

Abstract
The past few decades have seen a significant rise in research into alternative polymer based nanosized unilamellar drug delivery systems, termed polymersomes. The reported benefits of polymersomes over the more traditional liposomes include increased stability, higher encapsulation efficacies, better adaptability and reduced water permeation due to an increased bilayer thickness. Together, these advantages render them suitable for a plethora of therapies. The work presented in this manuscript creates and compares four such drug delivery systems, two based on the traditional liposome and two prepared from amphiphilic polymers. From there we assess these systems in terms of size, stability, encapsulation efficiency, drug release, cellular toxicity and cellular uptake. We can confirm from this comprehensive investigation that the multi-functional synthetic polymersomes are undoubtedly a future contender in this expanding field of nanomedicines. Their ability to encapsulate a cocktail of different compounds, high stability as well as their ease of adaptability will ensure that they feature prominently in the future of advanced drug delivery systems.
Keywords: Polymersomes, Liposomes, Amphiphilic polymers, Comparative study, In-vitro investigation
Graphical abstract
1. Introduction
Since the development of liposomes in the 1960's, they have been considered for numerous applications ranging from bioimaging to gene and vaccine delivery, treatments of infections and inflammation, lung diseases and anticancer therapy (Allen and Cullis, 2013; Qu et al., 2014; Xu et al., 2019). Liposomes have proven useful in reducing the side effects of encapsulated drugs and passively accumulating in areas of high vasculature when the particle size is below 200 nm. In addition, the incorporation of PEG ensures that there is an extended circulatory time as the liposomes are not recognised by the RES. Since the first commercially available liposome containing doxorubicin (Doxil) over two decades ago, there have been seven additional cancer based liposomal therapies receiving FDA approval, including the most recently approved Vyxoes® for acute myeloid leukaemia, in 2017. A similar amount of liposomal based therapies are available commercially for non-cancer based treatments (Bulbake et al., 2017). In addition to those nanotherapies which have received FDA approval, there are many more in various phases of clinical trials.
This measured increase in the commercially available liposomal formulations may be due to several limitations reported; such as physical and chemical instability due to their biological nature and drug leakage over time (He et al., 2019; Qin et al., 2011), lack of control over the rate of drug release, difficulty in overriding barriers such as the blood brain barrier, and insufficient loading of drugs (Barenholz, 2001). These limitations have drove the exploration towards expanding the range of cancer nanotherapeutics available which possess similar properties of assembling into monolayer or bilayer NPs and can overcome some of the problems associated with liposomes.
Currently there is a wide range of nano sized drug delivery systems (DDS) for cancer therapy utilizing both biodegradable and non- biodegradable materials. These include but are not limited to; micelles (Torchilin, 2007), nanotubes (Manzur et al., 2017), nanocapsules (Musyanovych and Landfester, 2014), niosomes (Kazi et al., 2018) and polymersomes (Aibani et al., 2018; Levine et al., 2008).
Of all of variations of the self-assembling, usually amphiphilic moieties, polymersomes most closely resemble the liposomal structure (Rideau et al., 2018). Discher et al. demonstrated in the late 1990s, the ability of diblock copolymers to assemble into bilayer vesicles termed polymersomes (Discher et al., 1999; Discher and Eisenberg, 2000). Polymersomes have a vesicular structure similar to liposomes with a hydrophilic core and a hydrophobic bilayer allowing encapsulation of both hydrophilic and hydrophobic drugs with a hydrophilic corona. Polymersomes as nanoparticulate drug delivery systems have gained a lot of attention recently for having several advantages over liposomes such as higher stability, better control over membrane properties and an ability to encapsulate a large variety of drugs (Lee and Feijen, 2012). They have also been shown to ‘pop’ when prepared from suitable polymers that allow an increase in osmotic pressure to build, something that the more permeable bilayer of the liposomes will not allow (Peyret et al., 2017).
Polymersomes consist of amphiphilic block polymers capable of self-assembling into nanoscale vesicles, with PEG commonly used as the hydrophilic block (Bleul et al., 2015). These high molecular weight polymers possess similar amphiphilic properties to lipids but are comprised of polymer chains covalently linked as successions of two or more blocks offering a membrane which is more compact providing rigidity and better stability to these vesicles (Discher and Ahmed, 2006). In addition, the modular structure of the polymer and the ease of synthesis enables these NP's to be both versatile and adaptable with varying both monomers and side chains.
The research presented herein compares directly the two different bilayered nanoparticular structures by the creation of both DDSs with a similar composition. The liposomes were prepared from phosphatidylcholines extracted from egg or soybean as they are a major component of the cell membrane (Raicu and Popescu, 2008), cholesterol which helps achieve rigidity of the bilayer membrane and a PEG outer layer which gives stealth properties to liposomes. The polymersomes were formulated using a polymer containing cholesteryl and PEG. The lipid used in the liposome was replaced with both hexadecanoic and oleic alkyl chains, to mirror the composition of the lipid. The subsequent formulation into liposomes and polymersomes resulted in two DDSs with similar composition, the only variation being the lipid in the liposome was replaced with a constitutionally similar polymer, Fig. 1. We then compared the two NP's in terms of hydrodynamic radius, PDI, stability, encapsulation of both hydrophobic and hydrophilic compounds, as well as drug release and surface charge. The results are reported below.
Fig. 1.

Illustration of liposomes and polymersomes with their corresponding components. Green depicts the PEG component, light blue the oleic chains, dark blue the charged acetyl choline, light grey the hexadecanoate chains and dark grey the cholesteryl. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2. Materials and methods
2.1. Materials
Cholesteryl Chloroformate, Ethylene diamine, 1-Octadecanol, oleic acid, (N,N′-Dicyclohexylcarbodiimide) DCC, 4-(Dimethylaminopyridine) DMAP, PEG-methacrylate (Mn 500), PEG-methylmethacrylate (Mn 2000), Methacrylic acid, methoxy PEG (550 & 2000), 1 1′-azobis (cyclohexanecarbonitrile) (AICN), L-α-Phosphatidylcholine from egg yolk, FITC-CM-Dextran (FCD) 4 kDa, FITC-DEAE-Dextran (FDD), FITC-Dextran (F-D), anthracene, Dialysis membrane (MWCO 14,000), PBS tablets, Fluorescence labelled TLC plates with aluminium backing were purchased from Sigma chemicals. Oleic acid, CDCl3 was purchased from TCI, Japan. DMEM, RPMI 1640, Hams F12, Trypsin-EDTA, PenStrep and Foetal bovine serum were sourced from Thermofisher Scientific, UK. All synthesis were carried out in inert conditions under nitrogen gas unless otherwise stated.
2.2. Synthesis of monomers and polymers
2.2.1. Synthesis of cholesteryl monomer (1)
Following a procedure previously published (Martin et al., 2016), 1.044 ml (15.61 mol) of Ethylene Diamine was dissolved in 5 ml dry DCM and cooled on an ice bath. 0.5007 g (1.11 mmol) Cholesteryl Chloroformate dissolved in 5 ml dry DCM was added slowly to above solution and stirred overnight. Product was washed 3 times with water and brine and dried to leave the intermediate compound, cholesteryl ethylene amine carbamate. 0.9889 g (2.09 mmol) of the intermediate was dissolved in 20 ml dry DCM with 0.1811 g (2.09 mmol) methacrylic Acid and 0.0522 g (0.20 mmol) DMAP and kept on an ice bath. 0.5175 g (2.09 mmol) DCC dissolved in 20 ml DCM was added dropwise and stirred for 24 h. Product was filtered and filtrate collected and purified using silica gel column with Chloroform: Methanol 18:2 as mobile phase and characterised using 1H NMR and mass spectroscopy.
