

Abstract
Motivation
The human leukocyte antigen (HLA) locus plays a critical role in tissue compatibility and regulates the host response to many diseases, including cancers and autoimmune di3orders. Recent improvements in the quality and accessibility of next-generation sequencing have made HLA typing from standard short-read data practical. However, this task remains challenging given the high level of polymorphism and homology between HLA genes. HLA typing from RNA sequencing is further complicated by post-transcriptional modifications and bias due to amplification.
Results
Here, we present arcasHLA: a fast and accurate in silico tool that infers HLA genotypes from RNA-sequencing data. Our tool outperforms established tools on the gold-standard benchmark dataset for HLA typing in terms of both accuracy and speed, with an accuracy rate of 100% at two-field resolution for Class I genes, and over 99.7% for Class II. Furthermore, we evaluate the performance of our tool on a new biological dataset of 447 single-end total RNA samples from nasopharyngeal swabs, and establish the applicability of arcasHLA in metatranscriptome studies.
Availability and implementation
arcasHLA is available at https://github.com/RabadanLab/arcasHLA.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Human leukocyte antigen (HLA) genes encode the proteins that make up the major histocompatibility complex (MHC). MHC Class I (HLA-A, -B and -C), produced in all nucleated somatic cells, binds to and presents intracellular antigens on the cell surface for cytotoxic T-cells, which trigger apoptosis if non-selfpeptides are detected. MHC Class II (including HLA-DPB1, -DQB1 and -DRB1), on the other hand, constitutively expressed only by specialized immune and epithelial cells, present extracellular proteins to helper T-cells which mediate the adaptive immune response (Meyer and Thomson, 2001).
HLA genes are the most polymorphic regions in the human genome, with over 12 000 known alleles across 38 genes (Robinson et al., 2015). Pathogen-driven selection may explain this level of HLA diversity: variation of residues in the binding region allows for a greater variety of immunogenic peptides. Populations in pathogen-rich areas exhibit increased HLA diversity (Prugnolle et al., 2005), and heterozygous individuals show both greater resistance towards infectious agents and greater fitness than homozygotes (Carrington et al., 1999; Penn et al., 2002; Thursz et al., 1997).
With the advent of immunotherapy, HLA typing and expression level quantification is increasingly important for cancer research. Immunotherapy depends on the ability of the patient’s HLAs to effectively bind and present tumor neoantigens on the cell surface (Chowell et al., 2018). Following immunotherapy, clonal selection may favor tumor cells with a loss of HLA heterozygosity (LOH) or silencing of the HLA loci, highlighting the importance of LOH detection from gene expression data (McGranahan et al., 2017). Indeed, although past methods look at copy number variation to determine LOH using microsatellites or NGS, RNA sequencing may give a more accurate picture of HLA expression in tumor cells, particularly if HLA expression is altered as a result of interruptions in HLA regulatory pathways due to post-transcriptional or epigenetic modifications.
High-resolution typing of HLA alleles is also imperative for the determination of tissue compatibility. HLA nomenclature (e.g. A*02: 01: 01: 02) consists of four successive fields: allele group, protein type, followed by synonymous changes in coding regions, and changes in non-coding regions. High-resolution genotyping is used to determine histocompatibility, resolving sequencing ambiguities in the peptide-binding region (Exons 2 and 3 for Class I and Exon 2 for Class II). Certain amino acid residue mismatches within this region correlate with increased risk of rejection (Petersdorf et al., 2014). Consequently, the majority of HLA alleles are partially sequenced (Supplementary Fig. S1), covering at least the peptide-binding regions.
Specialized methods of typing HLAs, including Sanger sequencing and PCR enrichment of the HLA loci, are expensive and time-consuming, given the sample size necessary for effective donor banks and association studies. Thus, methods using standard NGS reads with minimal loss of accuracy and resolution are useful. However, typing with short reads is complicated by the high level of homology between both HLA genes and alleles, some of which can differ by a single base. In addition, there exist HLA pseudogenes which can have detectable expression levels and interfere with typing from genomic sequencing (Kawaguchi et al., 2017; Lonsdale et al., 2013).
In the last few years, multiple tools that type HLAs from whole-genome sequencing, whole exome sequencing (WES), and RNA sequencing have been published, with improving benchmark performance and resolution (see Supplementary Table S1). These HLA typing tools attempt to find the one or two alleles that best explain the sampled reads, either by comparing assembled contigs or aligning reads directly to an HLA reference. Most current tools for RNA sequencing, including seq2HLA (Boegel et al., 2012), OptiType (Szolek et al., 2014) and PHLAT (Bai et al., 2014), are alignment-based. The latest RNA-dedicated HLA typing tool, HLAProfiler, takes a novel approach to graph-based alignment, breaking HLA transcripts into k-mers and constructing a taxonomic tree used to filter reads (Buchkovich et al., 2017). Tools also differ in the construction of their HLA reference: some tools, such as seq2HLA and OptiType, limit their reference to peptide-binding exons and flanking regions while others use a combination of coding and genomic sequences. For the purpose of serotyping, it is important to not only consider the peptide-binding region because it is possible for two alleles to have identical peptide-binding sequences but different protein types. In addition, limiting the number of exons considered increases the occurrence of ambiguous typing.
