

Abstract
Motivation
Huntington’s disease (HD) may evolve through gene deregulation. However, the impact of gene deregulation on the dynamics of genetic cooperativity in HD remains poorly understood. Here, we built a multi-layer network model of temporal dynamics of genetic cooperativity in the brain of HD knock-in mice (allelic series of Hdh mice). To enhance biological precision and gene prioritization, we integrated three complementary families of source networks, all inferred from the same RNA-seq time series data in Hdh mice, into weighted-edge networks where an edge recapitulates path-length variation across source-networks and age-points.
Results
Weighted edge networks identify two consecutive waves of tight genetic cooperativity enriched in deregulated genes (critical phases), pre-symptomatically in the cortex, implicating neurotransmission, and symptomatically in the striatum, implicating cell survival (e.g. Hipk4) intertwined with cell proliferation (e.g. Scn4b) and cellular senescence (e.g. Cdkn2a products) responses. Top striatal weighted edges are enriched in modulators of defective behavior in invertebrate models of HD pathogenesis, validating their relevance to neuronal dysfunction in vivo. Collectively, these findings reveal highly dynamic temporal features of genetic cooperativity in the brain of Hdh mice where a 2-step logic highlights the importance of cellular maintenance and senescence in the striatum of symptomatic mice, providing highly prioritized targets.
Availability and implementation
Weighted edge network analysis (WENA) data and source codes for performing spectral decomposition of the signal (SDS) and WENA analysis, both written using Python, are available at http://www.broca.inserm.fr/HD-WENA/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Understanding the progression of neurodegenerative diseases (NDs) on a molecular genetic system level may enhance therapeutic innovation through rule discovery and gene prioritization. The problems in question include those about the role of specific gene targets in modulating selective phases of ND processes and about the relations between these targets and the brain regions or cell types in which they may operate. In particular, the temporal order in which selective genes may come together into tight interaction (genetic cooperativity) for the purpose of responding to a specific phase of a ND process in a specific tissue is of high interest as this information might elucidate the rules underpinning the temporal remodeling of signaling networks in the course of ND progression, fostering a strong level of target prioritization. Inherited forms of neurodegeneration such as Huntington’s disease (HD) provide useful models in which to investigate these questions. HD is a neurodegenerative disorder associated with CAG expansion in huntingtin (HTT) (Zuccato et al., 2010). HD may arise from the cytotoxicity of misfolded, polyglutamine (polyQ)-expanded huntingtin (mHTT) in conjunction with production of misfolded HTT mRNAs and disruption of mRNA processing (Rue et al., 2016), mostly affecting the cortex and caudate nucleus through alteration of key regulatory mechanisms such as transcriptional regulation (Labbadia and Morimoto, 2013; Zuccato et al., 2010). Transcriptomic data obtained in models of HD pathogenesis have been analyzed using various network-based approaches such as spectral decomposition of the signal (SDS) against bio-networks (Rapaport et al., 2007; Tourette et al., 2014) and Weighted Gene Co-expression Network Analysis (WGCNA) (Langfelder et al., 2016). However, single network analysis may show limitations. For instance, methods such as WGCNA (Langfelder et al., 2016) generate gene co-expression networks but could lack the biological precision of SDS (e.g. protein–protein interactions) (Rapaport et al., 2007). However, the use of prior network knowledge in SDS could miss some of the correlations detected by WGCNA. Both SDS and WGCNA analysis generates undirected networks. In contrast, Bayesian causal inference (Hayete et al., 2017) generates causal networks, however with a bias towards generating a large number of predictions around strongly deregulated genes (Gligorijevic and Przulj, 2015) and with no insight provided into the biochemistry of causal relationships. Thus, taking advantage of the complementarity and consistency of edge data across network layers (Yan et al., 2017) could enrich the basis for modeling HD on a system level. Here, we hypothesized that applying a multi-layer network approach might reduce data complexity in robust ways while providing biologically precise information on how the architecture of genetic cooperativity may evolve in the course of HD. To test for this hypothesis, we applied weighted edge network analysis (WENA) for reconstructing the dynamics of genetic cooperativity in HD based on the integration of source network families into dense weighted networks, quantification of the temporal dynamics of shortest path lengths in weighted edges and assessment of robustness using randomization tests. We used WENA for integrating HD networks that describe the RNA-Seq time series data in the striatum and cortex of the allelic series of HD knock-in mice (Hdh mice), currently the largest dataset available for modeling molecular pathogenesis in HD (six CAG-repeat lengths, three age-points, several tissues) (Langfelder et al., 2016). These HD mice networks include causal networks obtained by using Bayesian causal inference (Hayete et al., 2017), WGCNA networks generated using settings similar to those defined by Langfelder et al. (2016) and deregulated bio-networks obtained by using SDS against probabilistic functional networks (Lejeune et al., 2012; Tourette et al., 2014). The resulting model identifies critical (i.e. tight interaction network enriched in HD-deregulated genes) and functionally distinct phases of genetic cooperativity in the brain of Hdh mice, pre-symptomatically in cortex and symptomatically in striatum. Noticeably, WENA analysis identifies highly prioritized targets that are known to regulate cell maintenance, cell proliferation, DNA damage repair and cellular senescence and that may be central to striatal homeostasis in Hdh mice as they show major symptoms. Moreover, top striatal weighted-edges are enriched in suppressors and enhancers of neuronal dysfunction and defective behavior in C.elegans (Lejeune et al., 2012) and Drosophila (Al-Ramahi et al., 2018) models of HD pathogenesis, validating their relevance to neuronal dysfunction in vivo.
2 Materials and methods
2.1 Source data
The RNA-Seq data analyzed herein originate from the striatum and cortex of the allelic series of Hdh knock-in mice (Q20, Q80, Q92, Q111, Q140 and Q175 at 2, 6 and 10 months of age, encompassing the presymptomatic to symptomatic phases of HD in these mice, with four males and four females per point) as previously reported (Langfelder et al., 2016).
2.2 Construction of source network families
2.2.1 Causal networks
Causal networks generated by GNS Healthcare using a Bayesian approach for analysis of RNA-Seq data in Hdh mice were kindly provided by the CHDI Foundation. Causal networks are available for three age points (2, 6, 10 months) for the striatum and for two age points (6 and 10 months) for the cortex. For integration with other families of source networks (see below), we removed edges involving phenotypic information, miRNAs and targets, mitochondrial genes, pseudogenes and genes whose name was not in the Ensembl mus musculus gene symbol list. To ensure homogeneity of network-data integration, we did not take information on the direction of gene–gene interactions into account. The resulting networks are herein referred to as GNS networks. Data on the direction of gene–gene or gene-phenotype interactions were re-introduced at the stage of data interpretation as a bona fide information that could help with reasoning on target prioritization.
2.2.2 WGCNA networks
The WGCNA package in R (https://www.r-project.org) was used to generate WGCNA modules (WGCNA package) from RNA-Seq data in the allelic series of Hdh mice at 2, 6 and 10 months of age, for the striatum and cortex. Before performing WGCNA analysis we used multidimensional scaling analysis in order to remove outlier samples in the 18 data points defined by tissue and age, retaining 256 samples out of a total of 289 samples. We then computed the correlation coefficient across the various CAG-repeat lengths, and only retaining gene pairs having a correlation higher than 0.25 in absolute value (ignoring the correlation sign), similarly to previous WGCNA analyses (Langfelder et al., 2016). For comparison with the networks obtained by using spectral decomposition of the signal (see below) which only involves genes, we removed edges involving non coding RNAs and pseudogenes, as well as genes whose name was not in the Ensembl Mus musculus gene symbol list.