2.2.2. Synthesis of octadecyl monomer (2)
1-Octadecanol (2.7 g, 9.98 mmol) was dissolved in 30 ml dry DCM with 0.244 g (0.002 mmol) DMAP and 0.86 g (0.01 mmol) methacrylic acid. 2.06 g (0.01 mmol) DCC solution was prepared in 20 ml dry DCM and added drop wise to this solution and maintained overnight under constant nitrogen gas. Resultant product was filtered and purified using Silica gel with Chloroform: Methanol 18:2 as mobile phase and further washed 3 times using 0.1 M Hydrochloric acid using a separating funnel and dried to obtain monomer 2.
2.2.3. Synthesis of oleate monomer (3)
Ethylene diamine (3.088 ml, 48.0 mol) was dissolved in 10 ml DCM. The following solution, 2.208 g (7.81 mmol) of Oleic Acid dissolved in 5 ml DCM with 1.609 g (7.81 mmol) DCC and 0.095 g (0.78 mmol) DMAP, was added slowly and stirred for 48 h at 40 °C. The resulting product was washed 3 times with brine and dried to obtain the intermediate compound, oleic carbamate- ethylene amine. 2.532 g (7.81 mmol) of the intermediate compound was dissolved in 20 ml DCM with 0.6729 g (7.81 mmol) of methacrylic acid and 0.09550 g (0.78 mmol) DMAP in ice bath. 1.612 g (7.81 mmol) DCC dissolved in 20 ml DCM solution was added drop wise and stirred for 24 h. Monomer 3 was purified as mentioned in above procedure.
2.2.4. Preparation of polymers (5a/b)
Monomer 1, 2, 3 and polyethylene glycol methacrylate 4a (Mn 500) or Polyethylene glycol methyl ether methacrylate (Mn 2000) 4b were taken in a reaction vessel in (1:1:1:1 M ratio) for P500 and (1:1:1: 0.25) for P2000 in 20 ml anhydrous Tetrahydrofuran with 5 mg 1 1′-azobis (cyclohexanecarbonitrile), freeze-thawed three times under vacuum and kept at 80 °C for 72 h. Polymer was precipitated and washed with hexane with centrifugation at 6000 rpm for 5 min three times. Final polymer was obtained as yellow thick viscous liquid (5a) or white powder (5b).
2.2.5. Synthesis of cholesteryl mPEG carbonate (6a/b)
2 g methoxy PEG Mn2000 (0.001 mol) or 0.55 g methoxy PEG Mn550 (0.001 mol) and 0.449 g (0.001 mol) cholesteryl chloroformate were dissolved in THF in an ice bath with a constant influx of nitrogen. Solution was stirred at 45–50 °C for 48 h, dried and washed with hexane to obtain final product, cholesteryl PEG (500) carbonate (6a) or cholesteryl PEG (2000) carbonate (6b).
2.2.6. Characterisation of compounds
Molecular weight was determined by Mass spectroscopy (1 mg/ml in methanol) using Thermo Finnigan LCQ Classic Ion Trap LC-MS in positive electron mode. Spectra were analysed using Tuneplus Version 1.0 SR1. 1H NMR spectroscopy was conducted using Varian (500 MHz) NMR spectroscope in deuterated chloroform and analysed on VNMRj 2.2 and Topspin 3.5 pl6 software. Fixed aqueous layer thickness was determined using 2 mg ml−1 liposomes and polymersomes loaded with 20 μg ml−1 Rhodamine 6G and prepared by the reverse phase evaporation method as described in section 2.3.1. Fixed Aqueous layer thickness (FALT) was calculated by zeta potential (L) of liposomes and polymersomes measured in different concentrations of NaCl (0 mM, 10 mM, 50 mM, 100 mM), Log L was plotted against k, where k = √C/0.3 for univalent salts and C is molality of NaCl, giving slope which is the fixed aqueous layer thickness in nm (Shimada et al., 1995).
2.3. Formulation of liposomes and polymersomes
2.3.1. Reverse phase evaporation method (RVE)
An aliquot of 1 ml of egg phosphatidylcholine and 6a/b (500/2000) (2 mg ml−1 in 9:1 M ratio) or 0.5 ml polymer 5a/b (500/2000) (2 mg ml−1) in chloroform were evaporated in a round bottom flask to form a thin film. Encapsulated compounds (FCD, FDD or F-D (100 μl of 2 mg ml−1 in PBS)) were added, if required, and evaporated to dryness. 0.5 ml chloroform was added and the solution was sonicated for 15 min. If anthracene loaded NPs were required, anthracene (66 μl of 1 mg ml−1 in ethanol) was added at this stage (prior to sonication). 1 ml PBS was added to chloroform solution for liposomes or 0.5 ml (2 mg ml−1) polymer 5a/b in PBS with 0.5 ml plain PBS was added for polymersomes and sonicated for 30 min after which the chloroform was evaporated to form 1 ml liposomes/polymersomes. Resultant nanoparticles were sonicated in a bath sonicator for 10 min after particle formation.
2.3.2. Emulsion evaporation method (EM-EV)
An aliquot of 1 ml of egg phosphatidylcholine and 6a/b (550/2000) (2 mg ml−1 in 9:1 M ratio) or 0.5 ml polymer 5a/b (2 mg ml−1) in chloroform was mixed with 1 ml PBS for liposomes and 0.5 ml (2 mg ml−1) polymer in PBS with 0.5 ml PBS for polymersomes. Anthracene (66 μl of 1 mg ml−1 in ethanol) was added in chloroform. Hydrophilic dyes (100 μl of 2 mg ml−1) were added in PBS. Resultant chloroform-PBS mixture was sonicated for 30–40 min to form an emulsion. Chloroform layer was evaporated to form nanoparticles instantly and bath sonicated for 10 min to form unilamellar particles.
2.4. Characterisation of nanoparticles
Encapsulation efficiency of polymersomes and liposomes loaded with hydrophilic dyes was measured by centrifugal filtration, using a method developed in house. Polymersomes/liposomes were placed in a dialysis tubing (MWCO 14,000 Da) tied at both ends and placed suspended in a centrifuge tube. These tubes were then centrifuged at 3000rcf at 4 °C for 2 h. Resultant filtrate was collected and analysed for unencapsulated drug using Varian Cary Eclipse Fluorescence spectrophotometer at Ex 490/Em 517 for FCD (y = 32.318x, R2 = 0.9982), FDD (y = 18.063x, R2 = 0.9997) and F-D (y = 33.788x, R2 = 0.999). For anthracene encapsulation efficiency, 100 μl of Anthracene loaded nanoparticle suspension was diluted to 2 ml with ethanol and measured for anthracene concentration at Ex 355/Em 400 (y = 221.14x, R2 = 0.9987). Particle size was measured by adding 100 μl of nanoparticles in 1 ml PBS were evaluated for size and PDI using Malvern Nano-ZS Zetasizer, softer version 7.03. In each case the most abundant peak is quoted with n = ≥ 3. Zeta potential was observed using Universal Dip cell electrode. Particle morphology was observed using scanning electron microscopy (SEM), nanoparticle suspensions were air dried overnight on aluminium stubs and coated with ultra-thin Gold/Palladium layer at 18 mA for 3 min using Polaron Equipment Ltd. E5100 Sputter coater and observed under FEI Quanta 200 ESEM in high vacuum mode.