arcasHLA takes an alignment-based approach, using two separate coding transcript references: one with alleles with complete sequences, and a second reference containing all possible combinations of exons including the binding region of all known alleles for typing partial alleles. This tool uses Kallisto (Bray et al., 2016), an RNA quantifier with a graph-based pseudo-alignment feature, to assign reads to their compatible HLA transcripts. Tools that perform graph-based alignment followed by expectation–maximization transcript quantification are used to quantify isoform expression. Thus, these methods extend naturally to highly polymorphic loci such as the HLA family. Allele abundance for each gene is quantified separately and the genotype that maximizes the number of reads aligned is selected from the most abundant alleles. Finally, homozygosity is determined using the ratio of minor to major non-shared read counts. As an optional step, partial alleles are typed in a similar fashion. Unlike other tools, population-specific allele frequencies are used as priors to distribute sampled reads within HLA compatibility classes in addition to breaking ties between ambiguous alleles (see Section 2). arcasHLA outperforms other popular HLA RNA-sequencing typing tools such PHLAT, OptiType, seq2HLA and HLAProfiler on paired-end benchmark samples (see Table 1).
Table 1.
Concordance with 1000 genomes gold-standard HLA typing for 358 RNA-sequencing samples for arcasHLA along with concordance rates for other tools reported in (Buchkovich et al., 2017)
| Gene | OptiType | seq2HLA | PHLAT | HLAProfiler | arcasHLA |
|---|---|---|---|---|---|
| A | 99.6% | 98.6% | 99.4% | 99.9% | 100.0% |
| B | 99.4% | 94.8% | 93.4% | 99.0% | 100.0% |
| C | 100.0% | 95.1% | 94.3% | 99.6% | 100.0% |
| DQB1 | — | 96.0% | 96.0% | 99.9% | 99.9% |
| DRB1 | — | 98.5% | 98.5% | 99.6% | 99.7% |
Note: Bold denotes maximized concordance.
2 Materials and methods
2.1 Database construction
HLA and related sequences were obtained from the ImMunoGeneTics (IMGT)/HLA database, IMGT/HLA, compiled by the Immuno Polymorphism Database project (Robinson et al., 2015). These sequences include both classical and non-classical MHC Classes I, II genes, HLA pseudogenes and some related non-HLA genes.
Due to post-transcription splicing, variants in intronic regions (indicated by the fourth field) cannot be confidently determined from mature messenger RNA. Excluding introns, we constructed coding DNA databases for all the HLA alleles in IMGT/HLA using the sequences and exon coordinates provided in the hla.dat file. Sequenced untranslated regions (UTRs), missing for many alleles, were included as noncontiguous sequences. Reference alleles with insertions or deletions causing a stop loss in the final exon were truncated if the sequence continuing into the UTR contained no changes with respect to the reference allele. Without these alterations, reads containing the UTR would be attributed only to transcripts containing the stop loss.
A majority of the alleles archived in IMGT/HLA are missing one or more exons. Some HLA typing tools include partial alleles by extending the sequence with an allele’s nearest neighbor (Optitype) or looks at each exon individually. The method described here uses two separate references for typing non-partial and partial alleles. The former contains only transcripts for alleles with complete sequences, while the latter contains transcripts for all possible contiguous combinations of exons for all known alleles (e.g. 2-3, 1-2-3 etc).
Two-field allele frequencies were retrieved from AlleleFrequencies Net Database (AFND) (González-Galarza et al., 2015). Only populations considered to be gold-standard, with allele frequencies that sum to 1 and a sample size ≥ 50, were used to build the database. These sample populations were grouped into broad population categories following the categorization laid out by The National Marrow Donor Program (Gragert et al., 2013). To account for alleles not seen in the selected population and those not reported on AFND, Dirichlet smoothing was applied to the allele frequencies, treating the entirety of the AFND data and IMGT/HLA database reference as priors.
2.2 Genotyping
2.2.1. Read alignment
arcasHLA takes as input a mapped RNA-seq BAM file. After extracting Chromosome 6 reads (and when applicable, extracting any additional reads aligned to HLA decoys or to Chromosome 6 alternate sequences) from input, we perform a pseudoalignment of the extracted reads with Kallisto, a graph-based RNA-seq quantifier selected primarily for its improved speed, accuracy and flexibility as compared with other local or graph-based aligners. Kallisto builds a de Bruijn graph from the reference transcriptome, in which k-mers (or k-length subsequences) represent the nodes, and edges add an additional base to left-shifted (k–1)-mers connecting between consecutive k-mers. Each read is decomposed into k-length sequences and hashed into a reference index. The compatibility class of a given read is then defined as the set of reference transcripts that are compatible with every one of its constituent k-mers. This method avoids base-by-base alignment in favor of speed; thus the moniker ‘pseudoalignment’. Because Kallisto skips k-mers that provide no new information on the compatibility class of a read, it is less sensitive to sequencing errors in the sampled reads if they happen to align within any one of these redundant k-mers. Of note: this method is also insensitive to novel alleles if the corresponding new variants lie along one of these conserved, skipped k-mers.