2.2.3 Spectral decomposition networks
These networks were generated by using an adapted version of the SDS analysis (Rapaport et al., 2007) as previously described (Lejeune et al., 2012; Tourette et al., 2014). This analysis was performed against Mousenet v2 (Kim et al., 2016). SDS is part of the BioGemix (BGM) framework that we developed for modeling ND datasets. Briefly, mRNA read-count data as available in the RNA-Seq data in the allelic series of Hdh mice was fed into DESeq2 1.14.1 in order to generate log-fold-change (LFC) values (given age and CAG-repeat length). The dataset at 2 months of age in Hdh/Q20 mice was used as a reference. This analysis was performed on the samples retained upon removal of outlier samples as above mentioned (see Section 2.2.2). Spectral decomposition was then used to remove noise from the LFCs, using prior knowledge in Mousenet v2, filtering out the 25% highest frequencies of the spectral decomposition. Edges of Mousenet v2, which contains signed only co-expression edges, involving any two such deregulated genes were retained to generate one network per condition considered (CAG-repeat length and age point). A normalized Hamming distance metric was then used to aggregate the 17 conditions (six CAG-repeat lengths x three age-points, minus the Q20/2-month reference) in the form of three clusters corresponding a long, intermediate and short time-to-(major)symptoms. For a given cluster, the resulting network was taken as the union of networks corresponding to the conditions unified by this cluster. The resulting networks are herein referred to as BioGemix (BGM) networks.
2.3 Construction, biological annotation, target prioritization and functional relevance of weighted edge networks
The cross-integration of source networks was performed by using weighted edge network analysis (WENA), which is part of the BioGemix framework. WENA analysis may be used for modeling any type of disease and integrating any type of network families. The current version of WENA may be applied to the cross-integration (CI) of up to three layers of networks across up to three conditions, e.g. time points (WENA-CI). WENA-CI (Supplementary Fig. S1) is able to quantify all types of weighted-edge dynamics, i.e. linear and non-linear changes of edge dynamics across conditions. WENA analysis, randomization tests, biological content analysis, comparison to WGCNA data, target prioritization and comparison to functional datasets are fully described in the Supplemental Information in the context of integrating the three above mentioned families of HD networks across three time points.
3 Results
We applied WENA to the cross-integration of three families of networks that describe RNA-seq time series data in Hdh mice, including networks generated by spectral decomposition of the RNA-seq signal (BioGemix or ‘BGM’ networks), WGCNA analysis and Bayesian causal inference.
3.1 Construction of BioGemix networks
To generate bio-networks that describe (CAG)n-dependent gene expression for each phase of the disease in the cortex and striatum of Hdh mice, we performed spectral decomposition of the signal against Mousenet v2 (Kim et al., 2016). Settings were similar to those we previously reported for modeling RNAi and microarray data in human HTT exon-1 nematodes (Lejeune et al., 2012; Tourette et al., 2014) (see Section 2). We identified three clusters of bio-networks for cortex or striatum, including one cluster for pre-symptomatic Hdh mice or Hdh mice with a long time-to-symptoms, one cluster for Hdh mice with an intermediary time-to-symptoms and one cluster for symptomatic mice or mice with a short time-to-symptoms (Supplementary Fig. S2, Tables S1 and S2).
3.2 Construction of WGCNA networks
To generate WGCNA edges that describe (CAG)n-dependent gene expression in the cortex and striatum of Hdh mice at 2, 6 and 10 months of age, we used settings similar to those previously reported (Langfelder et al., 2016) (see also Section 2) (Supplementary Tables S3 and S4).
3.3 Construction of dense weighted networks
Next, we used WENA analysis to integrate BGM, WGCNA and causal networks. The latter class of networks was generated by GNS Healthcare using Bayesian causal inference and data are fully available in the HDinHD database (https://www.hdinhd.org/gns-biological-networks/). To ensure data homogeneity, we removed information on directionality from causal networks, generating data herein referred to as GNS networks (Supplementary Tables S5 and S6). After transformation of these sparse networks into dense weighted networks, that is weighted networks sharing a significant number of edges (see Supplementary Methods, Section S1.1), WENA identified weighted edges that are common to source networks including 4.6 millions edges for striatum and 33 724 edges for cortex (Supplementary Fig. S3, Table S7; see http://www.broca.inserm.fr/HD-WENA/ for the lists of weighted edges).
3.4 Construction of WENA meta-networks
Weighted edges are characterized by the WENA score (product-P: see Section 2), which measures the change of shortest path length (SPL) across conditions (time-points), indicating the direction (expansion or contraction) and magnitude (or strength) of SPL variation. Weighted edges characterized by product-P values greater than 0.15 are rather dynamic over time, as illustrated for cortex. Weighted edges with an absolute product-P value above the inflexion point (here 0.26) of the log logistic curve describing the cumulative distribution of most dynamic edges are of primary interest (Fig. 1), as illustrated for the striatum. We used a threshold set at 0.3 to select for striatal weighted edges of high interest in terms of gene prioritization (Fig. 1).
Fig. 1.

Quantification of age-dependent strength of gene cooperativity in dense weighted networks. (A) Cumulative distribution of the number of Class-I or Class-II weighted edges as a function of product-P where product-P = ΔwGNS x ΔwWGCNA x ΔwBGM, for the cortex. (B) Cumulative distribution of the number of Class-I or Class-II weighted edges (left panel) and of the number of most dynamic weighted edges (right panel) as a function of product-P, for the striatum. Most dynamic edges are edges for which Δw = 1 in at least 2 dense weighted networks where Δw = 1 means a direct edge at 2 months and no edge at 10 months, and vice versa (see Supplementary Methods). The choice of threshold for product-P is based on the inflection point e of the log logistic cumulative distribution curve for most dynamic edges, here 0.26 and above (e.g. 0.3)
3.5 Robustness of weighted edge ranking in WENA
To evaluate the robustness of edge and gene prioritization in WENA, we performed randomization tests (see Section 2 and Supplementary Methods) on source networks. This analysis was performed for the striatum, a brain area where gene deregulation may be the most pronounced (Langfelder et al., 2016) and node degree in source networks may be high. Node degree distribution in the source networks was preserved while permutating gene node neighbors. We observed that, on a global level, product-P based ranking of edges in striatal meta-networks upon randomization is statistically different from that in meta-networks before randomization (Supplementary Fig. S4A). This observation strongly applies to the top 25 weighted edges (product-P > 0.3) retained by WENA analysis, where 17 edges did not categorize as top edges upon randomization, 7 edges remained top edges in 1/10 trials and 1 edge (containing nodes super-connected in source networks) remained a top edge in 3/10 trials (Supplementary Fig. S4B). The same applies to edges with lower product-P values, i.e. values between 0.15 and 0.3 (see edge groups outside the red line squares, Supplementary Fig. S4A). As expected, significant numbers of edges retained the same ranking when they have a product-P value below 0.15 (see edge groups within the red line squares, Supplementary Fig. S4A).
These observations suggest that weighted edge ranking and gene prioritization in WENA analysis is robust, generating top edges that are specific to the data contained in the source networks.