In-vitro release studies of polymersomes and liposomes were conducted by placing liposomes/polymersomes in dialysis tubes tied at both ends and placing them in PBS maintained at 37 °C and stirred using a magnetic stirrer. Aliquots were taken out at regular intervals of time and analysed by a relevant analytic method. The amount of sample removed was replaced by fresh PBS to maintain sink conditions.
2.5. Cell culture studies
All cells were obtained from in-house cell line repository. HeLa cells were cultured in DMEM medium with 10% FBS and 1% Penstrep and 1% NEAA (non-essential amino acids). BxPC-3 cells were cultured in RPMI 1650 medium with 10% FBS and 1% Penstrep whereas CHO cells were grown in Hams F12 media with 10% FBS and 1% Penstrep, 1% Glutamine and 1% NMEAA. Cells were grown in HeraCell incubators at 37 °C with constant influx of 5% CO2. All cells were counted using an Invitrogen Countess Automated cell counter.
2.5.1. Cellular uptake studies
1 × 105 cells ml−1 (100 μl) HeLa cells were seeded into 96 well plates and incubated overnight after which FCD (250 μg ml−1) or anthracene (50 μg ml−1) liposomes/ polymersomes, sterile filtered using 0.45 μm Millex MCE filters, were added at a volume of 100 μl in PBS and incubated for 4 h at 37 °C. After incubation cells were washed twice with PBS and measured using Fluostar Omega microplate reader at Ex 480 nm/Em 520 nm for FCD and Ex 355 nm/Em 460 nm for anthracene. After fluorescence measurements, protein estimation of cells was done by lysing the cells and adding 25 μl 0.1% Triton-X100 into each well with 15 min incubation at 37 °C. Protein estimation was done using BCA Protein assay kit after incubation at 37 °C for 30 min and measuring absorbance at 562 nm. Cellular uptake results are reported as fluorescence per mg of protein.
2.5.2. Mechanism of cell uptake by inhibition of endocytosis
HeLa cells were incubated with 100 μl of 30 μM Chlorpromazine Hydrochloride in media for 30 min. After which the supernatant medium was removed and cells were incubated with FCD loaded polymersomes/liposomes for 4 h as described above. Cells were then washed twice with PBS and analysed for FCD fluorescence as mentioned before.
2.5.3. Cellular toxicity studies
Cell toxicity of blank PS2000 and LS2000 was observed in 3 cell lines namely HeLa cells, BxPC-3 cells and CHO cells. 100 μl 5 × 104 cells/ml were seeded in 96 well plates and incubated overnight before being treated with blank polymersomes and liposomes at concentrations 250 and 500 μg/ml and incubated overnight. The number of live cells after treatment was determined using an MTT assay measured at 570 nm for intensity of colour relative to the concentration of live cells.
2.6. Stability studies
Nanoparticle suspensions were subjected to stability studies at 5 °C (stored in refrigerator) and 25 °C (incubator) for 8 weeks. Samples were taken at regular intervals and analysed for encapsulation efficiency and size.
2.7. Statistical analysis
All data is reported with SEM and n = 3 unless otherwise stated. Statistical significance of groups was determined using two tailed Unpaired Student's t-test in Graphpad Prism Version 5.01.
3. Results
3.1. Monomer and polymer synthesis
Polymers were prepared to mimic the composition regularly found in liposomes with the conformation of each NP displayed in Fig. 1. The monomers prepared for polymerisation included; cholesteryl, PEG, oleate and octadecyl with modification (if necessary) to ensure they contained an olefinic group essential for the Michael addition in the polymerisation reaction. The monomers used in the synthesis of polymer 5 are displayed in Fig. 2 (compounds 1–4), with compound 6 the cholesteryl-PEG component used in the formation of the liposomes. Polymer 5a was prepared using 4a, with PEG 500, and polymer 5b prepared using the higher molecular weight PEG 2000 (4b).
Fig. 2.
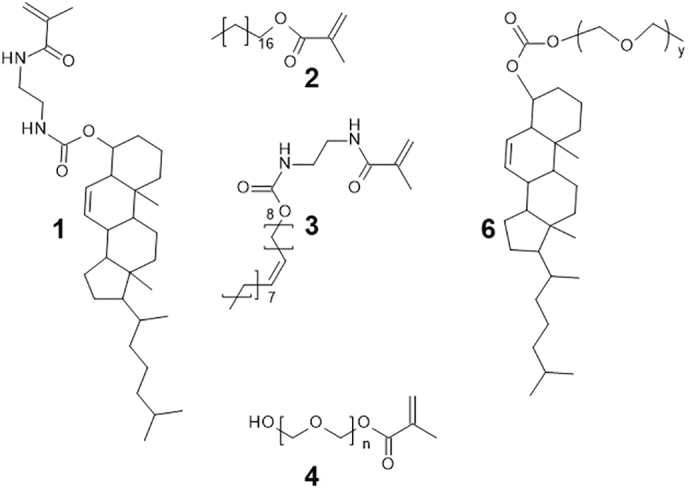
Monomers 1–4 utilised in the polymerisation reaction and compound 6, prepared for the incorporation within liposomes.
All monomers were characterised by both MS and 1H NMR (see supporting information S1-S4) and confirmed. Polymerisation occurred via free radical initiation using AICN, with the production of a random co-polymer. Content was tightly controlled using molar ratios, with an illustration of a potential product shown in Fig. 1. NMR was used to confirm that the Michael addition had taken place with the disappearance of the olefinic protons (S5).
The hydrophilic block fraction, f value, is a useful measure of hydrophilic volume with respect to overall polymer, especially in PEG based polymers. It is calculated by the formula f = Hp/Hp + Hn where Hp is the volume of PEG and Hn is the overall volume of polymer fraction. The f value should be 25% < f < 40% to obtain spherical uni-lamellar vesicles (Christian et al., 2009; Guan et al., 2015; Letchford and Burt, 2007). The f value of 5a was determined to be 25% (1:4 M ratio) and 5b was 30% (1:3.25 M ratio) indicating that both the polymers are capable of forming bi-layered spherical polymersomes. Given that the PEG moiety is the sole hydrophilic entity in the polymersomes, the amount present is essential for self-assembly. This is not the case for the liposomes due to the polar phosphorylcholine head group on the lipids, and so the lower value of 10% was chosen as it has previously been shown to have an increased circulation time (Doi et al., 2019; Ren et al., 2019), while at the same time allowing a 10% cholesteryl incorporation.