2.2.2. Transcript quantification
Like most HLA typing tools, arcasHLA seeks to find the pair of alleles with maximal support among the observed reads originating from the HLA locus. Given the thousands of possible alleles for a single gene, pairwise comparisons; however, are computationally expensive and they fail to account for the similarity between different alleles. To reduce the number of alleles considered, arcasHLA exploits the k-mer structure in transcript quantification and repeatedly culls low-support allele transcripts. The output of arcasHLA is the allele pair (or possibly a single allele) that best explains the observed reads.
Division of counts. Traditionally, graph-based transcript quantifiers (Bray et al., 2016; Patro et al., 2014) assign reads to equivalence classes of reference transcripts, further sub-dividing reads within each compatibility class with equal weights among all the alleles in a given class. This approach may be beneficial when calculating differential expression of genes with many possible, equally likely isoforms present in a single sample. arcasHLA sub-divides reads differently, accounting for the relative frequencies of HLA alleles in different human populations. Below, we formalize the setup for graph-based transcript quantifiers.
Let A be a set of reference alleles with lengths la for , and C a set of observed compatibility classes (where each compatibility class is a subset of A). For a given allele , we define as the set of compatibility classes which contain allele i. Thus, each element is a compatibility class consisting of alleles in A with . As such, the read count attributed to an allele with equal weights sub-division is then simply:
| (1) |
where denotes the number of alleles contained in the equivalence class ω, and rω is the total count assigned to class ω.
arcasHLA performs genotyping calls with an iterative procedure that optimizes the read assignment to individual alleles. At the first step, our genotyping algorithm gives the option to distribute reads between alleles with weights proportional to population-specific allele frequencies. The largest benefit of this approach is narrowing the pool of possible alleles as well as breaking ties between alleles that are indistinguishable given the sampled reads. Given such priors , the count attributed to allele i is thus
| (2) |
Subsequently, these counts are normalized by the allele length and converted into transcript abundances for each allele i:
| (3) |
Maximizing the proportion of explained reads. As with Kallisto, the likelihood of a specific attribution of reads to alleles given by is proportional to
| (4) |
In order to find the allocation of reads to alleles that maximizes the likelihood function (Equation 4), we follow an iterative procedure similar to Kallisto’s, with some essential differences.
First, we restrict the equivalence classes obtained from the reference de Bruijn graph construction gene by gene, and perform genotyping independently for each gene (namely, using our notation, we consider reference alleles for each HLA gene separately: ). Second, instead of numerically solving for the maximum likelihood of (Equation 4), we adopt a strong constrained approach consistent with our goal of outputting at most two alleles for each HLA gene. Reads in each class are iteratively reallocated based on abundances from the previous iteration, but after an empirically optimized 10 and 4 iterations for paired- and single-end respectively, alleles with abundances lower than one-tenth of the maximum observed abundance are dropped according to the following constraint:
| (5) |
for each gene G in MHC Classes I and II.
The 10% threshold, previously determined by HISAT-genotype (Kim et al., 2018) for use with whole-genome sequencing, assumes that the abundance of the minor allele does not fall below a tenth of the major allele’s abundance. When applied to RNA sequencing, this allows for a large range in the natural variation between major and minor allele expression as well as differences in read counts due to sequencing and amplification.
The iterative read re-allocation in arcasHLA is as follows:
| (6) |
for all iterations t until convergence. Here, the upper indices denote the respective allele abundances or reads at the specified iteration. Next, these counts are normalized by transcript lengths and converted back into abundances:
| (7) |
With each updated estimate, a higher proportion of reads are distributed to the alleles with the highest abundances and the lowest abundance alleles are culled, per (Equation 5).
Like HISAT-genotype (Kim et al., 2018) and Sailfish (Patro et al., 2014), we use SQUAREM (Varadhan and Roland, 2008) to accelerate the convergence. The read allocation is considered to converge when the difference in abundance from the previous iteration to the current one is below 10−7, with a maximum of 1000 iterations allowed. Indeed, arcasHLA has been shown to always meet the convergence criterion in both of our test datasets. At the end of the arcasHLA iteration procedure, the remaining alleles are those that explain the highest proportion of aligned reads for each gene.
2.2.3. Selecting the most likely genotype
Ideally, after convergence and culling, a single allele is left for homozygotes and two alleles for the heterozygotes. However, due to high levels of homology between certain alleles, particularly beyond the two field resolution, alleles may be indistinguishable given the observed reads and more than two likely alleles may be returned. In order to further narrow down the pool to exactly two alleles, the pair that explains the greatest proportion of reads is selected. Finally, we include a check for homozygosity by assessing the top two alleles’ non-shared read counts. If the minor-to-major ratio of non-shared allele counts lies below an empirically optimized threshold of 15%, the individual is called as homozygous for the major allele. Otherwise, the individual is called as heterozygous for the top ranking pair.