3.6 Weighted networks identify critical phases of genetic cooperativity in Hdh mice
In Hdh mice, WENA scores (see Fig. 1) enable to distinguish weighted edges with SPL increase, meaning genetic cooperativity is relaxed over time (Class-I edges and genes) and those with SPL decrease, meaning genetic cooperativity is reinforced (Class-II edges and genes). Depending on data richness, WENA also detects Class-III genes, i.e. those involved in both Class-I and Class-II edges. In Hdh mice, this means Class-III genes preferentially cooperate with different partners at 2 months versus 10 months of age (Supplementary Tables S8 and S9), documenting node rewiring. The discriminative power of this model is increased by integrating information from WENA meta-networks (subset of genes cooperating the most in a specific condition) and information about gene deregulation. In HD, the genes of particular interest are those deregulated in a (CAG)n length- and age-dependent manner, herein referred to as HD-and age-filtered (HDAF) genes. To test whether WENA meta-networks are enriched in HDAF genes, we calculated the log-fold-change (LFC) value between Q175 mice at 10 months of age and Q20 mice at 2 months of age. This provides a good estimate of HD- and age-dependence as, overall, there is no or little change of gene expression at 6–10 months of age in Q20 mice (see Supplementary Methods). Interestingly, in the cortex, Class-I genes are enriched for HDAF genes, a feature not shown by Class-II genes (Fig. 2A), whereas in the striatum enrichment for HDAF genes is true for Class-II genes and not for Class-I genes (Fig. 2B). Regarding Class-III genes, WENA did not retain any such genes for cortex due to the lower level of gene deregulation compared to striatum (Langfelder et al., 2016). For striatum, Class-III genes show some enrichment for HDAF genes, however to a lower extent compared to Class-II genes (Fig. 2B, lower panel). These results highlight two critical phases of genetic cooperativity in which HDAF genes cooperate first in the cortex of pre-symptomatic Hdh mice, then in the striatum of symptomatic Hdh mice. As developed below, these two critical phases are biologically distinct.
Fig. 2.

Distribution of log fold change values in Q175 Hdh mice at 10 months for gene nodes retained in WENA meta-networks. Shown is the distribution of log-fold-change (LFC) at 10 months in Q175 mice versus 2 months in Q20 mice (Q175/10 mo-LFC) for Class-I and Class-II genes in cortex and for Class-I, Class-II and Class-III genes in striatum. In all cases, the reference is the distribution of LFC (Q175, 10 months/Q20, 2 months) values for all genes with a statistically significant deregulation as inferred using DESeq2. (A) In the cortex (unfiltered dense weighted networks), the distribution of Q175/10 mo-LFCs for Class-I genes is statistically different (upper panel: P = 5.67 × 10−13) from that for reference genes whereas the LFC distribution for Class-II genes is similar to the reference (lower panel). Bin size is 0.05. There are 34 Class-I genes, 86 Class-II genes and 18948 reference genes in the LFC distributions. (B) In the striatum (unfiltered dense weighted networks), the Q175/10 mo-LFC distribution for Class-I genes is similar to the reference (upper panel) whereas the LFC distribution is statistically different for Class-II genes (middle panel: P = 1.01 × 10−4) compared to the reference. The distribution of Q175/10 mo-LFCs for Class-III genes (as instructed by unfiltered dense weighted networks) is statistically different (P = 6.35 × 10−3) compared to the reference (lower panel). Bin size is 0.1 for the Class-I, Class-II and Class-III meta-networks. There are 306 Class-I genes, 1103 Class-II genes, 26 Class-III and 19051 reference genes in the LFC distributions
3.7 Key regulators of cell stress response tightly cooperate in the striatum of Hdh mice synchronous to symptoms
Next, we assessed the biological significance of the WENA model of genetic-cooperativity in Hdh mice. To this end, we tested meta-networks for enrichment in biological annotations. Additionally, we used WENA scores (product-P), network concepts and source-network data (see Supplementary Methods: edge-based feature selection) to enhance biological precision. Disease relevance was assessed for Class-II meta-networks only, i.e. those in which genes come together for tight interaction in symptomatic Hdh mice (see Supplementary Methods). Together, these analyses showed the added value of WENA for modeling the dynamics of genetic cooperativity at high resolution, indicating when the tightness of a gene–gene interaction is maximal (temporal precision) and whether this is biologically homogeneous, involving selective pathways (biological precision).
In the cortex, we kept all weighted edges as their number is limited. Top annotations for the Class-I meta-network (recruiting 34 genes; critical phase of genetic cooperativity as shown in Fig. 2A) correspond to GPCR signaling and synaptic (e.g. glutamatergic) activity (Fig. 3, Supplementary Tables S10 and S11). Edge-based feature selection highlights cooperation between carboxypeptidase Cpa6 and double-C2 protein Doc2g, which also involves Doc2b and oxysterol 7-α-hydroxylase 2 Cyp39a1 (BGM-network data), as the interaction most dynamically and centrally associated to the response of the cortex to HD at 2 months of age for putative regulation of synaptic vesicle trafficking (see legend of Fig. 3). Top annotations for the Class-II meta-network (recruiting 86 genes) correspond to proteasome and to mRNA processing (Fig. 3, Supplementary Tables S10 and S11). However, edge-based feature selection highlights cooperation between tjp3 (Zonula occludens-3) and claudin Cldn1 (a component of tight junctions), also involving Cybrd1 and Lppr1 (BGM-network data), as the interaction most dynamically and centrally associated with the response of the cortex to HD at 2 months of age for putative regulation of synaptic plasticity (see legend of Fig. 3). Gene-phenotype interactions in causal networks (Supplementary Table S10) indicate WENA node acyl-CoA oxidase 2 (Acox2) may negatively influence locomotion bouts and duration of locomotion, predicting that the variation of Acox2 expression levels may be relevant to motor phenotypes.
Fig. 3.
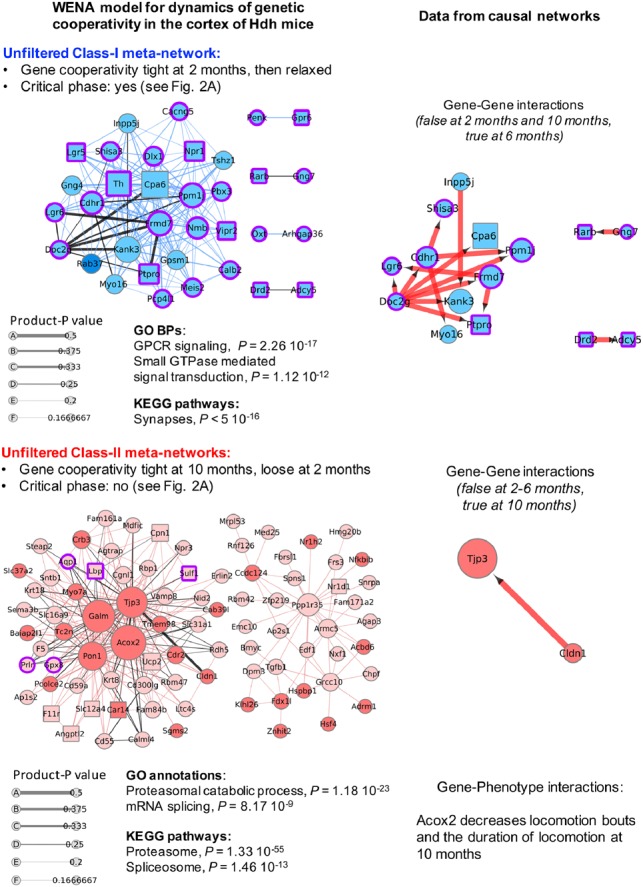