3.2. Preparation and characterisation of liposomes and polymersomes
Four NPs were prepared using two different literature methods. They include a liposome and polymersome containing a short (Mn500) PEG and are referred to as LS500 (liposome) and PS500 (polymersome). In addition, two NP's containing a larger Mn PEG (2000) are referred to as LS2000 (liposome) and PS2000 (polymersome). There are a number of different methods available within the literature regarding the preparation of NPs (Joshi et al., 2016; Sadzuka et al., 2002). The two most commonly used methods for preparation of liposomes (without the aid of a microfluidics device) are the reverse phase evaporation method (RPE), sometimes referred to as the thin film hydration (TFH) and the emulsion evaporation method (EM-EV). The RPE method involves hydration of the thin polymer/lipid film formed after vacuum evaporation in a round bottom flask using an aqueous medium such as PBS (Cortesi et al., 1999; Pattni et al., 2015). The EM-EV is a simplified method where the NPs are formed by evaporation of the organic phase from the emulsion under reduced pressure in one step (Elorza et al., 1993; Patil and Jadhav, 2014).
3.2.1. Effect of encapsulated compound and formulation method on hydrodynamic diameter, PDI, zeta potential and encapsulation efficiency
Table 1 displays the hydrodynamic diameter, PDI and zeta potential achieved from all four formulations using the two different methods of preparation. In addition, two compounds with vastly different logP values were encapsulated, the hydrophilic and negatively charged compound, fitc-cm-dextran (FCD), and the hydrophobic compound anthracene. As can be seen from the results there some variation between, not only the methods utilised to prepare the NPs, but also the effect of the encapsulated compound. In general, the smallest particle sizes were recorded for the reverse phase evaporation method which requires the hydration of the film over a period of time with the additional step of probe sonication converting multilamellar particles to unilamellar and thus helping to reduce size. The average size of the liposomes was found to be higher than that of their corresponding polymersomes. Thus suggesting that the charge on the lipid head group is responsible for this larger hydrodynamic radius. An increase in size was recorded for the anthracene encapsulating liposomes, most likely due to increasing the hydrophobic bilayer in this case, as liposomes are known to have thinner bilayers than polymersomes however, a similar increase was also observed for the polymersomes. This is possibly due to the similarity in PEG moieties.
Table 1.
Hydrodynamic diameter, PDI and Zeta potential of liposomes (LS500 and LS2000) and polymersomes (PS500 and PS2000) encapsulating hydrophobic and hydrophilic compounds.
| FCD | LS500 | PS500 | LS2000 | PS2000 | |
|---|---|---|---|---|---|
| Size (nm) | EMEV | 305.9 ± 14.6 | 174.2 ± 45.4 | 318.5 ± 84.6 | 223.4 ± 50.3 |
| RPE | 266.1 ± 31.2 | 163.4 ± 66.8 | 239.2 ± 55.4 | 162.9 ± 20.1 | |
| PDI | EMEV | 0.5 ± 0.1 | 0.8 ± 0.1 | 0.4 ± 0.07 | 0.4 ± 0.1 |
| RPE | 0.5 ± 0.09 | 0.7 ± 0.2 | 0.4 ± 0.1 | 0.3 ± 0.05 | |
| Zeta (mV) | EMEV | −10.2 ± 4.09 | 1.8 ± 1.2 | −10.05 ± 1.8 | 2.2 ± 1.6 |
| RPE | −8.1 ± 2.1 | 1.6 ± 1.7 | −10.8 ± 1.8 | 0.9 ± 1.5 | |
| Anthracene | LS500 | PS500 | LS2000 | PS2000 | |
|---|---|---|---|---|---|
| Size (nm) | EMEV | 174.6 ± 5.6 | 316.2 ± 76.0 | 442.9 ± 48.1 | 419.4 ± 69.7 |
| RPE | 348.2 ± 18.5 | 184.8 ± 23.5 | 300.2 ± 71.0 | 261.9 ± 82.6 | |
| PDI | EMEV | 0.3 ± 0.09 | 1 ± 0 | 0.4 ± 0.03 | 0.4 ± 0.03 |
| RPE | 0.4 ± 0.05 | 0.9 ± 0.1 | 0.4 ± 0.09 | 0.5 ± 0.1 | |
| Zeta (mV) | EMEV | −11.9 ± 0.7 | 3.5 ± 1.0 | −2.6 ± 1.3 | 4.8 ± 0.1 |
| RPE | −9.8 ± 1.8 | 3.1 ± 0.3 | −3.6 ± 0.7 | 4.5 ± 0.2 |
LS500, LS2000 and PS2000 all had appropriate PDIs ranging from 0.3–0.5 with PS500 having a higher PDI of approx. 0.7–1.0. The lack of monodispersity in the PS500 is most likely due to the hydrophilic block fraction, f value, being at the lower end of the required amount for the generation of a bilayered systems. Discher et al. (2007) reported that block copolymers having a hydrophilic fraction (f) of 35% ± 10% by weight of the total polymers generally tend to self-assemble into polymersomes, whereas polymers with f > 45% tend to form micelles and those with f < 25% form inverted microstructures, so the PS500 being at the lower end of the f values could suggest that an alternative NP formation is present in addition to the polymersomes. Evaluation of zeta potential indicated that all the polymersomal loaded systems were almost neutral, having a positive charge up to 5 mV, whereas liposomes had a net negative charge up to 12 mV. LS2000 and PS2000 were slightly more positively charged than LS500 and PS500 respectively owing to the higher density PEG moiety. In general, anthracene loaded liposomes and polymersomes were slightly more positive than FCD loaded systems. The method of preparation did not have any effect on the zeta potentials of the both types of NPs.
A representative size distribution curve of PS2000 is displayed in Fig. 3a. Scanning Electron microscopic images (Fig. 3b) of PS2000 confirm the spherical shape of the particles, with uniformity of size. Also suggested from the DLS results shown in Fig. 3a is the formation of smaller particles, possibly micelles, shown at around 10 nm in diameter. This is not particularly surprising given the high percentage of PEG included within the formulations, however, as the PEG operates as the only hydrophilic entity in the Ps formulations, it is essential that the amount included is sufficient to generate a balance between the hydrophobic and hydrophilic moieties.
Fig. 3.

(a) Representative size distribution graph of polymersomes showing formation of bilayer polymersomes of size approx. 150 nm and a small number of micelles formed in the process. (b) SEM image of PS2000 polymersomes, with highlighted diameters, from top tobottom, of 412, 359, 298, 356 and 606 nm.
There was almost no variation in encapsulation efficiency of FCD liposomes and polymersomes with PEG 500 and 2000 for both methods of preparation (S6). Reverse phase evaporation method indicated that the encapsulation efficiency of FCD in both liposomes and polymersomes was high (up to approximately 75%). This relates to the findings of Sardan et al. who have shown that up to 75% encapsulation efficiency of a hydrophilic drug such as Dox in liposomes can be achieved when prepared by reverse phase evaporation method (Sardan et al., 2013). In other cases there has been up to 40% encapsulation of highly water soluble proteins such as Drosophilia AChE and hydrophilic potassium chromate in liposomes prepared by thin film hydration method (Colletier et al., 2002; Thompson et al., 2009). A slightly lower encapsulation efficiency of FCD (up to 60%) was observed in liposomes LS500 having smaller PEG chain length but there was good encapsulation efficiency in polymersomes for both PS500 and PS2000. Thus suggesting that a high encapsulation efficacy can be achieved for all formulations regardless of PEG chain size for hydrophilic compounds such as FCD. Similarly, there was high encapsulation of hydrophobic anthracene in both LS2000 and PS2000 and slightly less in LS500 and PS500 considering the denser nature of the PEG 2000, it stands to reason that better entrapment of molecules is achieved.
Hence, from the observations of the above characteristics, it was concluded that the RPE method provides smaller sized homogenous NPs as compared to the other two methods of preparation for the formulations examined and was used for all subsequent investigations.
3.2.2. Fixed aqueous layer thickness (FALT)
The hydrophilic layer of PEG surrounding NPs is attributed to the prevention of interaction with serum protein. It is referred to as the fixed aqueous layer thickness (FALT) and can be measured by evaluating the zeta potential of the NPs in different concentrations of NaCl. The FALT of liposomes and polymersomes with PEG 500 (LS500 and PS500 respectively) and PEG 2000 (LS2000 and PS2000 respectively) are specified in Table 2 and suggest that the extent of the surrounding PEG layer of both polymersomes and liposomes are comparable.
Table 2.
Fixed aqueous layer thickness (nm) of different PEG chain lengths.
| Formulation | LS500 | PS500 | LS2000 | PS2000 |
| FALT (nm) | 0.78 ± 0.12 | 0.63 ± 0.46 | 1.40 ± 0.46 | 1.71 ± 0.20 |
3.2.3. Effect of encapsulating charged compounds on hydrodynamic radius, PDI, zeta potential and encapsulation efficiency
In order to establish the effect of any charges on the encapsulated compound, LS2000 and PS2000 encapsulating FDD (positive charge) F-D (Neutral) and FCD (negative charge) were prepared by the RPE method and evaluated for particle characteristics (Table 3). The sizes were as expected from our previous experiments and confirmed that in all cases the liposomes were larger than their corresponding polymers. The particle size and PDI of LS2000 and PS2000 was not affected by the charge of encapsulating compound. Perhaps not surprisingly, the largest difference was recorded in the zeta potentials with the charge on the encapsulating compound greatly affecting the overall surface charge. Although all formulations had any unencapsulated drug removed following preparation, this substantial change in surface charge would indicate that the interaction between the surface and any unencapsulated compounds was a greater attraction than the concentration gradient, and so some compound was remaining close to the NP surface and thus neutralising the charge. The encapsulation efficiencies were also similar leading to the conclusion that charge did not have any effect on the encapsulation and size or PDI of liposomes and polymersomes, this can again be attributed to the dense PEG layer coating the surface of liposomes and polymersomes.
Table 3.
Characterisation of LS2000 and PS2000 loaded with FDD (Positive), F-D (Neutral) and FCD (negative) compounds.
|
LS2000 |
PS2000 |
LS2000 |
PS2000 |
LS2000 |
PS2000 |
|
|---|---|---|---|---|---|---|
| FDD loaded | F-D loaded | FCD loaded | ||||
| Size (nm) | 286 ± 22 | 154 ± 14 | 242 ± 40 | 147 ± 46 | 239 ± 55 | 163 ± 20 |
| EE (%) | 81 ± 2 | 87 ± 2 | 68 ± 2 | 72 ± 19 | 75 ± 7 | 76 ± 2 |
| PDI | 0.4 ± 0.07 | 0.2 ± 0.01 | 0.3 ± 0.05 | 0.3 ± 0.09 | 0.4 ± 0.1 | 0.3 ± 0.05 |
| Zeta (mV) | −0.9 ± 1.9 | 4.0 ± 0.8 | −7.1 ± 1.1 | 2.1 ± 1.3 | −10.8 ± 1.8 | 0.9 ± 1.5 |
Given the similarities in encapsulation efficiency, the release profile of the different compounds from each formulation was established and displayed in Fig. 4.
Fig. 4.
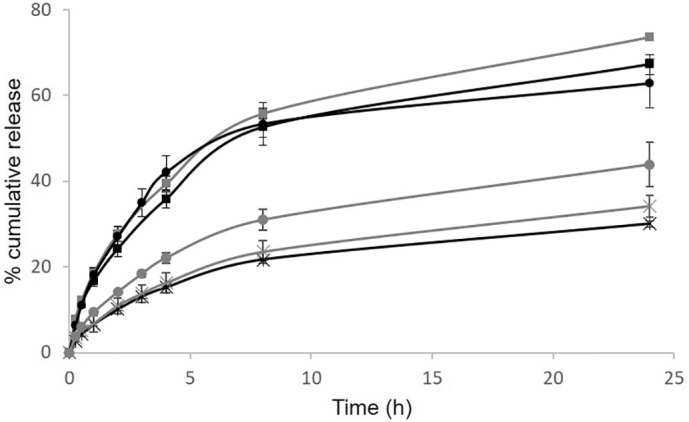
Release profiles of LS2000 (black lines) and PS2000 (grey lines) encapsulating the neutral charged FD (circles) positively charged FDD (crosses) and -negatively charged FCD (squares).
Polymersomes have been reported to have slower cargo release compared with liposomes (Ahmed and Discher, 2004; Chiang et al., 2013) with our results for the neutral compounds (FD) confirming this outcome, Fig. 4. This is most likely due to the increased thickness of the hydrophobic bilayer within the polymersomes. The negatively charged compound (FCD) has little effect on the release kinetics, with a profile almost identical to that of the neutral compound. Not surprisingly the positively charged compound had a slowest release from liposomes, most likely due to electrostatic interactions delaying transfer from the negatively charged NP. A comparison between the zeta potentials of the liposome containing the neutral compound (−7.1 mV) to that of the positive compound (−0.9 mV) (Table 3) is further confirmation of this interaction, and in fact it is suggesting that the FDD may be transcending the liposome as normal, but remaining at the corona and neutralising the negative surface charge. The release profiles from the polymersomes are somewhat more thought-provoking. The negatively charged FCD had a similar release profile from the polymersome as from the liposome, it did not seem to be hindered by the increased thickness of the hydrophobic bilayer, suggesting that there is less Van der Waals attraction between this compound compared to the neutral compound within the bilayer. In addition, the positively charged compound, FDD, had a substantially retarded release profile, similar to the electrostatically attracted liposome with the FDD. One possible explanation for this may be cross linking between the compound and potentially hydrolysed polymer chains, however, further analysis would be required to confirm.
3.3. Cellular uptake of FCD and anthracene loaded polymersomes and liposomes
In an attempt to establish the cellular uptake of the 4 NP's, each DDS was loaded with a hydrophilic dye (FCD) or a hydrophobic dye (anthracene) and both were incubated with HeLa cells for 4 h at 37 °C at concentrations of 250 μg ml−1 FCD and 50 μg ml−1 anthracene. As seen in Fig. 5, PS500 containing FCD had higher uptake (up to 0.9 ± 0.3 μg/mg protein) when compared to the same concentration of compound within either PS2000 (0.6 ± 0.08), LS500 (0.5 ± 0.1) or LS2000 (0.4 ± 0.09). When considering the hydrodynamic radius of each of these formulations, PS500 displayed the smallest size, so it is likely that the size of the NPs is the most significant factor in this case, especially when comparing PS500 to either of the liposomal formulations. This would suggest therefore that the PS2000 formulation resulted in a lower than expected value, one possible suggestion is that the PS500had a wider range of particles, given the higher PDI value, a phenomenon introduction of an extrusion method could eradicate. The uptake of anthracene in polymersomes PS500 and PS2000 (1.01 ± 0.2 and 0.5 ± 0.002 respectively) was significantly higher than their corresponding liposomes. Due to the negative charge character of the plasma membrane, small sized positively charged and neutral particles are better absorbed and endocytosed than negatively charged particles especially through clathrin mediated endocytosis (Hillaireau and Couvreur, 2009; Sadat et al., 2016). Due to their smaller size and neutral charge, polymersomes can prove advantageous for rapid uptake into cells. Hence it can be concluded that polymersomes have at least as good and in many cases superior uptake for both FCD and anthracene than liposomes. Random copolymers having cholesterol with PEG 500 and decyl side chains have been reported to have high uptake of FCD in HeLa cells (Martin et al., 2016).
Fig. 5.
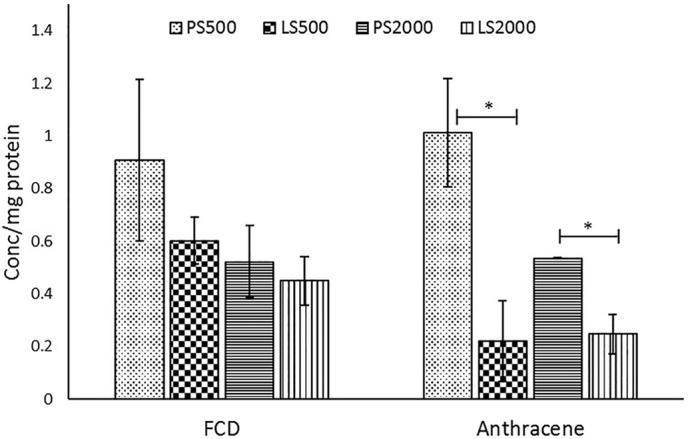
Cellular uptake within Hela cells of NPs encapsulating a hydrophobic or hydrophilic cargo.
The mechanism of cellular uptake was evaluated by chemical inhibition of endocytosis using chlorpromazine HCl and the FCD encapsulated NPs. Chlorpromazine inhibits clathrin coated pit formation by reversible displacement of clathrin and its adapter proteins from cell membrane to intracellular vesicles thereby inhibiting endocytosis by this pathway (Swaminathan et al., 2014; Vercauteren et al., 2010).
Cellular uptake of PS500 and PS2000 decreased significantly by 37.6 ± 19.7% and 46.3 ± 21.0% respectively on inhibition of endocytosis thus indicating that uptake of polymersomes is predominantly by clathrin- mediated endocytosis (S7). Miller et al. have shown that sterically stabilised liposomes with PEG 2000 undergo significantly less endocytosis than conventional non pegylated liposomes and it is possible that they are taken up by other mechanisms (Miller et al., 1998).
3.4. Cell toxicity using MTT assay
Liposomes prepared with PEG 2000 have been extensively used in research for anticancer therapy for their stealth properties and providing effective masking from serum proteins (Marqués-Gallego and Kroon, 2014; Van Den Hoven et al., 2013; Zhang et al., 2016). Therefore, only the two NP's using the higher weight PEG were analysed further within this study. Cell toxicity studies of blank liposomes and polymersomes at different concentrations in three cell lines after overnight incubation were observed using MTT assay. Fig. 6a presents the cell viability of polymersomes and liposomes at 0.25 mg/ml in HeLa cells, CHO cells and BxPC-3 cells. There was no statistically significant difference in the cell toxicities of blank liposomes and polymersomes in Hela and BxPC-3 cells with viabilities of 85.5 ± 4.6% and 87.2 ± 9.4% for liposomes in HeLa and BxPC-3 cells and 73.0 ± 3.9% and 86.8 ± 6.2% for polymersomes in HeLa and BxPC-3 cells respectively. Polymersomes were statistically slightly more toxic to CHO cells at 0.25 mg/ml demonstrating cell viability of 77.3 ± 2.6% viability as compared to liposomes having 88.0 ± 1.4% viability.
Fig. 6.

(a) Cell viability of 0.25 mg/ml NPs incubated for 16 h with various cell lines. (b) Cell viability of 0.5 mg/ml NPs incubated for 16 h with various cell lines.
When compared to cell toxicity at 0.5 mg/ml concentration (Fig. 6b), it was found that polymersomes were significantly more toxic to cells at higher concentration. There was 55.4 ± 1.7% cell viability of HeLa cells when treated with polymersomes and 71.5 ± 2.5% cells were viable in CHO cells whereas 83.6 ± 8.2% cells were viable in BxPC-3 cell line as compared to liposomes having 81.7 ± 4.4, 91.0 ± 0.8 and 98.5 ± 5.9% viability in HeLa, CHO and BxPC-3 cells respectively.
The increased toxicity of the polymersomes compared liposomes could be a direct result of the increased cellular uptake as seen in Fig. 5.
3.5. Physical stability studies
Physical stability of liposomes has always been a matter of concern especially for practical purposes of storage and handling. We compared the stability studies of PS2000 to LS2000 loaded with FCD and anthracene under refrigerated conditions and 25 °C for 8 weeks and evaluated them for size and encapsulation efficiency (to determine drug retention). As seen in Fig. 7a, drug retention of LS2000 and PS2000 of FCD and anthracene was not significantly affected at refrigerated conditions, however as the amount of drug retained decreased slightly at 25 °C for FCD in liposomes and polymersomes, there was drastic decrease in drug retention of anthracene liposomes.
Fig. 7.

Long term studies on both drug retention and size with storage at either 5 or 25 °C using PS2000 and LS2000 loaded with either anthracene or FCD. a. % drug retention over time at 5 °C and b. 25 °C. c. Change in hydrodynamic diameter over time at 5 °C and d. 25 °C, key given in top right hand corner.
This decrease in drug retention is reflected in the increase in size of liposomes after 8 weeks (Fig. 7 bottom left, bottom right). Drug retention of anthracene in PS2000 was not affected compared to LS2000 and also the size of PS2000 was more stable after 8 weeks at 25 °C than LS2000 for both FCD and anthracene. There was a slight increase in size of polymersomes with FCD after 8 weeks at 5 °C. Size of anthracene liposomes were not affected at 5 °C. Hence our studies conclude that liposomes are more stable under refrigerated condition but they can be unstable when stored at room temperature which is in keeping with previous observations in literature (Muppidi et al., 2012; Thompson et al., 2009), whereas the size and encapsulation efficiency of PS2000 of both FCD and anthracene was stable at 25 °C hence concluding that polymersomes were more stable than liposomes giving them an advantage over liposomes.
4. Conclusions
Polymersomes are multipurpose polymeric nanoparticle carriers which can be adapted for numerous applications in nanomedicine. Polymersomes made with random copolymers can prove advantageous over block copolymers because of their ease of preparation. In this study we have synthesized random copolymers using octadecanol, oleic acid, cholesterol and PEG 500 (PS500) or PEG2000 (PS2000) to obtain a chemical composition similar to liposomes (LS500 and LS2000). These polymers were capable of self-assembling to form bilayer polymersomes when prepared by reverse phase evaporation method and compared to liposomes. The polymersomes had good encapsulation efficiency of both hydrophilic (FCD) and hydrophobic anthracene dyes. The hydrodynamic radius of the polymersomes was smaller than their liposome counterpart and had better cellular uptake of FCD and anthracene than liposomes. Polymers were non-toxic to cells at 0.25 mg/ml concentration and had a release profile of their cargo comparable to that of liposomes for charged compounds, but slower for neutral compounds. Physical stability of polymersomes was better than liposomes when stored at 25 °C for 8 weeks. Thus, we have successfully synthesized biomimetic, versatile, biocompatible and stable polymersomes imitating liposomes encapsulating different types of compounds and having good cellular uptake. The work presented herein confirms that, with comparable components, the amphiphilic polymer is indeed a capable contender to the liposome. The full potential of polymersomes is emerging as their versatility and innovation grows, such as the possibility of gas transportation (Kim et al., 2019) or bubble generating (Zhu et al., 2018) it will be exciting to watch their use expand as multifunctional and innovative therapy systems.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
TNK and NA would like to thank Ulster University for their Vice-Chancellor research studentships
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijpx.2019.100040.
Appendix A. Supplementary data
Supplementary material
References
- Ahmed F., Discher D.E. Self-porating polymersomes of PEG-PLA and PEG-PCL: hydrolysis-triggered controlled release vesicles. J. Control. Release. 2004 doi: 10.1016/j.jconrel.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Aibani N., Nesbitt H., Marino N., Jurek J., O’Neill C., Martin C., di Bari I., Sheng Y., Logan K., Hawthorne S., McHale A., Callan J.F., Callan B. Electroneutral polymersomes for combined cancer chemotherapy. Acta Biomater. 2018 doi: 10.1016/j.actbio.2018.09.005. [DOI] [PubMed] [Google Scholar]
- Allen T.M., Cullis P.R. Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Deliv. Rev. 2013 doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- Barenholz Y. Liposome application: Problems and prospects. Curr. Opin. Colloid Interface Sci. 2001 [Google Scholar]
- Bleul R., Thiermann R., Maskos M. Techniques to control polymersome size. Macromolecules. 2015 [Google Scholar]
- Bulbake U., Doppalapudi S., Kommineni N., Khan W. Liposomal formulations in clinical use: an updated review. Pharmaceutics. 2017 doi: 10.3390/pharmaceutics9020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang W.H., Huang W.C., Chang C.W., Shen M.Y., Shih Z.F., Huang Y.F., Lin S.C., Chiu H.C. Functionalized polymersomes with outlayered polyelectrolyte gels for potential tumor-targeted delivery of multimodal therapies and MR imaging. J. Control. Release. 2013 doi: 10.1016/j.jconrel.2013.03.029. [DOI] [PubMed] [Google Scholar]
- Christian D.A., Cai S., Bowen D.M., Kim Y., Pajerowski J.D., Discher D.E. Polymersome carriers: from self-assembly to siRNA and protein therapeutics. Eur. J. Pharm. Biopharm. 2009 doi: 10.1016/j.ejpb.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colletier J.P., Chaize B., Winterhalter M., Fournier D. Protein encapsulation in liposomes: efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol. 2002 doi: 10.1186/1472-6750-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesi R., Esposito E., Gambarin S., Telloli P., Menegatti E., Nastruzzi C. Preparation of liposomes by reverse-phase evaporation using alternative organic solvents. J. Microencapsul. 1999 doi: 10.1080/026520499289220. [DOI] [PubMed] [Google Scholar]
- Discher D.E., Ahmed F. Polymersomes. Annu. Rev. Biomed. Eng. 2006 doi: 10.1146/annurev.bioeng.8.061505.095838. [DOI] [PubMed] [Google Scholar]
- Discher D.E., Eisenberg A. J. Coll; Interface Sci: 2000. Polymer Vesicles. [DOI] [PubMed] [Google Scholar]
- Discher B.M., Won Y.Y., Ege D.S., Lee J.C.M., Bates F.S., Discher D.E., Hammer D.A. Polymersomes: tough vesicles made from diblock copolymers. Science (80-.) 1999 doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
- Discher D.E., Ortiz V., Srinivas G., Klein M.L., Kim Y., Christian D., Cai S., Photos P., Ahmed F. Emerging applications of polymersomes in delivery: from molecular dynamics to shrinkage of tumors. Progress in Polymer Science. 2007 August 1;32(8–9):838–857. doi: 10.1016/j.progpolymsci.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi Y., Shimizu T., Ishima Y., Ishida T. Long-term storage of PEGylated liposomal oxaliplatin with improved stability and long circulation times in vivo. Int. J. Pharm. 2019 doi: 10.1016/j.ijpharm.2019.04.042. [DOI] [PubMed] [Google Scholar]
- Elorza B., Elorza M.A., Frutos G., Chantres J.R. Characterization of 5-fluorouracil loaded liposomes prepared by reverse-phase evaporation or freezing-thawing extrusion methods: study of drug release. BBA - Biomembr. 1993 doi: 10.1016/0005-2736(93)90398-j. [DOI] [PubMed] [Google Scholar]
- Guan L., Rizzello L., Battaglia G. Polymersomes and their applications in cancer delivery and therapy. Nanomedicine. 2015 doi: 10.2217/nnm.15.110. [DOI] [PubMed] [Google Scholar]
- He H., Lu Y., Qi J., Zhu Q., Chen Z., Wu W. Adapting liposomes for oral drug delivery. Acta Pharm. Sin. B. 2019 doi: 10.1016/j.apsb.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillaireau H., Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell. Mol. Life Sci. 2009 doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S., Hussain M.T., Roces C.B., Anderluzzi G., Kastner E., Salmaso S., Kirby D.J., Perrie Y. Microfluidics based manufacture of liposomes simultaneously entrapping hydrophilic and lipophilic drugs. Int. J. Pharm. 2016 doi: 10.1016/j.ijpharm.2016.09.027. [DOI] [PubMed] [Google Scholar]
- Kazi K.M., Mandal A.S., Biswas N., Guha A., Chatterjee S., Behera M. Niosome: a future of targeted drug delivery systems. J. Adv. Pharm. Technol. Res. doi. 2018 doi: 10.4103/0110-5558.76435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Jeong S., Korneev R., Shin K., Kim K.T. Cross-Linked Polymersomes with Reversible Deformability and Oxygen Transportability. Biomacromolecules. 2019 doi: 10.1021/acs.biomac.9b00485. [DOI] [PubMed] [Google Scholar]
- Lee J.S., Feijen J. Polymersomes for drug delivery: design, formation and characterization. J. Control. Release. 2012 doi: 10.1016/j.jconrel.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Letchford K., Burt H. A review of the formation and classification of amphiphilic block copolymer nanoparticulate structures: micelles, nanospheres, nanocapsules and polymersomes. Eur. J. Pharm. Biopharm. 2007 doi: 10.1016/j.ejpb.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Levine D.H., Ghoroghchian P.P., Freudenberg J., Zhang G., Therien M.J., Greene M.I., Hammer D.A., Murali R. Polymersomes: a new multi-functional tool for cancer diagnosis and therapy. Methods. 2008 doi: 10.1016/j.ymeth.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzur A., Oluwasanmi A., Moss D., Curtis A., Hoskins C. Nanotechnologies in pancreatic cancer therapy. Pharmaceutics. 2017 doi: 10.3390/pharmaceutics9040039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqués-Gallego, P., de Kroon, A.I.P.M., 2014. Ligation strategies for targeting liposomal nanocarriers. Biomed. Res. Int. doi: 10.1155/2014/129458. [DOI] [PMC free article] [PubMed]
- Martin C., Marino N., Curran C., McHale A.P., Callan J.F., Callan B. Cholesteryl to improve the cellular uptake of polymersomes within HeLa cells. Int. J. Pharm. 2016 doi: 10.1016/j.ijpharm.2016.07.036. [DOI] [PubMed] [Google Scholar]
- Miller C.R., Bondurant B., McLean S.D., McGovern K.A., O’Brien D.F. Liposome-cell interactions in vitro: effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry. 1998 doi: 10.1021/bi980096y. [DOI] [PubMed] [Google Scholar]
- Muppidi K., Pumerantz A.S., Wang J., Betageri G. Development and stability studies of novel liposomal vancomycin formulations. ISRN Pharm. 2012 doi: 10.5402/2012/636743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musyanovych A., Landfester K. Polymer micro- and nanocapsules as biological carriers with multifunctional properties. Macromol. Biosci. 2014 doi: 10.1002/mabi.201300551. [DOI] [PubMed] [Google Scholar]
- Patil Y.P., Jadhav S. Novel methods for liposome preparation. Chem. Phys. Lipids. 2014 doi: 10.1016/j.chemphyslip.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Pattni B.S., Chupin V.V., Torchilin V.P. New developments in liposomal drug delivery. Chem. Rev. 2015 doi: 10.1021/acs.chemrev.5b00046. [DOI] [PubMed] [Google Scholar]
- Peyret A., Ibarboure E., Tron A., Beauté L., Rust R., Sandre O., McClenaghan N.D., Lecommandoux S. Polymersome popping by light-induced osmotic shock under temporal, spatial, and spectral control. Angew. Chemie - Int. Ed. 2017 doi: 10.1002/anie.201609231. [DOI] [PubMed] [Google Scholar]
- Qin G., Li Z., Xia R., Li F., O’Neill B.E., Goodwin J.T., Khant H.A., Chiu W., Li K.C. Partially polymerized liposomes: stable against leakage yet capable of instantaneous release for remote controlled drug delivery. Nanotechnology. 2011 doi: 10.1088/0957-4484/22/15/155605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu M.H., Zeng R.F., Fang S., Dai Q.S., Li H.P., Long J.T. Liposome-based co-delivery of siRNA and docetaxel for the synergistic treatment of lung cancer. Int. J. Pharm. 2014 doi: 10.1016/j.ijpharm.2014.08.019. [DOI] [PubMed] [Google Scholar]
- Raicu V., Popescu A. Integrated Molecular and Cellular Biophysics. 2008. The composition and architecture of the cell. [Google Scholar]
- Ren H., He Y., Liang J., Cheng Z., Zhang M., Zhu Y., Hong C., Qin J., Xu X., Wang J. Role of liposomes size, surface charge and PEGylation on rheumatoid arthritis targeting therapy. ACS Appl. Mater. Interfaces. 2019 doi: 10.1021/acsami.8b22693. [DOI] [PubMed] [Google Scholar]
- Rideau E., Dimova R., Schwille P., Wurm F.R., Landfester K. Liposomes and polymersomes: a comparative review towards cell mimicking. Chem. Soc. Rev. 2018 doi: 10.1039/c8cs00162f. [DOI] [PubMed] [Google Scholar]
- Sadat S.M.A., Jahan S.T., Haddadi A. Effects of size and surface charge of polymeric nanoparticles on in vitro and in vivo applications. J. Biomater. Nanobiotechnol. 2016 [Google Scholar]
- Sadzuka Y., Nakade A., Hirama R., Miyagishima A., Nozawa Y., Hirota S., Sonobe T. Effects of mixed polyethyleneglycol modification on fixed aqueous layer thickness and antitumor activity of doxorubicin containing liposome. Int. J. Pharm. 2002 doi: 10.1016/s0378-5173(02)00075-3. [DOI] [PubMed] [Google Scholar]
- Sardan M., Kilinc M., Genc R., Tekinay A.B., Guler M.O. Cell penetrating peptide amphiphile integrated liposomal systems for enhanced delivery of anticancer drugs to tumor cells. Faraday Discuss. 2013 doi: 10.1039/c3fd00058c. [DOI] [PubMed] [Google Scholar]
- Shimada K., Miyagishima A., Sadzuka Y., Nozawa Y., Mochizuki Y., Ohshima H., Hirota S. Determination of the thickness of the fixed aqueous layer around polyethyleneglycol-coated liposomes. J. Drug Target. 1995 doi: 10.3109/10611869509015957. [DOI] [PubMed] [Google Scholar]
- Swaminathan S., Fowley C., McCaughan B., Cusido J., Callan J.F., Raymo F.M. Intracellular guest exchange between dynamic supramolecular hosts. J. Am. Chem. Soc. 2014 doi: 10.1021/ja500285p. [DOI] [PubMed] [Google Scholar]
- Thompson A.K., Couchoud A., Singh H. Comparison of hydrophobic and hydrophilic encapsulation using liposomes prepared from milk fat globule-derived phospholipids and soya phospholipids. Dairy Sci. Technol. 2009 [Google Scholar]
- Torchilin V.P. Micellar nanocarriers: pharmaceutical perspectives. Pharm. Res. 2007 doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- Van Den Hoven J.M., Nemes R., Metselaar J.M., Nuijen B., Beijnen J.H., Storm G., Szebeni J. Complement activation by PEGylated liposomes containing prednisolone. Eur. J. Pharm. Sci. 2013 doi: 10.1016/j.ejps.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Vercauteren, D., Vandenbroucke, R.E., Jones, A.T., Rejman, J., Demeester, J., De Smedt, S.C., Sanders, N.N., Braeckmans, K., 2010. The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol. Ther. doi: 10.1038/mt.2009.281. [DOI] [PMC free article] [PubMed]
- Xu H., Ohulchanskyy T.Y., Yakovliev A., Zinyuk R., Song J., Liu L., Qu J., Yuan Z. Nanoliposomes co-encapsulating CT imaging contrast agent and photosensitizer for enhanced, imaging guided photodynamic therapy of cancer. Theranostics. 2019 doi: 10.7150/thno.31079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Lu L., Zhang L., Shi K., Cun X., Yang Y., Liu Y., Gao H., He Q. Dual-functionalized liposomal delivery system for solid tumors based on RGD and a pH-responsive antimicrobial peptide. Sci. Rep. 2016 doi: 10.1038/srep19800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D., Fan F., Huang C., Zhang Z., Qin Y., Lu L., Wang H., Jin X., Zhao H., Yang H., Zhang C., Yang J., Liu Z., Sun H., Leng X., Kong D., Zhang L. Bubble-generating polymersomes loaded with both indocyanine green and doxorubicin for effective chemotherapy combined with photothermal therapy. Acta Biomater. 2018 doi: 10.1016/j.actbio.2018.05.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material