2.2.4. Partial allele typing
Partial allele typing is included as an optional step. Extracted reads are aligned to the reference containing the allele transcripts from the database. Possible partial alleles are first identified by running transcript quantification on the peptide-binding exon transcripts. Next, arcasHLA iterates through the set of exon combinations represented in the returned partial alleles. If a partial allele does not exceed by more than 10 reads a non-partial minor allele in a given region, then it is discarded as it cannot be confidently called as a valid allele. Next, all combinations of remaining partial alleles and the previously called non-partial alleles are considered. If a pair with one or more partial alleles explains a greater proportion of reads in any of these exon regions than the original genotype, then it is returned. If more than one partial-containing pairs explains the same amount of reads, allele frequencies are used to break the tie.
2.3 Datasets
2.3.1. Benchmark dataset: 1000 genomes
HLA-A, -B, -C, -DRB1 and -DQB1 genes for 1267 of the 1000 Genomes individuals were typed using Sanger sequencing based on the IMGT/HLA database from 2009 (Gourraud et al., 2014). Only the peptide-binding region for each gene was sequenced. As previously stated, multiple alleles can share the same binding region sequence, and thus a list of equivalent alleles is reported. Since 2009, IMGT/HLA has expanded their database to more than four times as many alleles, and, like HLAProfiler (Buchkovich et al., 2017), we used the latest list of ambiguous alleles provided by IMGT/HLA to update the ground truth.
mRNA sequencing for 358 of these samples is provided by the Geuvadis project, representing five of the 1000 Genomes populations (CEU, FIN, GBR, TSI and YRI) (Lappalainen et al., 2017). These samples are generally high in quality with a mean RNA integrity number or RIN (Schroeder et al., 2006), of 9.1 (ranging from 6.2 to 10), and a mean of 58.5 million reads mapped to the hg19 reference (ranging from 17 to 163.5M reads). Reads are paired-end, and 75 base pairs (bp) in length. 25.1 and 14.8% of these individuals are homozygous for at least one gene at two fields in resolution for MHC Classes I and II, respectively.
We ran arcasHLA on these samples with IMGT/HLA v3.24.0, the version also used by HLAProfiler, for comparison purposes. In addition to updating the ground truth with allele ambiguities, calls were updated with the high-resolution typing using Ilumina TruSight provided by HLAProfiler. In order to test arcasHLA’s performance on high quality single-end samples too, we also treated the 1000 Genomes samples as being single-end, following PHLAT’s (Bai et al., 2014) methodology.
2.3.2. New biological dataset: the Virome of Manhattan
We ran arcasHLA on a set of 447 single-end total RNA-sequencing samples collected from nasopharyngeal swabs from 69 healthy individuals enrolled as part of a DARPA-funded project entitled ‘The Virome of Manhattan: a Testbed for Radically Advancing Understanding and Forecast of Viral Respiratory Infections’ (Birger et al., 2018; Galanti et al., 2019).
Sample collection and preparation. Nasopharyngeal samples were collected using minitip flock swabs and stored in tubes with 2 ml DNA/RNA Shield (Zymo Research, R1100-250) at 4–25°C for up to 30 days and then aliquoted into two 2 ml cryovials and stored at −80°C. RNA was extracted from 200 μl of each stored sample using the Quick-RNA MicroPrep Kit (Zymo Research, Irvine, CA). Eluted RNA was then quantified and assessed for quality using Agilent Bioanalyzer (Santa Clara, CA), and the remaining quantity was sequenced with Illumina following the Ribo-Zero rRNA Removal Kit, target 30 M single-end 100 bp reads.
Sample processing. The individuals in the Virome study represent a heterogeneous cohort with self-reported and SNP-validated race/ethnicity (using the population clusters from the Exome Aggregation Consortium (ExAC) dataset (Lek et al., 2016)) from African-American, Caucasian, Asian, Hispanic and Native American groups. As such, known population-specific allele frequency priors were passed to arcasHLA on this dataset executed in single-end mode from input BAM files mapped with STAR v.2.5.2b (Dobin et al., 2013) to human reference GRCh37 (Aken et al., 2016). In contrast to the high quality, homogeneous samples from the benchmark set, the Virome samples have a mean RIN (Schroeder et al., 2006) of 7.0 (ranging from 1.0 to 9.9), and a mean of 22.2 M reads mapped to the human GRCh37 reference (ranging from 5.9 to 68.2 M reads).
Ground truth for typing comparison. We established the HLA genotyping ground truth for the Virome dataset using an assortment of in silico tools which attain high concordance with deep targeted sequencing validation protocols: xHLA (Xie et al., 2017), HISAT-genotype (Kim et al., 2018) and OptiType Szolek et al. (2014)—that we ran on WES data processed with the xGEN-Illumina platform (at 60 × 25 M target PE 100 bp reads) and extracted from saliva samples drawn independently from the nasopharyngeal swabs in our cohort.
Since we required both MHC Classes I and II calls to test the full capability of arcasHLA, we resorted to setting xHLA’s two-field calls as the true Virome genotypes. On average, for HLA-A, -B and -C genes, two additional tools showed good agreement with xHLA (Table 2). Further, in order to optimize speed and memory usage, we first used the HISAT-genotype extract_reads function, which builds on the HISAT aligner (Kim et al., 2015), to extract reads mapping to the HLA locus before genotyping with xHLA. xHLA calls MHC Class I HLA-A, -B and -C and MHC Class II HLA-DPB1, -DQB1 and -DRB1. According to xHLA calls, the Virome individuals show lower rates of homozygosity than in the benchmark set with rates of homozygosity of 14.5 and 10.1% for MHC Classes I and II, respectively. HISAT-genotype and arcasHLA were run using IMGT/HLA database v. 3.26.0. For arcasHLA typing, we extracted the unmapped reads in addition to those from Chromosome 6.
Table 2.
Concordance of calls for arcasHLA, OptiType and HISAT-genotype with xHLA for 447 RNA samples from 69 individuals
| Input (#) | RNA (447) |
WES (69) |
||
|---|---|---|---|---|
| Tool | arcasHLA | OptiType | OptiType | HISAT |
| A | 97.5% | 95.2% | 98.6% | 99.4% |
| B | 98.0% | 94.5% | 96.4% | 98.6% |
| C | 97.7% | 97.4% | 98.6% | 100.0% |
| DPB1 | 94.2% | — | — | — |
| DQB1 | 93.3% | — | — | 94.9% |
| DRB1 | 94.9% | — | — | 94.2% |
Note: Bold denotes maximized concordance for RNA sequencing results.
2.4 Implementation and availability
arcasHLA is as a command line tool written in Python 3.6 available on the public GitHub repository https://github.com/RabadanLab/arcasHLA. This software is divided into four steps (Fig. 1). (i) Database construction takes fewer than 3 min on average and allows for the selection of a specific IMGT/HLA version. (ii) Reads are extracted from previously sorted BAM files. (iii) Reads are pseudoaligned and allele abundances are quantified, followed by selection of the most likely genotype. Although it is possible to do so, we do not recommend genotyping with arcasHLA directly from raw FASTQ files. (iv) (Optional) Reads are aligned to a reference containing partial alleles. Possible partial alleles are selected and compared with the complete genotype from Step iii.
Fig. 1.

Overview of arcasHLA pipeline from alignment to genotyping. The HLA de Bruijn graph was generated with Velvet (Zerbino and Birney, 2008) and visualized with Bandage (Wick et al., 2015)
The Geuvadis RNA sequencing of the 1000 Genomes individuals was used for benchmarking the performance of arcasHLA, and the data are available from ArrayExpress (E-GEUV-1).
3 Results
3.1 Benchmark performance
When run on the 1000 Genomes benchmark set, arcasHLA achieves 100% accuracy for Class I and above 99.7% accuracy for Class II genes, outperforming other established tools (Table 1). Overall, arcasHLA provides high levels of concordance for the HLA region using this benchmark set. 99.9% of A, B and C alleles have complete sequences in the gold-standard (reference version 3.24.0), while only 96.8% of DQB1 and 98.6% of DRB1 alleles are not partial, which accounts for the lower accuracy of seq2HLA and PHLAT (they do not type partial alleles). arcasHLA’s accuracy dropped slightly when genotyping complete alleles for the ‘single-end’ samples with an average accuracy of 98.5 and 97.3% for Classes I and II, respectively (Supplementary Table S2). However, the current method of partial typing is prone to false positives for partial alleles when typing Class I from single-end reads, with an accuracy of 90.1%. Surprisingly, typing improves for DQB1, with an accuracy of 99.3% for Class II.
For computational analysis of arcasHLA, we randomly selected 30 samples from the 1000 Genomes benchmark dataset (Fig. 2). These samples, typed without the optional partial allele typing step, were analyzed on a Linux instance with 16 vCPUs and 64 GiB of memory using 8 threads per sample. All samples were genotyped in <2 min. arcasHLA effectively achieves an order of magnitude runtime improvement over HLAProfiler (without its additional refinement and partial typing steps) when mapped RNA-seq reads are readily available. This runtime improvement should be interpreted by practitioners as an advantage to be gained from integrating arcasHLA as part of an existing pipeline (with BAM file intermediates) for typing HLA as compared with standalone HLA typing pipeline (HLAProfiler).
Fig. 2.
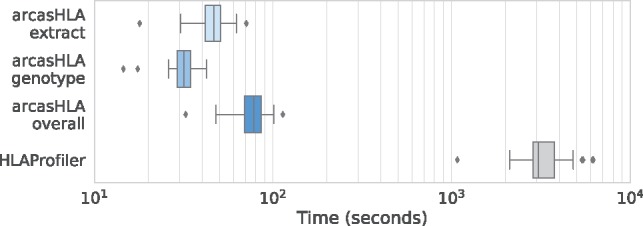
Runtime analysis on 30 randomly selected samples from 1000 Genomes dataset for arcasHLA (extract and genotype steps, and overall runtime) and HLAProfiler
3.2 Performance on the Virome of Manhattan dataset
In spite of the lower quality metrics in the Virome dataset, arcasHLA yields high accuracy (Table 2): 97.7% for Class I and 94.1% for Class II. Given the lower accuracy for partial typing using single-end reads in the benchmark set, we did not perform partial typing for this set.
Expression of MHC Class II in the Virome samples (extracted from nasopharyngeal swabs) can likely be attributed to the upper airway epithelial cells which are known to constitutively express MHC Class II (Wosen et al., 2018), and to the infiltration of leukocytes within the tissue lining the turbinates. Previous transcriptome analyses have shown that leukocyte markers are indeed expressed at low but detectable levels in samples from nasopharyngeal swabs (Chu et al., 2016). Such specialized epithelial and immune cells are likely in the minority, which may explain our tool’s lower accuracy result for Class II genes. In fact, although arcasHLA was able to correctly call MHC Class II alleles for a majority of the samples, it failed to call HLA-DQB1 for several samples with an RIN of 1 where there were no reads at all mapping to DQB1 alleles. We highlight the fact that the Virome samples contain variable mixtures of human, bacterial and viral RNA (as detected by a BLAST search of the un-mapped reads Altschul et al., 1990), which can impact the RIN score. The variable sampling depth of the nasal cavity is another source of RIN variation and it can have a considerable effect, as mentioned earlier, on the coverage of HLA genes.
Another source of error likely stems from the single-end sequencing used in the Virome study which is known to generate a less accurate mapping due, in particular, to the inability to resolve single-base ambiguities. In spite of these study limitations, we report that HLA calling can still be successfully performed in silico from low RIN samples with relatively low coverage of the HLA locus (Fig. 3).
Fig. 3.

Concordance rates restricted to Virome samples below a threshold for (a) RIN and (b) log-scaled reads by HLA gene, truncated when the number of samples dropped below 55, approximately one-eighth the total sample size. Panels (c) and (d) show the number of samples remaining
4 Discussion
Accurate high-resolution HLA typing is imperative for determining tissue and hematopoietic compatibility. Typing from NGS reads is a boon to large-scale association studies where specialized assays prove too time-consuming and expensive. However, typing from short reads is complicated by the high level of homology between HLA alleles and genes. Typing tools must be able to resolve ambiguities given limited information, low read counts, short length reads, or single-end sequencing.
With our new tool, arcasHLA, we have adapted transcript quantification algorithms to aid in typing of HLAs, a method which could be extended to type other highly polymorphic regions. arcasHLA performs at or near 100% accuracy on the gold-standard benchmark set, outperforming all other tools that run on RNA-sequencing data. We have also validated our tool on a new biological dataset from a metatranscriptomic study of human nasopharyngeal swabs, showing how low read counts and low quality reads (as measured by the RIN) can affect the ability to type the MHC locus in silico.
Given the high level of homology between alleles, it is unlikely that reads align solely to a single allele. Although it may be possible to select the top pair from all observed alleles, it often improves accuracy and runtime to filter out low-support alleles before the final scoring. For example, OptiType drops alleles that are not present in HLA databases, and HLAProfiler only selects the top n pairs by proportion of explained reads. Taking inspiration from HISAT-genotype, arcasHLA uses allele transcript quantification combined with culling of low-support alleles to narrow down the pool of possible alleles, thereby improving on previous filtering methods. Like OptiType, arcasHLA uses allele priors to influence results; however, the method presented here uses these to nudge the transcript quantification in the direction of more common alleles without disregarding alleles rare enough to not be cataloged in HLA databases. Indeed, the use of allele priors does not change the high level of accuracy on the 1000 Genomes set, which consists of high quality samples only (in read count and RIN). The use of population-specific allele frequencies does, however, consistently improve concordance between arcasHLA and calls from WES in the Virome dataset. This may be due to lower quality metrics and the single-end protocol used in the Virome study. Indeed, in the Virome cohort, using an individual’s specific population improved results; as did simply using the prior used for the Dirichlet smoothing. Thus, for high quality samples of unknown origin no priors are required; for low quality samples; however, a prior based on existing populations is likely to improve the accuracy of calls (see Supplementary Table S3).
After this initial filtering step, HLA typing tools must resolve ambiguities between alleles to select the most likely genotype. Most tools use some scoring function to select the pair of alleles that explains the greatest proportion of observed reads, at times including consideration for noise, allele priors, or other factors. arcasHLA, at present, takes a relatively simple approach, selecting the pair of alleles that explains the greatest proportion of reads for each gene. Like Polysolver (Shukla et al., 2015), arcasHLA uses allele frequencies to break ties between pairs with the same read count.
As tools approach 100% accuracy for standard benchmark tests, other criteria must be used to distinguish between them. With increased use of HLA typing in large-scale association studies, runtime becomes a more important factor. In such studies, RNA sequencing is likely already aligned for other purposes. Given that alignment is usually the most time consuming part of the HLA typing process, running from pre-aligned input cuts down on overall runtime. As such, tools like arcasHLA and xHLA can boast runtimes that are orders of magnitude smaller than their competitors. When typing thousands of samples, such a difference can have a significant impact. When genotyping from a pre-aligned BAM file, it is possible that HLA reads may be lost (either unmapped or discarded by the aligner) if the individual’s HLA genotype differs significantly from the reference sequence. However, given the competitive levels of accuracy between tools that use raw FASTQs versus BAMs, it is unlikely that lost reads significantly affect performance. In addition, the scales may tip in favor of BAM-based tools that extract HLA sequences from BAMs aligned to GRCh38 with alternative sequences (decoys and HLAs).
Another distinguishing factor between tools is their ability to perform under suboptimal conditions. Single-end sequencing makes resolving ambiguities between similar alleles a harder task; as such, many HLA typing tools for RNA sequencing, such as Seq2HLA and HLAProfiler, only accept paired-end data. When typing non-partial alleles for the ‘single-end’ benchmark dataset, arcasHLA performs competitively with other tools as well as outperforming OptiType for the Virome dataset. However, arcasHLA’s higher agreement with xHLA than Optitype for this set may be due to the nature of OptiType’s calls which is limited to resolving ambiguities in the antigen-binding region. In addition, it is possible that both xHLA and arcasHLA make the same typing errors, limiting to some extent this type of comparison without a true ground truth. Nonetheless, without the resources to perform specialized HLA typing assays, taking a consensus between results from different tools may be the best approach.
In recent years, a whole body of work has begun to map out the critical importance of our microbiome in systemic immunity, development, homeostasis, disease and patient responses to immunotherapy (Grice and Segre, 2012; Obata and Pachnis, 2016; Zitvogel et al., 2018). As in our project on the Virome of Manhattan, we expect that future metatranscriptomic studies of the human host will rely on in silico methods to disentangle human from bacterial reads and maximally extract biological signal from low quality and highly heterogeneous bulk samples. arcasHLA has been validated here for use with bulk total RNA samples that contain eukaryotic, prokaryotic and viral mixtures, showing high concordance for MHC Classes I and II with the top HLA calling tools. Indeed, arcasHLA is minimally impacted by low read counts, low quality and the single-end sequencing protocol.
In the future, we plan on adding confidence scores for the most likely genotype calls, as well as a more robust check for zygosity that takes expected levels of noise into account. We also plan to build a pipeline to detect novel alleles using de novo assembly of reads mapping to the HLA locus. In order to identify novel alleles, we would compare the difference in coverage at mismatch sites between these novel contigs and the original genotype produced by arcasHLA. Another feature in the works for the upcoming arcasHLA version is allele-specific expression quantification post genotyping. Expression levels for HLA-C have been shown to correlate with certain allele groups that share an upstream variant; such differences have shown clinically significant implications in HIV and tissue-compatibility (Petersdorf et al., 2014; Thomas et al., 2009). Along these lines, expression-level data may enable us to detect loss or silencing of the HLA loci as a possible mechanism of immune evasion. We are developing two new applications of arcasHLA: a test for HLA allele imbalance in tumors, and the verification of mutations called from genomic sequencing.
The development of HLA typing tools from DNA and RNA sequencing is limited by the availability of gold-standard, benchmark datasets. These sets, used to both develop and test these tools, have only two field resolution typing for six HLA loci as well as unresolved ambiguity between alleles beyond the peptide-binding region. As such, tools often call different alleles for individuals from the 1000 Genomes set, yet they report near perfect accuracy. Although grouping by identical antigen-binding region is useful from a serological standpoint, this may hinder association studies as differences outside this region may influence expression, antigen-binding ability and interaction with T-cell receptors. The development of tools with accurate calling beyond the second field is hampered by the lack of public datasets with quality NGS samples and highest resolution typing.
Although arcasHLA is capable of genotyping non-classical HLA loci as well as HLA pseudogenes if they are expressed at sufficiently high levels, there exists no benchmark set to validate these calls at any level of resolution. In addition, validation of in silico HLA tools with publicly available data is limited to a small subset of populations. RNA sequencing for the 1000 Genomes project is available for only four Caucasian populations and one African population, and thus is far from representative of a global population. Under-representation is not limited to these benchmark datasets; there are likely many gaps in IMGT/HLA and AFND, both of which are dependent on independent researcher contributions. Finally, HLA test sets with lower quality and single-end reads or sourced from tissue samples rather than lymphocyte cell lines would aid in the development of tools that perform well in challenging, real-world conditions.
Supplementary Material
Acknowledgements
We thank Andrew Chen and Benjamin Schweinhart for insightful comments and suggestions on an early version of the draft. Additionally, we thank Ruthie Birger, Marta Galanti, Erik Ladewig, Haruka Morita and Minhaz Ud-Dean for useful discussions. We would also like to thank Adithya Paramasivam for his help with testing the code.
Funding
This work was partially supported by DARPA [grant number W911NF-16-2-0035], National Institutes of Health [grant number U54CA193313] and the Phillip A. Sharp award. Additionally, I.F. acknowledges funding from [grant number R01-GM117591].
Conflict of Interest: none declared.
References
- Aken B.L. et al. (2016) The Ensembl gene annotation system. Database, doi: 10.1093/database/baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Bai Y. et al. (2014) Inference of high resolution HLA types using genome-wide RNA or DNA sequencing reads. BMC Genomics, 15, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birger R. et al. (2018) Asymptomatic shedding of respiratory virus among an ambulatory population across seasons. mSphere, 3, e00249–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boegel S. et al. (2012) HLA typing from RNA-Seq sequence reads. Genome Med., 4, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N.L. et al. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Buchkovich M.L. et al. (2017) HLAProfiler utilizes k-mer profiles to improve HLA calling accuracy for rare and common alleles in RNA-seq data. Genome Med., 9, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington M. et al. (1999) HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science, 283, 1748–1752. [DOI] [PubMed] [Google Scholar]
- Chowell D. et al. (2018) Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science, 359, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.Y. et al. (2016) The healthy infant nasal transcriptome: a benchmark study. Sci. Rep., 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A. et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanti M. et al. (2019) Longitudinal active sampling for respiratory viral infections across age groups. Influenza. Other Respir. Viruses, 13, 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Galarza F.F. et al. (2015) Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res., 43, D784–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourraud P.A. et al. (2014) HLA diversity in the 1000 genomes dataset. PLoS One, 9, e97282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gragert L. et al. (2013) Six-locus high resolution HLA haplotype frequencies derived from mixed-resolution DNA typing for the entire US donor registry. Hum. Immunol., 74, 1313–1320. [DOI] [PubMed] [Google Scholar]
- Grice E.A., Segre J.A. (2012) The human microbiome: our second genome. Ann. Rev. Genomics Hum. Genet., 13, 151–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S. et al. (2017) HLA-HD: an accurate HLA typing algorithm for next-generation sequencing data. Hum. Mut., 38, 788–797. [DOI] [PubMed] [Google Scholar]
- Kim D. et al. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2018) HISAT-genotype: next generation genomic analysis platform on a personal computer. bioRxiv, 266197.
- Lappalainen T. et al. (2017) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M. et al. (2016) Analysis of protein-coding genetic variation in 60, 706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J. et al. (2013) The genotype-tissue expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N. et al. (2017) Allele-specific HLA loss and immune escape in lung cancer evolution. Cell, 171, 1259–1271.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D., Thomson G. (2001) How selection shapes variation of the human major histocompatibility complex: a review. Ann. Hum. Genet., 65(Pt 1), 1–26. [DOI] [PubMed] [Google Scholar]
- Obata Y., Pachnis V. (2016) The effect of microbiota and the immune system on the development and organization of the enteric nervous system. Gastroenterology, 151, 836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R. et al. (2014) Seq reads using lightweight algorithms. Nat. Biotechnol., 32, 462–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn D.J. et al. (2002) MHC heterozygosity confers a selective advantage against multiple-strain infections. Proc. Natl. Acad. Sci. USA, 99, 11260–11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersdorf E.W. et al. (2014) HLA-C expression levels define permissible mismatches in hematopoietic cell transplantation. Blood, 124, 3996–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prugnolle F. et al. (2005). Pathogen-driven selection and worldwide HLA class I diversity. Curr. Biol., 15, 1022–1027. [DOI] [PubMed] [Google Scholar]
- Robinson J. et al. (2015) The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res., 43, D423–D431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder A. et al. (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol., 7, 3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S.A. et al. (2015) Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol., 33, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolek A. et al. (2014) OptiType: Precision HLA typing from next-generation sequencing data. Bioinformatics, 30, 3310–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R. et al. (2009) HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat. Genet., 41, 1290–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thursz M.R. et al. (1997) Heterozygote advantage for HLA class-II type in hepatitis B virus infection. Nat. Genet, 17, 11. [DOI] [PubMed] [Google Scholar]
- Varadhan R., Roland C. (2008) Simple and globally convergent methods for accelerating the convergence of any EM algorithm. Scand. J. Stat., 35, 335–353. [Google Scholar]
- Wick R.R. et al. (2015) Bandage: interactive visualization of de novo genome assemblies. Bioinformatics, 31, 3350–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosen J.E. et al. (2018) Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front. Immunol., 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C. et al. (2017) Fast and accurate HLA typing from short-read next-generation sequence data with xHLA. Proc. Natl. Acad. Sci. USA, 114, 8059–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D.R., Birney E. (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res, 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L. et al. (2018) The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science, 359, 1366–1370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.