Temporal dynamics of genetic cooperativity in the cortex of Hdh mice. Shown are the unfiltered Class-I meta-network (blue nodes) containing 162 weighted edges and Class-II meta-network (red nodes) containing 271 weighted edges such that |product-P| > 0. Node size is scaled with node degree (number of neighbors). Edge thickness is scaled with product-P value. Dark (red or blue) colors indicate up-regulated genes. Light (red or blue) colors indicate down-regulated genes. Purple borders indicate genes with |LFC| > 0.5, i.e. the LFC value that distinguishes critical from non-critical phases of genetic cooperativity (see Fig. 2). Squared nodes are druggable genes. Black edges indicate that the edge is direct in at least one the source network families. Corresponding information in causal networks shows edge orientation as predicted by Bayesian causal inference with indication of the effect (blue: decrease; red: increase) of mRNA abundance of the upstream gene on that of the downstream gene. Gene-phenotype interactions are also indicated for Class-II genes, i.e. the genes associated with the most symptomatic phase of the disease process. Biological annotations (n > 10 genes, P < 1 × 10−4) are inferred from STRING analysis, using high-confidence neighbors (see Supplementary Methods for settings). In the Class-I meta-network, the direct weighted edge with the highest combined values for product-P and betweenness involves Cpa6 (also the hub gene with the highest degree: see Supplementary Table S10), a carboxypeptidase that processes several neuropeptides, and Doc2g, a double-C2 protein involved in synaptic vesicle exocytosis (Yao et al., 2011). Gene–gene interaction data in causal networks (Supplementary Table S11) indicates that Doc2g may act upstream of most of the other nodes in this meta-network, including Cpa6. Consistently with the outcome of WENA, this prediction is true for Hdh mice at 6 months of age and was not detected for mice at 10 months of age. WENA further indicates that the tightness of genetic cooperativity between Doc2g and Cpa6 is maximal at 2 months of age, then is relaxed. The same applies to the other causal gene-to-gene relationships relevant to cortex at 6 months of age. Although the Cpa6-Doc2g edge is direct as predicted by WGCNA and Bayesian network analysis, BGM network data indicate that the path linking these two genes could involve Cyp39a1 and Doc2b. In the Class-II meta-network, the direct weighted edge with the highest combined values for product-P and betweenness recruits Tjp3 (also known as Zonula occludens-3; hub gene with the third highest degree value), a tight junction protein that interacts with connexins, and claudin Cldn1, a component of tight junctions, suggesting this meta-network might also regulate the plasticity of electrical synapses (Flores et al., 2008). Gene–gene interaction data in causal networks (Supplementary Table S11) indicates that Cldn1 may act upstream of Tjp3. Consistently with the WENA model, this prediction was only detected for Hdh mice at 10 months of age. WENA further indicates that cooperativity between these two genes is loose at 2 months of age, becoming tight at 10 months of age. WENA also indicates that the path linking these two genes may involve cytochrome B reductase Cybrd1 and phospholipid phosphatase Lppr1 as provided in BGM networks (see Supplementary Table S11). Gene-phenotype interactions in causal networks (see Supplementary Table S10) indicate that acyl-CoA oxidase 2 (Acox2) may negatively influence two disease phenotypes at 10 months, including locomotion bouts and the duration of locomotion
In the striatum, weighted edges show a wide range of loose to tight levels of genetic cooperativity (Fig. 1B). To select for most dynamic edges, we applied a threshold of 0.3 on product-P values. This significantly reduces the number of weighted edges (Fig. 4, Supplementary Tables S12 and S13) and translates into a SPL value of 1 (direct edge) in at least one of the source networks (see Section 2 and Supplementary Methods). The outcome of a threshold set at ≥0.25 is also shown to provide a larger view of genetic cooperativity (Supplementary Fig. S5). Top annotations for the Class-I meta-network with threshold set at 0.3 (9 genes) correspond to metabolism of neurotransmitters (Fig. 4). Edge-based feature selection further indicates this meta-network may correspond to a glial response targeting axonal integrity as supported by key regulators of myelination, also involving regulators of cell movement (see legend of Fig. 4). Top annotations for the Class-II meta-network (17 genes; critical phase of genetic cooperativity as shown in Fig. 2B) almost uniquely correspond to regulation of cell cycle arrest, DNA repair and cell division by regulatory proteins in the p16INK4a and p19ARF (Cdkn2a products), p53 and FOXO signaling pathways (Fig. 4). This could be relevant to the proliferation of striatal dividing cells as well as stress response (e.g. DNA repair, autophagy) of post-mitotic striatal neurons. Another outcome may be the modulation of axonal transport of mitochondria (via Mgarp). Edge-based feature selection indicates this is primarily achieved through genetic cooperativity centered onto cAMP-regulated phosphoproteins Arpp21 and Arpp19, the sodium voltage-gated channel beta subunit and metastasis suppressor Scn4b, and the homeodomain-interacting (HIP) kinase Hipk4. Interestingly, this also involves the Cdkn2a locus, and regulators of splicing and translation as indicated by BGM network data (see legend of Fig. 4). Gene-phenotype interaction data in causal networks (Supplementary Table S12) link Arpp19 and Arpp21 to disease phenotypes, predicting that the variation of Arpp expression levels may be relevant to behavioral phenotypes.
Fig. 4.
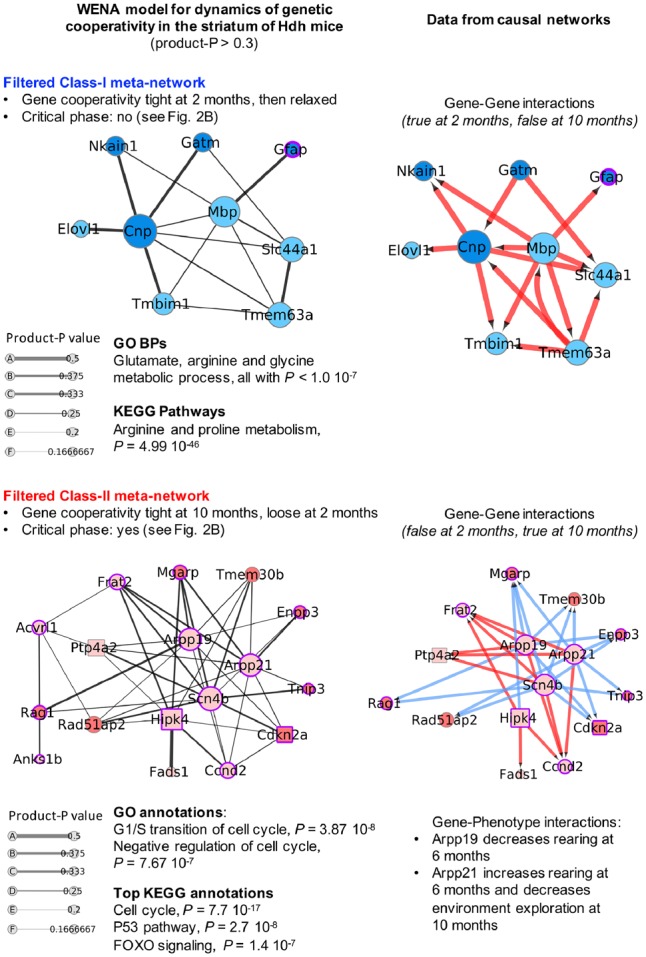
Temporal dynamics of genetic cooperativity in the striatum of Hdh mice. Shown are the Class-I meta-network (blue nodes) containing 15 weighted edges and Class-II meta-network (red nodes) containing 44 weighted edges such that |product-P| > 0.3, which selects for highly dynamic weighted edges in which there is a direct gene-to-gene interaction (SPL value of 1) in at least one of the source networks. The legend of nodes and edges and method for inference of biological annotations are the same as in Figure 3. Meta-networks are also shown for |product-P| > 0.25, providing a larger though less-selective model of the temporal dynamics of genetic cooperativity (see Supplementary Fig. S4). In the Class-I meta-network, the direct weighted edges with the highest product-P values involve two hub genes including (i) phosphodiesterase Cnp in direct interaction with four genes [mitochondrial glycine amidinotransferase Gatm, Na(+)/K(+)-transporting ATPase subunit Beta-1-interacting protein Nkain1, fatty acid elongase elov1 and transmembrane BAX inhibitor motif-containing protein Tmbim1] and (ii) myelin basic protein MBP in direct interaction with glial fibrillary acidic protein Gfap, the latter a marker of astrocytes. Cnp is an important regulator of myelination (Aguirre et al., 2007) and Mbp is a well-known and major constituent of the myelin sheath of oligodendrocytes. Gene–gene interaction data in causal networks indicate that Cnp and Mbp may act upstream of most of the other nodes in this meta-network. Consistently with the outcome of WENA, this prediction is only detected for Hdh mice at 6 months of age. WENA further indicates that the tightness of genetic cooperativity between Cnp, Mbp and their neighbors is maximal at 2 months of age then relaxed, and that causal relationships such as the one between Mbp and Cnp or between Mbp and Tmbim1 are poorly dynamic over time (product-P value less than 0.25). Finally, BGM networks indicate that this response may involve genes associated to RNA degradation and cell projection morphogenesis (see Supplementary Table S13). In the Class-II meta-network, the direct weighted edges with the highest product-P values and gene nodes with the highest degree suggest this may be primarily achieved through genetic cooperativity centered onto cAMP-regulated phosphoproteins Arpp21 and Arpp19, the sodium voltage-gated channel beta subunit Scn4b, a protein that may act as a metastasis suppressor (Bon et al., 2016) and Hipk4, a member of the homeodomain interacting kinase family, the latter a class of kinases that are associated with DNA damage response (Kuwano et al., 2016) and that is involved in cellular proliferation, differentiation and apoptosis (Kovacs et al., 2015), including in neurons (Chalazonitis et al., 2011). These four hub genes may tightly cooperate with proximal interactors such as cyclin D2 (Ccnd2), cell cycle regulators encoded by Cdkn2a (i.e. p16INK4a, p19ARF), phosphatase Ptp4a2 (also known as Prl2) which regulates the proliferation of hematopoietic stem cells (Kobayashi et al., 2014), fatty acid desaturase Fads1 which expression is modulated during neuronal differentiation (Park et al., 2012), Mgarp which is a regulator of mitochondrial distribution and motility in neurons and may contribute to dendritic atrophy when in excess (Jia et al., 2014), Frat2 which is a member of canonical Wnt signaling, ectonucleotide pyrophosphatase/phosphodiesterase Enpp3 which achieves an enzymatic activity (Gomez-Villafuertes et al., 2014) associated with neuronal differentiation and two genes associated with inflammation including Tnip3 and Rag-1, the latter an immunoglobulin recombination activation gene with a role in neuronal death (Hirano et al., 2015). Causal networks (gene–gene interactions: see Supplementary Table S13) indicate that hub genes Arpp 19, Arpp21, Hipk4 and Snc4b could act upstream to the other members in this Class-II meta-network at 10 months of age. WENA analysis further indicates that the tightness of cooperativity between these hub genes and neighbors such as Cdkn2a and Prl2 becomes maximal at 10 months of age. BGM networks indicate this may also involve regulators of splicing and (mitochondrial) translation and other genes, such as for example cyclin D2 and ion channel Trpa1 in the Hipk4-Cdkn2a path, PGC1ß and Host Cell Factor C1 Regulator 1 in the Hipk4-Mgarp path and scaffold protein Pdlim1 (CLP36) and Acyl-CoA thioesterase 1 (Acot1) in the Scn4b-Perl2 path (see Supplementary Table S13). Gene-phenotype interactions data in causal networks (see Supplementary Table S12) indicate that Arpp19 and Arpp21, two central nodes, modulate disease phenotypes at 6–10 months. Noticeably, Class-II nodes are not associated with mouse phenotypes at 2–6 months and Class-I nodes are not associated with mouse phenotypes at 10 months (see Supplementary Table S12), showing that WENA can distinguish waves of gene cooperativity associated with specific phases of disease, phenotypically. As indicated by randomization tests, the Frat2-Scn4b weighted edge may have limited robustness (see Supplementary Fig. S4B)
3.8 WENA documents node-rewiring between waves of genetic cooperativity
Class-III nodes are connected to both Class-I and Class-II nodes. WENA detected Class-III genes in the striatum (Supplementary Tables S8 and S9). These genes may participate into different biological processes over time because they change neighbors (node-rewiring) or because their most-proximal Class-I neighbors belong to the wave of genetic cooperativity (i.e. group of genes linked by edges with the most dynamic change of SPL over time) that is relaxed over time while most-proximal Class-II neighbors belong to the wave that is reinforced (role changing). Striatal Class-III genes are engaged into weighted edges that, overall, are poorly dynamic over time. Nonetheless, retaining this information (neighbors gained or lost regardless of product-P value, proximal neighbors with product-P > 0.1) delineates a group of highly connected Class-III nodes (8 genes) associated with lipid metabolism, cell projection, membrane dynamics and stress response (see Legend of Fig. 5). Interestingly, Class-I and Class-II neighbors suggest that these Class-III genes are initially involved in synaptic vesicle cycle and phagocytosis and that they proximally interact with Class-I central nodes such as the constituent of the myelin sheath Mbp (Fig. 5). These Class-III genes then become more closely involved in stress resistance and cell survival, proximally interacting with highly deregulated Class-II nodes such as cyclin-dependent-kinase inhibitors encoded by Cdkn2a (Fig. 5; see also Fig. 4). Information from causal networks suggests that (i) Class-I neighbors may act upstream to Class-III genes (Supplementary Table S9) and (ii) that Inadl, a Class-III gene encoding a tight junction protein, modulates several gait phenotypes at 2 months of age (Supplementary Table S8), illustrating the phenotypic relevance of Class-III genes. However, causal gene–gene interaction data were not available for Class-II neighbors (Fig. 5). In contrast, BGM network data identify additional genes that may participate into interactions between Class-III genes and their neighbors, including for Class-II neighbors (see Supplementary Table S9, last two columns). Together, these results further highlight the value of WENA for characterizing the dynamics of genetic cooperativity as a sum of effects, showing how successive phases of molecular genetic response may be established over time.
Fig. 5.
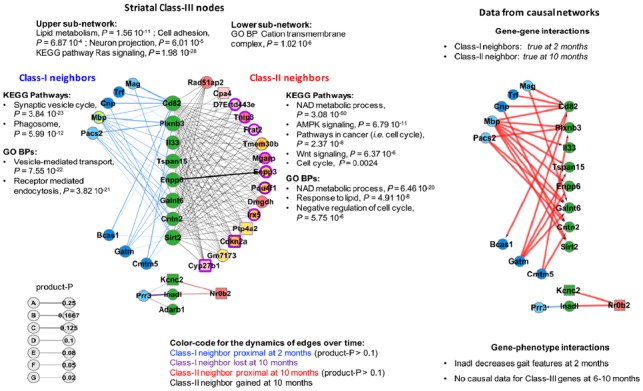
Class-III genes and their Class-I and Class-II neighbors in the striatum of Hdh mice
The left panel shows 11 striatal Class-III genes (green nodes) involved in weighted class-I (blue nodes) and Class-II (red nodes) edges for |product-P| > 0.1. A total of 26 Class-III genes were detected regardless of product-P values (see Supplementary Table S8). The legend of nodes and edges and method for inference of biological annotations are the same as in Figure 3, except that we allowed information from ‘Text mining’ to be used by STRING for assessing the biological content of Class-II neighbors. Yellow circles indicate the Class-I or Class-II genes connected to all of the Class-III genes in their respective sub-network. Neighbors with a yellow circle are also found in either the Class-I or Class-II striatal meta-networks (see Fig. 4). A specific color code is provided for the dynamics of the edges over time in which nodes lost or gained at 10 months indicate node-rewiring. The larger sub-network contains 8 genes associated with alpha lipid metabolism (enpp6), cell projection (cntn2: GPI-anchored protein contactin-2), stress response (sirtuin Sirt2, plexin plxnb3, glycosyltransferase Galnt6), membrane dynamics (tetraspanins Tspan15 and CD82) and innate immunity (interleukin Il33). The histone deacetylase Sirt2 is a target of cyclin-dependent kinase inhibitors (Pandithage et al., 2008) that are encoded by Cdkn2a and found in the Class-II striatal meta-network (see Fig. 4). The right panel shows data from causal networks, which is available for Class-I neighbors and not for Class-II neighbors, including (i) edge orientation as predicted by Bayesian causal inference with indication of the effect (blue: decrease; red: increase) of mRNA abundance of the upstream gene on that of downstream gene(s) and (ii) data available on gene-phenotype interactions.
3.9. WENA multimodal information narrows and complements WGCNA information
To provide a comparison of WENA- and WGCNA-generated data, we performed overlap analyses between gene nodes in WENA networks (this study) and those previously identified in CAG-repeat and age-dependent WGCNA modules (Langfelder et al., 2016), followed by pathway enrichment analysis. As expected, these analyses indicate that WENA narrows the mouse gene space associated by WGCNA with the temporal dynamics of HD, especially in the striatum (Supplementary Fig. S6). These analyses also indicate that WENA may put forth target genes not prioritized by WGCNA analysis (Supplementary Fig. S6).
3.10 WENA multimodal information enhances target prioritization
WENA allowed to select a manageable number of genes (n = 100) that are central to the dynamics of genetic cooperativity in Hdh mice (Supplementary Table S14). Applying additional criteria such as druggability identifies 17 genes of high-interest (Table 1).
Table 1.
Druggable genes of high interest retained by WENA analysis of the dynamics of genetic cooperativity in the brain of Hdh mice
| Gene name | WENA node | WENA edge | Brain area | Critical phase (age) | Rewiring to Class-II nodes (1) | Intra-node in BGM paths of interest (2) | LFC (3) |
|---|---|---|---|---|---|---|---|
| Cdkn2a | Class-II | Class-II | Striatum | Yes (10 months) | NA | No | 1.018 |
| Kcng4 | Class-III | NA | Striatum | No | Yes | No | 0.473 |
| Nr2f2 | Class-III | NA | Striatum | No | Yes | No | 0.414 |
| Cyp39a1 | NA | Class-I | Cortex | Yes (2 months) | NA | Yes | 0.364 |
| Kcns3 | Class-III | NA | Striatum | No | Yes | No | 0.353 |
| Kcnc2 | Class-III | NA | Striatum | No | Yes | No | 0.331 |
| Hcn3 | NA | Class-II | Striatum | Yes (10 months) | NA | Yes | 0.228 |
| Sirt2 | Class-III | NA | Striatum | No | Yes | No | 0.094 |
| Chrna4 | Class-III | NA | Striatum | No | Yes | No | 0.091 |
| Ptp4a2 | Class-II | Class-II | Striatum | Yes (10 months) | NA | No | −0.102 |
| Car4 | Class-III | NA | Striatum | No | Yes | No | −0.107 |
| Car8 | NA | Class-I | Cortex | Yes (2 months) | NA | Yes | −0.126 |
| Cpa6 | Class-I | Class-I | Cortex | Yes (2 months) | NA | No | −0.402 |
| Pde2a | NA | Class-II | Striatum | Yes (10 months) | NA | Yes | −0.605 |
| Ptpro | Class-I | Class-I | Cortex | Yes (2 months) | NA | No | −1.285 |
| Hipk4 | Class2 | Class-II | Striatum | Yes (10 months) | NA | No | −1.485 |
| Ptpn7 | NA | Class-I | Cortex | Yes (2 months) | NA | Yes | −1.493 |
Note: (1) Striatum only. (2) i.e. most deregulated and part of a highly dynamic WENA edge: see node content in Supplementary Table S14/BGM network data. (3) Log-fold-change (Q175; 10 months/Q20; 2 months). These 17 genes match either one of the following criteria: (i) recruited into the most dynamic (product-P ≥ 0.25) Class-I and Class-II edges in the cortex, (ii) recruited into the most dynamic (product-P ≥ 0.3) Class-I and Class-II edges in the striatum, (iii) Class-III genes that interact with neighbor genes in the Class-I or Class-II meta-network for the striatum (see Fig. 5) and (iv) BGM nodes involved in the meta-networks that define critical phases (for weighted edges with a product-P value ≥ 0.25) and that are part of the most deregulated BGM path that link two WENA nodes. Profiles of gene deregulation across CAG repeat lengths and age points in Hdh mice can be visualized in the BioGemix-3D database (see https://www.hdinhd.org/; see also http://www.broca.inserm.fr/webportal/Biogemix_3D.php). The full list of genes of interest retained by WENA contains 100 genes (see Supplementary Table S14).
NA, not applicable.
This notably highlights tight interaction between Hipk4 (down-regulated: see the BioGemix-3D database at http://www.broca.inserm.fr/webportal/Biogemix_3D.php or at www.hdinhd.org) and Cdkn2a products (e.g. cell-senescence marker p16INK4a, up-regulated: see BioGemix-3D) as gene pairs of interest for targeting the critical phase of genetic cooperativity in the striatum. The same applies to cooperation between Hipk4 and Mgarp (axonal transport of mitochondria), where Hipk4 could act upstream of Mgarp as suggested by causal networks, and to cooperation between Scn4b and Mgarp, which involves the phosphodiesterase and candidate-target in Alzheimer’s disease Pde2a (down-regulated: see BioGemix 3D) as indicated by the BGM-network data. Regarding the critical phase of genetic cooperativity in the cortex, this applies to phosphatase Ptpn7, a member of MAPK signaling, where Ptpn7 may participate in the interaction between Ptpro, a synaptic adhesion molecule, and Frmd7, a key regulator of neuronal circuit asymmetry, as also indicated by BGM networks. Thus, WENA multimodal information greatly enriches the basis for target prioritization.
3.11 Striatal weighted edges are enriched in modulators of neuronal dysfunction and defective behavior in invertebrate models of HD pathogenesis
To test for the relevance of the WENA model of the dynamics of genetic cooperativity in the brain of Hdh mice to diseased neuron dysfunction in vivo, we tested for overlap between the list of (i) most interesting genes retained by WENA analysis in the striatum (227 genes recruited in edges with a product-P value above 0.25 including 119 genes with an ortholog in C.elegans or Drosophila) or (ii) 120 genes recruited in all WENA edges for the cortex (including 69 genes with an ortholog in C.elegans or Drosophila) and the modulators of neuronal dysfunction and defective behavior in invertebrate models of HD. Invertebrate data included data from a genome-scale gene perturbation screen that we performed for modification of defective touch response in C.elegans nematodes expressing human exon 1 huntingtin in touch receptor neurons (128Q nematodes) (Lejeune et al., 2012) and data from a similar type of screen for modification of motor impairment in flies expressing N-terminal human huntingtin in all neurons (Al-Ramahi et al., 2018). Overlap was not significant for cortical WENA nodes (P < 0.11), (Supplementary Fig. S7), which is expected considering the small number of dynamic edges retained by WENA for this tissue, which itself results from less gene deregulation in the cortex compared to striatum of Hdh mice (Langfelder et al., 2016). In contrast, overlap was significant for striatal WENA nodes (P < 0.03), involving 38/119 genes (31.9%) (Supplementary Fig. S6). For example, the striatal Class-II meta-network (critical phase at 10 months) contains central nodes such as down-regulated Hipk4, down-regulated Arpp21 and down-regulated Fads1 (see Fig. 4). In the Drosophila model (Al-Ramahi et al., 2018), reducing Hipk/Hipk4 suppresses whereas reducing Endos/arpp19 aggravates motor impairment, and reducing fat-3/Fads1 in 128Q; rff-3 nematodes enhances defective touch response (Lejeune et al., 2012). Finally, RNAi feeding of our 128Q nematodes (Parker et al., 2005), here stably overexpressing sid-1 in touch receptor neurons (for enhancing sensitivity of these neurons to RNAi), suggests that reducing the expression of other striatal Class-II meta-network gene nodes significantly (N > 90, P < 0.001 compared to empty vector) aggravates (Enpp3, Arpp21) or reduces (Tmem30b) defective touch response (Ilaria Suriano and Francesca Farina, personal communication). Together, invertebrate data suggest that WENA meta-networks contain or are enriched in modulators of neuronal dysfunction in vivo and that they recapitulate a combination of pathogenic and protective effects involved in the temporal response of the mouse brain to HD.
4 Discussion
The analysis of HD datasets using single network approaches has highlighted prominent alterations of gene expression in developmental pathways during neuronal differentiation as observed in polyQ nematodes (Tourette et al., 2014) and human HD neural stem cells (Ring et al., 2015). In the RNA-seq time series data from the allelic series of Hdh knock-in mice (Langfelder et al., 2016), WGCNA highlighted signatures suggestive of a loss of striatal neuron identity (Langfelder et al., 2016), a possibility supported by epigenetic data (Achour et al., 2015). Here, we used a multi-layer network approach to build a quantitative model of the dynamics of genetic cooperativity in the brain of the Hdh knock-in mice through the integration of three complementary families of networks, all inferred from the RNA-seq time series data in these mice. Our results suggest this integrative approach may greatly reduce data complexity and biases in source networks while allowing robust ranking of gene cooperativity networks, highlighting weighted gene-to-gene path lengths that are highly dynamic over time (based on the product-P value associated to each edge in WENA meta-networks). Our results also suggest this integrative approach may greatly reduce the biases associated with super-connectivity of gene nodes in source networks The novelty of our findings thus comes from the principle of the WENA method itself compared to using single-network analysis, where the rationale is to build-up on the consistency and complementary of edge data in different types of source networks for modeling the molecular and temporal dynamics of genetic cooperativity the HD process at high resolution.
Although some of the pathways highlighted by WENA might be detectable in source networks such as for example alteration of neuronal-identity and cell-division pathways (Langfelder et al., 2016), the added value of our approach is to provide an unbiased model for the dynamics of genetic cooperativity that is more comprehensive compared to source networks as shown herein by the identification of critical phases of genetic cooperativity in the cortex and striatum of Hdh mice. Biological precision and biological homogeneity are two major features of the waves of genetic cooperativity identified by WENA analysis. For example, it could be that a path linking two genes Ga and Gb is about the same overtime as indicated by WGCNA (e.g. path Ga-Gb becomes Ga-Gc-Gb from 2 to 6 months and remains the same at 10 months) whereas BGM data could show that Ga-Gb becomes Ga-Gc-Gb then Ga-Gc-Gd-Gb from 2 to 10 months. Edge fusion in WENA provides a quantification of this phenomenon on a global level. This analysis reveals the existence of successive and critical phases of genetic cooperativity that are biologically distinct and that in the brain of Hdh mice may peak pre-symptomatically in cortex and symptomatically in striatum. This suggests the cortex might sense HD before striatum. In the cortex, WENA highlights a discontinued-response model in which the early response is disrupted over time by the loss of genetic cooperativity and by increasingly pronounced gene deregulation. In contrast, WENA predicts the early and late responses tend to work as a continuum in the striatum (convergent-response model). The striatal genes defining the late-stage response are already deregulated, suggesting that the biological efficiency of this response is not going to be hampered by dramatic changes of gene expression. This also applies to the early-stage response, which involves genes that are not dramatically deregulated over time. Additionally, the early and late response may involve a common group of genes as suggested by Class-III gene and node rewiring data between 2 and 10 months of age. One of the possible explanations for a discontinued- versus convergent-response in cortex compared to striatum is that epigenetic alteration, which significantly modifies gene expression in HD mice (Achour et al., 2015), may impact on the dynamics of genetic cooperativity earlier, and in a more acute manner, in cortex compared to striatum. Collectively, these data are coherent with previous knowledge of HD pathogenesis while providing insight into the critical phases that may govern the evolution of the disease on a genetic system level. Our data identify the early alteration of gene cooperativity networks associated with neurotransmission in the cortex, which may significantly contribute to HD pathogenesis (Virlogeux et al., 2018), and they emphasize the alteration of gene cooperativity associated with stress response, cell maintenance and senescence at a later stage in the striatum, which may apply to mouse glial cells and neurons and which may also apply to the human HD caudate (Scarpa et al., 2016).
The cross-integration of three layers of source networks puts forth a small number of biologically precise rules where data enrichment in weighted edges enhances the basis for target prioritization in view of preclinical studies (Table 1, Supplementary Table S14). However, it should be noted that we integrate three layers of HD networks and therefore we eliminated all information that may relate to consistency between any two source networks. Nonetheless, along the line of maximizing the discriminative power of target prioritization approaches, our data provide new information for studying the mechanisms that modulate the progression of striatal neurons, which are highly susceptible to HD (Zuccato et al., 2010), through dysfunction towards cell death. Our data noticeably indicate that down-regulation of stress-activated (e.g. by DNA damage) kinases such as Hipk4 is central to the dynamics of gene cooperativity in the striatum of symptomatic Hdh mice. Interestingly, reducing Hipk3 decreases mHTT aggregation in mouse HD striatal cells and striata of knock-in HD mice (Yu et al., 2017), reducing Hipk2 protects mHtt-expressing cells from apoptosis via p53 (Grison et al., 2011) and reducing Hipk suppresses motor impairment in a Drosophila HD model (Al-Ramahi et al., 2018). Thus, downregulated Hipk4 in the striatal class-2 meta-network (see Fig. 4) could correspond to a protective response of the striatum of HD mice. Conversely, reducing fly cAMP-regulated phosphoprotein Arpp19 (Endos) enhances motor impairment in a Drosophila HD model (Al-Ramahi et al., 2018), suggesting that downregulation of Arpp19 in the striatum of HD mice could correspond to a pathogenic effect. A full view of the genes that are retained in weighted edges and that show activity in HD flies (Al-Ramahi et al., 2018) is provided in Supplementary Table S15, suggesting that weighted edge networks in Hdh mice may implicate pathogenic intertwined with compensatory responses. Our data also indicate that in the striatum of symptomatic Hdh mice, Hipk4 may tightly cooperate with Cdkn2a (up-regulated), a locus encoding key regulators of cell cycle arrest, DNA damage repair and cellular senescence (p16INK4a, p14ARF), suggesting that prosurvival and prosenescence features are tightly integrated in response to HD in the mouse striatum, and linking HD molecular pathogenesis with cellular senescence. Here, it is noticeable that cellular senescence of astrocytes or microglia has been associated with neurodegeneration and neuronal dysfunction in mouse models of tauopathies (Bussian et al., 2018; Musi et al., 2018) and Parkinson’s disease (Chinta et al., 2018). Finally, cortico-striatal communication is altered in HD (Deng et al., 2013; Unschuld et al., 2012; Veldman and Yang, 2018; Virlogeux et al., 2018), and WENA information about synchronous waves of genetic cooperativity in cortex and striatum (i.e. cortical neurotransmission and striatal neuroglial responses at 2 months; cortical proteostasis and striatal cell senescence response at 10 months) provides a molecular basis and manageable number of candidate genes for testing functional interplay between cellular responses to HD in the cortex and those in the striatum of Hdh mice.
In summary, our data highlight a 2-step logic for the dynamics of genetic cooperativity in the brain of HD knock-in mice where cell proliferation and cellular senescence responses may be centrally involved in the striatum, synchronous to major symptoms. The ability of the top genes retained by WENA analysis to modulate motor, behavioral or cognitive phenotypes in HD mice will be addressed in future studies. In this study, several of the top genes retained in WENA meta-networks are associated with behavioral phenotypes in Hdh mice (Alexandrov et al., 2016), as contributed by causal network data. Additionally, gene perturbation data obtained in nematode and Drosophila models of HD pathogenesis suggest that several WENA nodes are relevant to neuronal dysfunction in vivo, which notably applies (significant enrichment) to the genes retained in the most-dynamic striatal weighted edges. Collectively, our findings illustrate the value of combining several layers of network data for accurately modeling the temporal dynamics of genetic cooperativity in HD and for enriching the basis for target prioritization prior to preclinical testing.
Supplementary Material
Acknowledgements
We thank GNS Healthcare for sharing causal network data, and Ryan Lim and Leslie Thompson (University of California Irvine) for critical reading of the manuscript. We thank the members of the Working Group ‘Systems Modeling’ of the European Huntington’s Disease network (EHDN) (http://www.ehdn.org/systemsmodelling-wg/) for prospective discussion on network integration in HD research.
Author contributions
E.B. designed and performed WENA analysis, interpreted the data and wrote the manuscript. S.S.N. performed bioinformatic and network analyses, helped with randomization tests, interpreted the data and helped to draft the manuscript. F.X.L. performed spectral decomposition of the signal and WGCNA analysis, interpreted the data and helped to draft the manuscript. H.F. performed clustering of spectral decomposition networks, interpreted the data and helped to draft the manuscript. F.P. performed clustering of spectral decomposition networks, interpreted the data and helped to draft the manuscript. L.M. exemplified thresholds in construction of WENA networks and helped to draft the manuscript. M.V. interpreted the data and helped to draft the manuscript. J.A. provided knowledge databases and helped to draft the manuscript. J.R. provided knowledge databases, interpreted the data and wrote the manuscript. C.N. conceived and designed WENA analysis, analyzed and interpreted the data and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was funded by Institut National de la Santé et de la Recherche Médicale (INSERM), Sorbonne Université, Agence Nationale pour la Recherche ANR-12-BSV4-0023-01, France and the CHDI Foundation grant number A-12273, USA, to C.N. This work was also supported by a grant from the European Huntington’s Disease Network (EHDN), funded by CHDI Foundation, Inc, to C.N.
Conflict of Interest: none declared.
References
- Achour M. et al. (2015) Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington’s disease mice. Hum. Mol. Genet., 24, 3481–3496. [DOI] [PubMed] [Google Scholar]
- Aguirre A. et al. (2007) A functional role for EGFR signaling in myelination and remyelination. Nat. Neurosci., 10, 990–1002. [DOI] [PubMed] [Google Scholar]
- Al-Ramahi I. et al. (2018) High-throughput functional analysis distinguishes pathogenic, nonpathogenic, and compensatory transcriptional changes in neurodegeneration. Cell Syst., 7, 28–40.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov V. et al. (2016) Large-scale phenome analysis defines a behavioral signature for Huntington’s disease genotype in mice. Nat. Biotechnol., 34, 838–844. [DOI] [PubMed] [Google Scholar]
- Bon E. et al. (2016) SCN4B acts as a metastasis-suppressor gene preventing hyperactivation of cell migration in breast cancer. Nat. Commun., 7, 13648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian T.J. et al. (2018) Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature, 562, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalazonitis A. et al. (2011) Homeodomain interacting protein kinase 2 regulates postnatal development of enteric dopaminergic neurons and glia via BMP signaling. J. Neurosci., 31, 13746–13757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta S.J. et al. (2018) Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell. Rep., 22, 930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y.P. et al. (2013) Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington’s disease mice. Neurobiol Dis., 60, 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores C.E. et al. (2008) Interaction between connexin35 and zonula occludens-1 and its potential role in the regulation of electrical synapses. Proc. Natl. Acad. Sci. USA, 105, 12545–12550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligorijevic V., Przulj N. (2015) Methods for biological data integration: perspectives and challenges. J. R. Soc. Interface, 12. pii: 20150571. doi: 10.1098/rsif.2015.0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Villafuertes R. et al. (2014) Ectonucleotide pyrophosphatase/phosphodiesterase activity in Neuro-2a neuroblastoma cells: changes in expression associated with neuronal differentiation. J. Neurochem., 131, 290–302. [DOI] [PubMed] [Google Scholar]
- Grison A. et al. (2011) Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc. Natl. Acad. Sci. USA, 108, 17979–17984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayete B. et al. (2017) A Bayesian mathematical model of motor and cognitive outcomes in Parkinson’s disease. PLoS One, 12, e0178982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T. et al. (2015) Physiological significance of recombination-activating gene 1 in neuronal death, especially optic neuropathy. FEBS J., 282, 129–141. [DOI] [PubMed] [Google Scholar]
- Jia L. et al. (2014) MGARP regulates mouse neocortical development via mitochondrial positioning. Mol. Neurobiol., 49, 1293–1308. [DOI] [PubMed] [Google Scholar]
- Kim E. et al. (2016) MouseNet v2: a database of gene networks for studying the laboratory mouse and eight other model vertebrates. Nucleic Acids Res., 44, D848–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M. et al. (2014) PRL2/PTP4A2 phosphatase is important for hematopoietic stem cell self-renewal. Stem Cells, 32, 1956–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs K.A. et al. (2015) Complex regulation of CREB-binding protein by homeodomain-interacting protein kinase 2. Cell Signal., 27, 2252–2260. [DOI] [PubMed] [Google Scholar]
- Kuwano Y. et al. (2016) Homeodomain-interacting protein kinase-2: a critical regulator of the DNA damage response and the epigenome. Int. J. Mol. Sci., 17. doi: 10.3390/ijms17101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J., Morimoto R.I. (2013) Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci., 38, 378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P. et al. (2016) Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci., 19, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F.X. et al. (2012) Large-scale functional RNAi screen in C. elegans identifies genes that regulate the dysfunction of mutant polyglutamine neurons. BMC Genomics, 13, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musi N. et al. (2018) Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell, 17, e12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandithage R. et al. (2008) The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol., 180, 915–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park W.J. et al. (2012) A novel FADS1 isoform potentiates FADS2-mediated production of eicosanoid precursor fatty acids. J. Lipid Res., 53, 1502–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.A. et al. (2005) Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat. Genet., 37, 349–350. [DOI] [PubMed] [Google Scholar]
- Rapaport F. et al. (2007) Classification of microarray data using gene networks. BMC Bioinformatics, 8, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring K.L. et al. (2015) Genomic analysis reveals disruption of striatal neuronal development and therapeutic targets in human huntington’s disease neural stem cells. Stem Cell Rep., 5, 1023–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rue L. et al. (2016) Targeting CAG repeat RNAs reduces Huntington’s disease phenotype independently of huntingtin levels. J. Clin. Invest., 126, 4319–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa J.R. et al. (2016) Systems genetic analyses highlight a TGFbeta-FOXO3 dependent striatal astrocyte network conserved across species and associated with stress, sleep, and huntington’s disease. PLoS Genet., 12, e1006137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourette C. et al. (2014) The Wnt receptor Ryk reduces neuronal and cell survival capacity by repressing FOXO activity during the early phases of mutant huntingtin pathogenicity. PLoS Biol., 12, e1001895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unschuld P.G. et al. (2012) Impaired cortico-striatal functional connectivity in prodromal Huntington’s Disease. Neurosci. Lett., 514, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman M.B., Yang X.W. (2018) Molecular insights into cortico-striatal miscommunications in Huntington’s disease. Curr. Opin. Neurobiol., 48, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virlogeux A. et al. (2018) Reconstituting corticostriatal network on-a-chip reveals the contribution of the presynaptic compartment to Huntington’s disease. Cell Rep., 22, 110–122. [DOI] [PubMed] [Google Scholar]
- Yan J. et al. (2017) Network approaches to systems biology analysis of complex disease: integrative methods for multi-omics data. Brief Bioinform., 19, 1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J. et al. (2011) Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell, 147, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M. et al. (2017) Suppression of MAPK11 or HIPK3 reduces mutant Huntingtin levels in Huntington’s disease models. Cell Res., 27, 1441–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C. et al. (2010) Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev., 90, 905–981. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.