Google Scholar]
- 25. Sánchez‐Martín MJ, Ramon E, Torrent‐Burgués J, Garriga P. Improved conformational stability of the visual G protein‐coupled receptor rhodopsin by specific interaction with docosahexaenoic acid phospholipid. ChemBioChem. 2013;14:639‐644. [DOI] [PubMed] [Google Scholar]
- 26. Litman BJ, Niu SL, Polozova A, Mitchell DC. The role of docosahexaenoic acid containing phospholipids in modulating G protein‐coupled signaling pathways: visual transduction. J Mol Neurosci. 2001;16:237‐242; discussion 279‐284. [DOI] [PubMed] [Google Scholar]
- 27. Mitchell DC, Niu S‐L, Litman BJ. Enhancement of G protein‐coupled signaling by DHA phospholipids. Lipids. 2003;38:437‐443. [DOI] [PubMed] [Google Scholar]
- 28. Bazan NG. Docosanoids and elovanoids from omega‐3 fatty acids are pro‐homeostatic modulators of inflammatory responses, cell damage and neuroprotection. Mol Aspects Med. 2018;64:18‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lagali PS, Liu J, Ambasudhan R, et al. Evolutionarily conserved ELOVL4 gene expression in the vertebrate retina. Invest Ophthalmol Vis Sci. 2003;44:2841‐2850. [DOI] [PubMed] [Google Scholar]
- 30. Hopiavuori BR, Anderson RE, Agbaga M‐P. ELOVL4: Very long‐chain fatty acids serve an eclectic role in mammalian health and function. Prog Retin Eye Res. 2018;69:137–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suh M, Clandinin MT. 20:5n–3 but not 22:6n–3 is a preferred substrate for synthesis of n‐3 very‐long‐ chain fatty acids (C24–C36) in retina. Curr Eye Res. 2005;30:959‐968. [DOI] [PubMed] [Google Scholar]
- 32. Yu M, Benham A, Logan S, et al. ELOVL4 protein preferentially elongates 20:5n3 to very long chain PUFAs over 20:4n6 and 22:6n3. J Lipid Res. 2012;53:494‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Agbaga M‐P, Mandal MNA, Anderson RE. Retinal very long‐chain PUFAs: new insights from studies on ELOVL4 protein. J Lipid Res. 2010;51:1624‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oresti GM, Ayuza Aresti PL, Gigola G, Reyes LE, Aveldaño MI. Sequential depletion of rat testicular lipids with long‐chain and very long‐chain polyenoic fatty acids after X‐ray‐induced interruption of spermatogenesis. J Lipid Res. 2010;51:2600‐2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cameron DJ, Tong Z, Yang Z, et al. Essential role of Elovl4 in very long chain fatty acid synthesis, skin permeability barrier function, and neonatal survival. Int J Biol Sci. 2007;3:111‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Monroig Ó, Rotllant J, Cerdá‐Reverter JM, Dick JR, Figueras A, Tocher DR. Expression and role of Elovl4 elongases in biosynthesis of very long‐chain fatty acids during zebrafish Danio rerio early embryonic development. Biochimica Biophys Acta (BBA) ‐ Mol Cell Biol Lipids. 2010;1801:1145‐1154. [DOI] [PubMed] [Google Scholar]
- 37. Jun B, Mukherjee PK, Asatryan A, et al. Elovanoids are novel cell‐specific lipid mediators necessary for neuroprotective signaling for photoreceptor cell integrity. Sci Rep. 2017;7:5279–5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hollyfield JG, Bonilha VL, Rayborn ME, et al. Oxidative damage‐induced inflammation initiates age‐related macular degeneration. Nat Med. 2008;14:194‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mattapallil MJ, Wawrousek EF, Chan C‐C, et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012;53:2921‐2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ferguson LR, Dominguez JM, Balaiya S, Grover S, Chalam KV. Retinal thickness normative data in wild‐type mice using customized miniature SD‐OCT. PLoS ONE. 2013;8:e67265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liebisch G, Vizcaíno JA, Köfeler H, et al. Shorthand notation for lipid structures derived from mass spectrometry. J Lipid Res. 2013;54:1523‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013;41:D793‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hankin JA, Barkley RM, Murphy RC. Sublimation as a method of matrix application for mass spectrometric imaging. J Am Soc Mass Spectrom. 2007;18(9):1646‐1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mao X, Kikani CK, Riojas RA, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:516‐523. [DOI] [PubMed] [Google Scholar]
- 45. Won J, Smith RS, Peachey NS, et al. Membrane frizzled‐related protein is necessary for the normal development and maintenance of photoreceptor outer segments. Vis Neurosci. 2008;25:563‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hayward C, Shu X, Cideciyan AV, et al. Mutation in a short‐chain collagen gene, CTRP5, results in extracellular deposit formation in late‐onset retinal degeneration: a genetic model for age‐related macular degeneration. Hum Mol Genet. 2003;12:2657‐2667. [DOI] [PubMed] [Google Scholar]
- 47. Mandal MNA, Vasireddy V, Jablonski MM, et al. Spatial and temporal expression of MFRP and its interaction with CTRP5. Invest Ophthalmol Vis Sci. 2006;47:5514‐5521. [DOI] [PubMed] [Google Scholar]
- 48. National Library of Medicine (US) . Genetics Home Reference [Internet]. Bethesda, MD: The Library; November 12, 2019. C1QTNF5 gene; Available from: https://ghr.nlm.nih.gov/gene/C1QTNF5. Accessed July 24, 2019. [Google Scholar]
- 49. Mandal MNA, Vasireddy V, Reddy GB, et al. CTRP5 is a membrane‐associated and secretory protein in the RPE and ciliary body and the S163R mutation of CTRP5 impairs its secretion. Invest Ophthalmol Vis Sci. 2006;47:5505‐5513. [DOI] [PubMed] [Google Scholar]
- 50. Li Y, Wu W‐H, Hsu C‐W, et al. Gene therapy in patient‐specific stem cell lines and a preclinical model of retinitis pigmentosa with membrane frizzled‐related protein defects. Mol Ther. 2014;22:1688‐1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tu X, Palczewski K. The macular degeneration‐linked C1QTNF5 (S163) mutation causes higher‐order structural rearrangements. J Struct Biol. 2014;186:86‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dinculescu A, Min S‐H, Deng W‐T, Li Q, Hauswirth WW. Gene therapy in the rd6 mouse model of retinal degeneration. Adv Exp Med Biol. 2014;801:711‐718. [DOI] [PubMed] [Google Scholar]
- 53. Liu A, Chang J, Lin Y, Shen Z, Bernstein PS. Long‐chain and very long‐chain polyunsaturated fatty acids in ocular aging and age‐related macular degeneration. J Lipid Res. 2010;51:3217‐3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hunot S, Vila M, Teismann P, et al. JNK‐mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA. 2004;101:665‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sagulenko V, Thygesen SJ, Sester DP, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20:1149‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials